Heightened JNK Activation and Reduced XIAP Levels Promote TRAIL and Sunitinib-Mediated Apoptosis in Colon Cancer Models

 , , and
, , and 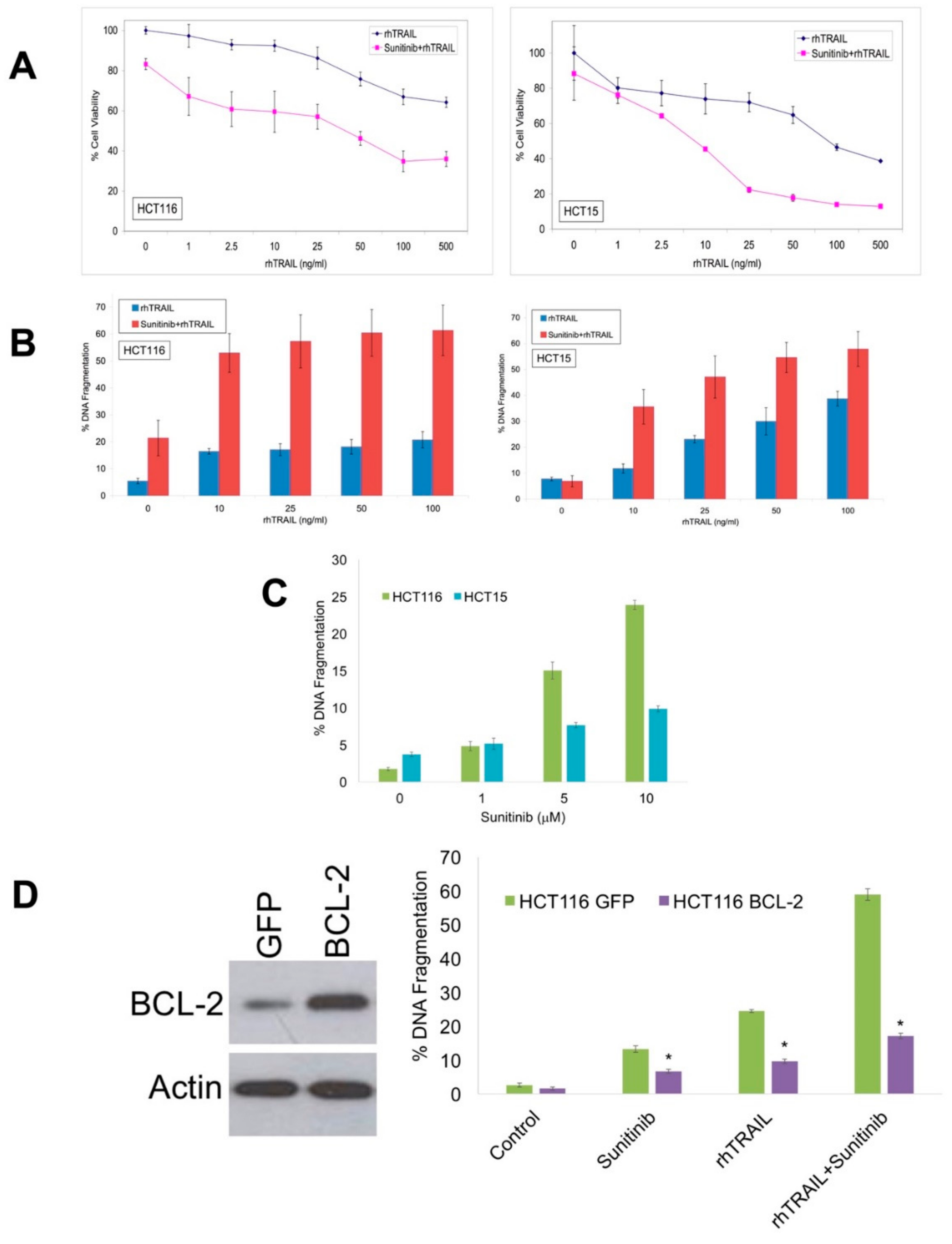{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Sunitinib Potentiates the Pro-Apoptotic Activity of Recombinant Human (rh)TRAIL in Colon Cancer Cell Models
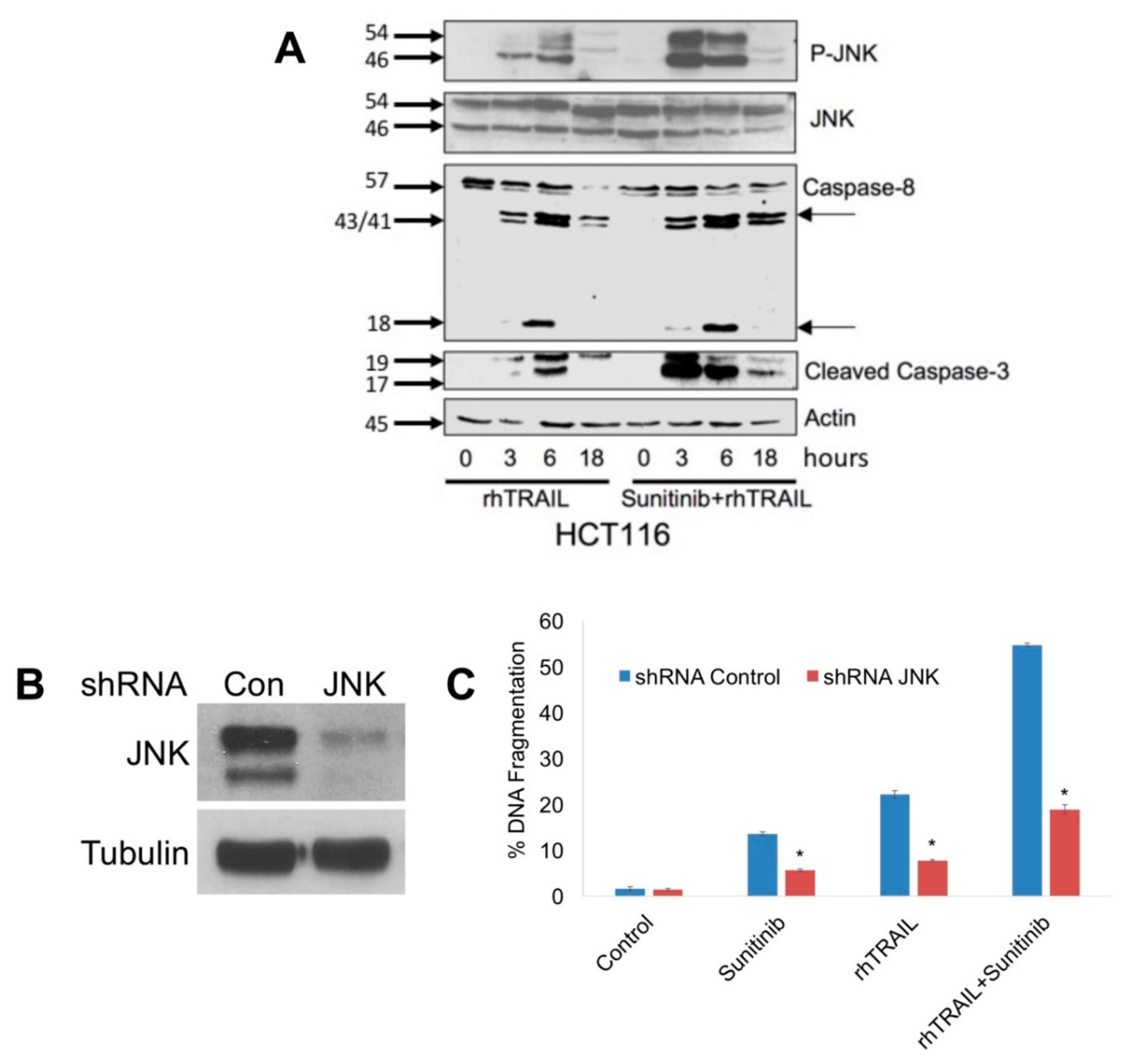
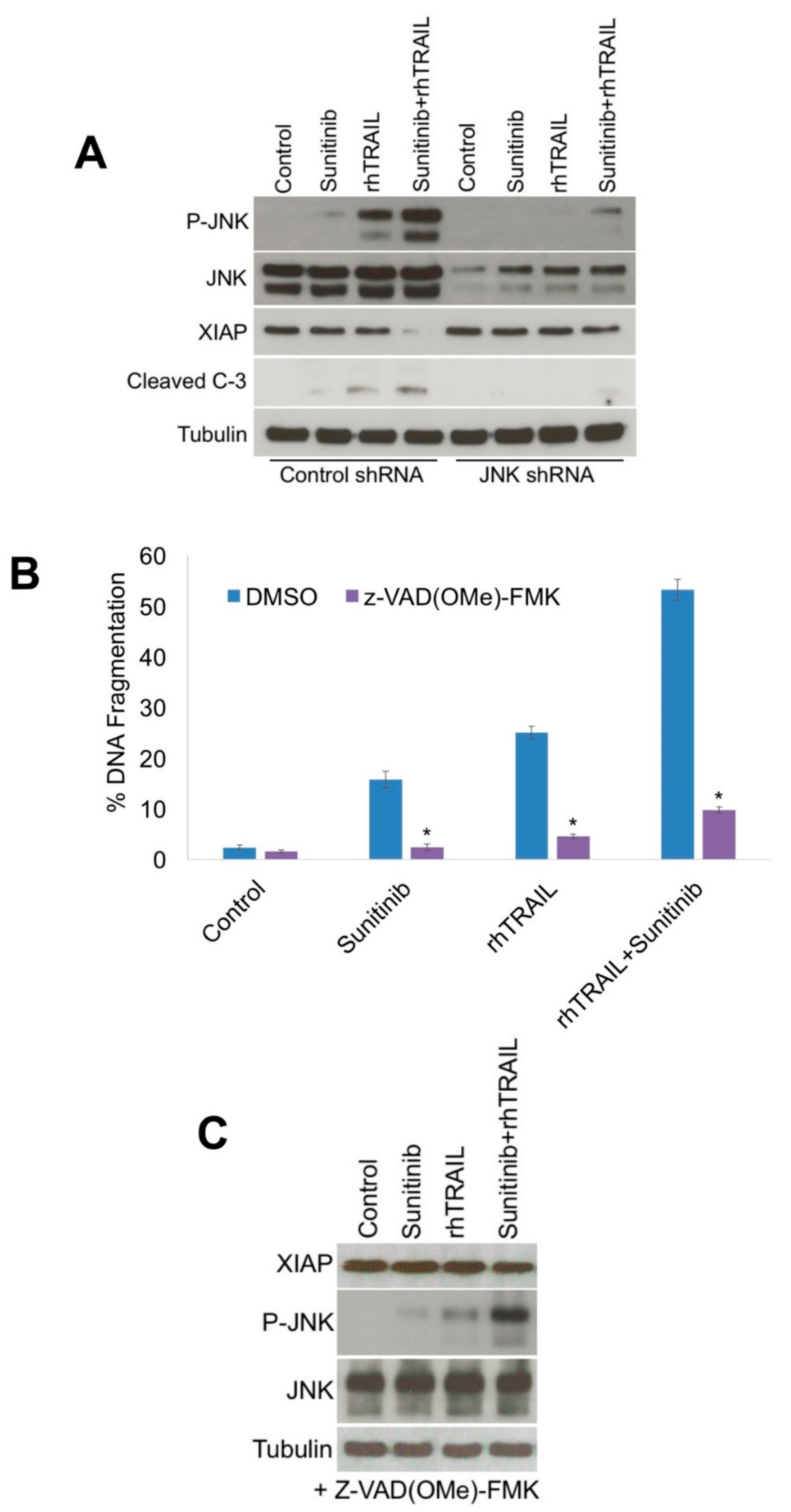
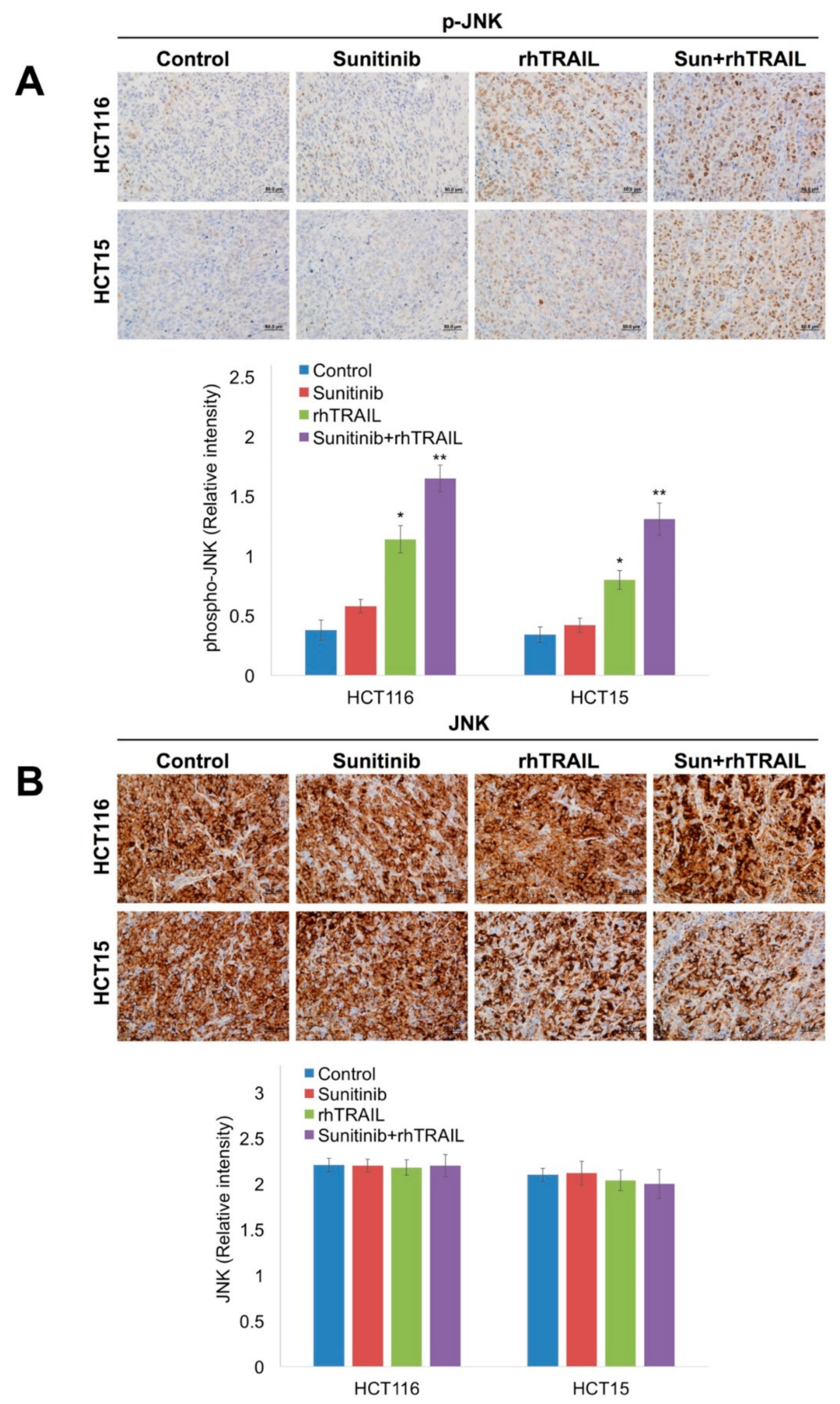
2.2. JNK Activation Is a Critical Mediator of Apoptosis Following Treatment with rhTRAIL and Sunitinib
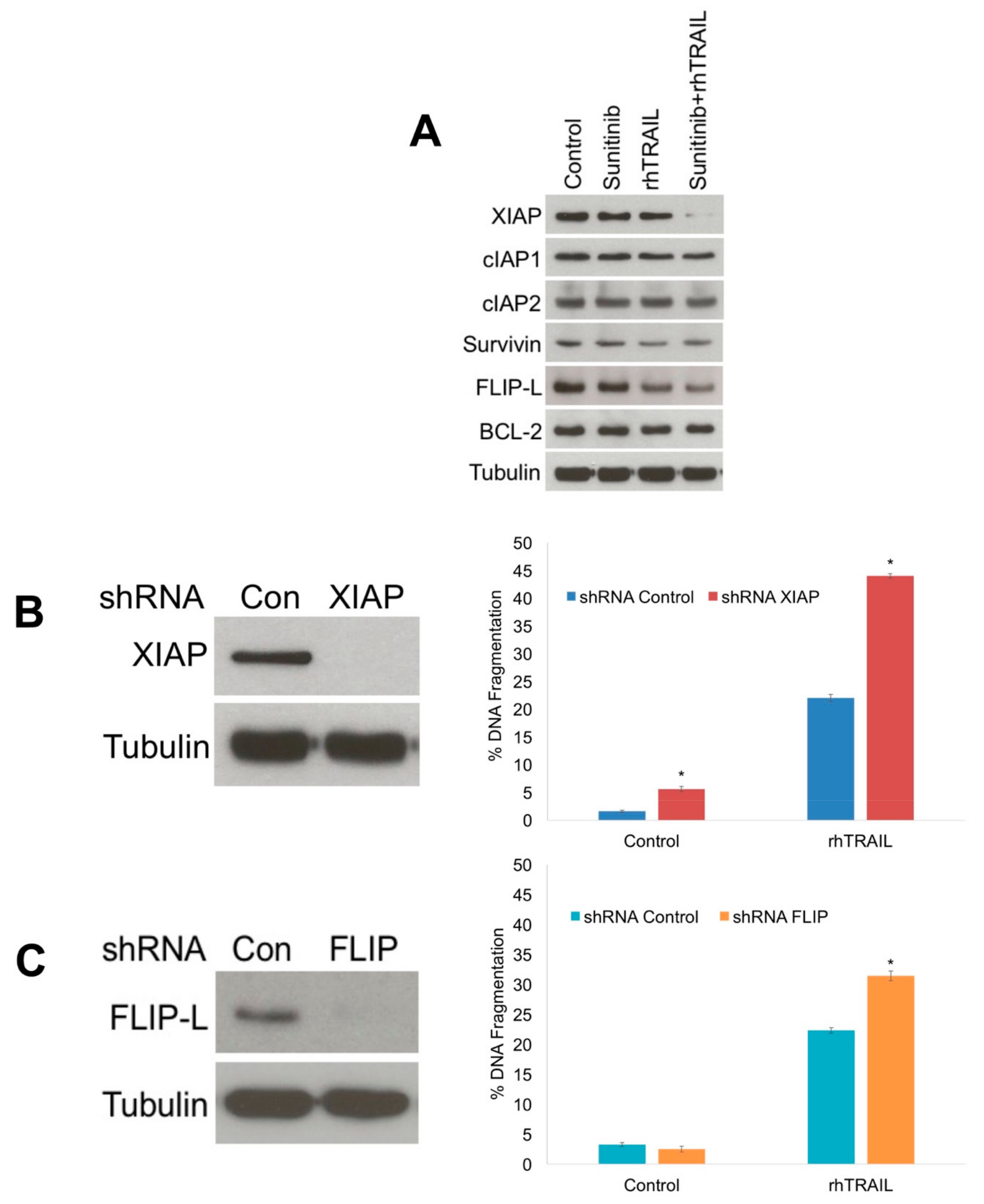
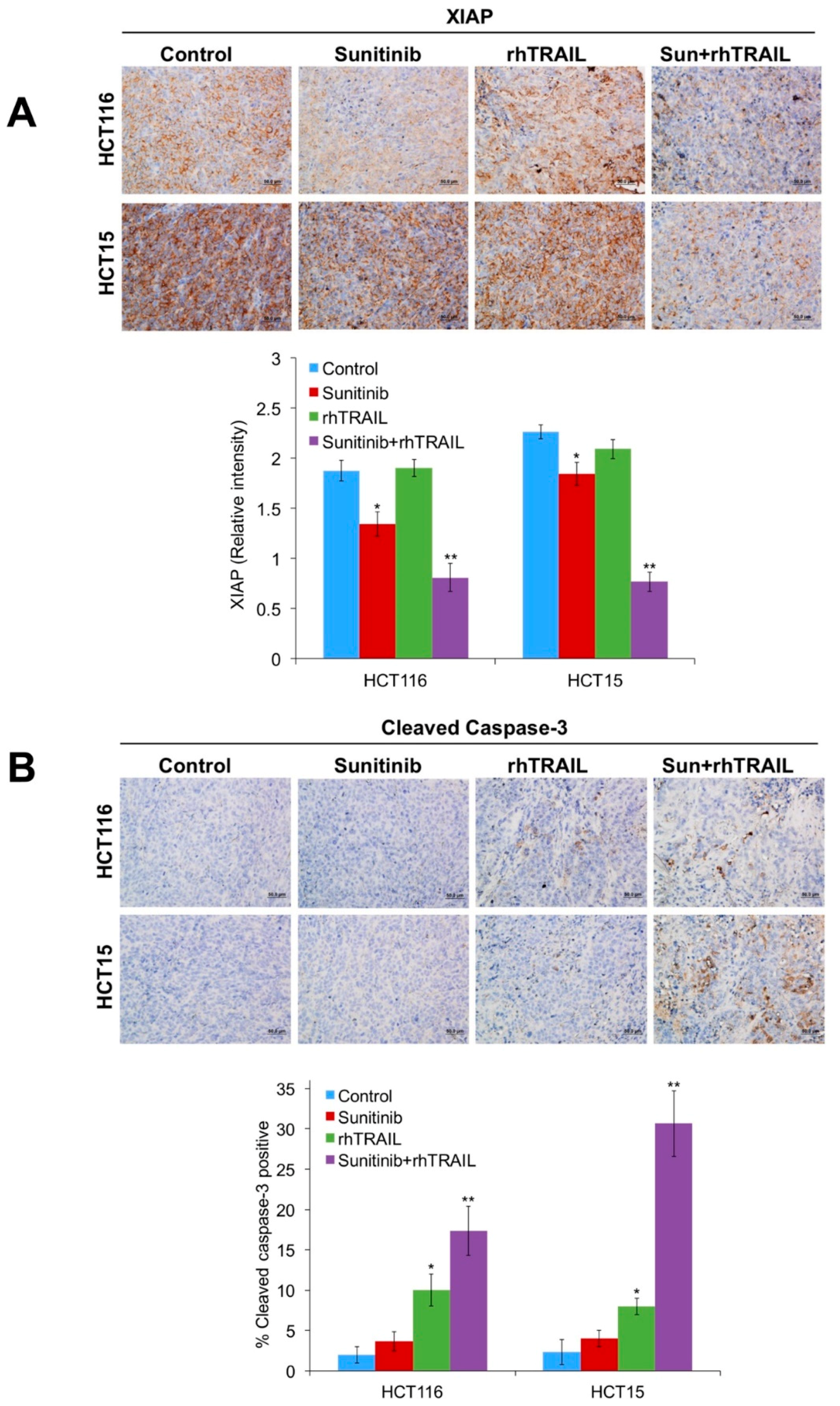
2.3. Diminished XIAP Expression Drives the Pro-Apoptotic Effects of the rhTRAIL and Sunitinib Combination
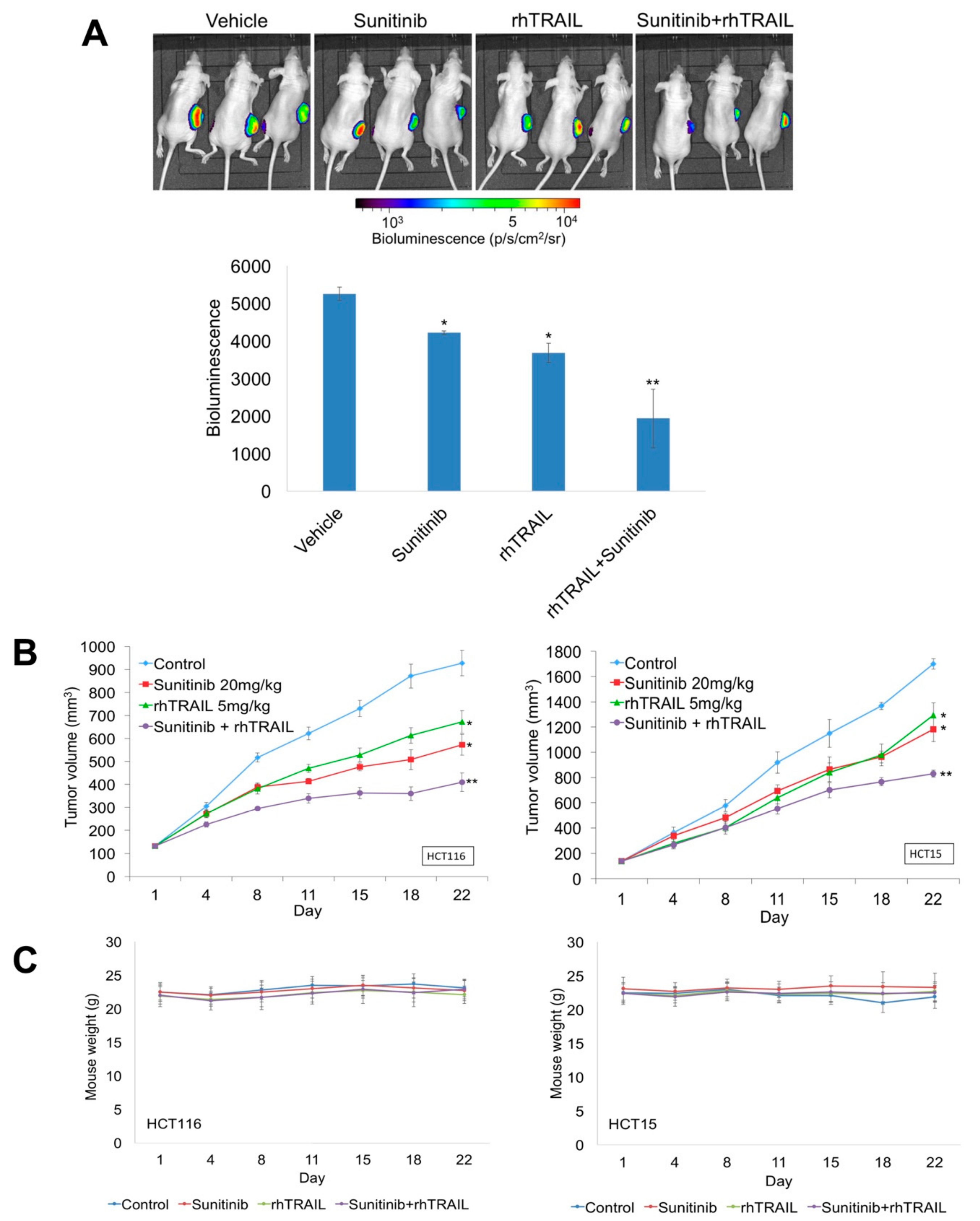
2.4. Sunitinib and rhTRAIL Reduce Tumor Burden in Colon Cancer Xenografts
2.5. The Combination of Sunitinib and rhTRAIL Significantly Enhances JNK Phosphorylation, Reduces XIAP Levels, and Induces Apoptosis In Vivo
3. Discussion
4. Materials and Methods
4.1. Cells and Cell Culture
4.2. Antibodies and Reagents
4.3. Quantification of Drug-Induced Cytotoxicity
4.4. Immunoblotting
4.5. Preparation and Transfection of shRNAs
4.6. Xenograft Studies
4.7. Immunohistochemistry
4.8. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2l/trail-dependent recruitment of endogenous fadd and caspase-8 to death receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar] [CrossRef]
- Sprick, M.R.; Weigand, M.A.; Rieser, E.; Rauch, C.T.; Juo, P.; Blenis, J.; Krammer, P.H.; Walczak, H. Fadd/mort1 and caspase-8 are recruited to trail receptors 1 and 2 and are essential for apoptosis mediated by trail receptor 2. Immunity 2000, 12, 599–609. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef]
- Mahalingam, D.; Szegezdi, E.; Keane, M.; Jong, S.; Samali, A. Trail receptor signalling and modulation: Are we on the right trail? Cancer Treat. Rev. 2009, 35, 280–288. [Google Scholar] [CrossRef]
