Hypoxia Downregulates LPP3 and Promotes the Spatial Segregation of ATX and LPP1 During Cancer Cell Invasion


Abstract
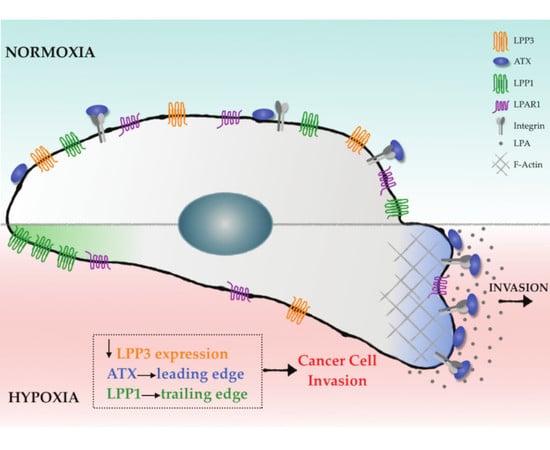
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
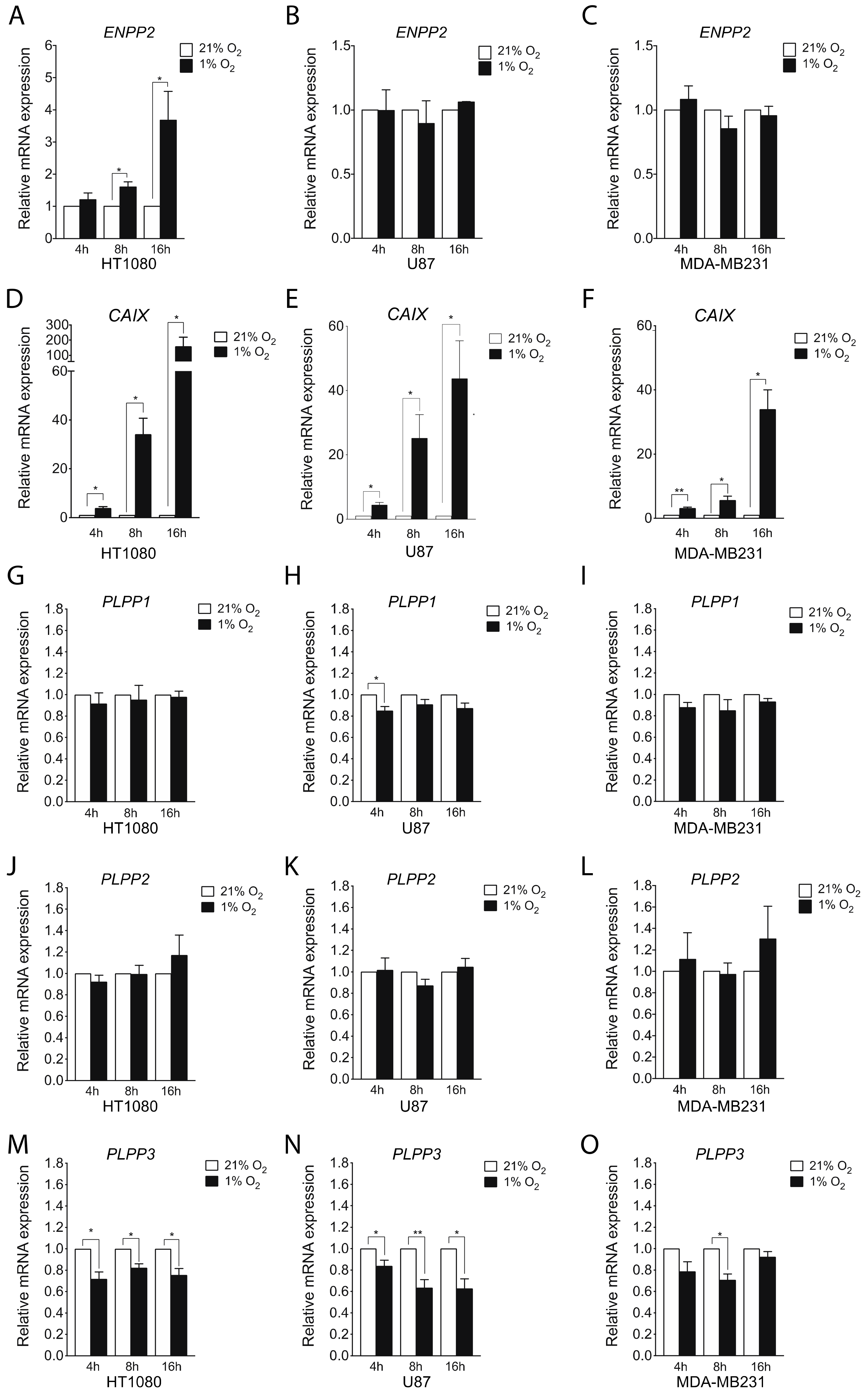
2.1. Hypoxia Induces ATX and Represses LPP Expression in Certain Cell Lines
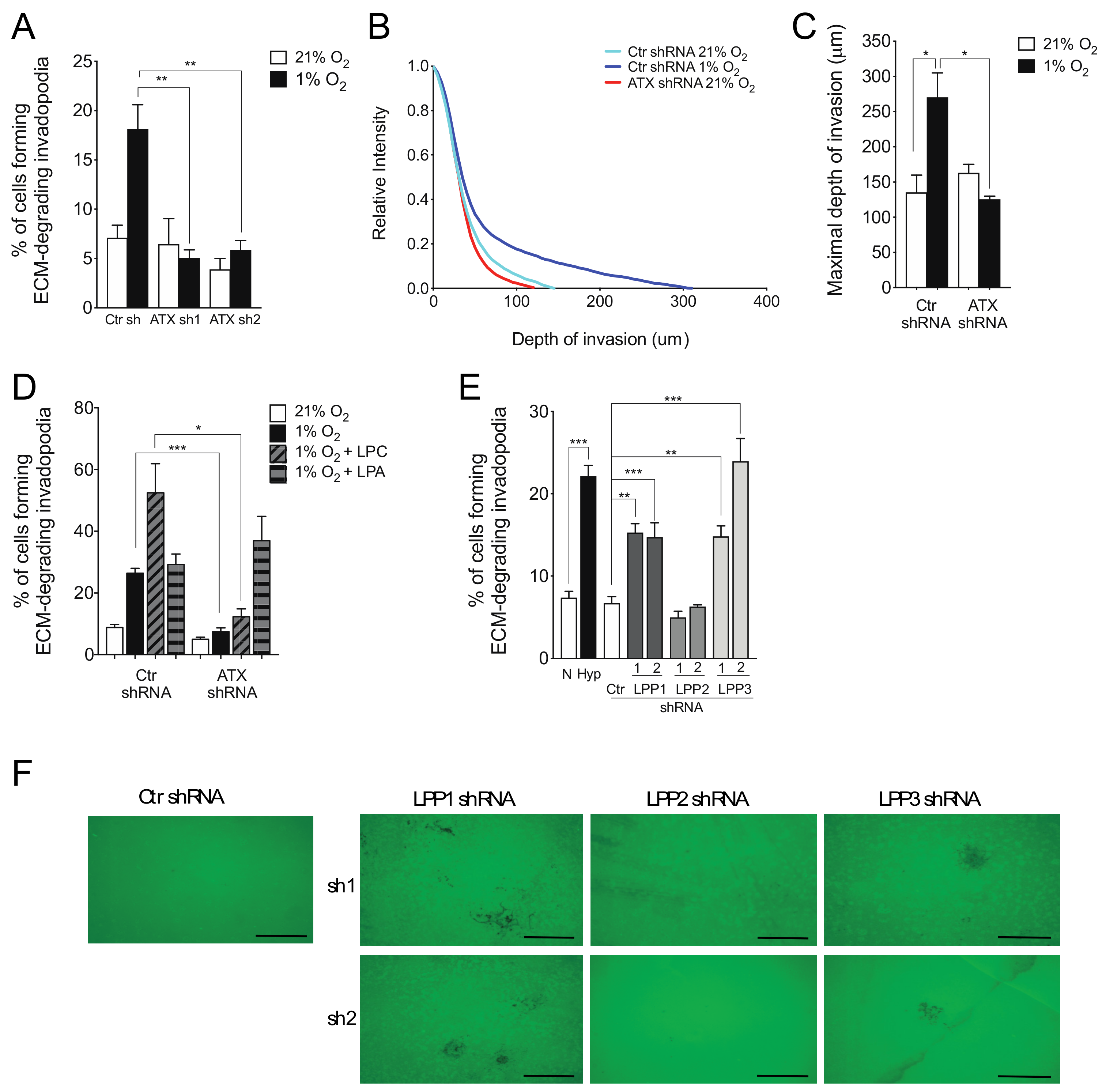
2.2. ATX and LPP1 or LPP3 Exert Opposite Effects on Cell Invasion
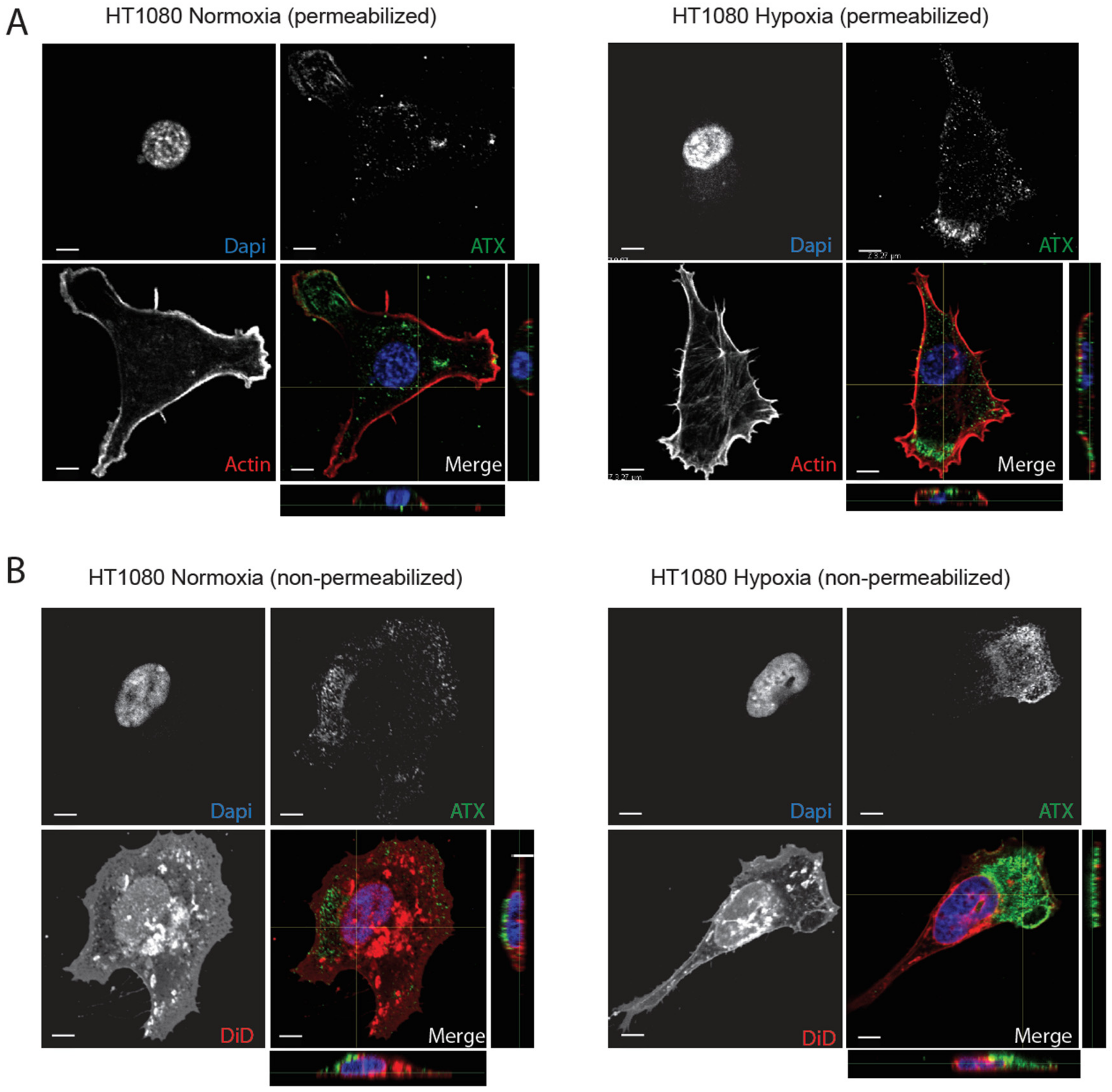
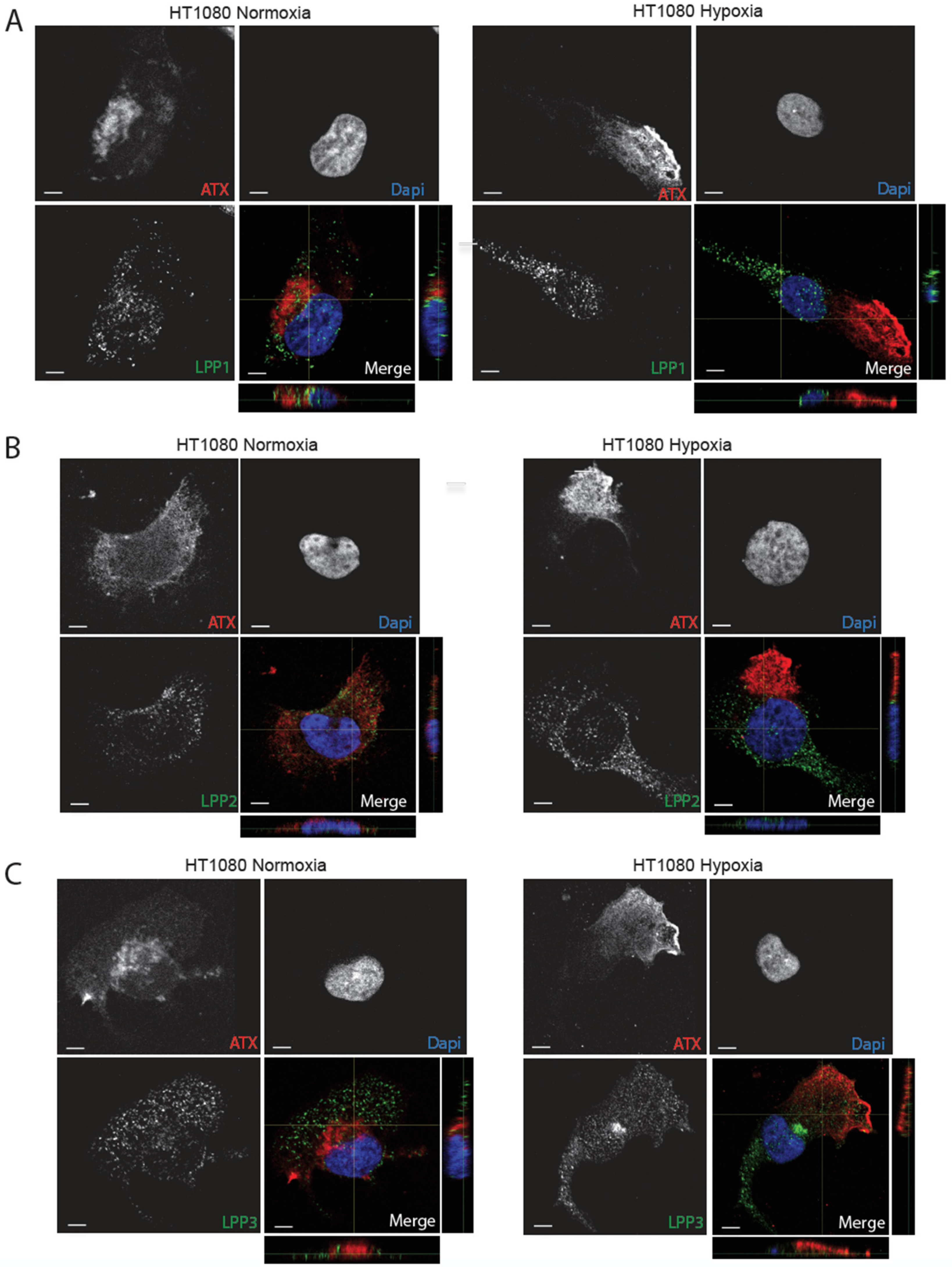
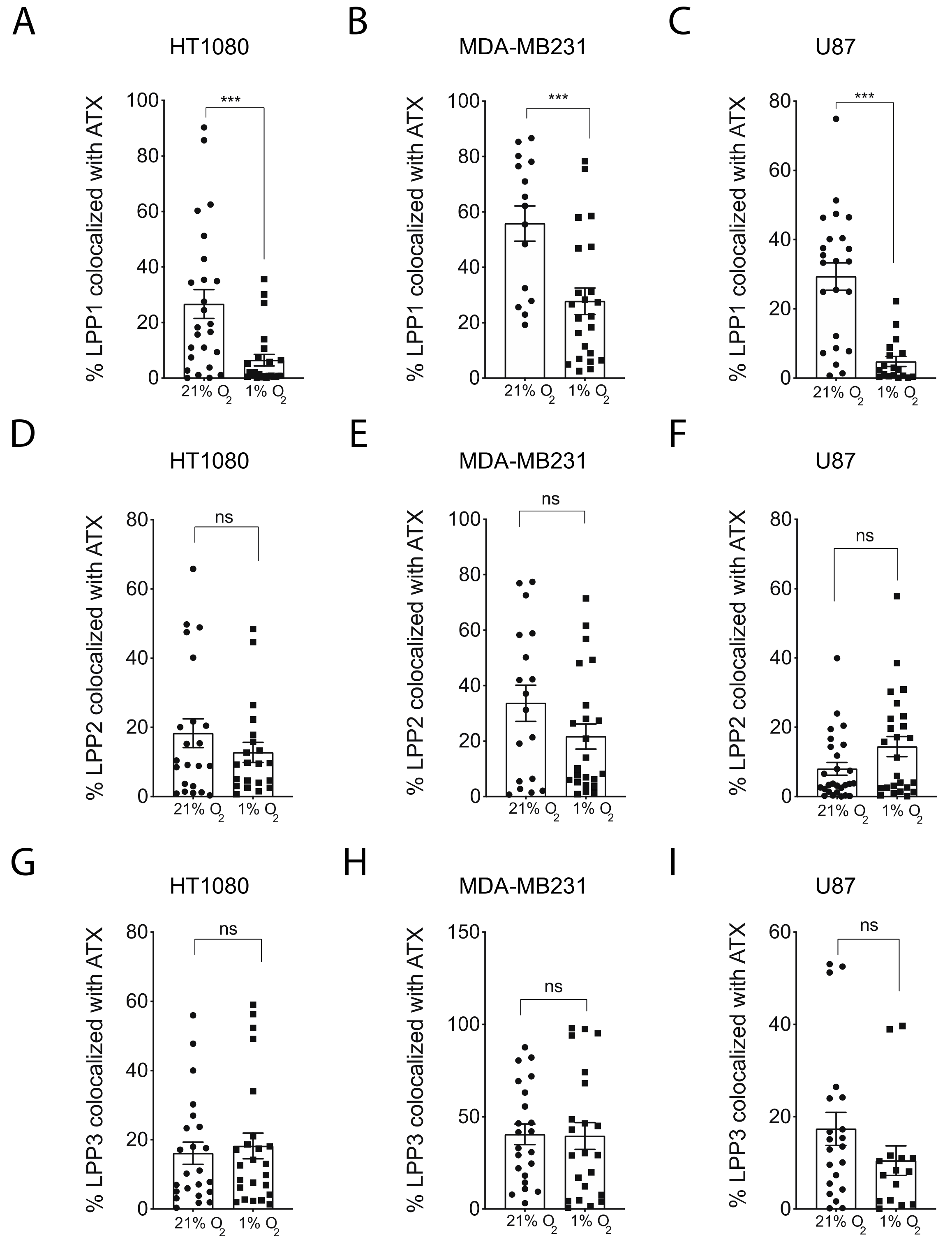
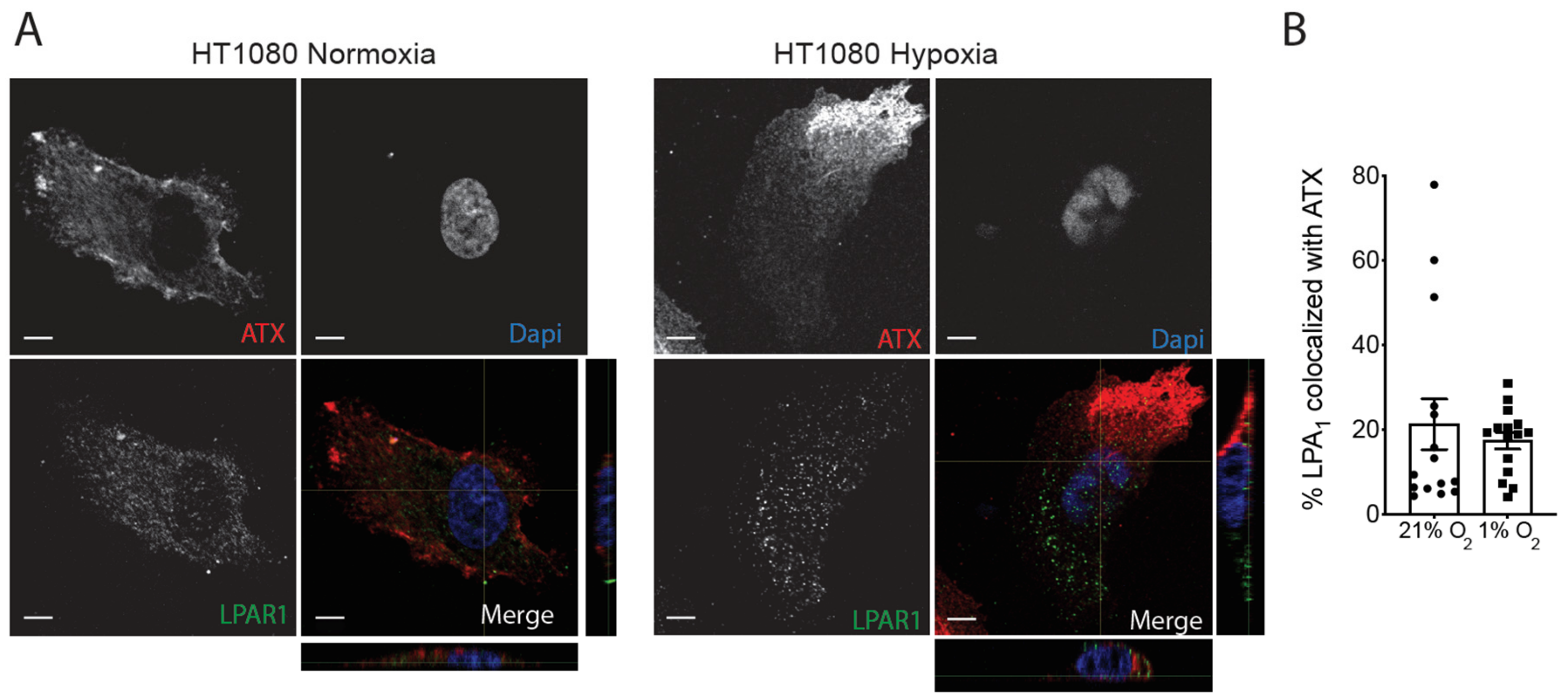
2.3. Hypoxia Induces Spatial Segregation of ATX and LPPs
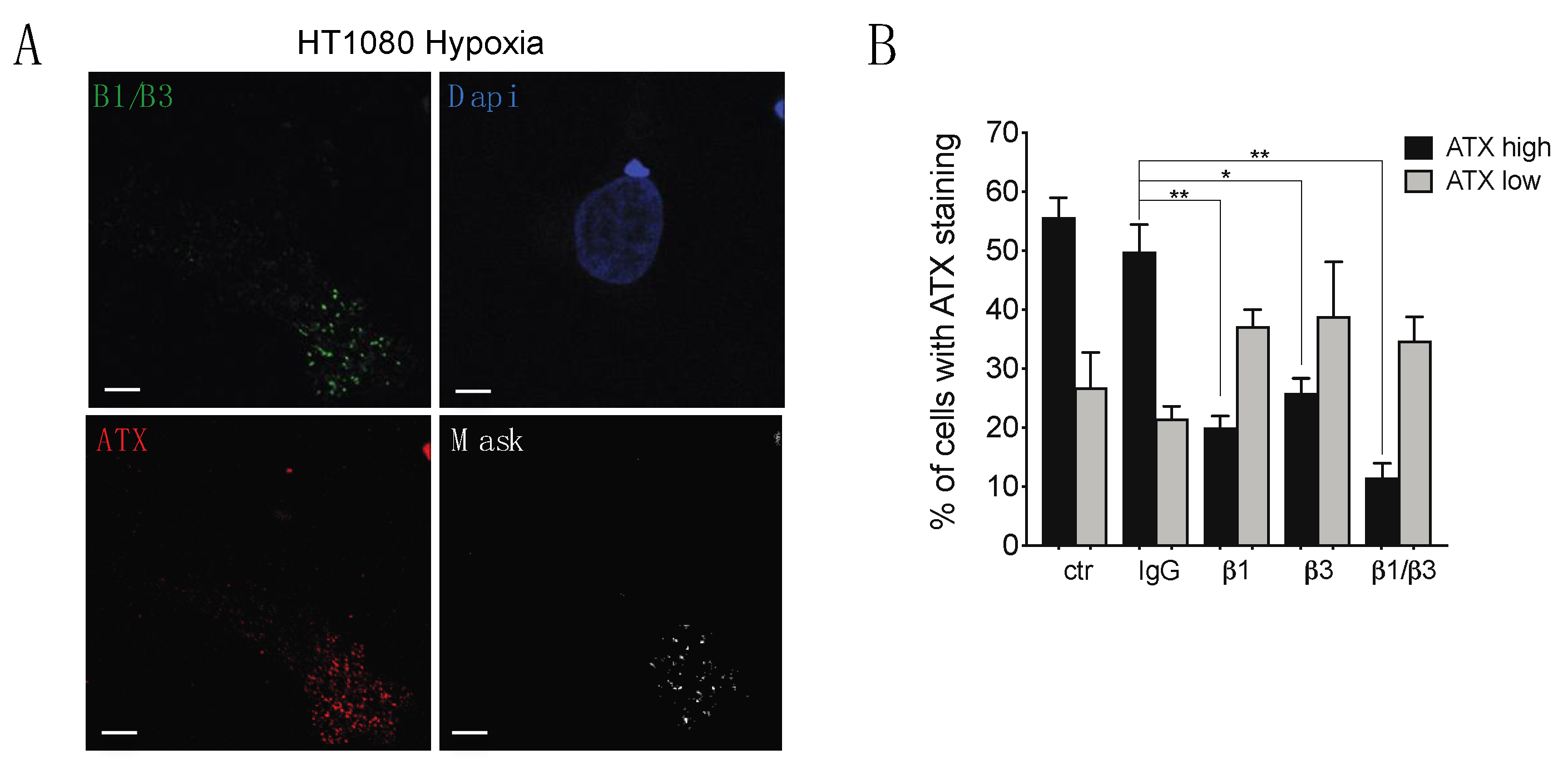
2.4. β1 and β3 Integrins Are Implicated in Cell-Surface ATX Localization
3. Discussion
4. Materials and Methods
4.1. Reagents
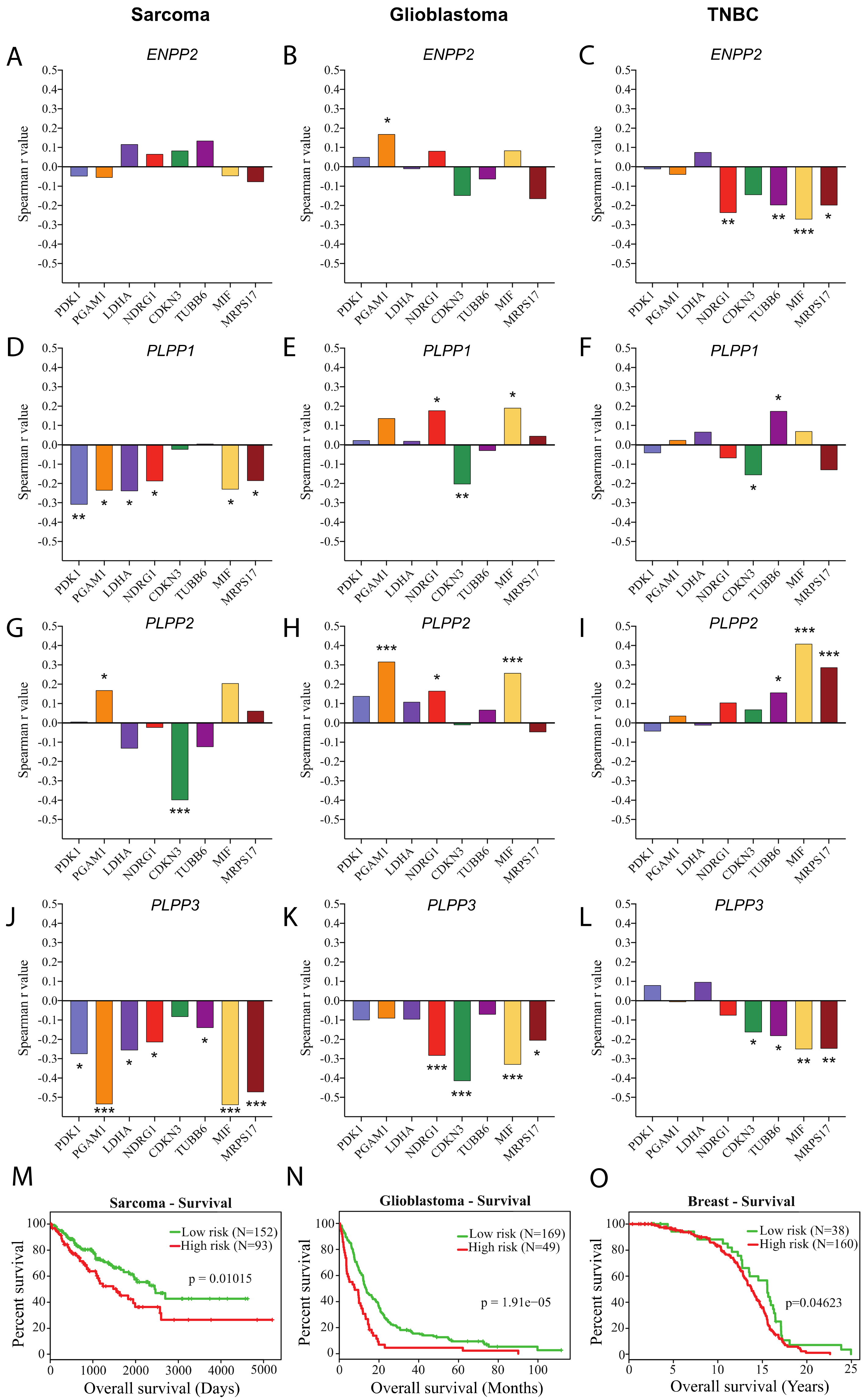
4.2. TCGA RNAseq Data Analysis
4.3. Cell Culture and Transfection
4.4. Real Time RT-PCR
4.5. Western Blotting
4.6. Invadopodia Assay
4.7. 3D Invasion Assay
4.8. Immunofluorescence
4.9. Integrin Blocking Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ray, U.; Roy, S.S. Aberrant lipid metabolism in cancer cells—The role of oncolipid-activated signaling. FEBS J. 2018, 285, 432–443. [Google Scholar] [CrossRef]
- Benesch, M.G.K.; MacIntyre, I.T.K.; McMullen, T.P.W.; Brindley, D.N. Coming of Age for Autotaxin and Lysophosphatidate Signaling: Clinical Applications for Preventing, Detecting and Targeting Tumor-Promoting Inflammation. Cancers 2018, 10, 73. [Google Scholar] [CrossRef]
- Blaho, V.A.; Chun, J. “Crystal” Clear? Lysophospholipid Receptor Structure Insights and Controversies. Trends Pharmacol. Sci. 2018, 39, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef]
- Kato, K.; Fukui, R.; Okabe, K.; Tanabe, E.; Kitayoshi, M.; Fukushima, N.; Tsujiuchi, T. Constitutively active lysophosphatidic acid receptor-1 enhances the induction of matrix metalloproteinase-2. Biochem. Biophys. Res. Commun. 2012, 417, 790–793. [Google Scholar] [CrossRef]