- Ng, C.P.; Bonavida, B. X-linked inhibitor of apoptosis (xiap) blocks apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis of prostate cancer cells in the presence of mitochondrial activation: Sensitization by overexpression of second mitochondria-derived activator of caspase/direct iap-binding protein with low pl (smac/diablo). Mol. Cancer Ther. 2002, 1, 1051–1058. [Google Scholar]
- Bratton, S.B.; Cohen, G.M. Death receptors leave a caspase footprint that smacs of xiap. Cell Death Differ. 2003, 10, 4–6. [Google Scholar] [CrossRef]
- Cummins, J.M.; Kohli, M.; Rago, C.; Kinzler, K.W.; Vogelstein, B.; Bunz, F. X-linked inhibitor of apoptosis protein (xiap) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (trail)-mediated apoptosis in human cancer cells. Cancer Res. 2004, 64, 3006–3008. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. The functional contrariety of jnk. Mol. Carcinog. 2007, 46, 591–598. [Google Scholar] [CrossRef]
- Lin, H.H.; Chen, J.H.; Huang, C.C.; Wang, C.J. Apoptotic effect of 3,4-dihydroxybenzoic acid on human gastric carcinoma cells involving jnk/p38 mapk signaling activation. Int. J. Cancer 2007, 120, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Keane, M.; Pirianov, G.; Mehmet, H.; Samali, A.; Szegezdi, E. Differential activation of jnk1 isoforms by trail receptors modulate apoptosis of colon cancer cell lines. Br. J. Cancer 2009, 100, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Minemoto, Y.; Zhang, J.; Liu, J.; Tang, F.; Bui, T.N.; Xiang, J.; Lin, A. Jnk suppresses apoptosis via phosphorylation of the proapoptotic bcl-2 family protein bad. Mol. Cell 2004, 13, 329–340. [Google Scholar] [CrossRef]
- Maundrell, K.; Antonsson, B.; Magnenat, E.; Camps, M.; Muda, M.; Chabert, C.; Gillieron, C.; Boschert, U.; Vial-Knecht, E.; Martinou, J.C.; et al. Bcl-2 undergoes phosphorylation by c-jun n-terminal kinase/stress-activated protein kinases in the presence of the constitutively active gtp-binding protein rac1. J. Biol. Chem. 1997, 272, 25238–25242. [Google Scholar] [CrossRef] [PubMed]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of su11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar] [PubMed]