- Yamada, T.; Sato, K.; Komachi, M.; Malchinkhuu, E.; Tobo, M.; Kimura, T.; Kuwabara, A.; Yanagita, Y.; Ikeya, T.; Tanahashi, Y.; et al. Lysophosphatidic acid (LPA) in malignant ascites stimulates motility of human pancreatic cancer cells through LPA1. J. Biol. Chem. 2004, 279, 6595–6605. [Google Scholar] [CrossRef]
- Shida, D.; Kitayama, J.; Yamaguchi, H.; Okaji, Y.; Tsuno, N.H.; Watanabe, T.; Takuwa, Y.; Nagawa, H. Lysophosphatidic acid (LPA) enhances the metastatic potential of human colon carcinoma DLD1 cells through LPA1. Cancer Res. 2003, 63, 1706–1711. [Google Scholar]
- Park, S.Y.; Jeong, K.J.; Panupinthu, N.; Yu, S.; Lee, J.; Han, J.W.; Kim, J.M.; Lee, J.-S.; Kang, J.; Park, C.G.; et al. Lysophosphatidic acid augments human hepatocellular carcinoma cell invasion through LPA1 receptor and MMP-9 expression. Oncogene 2011, 30, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Ribeiro, J.; Descotes, F.; Serre, C.-M.; Barbier, M.; Murone, M.; Clézardin, P.; Peyruchaud, O. Targeting lysophosphatidic acid receptor type 1 with Debio 0719 inhibits spontaneous metastasis dissemination of breast cancer cells independently of cell proliferation and angiogenesis. Int. J. Oncol. 2012, 40, 1133–1141. [Google Scholar] [CrossRef]
- Hope, J.M.; Wang, F.-Q.; Whyte, J.S.; Ariztia, E.V.; Abdalla, W.; Long, K.; Fishman, D.A. LPA receptor 2 mediates LPA-induced endometrial cancer invasion. Gynecol. Oncol. 2009, 112, 215–223. [Google Scholar] [CrossRef] [PubMed]
- So, J.; Wang, F.; Navari, J.; Schreher, J.; Fishman, D.A. LPA-induced epithelial ovarian cancer (EOC) in vitro invasion and migration are mediated by VEGF receptor-2 (VEGF-R2). Gynecol. Oncol. 2005, 97, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.M.; Havik, E.; Postma, F.R.; Beijnen, J.H.; Dalesio, O.; Moolenaar, W.H.; Rodenhuis, S. Malignant effusions contain lysophosphatidic acid (LPA)-like activity. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1998, 9, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shen, Z.; Wiper, D.W.; Wu, M.; Morton, R.E.; Elson, P.; Kennedy, A.W.; Belinson, J.; Markman, M.; Casey, G. Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. JAMA 1998, 280, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Susanto, O.; Koh, Y.W.H.; Morrice, N.; Tumanov, S.; Thomason, P.A.; Nielson, M.; Tweedy, L.; Muinonen-Martin, A.J.; Kamphorst, J.J.; Mackay, G.M.; et al. LPP3 mediates self-generation of chemotactic LPA gradients by melanoma cells. J. Cell Sci. 2017, 130, 3455–3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stracke, M.L.; Krutzsch, H.C.; Unsworth, E.J.; Arestad, A.; Cioce, V.; Schiffmann, E.; Liotta, L.A. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992, 267, 2524–2529. [Google Scholar] [PubMed]
- Gaetano, C.G.; Samadi, N.; Tomsig, J.L.; Macdonald, T.L.; Lynch, K.R.; Brindley, D.N. Inhibition of autotaxin production or activity blocks lysophosphatidylcholine-induced migration of human breast cancer and melanoma cells. Mol. Carcinog. 2009, 48, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Kishi, Y.; Okudaira, S.; Tanaka, M.; Hama, K.; Shida, D.; Kitayama, J.; Yamori, T.; Aoki, J.; Fujimaki, T.; Arai, H. Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidylcholine to lysophosphatidic acid. J. Biol. Chem. 2006, 281, 17492–17500. [Google Scholar] [CrossRef]
- Kehlen, A.; Englert, N.; Seifert, A.; Klonisch, T.; Dralle, H.; Langner, J.; Hoang-Vu, C. Expression, regulation and function of autotaxin in thyroid carcinomas. Int. J. Cancer 2004, 109, 833–838. [Google Scholar] [CrossRef]
- Yang, Y.; Mou, L.; Liu, N.; Tsao, M.S. Autotaxin expression in non-small-cell lung cancer. Am. J. Respir. Cell Mol. Biol. 1999, 21, 216–222. [Google Scholar] [CrossRef]
- Yang, S.Y.; Lee, J.; Park, C.G.; Kim, S.; Hong, S.; Chung, H.C.; Min, S.K.; Han, J.W.; Lee, H.W.; Lee, H.Y. Expression of autotaxin (NPP-2) is closely linked to invasiveness of breast cancer cells. Clin. Exp. Metastasis 2002, 19, 603–608. [Google Scholar] [CrossRef]
- Fulkerson, Z.; Wu, T.; Sunkara, M.; Kooi, C.V.; Morris, A.J.; Smyth, S.S. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J. Biol. Chem. 2011, 286, 34654–34663. [Google Scholar] [CrossRef] [PubMed]
- Houben, A.J.S.; van Wijk, X.M.R.; van Meeteren, L.A.; van Zeijl, L.; van de Westerlo, E.M.A.; Hausmann, J.; Fish, A.; Perrakis, A.; van Kuppevelt, T.H.; Moolenaar, W.H. The polybasic insertion in autotaxin α confers specific binding to heparin and cell surface heparan sulfate proteoglycans. J. Biol. Chem. 2013, 288, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, R.; Peyruchaud, O. New insights into the autotaxin/LPA axis in cancer development and metastasis. Exp. Cell Res. 2015, 333, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Brindley, D.N. Lipid phosphate phosphatases and related proteins: Signaling functions in development, cell division, and cancer. J. Cell. Biochem. 2004, 92, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Tomsig, J.L.; Snyder, A.H.; Berdyshev, E.V.; Skobeleva, A.; Mataya, C.; Natarajan, V.; Brindley, D.N.; Lynch, K.R. Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo. Biochem. J. 2009, 419, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Escalante-Alcalde, D.; Hernandez, L.; Le Stunff, H.; Maeda, R.; Lee, H.S.; Cheng, G., Jr.; Sciorra, V.A.; Daar, I.; Spiegel, S.; Morris, A.J.; et al. The lipid phosphatase LPP3 regulates extra-embryonic vasculogenesis and axis patterning. Development 2003, 130, 4623–4637. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Benesch, M.G.K.; Brindley, D.N. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J. Lipid Res. 2015, 56, 2048–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanyi, J.L.; Hasegawa, Y.; Lapushin, R.; Morris, A.J.; Wolf, J.K.; Berchuck, A.; Lu, K.; Smith, D.I.; Kalli, K.; Hartmann, L.C.; et al. Role of decreased levels of lipid phosphate phosphatase-1 in accumulation of lysophosphatidic acid in ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 3534–3545. [Google Scholar]
- Tang, X.; Benesch, M.G.K.; Dewald, J.; Zhao, Y.Y.; Patwardhan, N.; Santos, W.L.; Curtis, J.M.; McMullen, T.P.W.; Brindley, D.N. Lipid phosphate phosphatase-1 expression in cancer cells attenuates tumor growth and metastasis in mice. J. Lipid Res. 2014, 55, 2389–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kai, M.; Sakane, F.; Jia, Y.-J.; Imai, S.-I.; Yasuda, S.; Kanoh, H. Lipid phosphate phosphatases 1 and 3 are localized in distinct lipid rafts. J. Biochem. 2006, 140, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl) 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 71–103. [Google Scholar] [CrossRef] [PubMed]