- Abrams, T.J.; Lee, L.B.; Murray, L.J.; Pryer, N.K.; Cherrington, J.M. Su11248 inhibits kit and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol. Cancer Ther. 2003, 2, 471–478. [Google Scholar]
- O’Farrell, A.M.; Abrams, T.J.; Yuen, H.A.; Ngai, T.J.; Louie, S.G.; Yee, K.W.; Wong, L.M.; Hong, W.; Lee, L.B.; Town, A.; et al. Su11248 is a novel flt3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 2003, 101, 3597–3605. [Google Scholar] [CrossRef]
- Mahalingam, D.; Espitia, C.M.; Medina, E.C.; Esquivel, J.A., 2nd; Kelly, K.R.; Bearss, D.; Choy, G.; Taverna, P.; Carew, J.S.; Giles, F.J.; et al. Targeting pim kinase enhances the activity of sunitinib in renal cell carcinoma. Br. J. Cancer 2011, 105, 1563–1573. [Google Scholar] [CrossRef]
- Ding, W.; Cai, T.; Zhu, H.; Wu, R.; Tu, C.; Yang, L.; Lu, W.; He, Q.; Yang, B. Synergistic antitumor effect of trail in combination with sunitinib in vitro and in vivo. Cancer Lett. 2010, 293, 158–166. [Google Scholar] [CrossRef]
- Rosato, R.R.; Almenara, J.A.; Coe, S.; Grant, S. The multikinase inhibitor sorafenib potentiates trail lethality in human leukemia cells in association with mcl-1 and cflipl down-regulation. Cancer Res. 2007, 67, 9490–9500. [Google Scholar] [CrossRef]
- Shrader, M.; Pino, M.S.; Lashinger, L.; Bar-Eli, M.; Adam, L.; Dinney, C.P.; McConkey, D.J. Gefitinib reverses trail resistance in human bladder cancer cell lines via inhibition of akt-mediated x-linked inhibitor of apoptosis protein expression. Cancer Res. 2007, 67, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Mita, M.M.; Mita, A.C.; Nawrocki, S.T. Oncolytic reovirus inhibits angiogenesis through induction of cxcl10/ip-10 and abrogation of hif activity in soft tissue sarcomas. Oncotarget 2017, 8, 86769–86783. [Google Scholar] [CrossRef] [PubMed]
- Corazza, N.; Jakob, S.; Schaer, C.; Frese, S.; Keogh, A.; Stroka, D.; Kassahn, D.; Torgler, R.; Mueller, C.; Schneider, P.; et al. Trail receptor-mediated jnk activation and bim phosphorylation critically regulate fas-mediated liver damage and lethality. J. Clin. Investig. 2006, 116, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Herr, I.; Wilhelm, D.; Meyer, E.; Jeremias, I.; Angel, P.; Debatin, K.M. Jnk/sapk activity contributes to trail-induced apoptosis. Cell Death Differ. 1999, 6, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Muhlenbeck, F.; Haas, E.; Schwenzer, R.; Schubert, G.; Grell, M.; Smith, C.; Scheurich, P.; Wajant, H. Trail/apo2l activates c-jun nh2-terminal kinase (jnk) via caspase-dependent and caspase-independent pathways. J. Biol. Chem. 1998, 273, 33091–33098. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.S.; Duckett, C.S. Iap proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Minemoto, Y.; Dibling, B.; Purcell, N.H.; Li, Z.; Karin, M.; Lin, A. Inhibition of jnk activation through nf-kappab target genes. Nature 2001, 414, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Oldenhuis, C.N.A.M.; Szegezdi, E.; Giles, F.J.; de Vries, E.G.E.; de Jong, S.; Nawrocki, S.T. Targeting trail towards the clinic. Curr. Drug Targets 2011, 12, 2079–2090. [Google Scholar] [CrossRef]
- Allen, J.E.; Crowder, R.N.; El-Deiry, W.S. First-in-class small molecule onc201 induces dr5 and cell death in tumor but not normal cells to provide a wide therapeutic index as an anti-cancer agent. PLoS ONE 2015, 10, e0143082. [Google Scholar] [CrossRef]
- Huet, H.A.; Growney, J.D.; Johnson, J.A.; Li, J.; Bilic, S.; Ostrom, L.; Zafari, M.; Kowal, C.; Yang, G.; Royo, A.; et al. Multivalent nanobodies targeting death receptor 5 elicit superior tumor cell killing through efficient caspase induction. MAbs 2014, 6, 1560–1570. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Isaacs, R.; Bilic, S.; Kentsch, K.; Huet, H.A.; Hofmann, M.; Rasco, D.; Kundamal, N.; Tang, Z.; Cooksey, J.; et al. Unexpected hepatotoxicity in a phase i study of tas266, a novel tetravalent agonistic nanobody(r) targeting the dr5 receptor. Cancer Chemother. Pharmacol. 2015, 75, 887–895. [Google Scholar] [CrossRef]