- Hockel, M.; Schlenger, K.; Aral, B.; Mitze, M.; Schaffer, U.; Vaupel, P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996, 56, 4509–4515. [Google Scholar] [PubMed]
- Cairns, R.A.; Hill, R.P. Acute hypoxia enhances spontaneous lymph node metastasis in an orthotopic murine model of human cervical carcinoma. Cancer Res. 2004, 64, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Kalliomaki, T.; Hill, R.P. Acute (cyclic) hypoxia enhances spontaneous metastasis of KHT murine tumors. Cancer Res. 2001, 61, 8903–8908. [Google Scholar]
- Bourseau-Guilmain, E.; Menard, J.A.; Lindqvist, E.; Indira Chandran, V.; Christianson, H.C.; Cerezo Magaña, M.; Lidfeldt, J.; Marko-Varga, G.; Welinder, C.; Belting, M. Hypoxia regulates global membrane protein endocytosis through caveolin-1 in cancer cells. Nat. Commun. 2016, 7, 11371. [Google Scholar] [CrossRef]
- Cassavaugh, J.; Lounsbury, K.M. Hypoxia-mediated biological control. J. Cell. Biochem. 2011, 112, 735–744. [Google Scholar] [CrossRef]
- Wang, Y.; Ohh, M. Oxygen-mediated endocytosis in cancer. J. Cell. Mol. Med. 2010, 14, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Hanna, S.C.; Krishnan, B.; Bailey, S.T.; Moschos, S.J.; Kuan, P.-F.; Shimamura, T.; Osborne, L.D.; Siegel, M.B.; Duncan, L.M.; O’Brien, E.T.; et al. HIF1α and HIF2α independently activate SRC to promote melanoma metastases. J. Clin. Investig. 2013, 123, 2078–2093. [Google Scholar] [CrossRef] [PubMed]
- Md Hashim, N.F.; Nicholas, N.S.; Dart, A.E.; Kiriakidis, S.; Paleolog, E.; Wells, C.M. Hypoxia-induced invadopodia formation: A role for β-PIX. Open Biol. 2013, 3, 120159. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, D.; Brochu-Gaudreau, K.; Charbonneau, M.; Dubois, C.M. HDAC6 deacetylase activity is required for hypoxia-induced invadopodia formation and cell invasion. PLoS ONE 2013, 8, e55529. [Google Scholar] [CrossRef] [PubMed]
- Lucien, F.; Brochu-Gaudreau, K.; Arsenault, D.; Harper, K.; Dubois, C.M. Hypoxia-induced invadopodia formation involves activation of NHE-1 by the p90 ribosomal S6 kinase (p90RSK). PLoS ONE 2011, 6, e28851. [Google Scholar] [CrossRef] [PubMed]