- He, Y.; Hendriks, D.; van Ginkel, R.; Samplonius, D.; Bremer, E.; Helfrich, W. Melanoma-directed activation of apoptosis using a bispecific antibody directed at mcsp and trail receptor-2/death receptor-5. J. Investig. Dermatol. 2016, 136, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, G.A.; Condamine, T.; Mony, S.; Hashimoto, A.; Wang, F.; Liu, Q.; Forero, A.; Bendell, J.; Witt, R.; Hockstein, N.; et al. Selective targeting of myeloid-derived suppressor cells in cancer patients using ds-8273a, an agonistic trail-r2 antibody. Clin. Cancer Res. 2016, 23, 2942–2950. [Google Scholar] [CrossRef] [PubMed]
- Burvenich, I.J.; Lee, F.T.; Guo, N.; Gan, H.K.; Rigopoulos, A.; Parslow, A.C.; O’Keefe, G.J.; Gong, S.J.; Tochon-Danguy, H.; Rudd, S.E.; et al. In vitro and in vivo evaluation of 89zr-ds-8273a as a theranostic for anti-death receptor 5 therapy. Theranostics 2016, 6, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Forero, A.; Bendell, J.C.; Kumar, P.; Janisch, L.; Rosen, M.; Wang, Q.; Copigneaux, C.; Desai, M.; Senaldi, G.; Maitland, M.L. First-in-human study of the antibody dr5 agonist ds-8273a in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 298–306. [Google Scholar] [CrossRef]
- Jo, M.; Kim, T.H.; Seol, D.W.; Esplen, J.E.; Dorko, K.; Billiar, T.R.; Strom, S.C. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat. Med. 2000, 6, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Kretz, A.L.; Trauzold, A.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; von Karstedt, S.; Lemke, J. Trailblazing strategies for cancer treatment. Cancers 2019, 11, 456. [Google Scholar] [CrossRef]
- Ganten, T.M.; Koschny, R.; Sykora, J.; Schulze-Bergkamen, H.; Buchler, P.; Haas, T.L.; Schader, M.B.; Untergasser, A.; Stremmel, W.; Walczak, H. Preclinical differentiation between apparently safe and potentially hepatotoxic applications of trail either alone or in combination with chemotherapeutic drugs. Clin. Cancer Res. 2006, 12, 2640–2646. [Google Scholar] [CrossRef]
- Schneider, B.; Munkel, S.; Krippner-Heidenreich, A.; Grunwald, I.; Wels, W.S.; Wajant, H.; Pfizenmaier, K.; Gerspach, J. Potent antitumoral activity of trail through generation of tumor-targeted single-chain fusion proteins. Cell Death Dis. 2010, 1, e68. [Google Scholar] [CrossRef]
- Wang, H.; Davis, J.S.; Wu, X. Immunoglobulin fc domain fusion to trail significantly prolongs its plasma half-life and enhances its antitumor activity. Mol. Cancer 2014, 13, 643–650. [Google Scholar] [CrossRef]
- Ashkenazi, A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer 2002, 2, 420–430. [Google Scholar] [CrossRef]
- Hinz, S.; Trauzold, A.; Boenicke, L.; Sandberg, C.; Beckmann, S.; Bayer, E.; Walczak, H.; Kalthoff, H.; Ungefroren, H. Bcl-xl protects pancreatic adenocarcinoma cells against cd95-and trail-receptor-mediated apoptosis. Oncogene 2000, 19, 5477–5486. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Penington, R.C. Sulindac sulfide-induced apoptosis is enhanced by a small-molecule bcl-2 inhibitor and by trail in human colon cancer cells overexpressing bcl-2. Mol. Cancer Ther. 2005, 4, 1475–1483. [Google Scholar] [CrossRef]
- Clohessy, J.G.; Zhuang, J.; de Boer, J.; Gil-Gomez, G.; Brady, H.J. Mcl-1 interacts with truncated bid and inhibits its induction of cytochrome c release and its role in receptor-mediated apoptosis. J. Biol. Chem. 2006, 281, 5750–5759. [Google Scholar] [CrossRef]
- Ndozangue-Touriguine, O.; Sebbagh, M.; Merino, D.; Micheau, O.; Bertoglio, J.; Breard, J. A mitochondrial block and expression of xiap lead to resistance to trail-induced apoptosis during progression to metastasis of a colon carcinoma. Oncogene 2008, 27, 6012–6022. [Google Scholar] [CrossRef] [PubMed]
- Pennarun, B.; Kleibeuker, J.H.; Boersma-van Ek, W.; Kruyt, F.A.; Hollema, H.; de Vries, E.G.; de Jong, S. Targeting flip and mcl-1 using a combination of aspirin and sorafenib sensitizes colon cancer cells to trail. J. Pathol. 2013, 229, 410–421. [Google Scholar] [CrossRef]
- Piggott, L.; Silva, A.; Robinson, T.; Santiago-Gomez, A.; Simoes, B.M.; Becker, M.; Fichtner, I.; Andera, L.; Young, P.; Morris, C.; et al. Acquired resistance of er-positive breast cancer to endocrine treatment confers an adaptive sensitivity to trail through posttranslational downregulation of c-flip. Clin. Cancer Res. 2018, 24, 2452–2463. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yue, P.; Lonial, S.; Khuri, F.R.; Sun, S.Y. The nedd8-activating enzyme inhibitor, mln4924, cooperates with trail to augment apoptosis through facilitating c-flip degradation in head and neck cancer cells. Mol. Cancer 2011, 10, 2415–2425. [Google Scholar] [CrossRef]