- Harper, K.; Lavoie, R.R.; Charbonneau, M.; Brochu-Gaudreau, K.; Dubois, C.M. The Hypoxic Tumor Microenvironment Promotes Invadopodia Formation and Metastasis through LPA1 Receptor and EGFR Cooperation. Mol. Cancer Res. MCR 2018, 16, 1601–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.J.; Schwartz, B.; Washington, M.; Kennedy, A.; Webster, K.; Belinson, J.; Xu, Y. Electrospray ionization mass spectrometry analysis of lysophospholipids in human ascitic fluids: Comparison of the lysophospholipid contents in malignant vs nonmalignant ascitic fluids. Anal. Biochem. 2001, 290, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Yukiura, H.; Kano, K.; Kise, R.; Inoue, A.; Aoki, J. Autotaxin overexpression causes embryonic lethality and vascular defects. PLoS ONE 2015, 10, e0126734. [Google Scholar] [CrossRef]
- Nasa, I.; Kettenbach, A.N. Coordination of Protein Kinase and Phosphoprotein Phosphatase Activities in Mitosis. Front. Cell Dev. Biol. 2018, 6, 30. [Google Scholar] [CrossRef]
- Eustace, A.; Mani, N.; Span, P.N.; Irlam, J.J.; Taylor, J.; Betts, G.N.J.; Denley, H.; Miller, C.J.; Homer, J.J.; Rojas, A.M.; et al. A 26-gene hypoxia signature predicts benefit from hypoxia-modifying therapy in laryngeal cancer but not bladder cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4879–4888. [Google Scholar] [CrossRef]
- Harper, K.; Arsenault, D.; Boulay-Jean, S.; Lauzier, A.; Lucien, F.; Dubois, C.M. Autotaxin promotes cancer invasion via the lysophosphatidic acid receptor 4: Participation of the cyclic AMP/EPAC/Rac1 signaling pathway in invadopodia formation. Cancer Res. 2010, 70, 4634–4643. [Google Scholar] [CrossRef]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.H.G.; van Meeteren, L.A.; Houben, A.J.S.; van Zeijl, L.; et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, H.; Newton, R.; Klein, R.; Morita, Y.; Gunn, M.D.; Rosen, S.D. Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat. Immunol. 2008, 9, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, J.; Wang, R.; Zeng, X. Lipid rafts regulate the lamellipodia formation of melanoma A375 cells via actin cytoskeleton-mediated recruitment of β1 and β3 integrin. Oncol. Lett. 2018, 16, 6540–6546. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-S.; Sengupta, S.; Berk, M.; Kwak, Y.-G.; Escobar, P.F.; Belinson, J.; Mok, S.C.; Xu, Y. Hypoxia enhances lysophosphatidic acid responsiveness in ovarian cancer cells and lysophosphatidic acid induces ovarian tumor metastasis in vivo. Cancer Res. 2006, 66, 7983–7990. [Google Scholar] [CrossRef] [PubMed]