- Tong, Q.S.; Zheng, L.D.; Wang, L.; Zeng, F.Q.; Chen, F.M.; Dong, J.H.; Lu, G.C. Downregulation of xiap expression induces apoptosis and enhances chemotherapeutic sensitivity in human gastric cancer cells. Cancer Gene Ther. 2005, 12, 509–514. [Google Scholar] [CrossRef]
- Rosato, R.R.; Dai, Y.; Almenara, J.A.; Maggio, S.C.; Grant, S. Potent antileukemic interactions between flavopiridol and trail/apo2l involve flavopiridol-mediated xiap downregulation. Leukemia 2004, 18, 1780–1788. [Google Scholar] [CrossRef]
- Kim, E.H.; Kim, S.U.; Shin, D.Y.; Choi, K.S. Roscovitine sensitizes glioma cells to trail-mediated apoptosis by downregulation of survivin and xiap. Oncogene 2004, 23, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Walczak, H.; Stadel, D.; Haas, T.L.; Genze, F.; Jovanovic, M.; Gschwend, J.E.; Simmet, T.; Debatin, K.M.; Fulda, S. Targeting xiap bypasses bcl-2-mediated resistance to trail and cooperates with trail to suppress pancreatic cancer growth in vitro and in vivo. Cancer Res. 2008, 68, 7956–7965. [Google Scholar] [CrossRef] [PubMed]
- Fakler, M.; Loeder, S.; Vogler, M.; Schneider, K.; Jeremias, I.; Debatin, K.M.; Fulda, S. Small molecule xiap inhibitors cooperate with trail to induce apoptosis in childhood acute leukemia cells and overcome bcl-2-mediated resistance. Blood 2009, 113, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Walczak, H.; Stadel, D.; Haas, T.L.; Genze, F.; Jovanovic, M.; Bhanot, U.; Hasel, C.; Moller, P.; Gschwend, J.E.; et al. Small molecule xiap inhibitors enhance trail-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Res. 2009, 69, 2425–2434. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Natoni, A.; Keane, M.; Samali, A.; Szegezdi, E. Early growth response-1 is a regulator of dr5-induced apoptosis in colon cancer cells. Br. J. Cancer 2010, 102, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Cahill, M.A.; Peter, M.E.; Kischkel, F.C.; Chinnaiyan, A.M.; Dixit, V.M.; Krammer, P.H.; Nordheim, A. Cd95 (apo-1/fas) induces activation of sap kinases downstream of ice-like proteases. Oncogene 1996, 13, 2087–2096. [Google Scholar]
- Sluss, H.K.; Barrett, T.; Derijard, B.; Davis, R.J. Signal transduction by tumor necrosis factor mediated by jnk protein kinases. Mol. Cell. Biol. 1994, 14, 8376–8384. [Google Scholar] [CrossRef]
- Yoo, J.; Park, S.S.; Lee, Y.J. Pretreatment of docetaxel enhances trail-mediated apoptosis in prostate cancer cells. J. Cell. Biochem. 2008, 104, 1636–1646. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Carew, J.S.; Maclean, K.H.; Courage, J.F.; Huang, P.; Houghton, J.A.; Cleveland, J.L.; Giles, F.J.; McConkey, D.J. Myc regulates aggresome formation, the induction of noxa, and apoptosis in response to the combination of bortezomib and saha. Blood 2008, 112, 2917–2926. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Visconte, V.; Anwer, F.; Kelly, K.R.; Nawrocki, S.T. Rational cotargeting of hdac6 and bet proteins yields synergistic antimyeloma activity. Blood Adv. 2019, 3, 1318–1329. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahalingam, D.; Carew, J.S.; Espitia, C.M.; Cool, R.H.; Giles, F.J.; de Jong, S.; Nawrocki, S.T. Heightened JNK Activation and Reduced XIAP Levels Promote TRAIL and Sunitinib-Mediated Apoptosis in Colon Cancer Models. Cancers 2019, 11, 895. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11070895
Mahalingam D, Carew JS, Espitia CM, Cool RH, Giles FJ, de Jong S, Nawrocki ST. Heightened JNK Activation and Reduced XIAP Levels Promote TRAIL and Sunitinib-Mediated Apoptosis in Colon Cancer Models. Cancers. 2019; 11(7):895. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11070895
Chicago/Turabian StyleMahalingam, Devalingam, Jennifer S. Carew, Claudia M. Espitia, Robbert H. Cool, Francis J. Giles, Steven de Jong, and Steffan T. Nawrocki. 2019. "Heightened JNK Activation and Reduced XIAP Levels Promote TRAIL and Sunitinib-Mediated Apoptosis in Colon Cancer Models" Cancers 11, no. 7: 895. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11070895