- Shlyonsky, V.; Naeije, R.; Mies, F. Possible role of lysophosphatidic acid in rat model of hypoxic pulmonary vascular remodeling. Pulm. Circ. 2014, 4, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-G.; Yu, Z.-C.; Wang, D.-Z.; Ju, W.-P.; Zhan, X.; Wu, Q.-Z.; Wu, X.-J.; Cong, H.-M.; Man, H.-H. Influence of acetylsalicylate on plasma lysophosphatidic acid level in patients with ischemic cerebral vascular diseases. Neurol. Res. 2008, 30, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Samadi, N.; Bekele, R.; Capatos, D.; Venkatraman, G.; Sariahmetoglu, M.; Brindley, D.N. Regulation of lysophosphatidate signaling by autotaxin and lipid phosphate phosphatases with respect to tumor progression, angiogenesis, metastasis and chemo-resistance. Biochimie 2011, 93, 61–70. [Google Scholar] [CrossRef]
- Tang, X.; Zhao, Y.Y.; Dewald, J.; Curtis, J.M.; Brindley, D.N. Tetracyclines increase lipid phosphate phosphatase expression on plasma membranes and turnover of plasma lysophosphatidate. J. Lipid Res. 2016, 57, 597–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parent, C.A.; Devreotes, P.N. A cell’s sense of direction. Science 1999, 284, 765–770. [Google Scholar] [CrossRef]
- Mañes, S.; Mira, E.; Gómez-Moutón, C.; Lacalle, R.A.; Keller, P.; Labrador, J.P.; Martínez-A, C. Membrane raft microdomains mediate front-rear polarity in migrating cells. EMBO J. 1999, 18, 6211–6220. [Google Scholar] [CrossRef]
- Mañes, S.; Mira, E.; Gómez-Moutón, C.; Lacalle, R.A.; Martínez, C. Cells on the move: A dialogue between polarization and motility. IUBMB Life 2000, 49, 89–96. [Google Scholar] [PubMed]
- Gómez-Móuton, C.; Abad, J.L.; Mira, E.; Lacalle, R.A.; Gallardo, E.; Jiménez-Baranda, S.; Illa, I.; Bernad, A.; Mañes, S.; Martínez-A, C. Segregation of leading-edge and uropod components into specific lipid rafts during T cell polarization. Proc. Natl. Acad. Sci. USA 2001, 98, 9642–9647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Kooi, C.V.; Shah, P.; Charnigo, R.; Huang, C.; Smyth, S.S.; Morris, A.J. Integrin-mediated cell surface recruitment of autotaxin promotes persistent directional cell migration. FASEB J. 2014, 28, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-O.; Shin, S.; Mercurio, A.M. Hypoxia stimulates carcinoma invasion by stabilizing microtubules and promoting the Rab11 trafficking of the alpha6beta4 integrin. Cancer Res. 2005, 65, 2761–2769. [Google Scholar] [CrossRef] [PubMed]
- Caswell, P.T.; Spence, H.J.; Parsons, M.; White, D.P.; Clark, K.; Cheng, K.W.; Mills, G.B.; Humphries, M.J.; Messent, A.J.; Anderson, K.I.; et al. Rab25 associates with alpha5beta1 integrin to promote invasive migration in 3D microenvironments. Dev. Cell 2007, 13, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Beaty, B.T.; Sharma, V.P.; Bravo-Cordero, J.J.; Simpson, M.A.; Eddy, R.J.; Koleske, A.J.; Condeelis, J. β1 integrin regulates Arg to promote invadopodial maturation and matrix degradation. Mol. Biol. Cell 2013, 24, 1661–1675. [Google Scholar] [CrossRef] [PubMed]
- Peláez, R.; Morales, X.; Salvo, E.; Garasa, S.; Ortiz de Solórzano, C.; Martínez, A.; Larrayoz, I.M.; Rouzaut, A. β3 integrin expression is required for invadopodia-mediated ECM degradation in lung carcinoma cells. PLoS ONE 2017, 12, e0181579. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Murph, M.; Yu, S.; Tigyi, G.; Mills, G.B. Lysophosphatidic acid (LPA)-induced vasodilator-stimulated phosphoprotein mediates lamellipodia formation to initiate motility in PC-3 prostate cancer cells. Mol. Oncol. 2008, 2, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; He, D.; Berdyshev, E.; Zhong, M.; Salgia, R.; Morris, A.J.; Smyth, S.S.; Natarajan, V.; Zhao, Y. Autotaxin induces lung epithelial cell migration through lysoPLD activity-dependent and -independent pathways. Biochem. J. 2011, 439, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Yoshida, S.; Muroi, E.; Yoshida, N.; Kawamura, M.; Kouchi, Z.; Nakamura, Y.; Sakai, R.; Fukami, K. Phosphoinositide 3-kinase signaling pathway mediated by p110α regulates invadopodia formation. J. Cell Biol. 2011, 193, 1275–1288. [Google Scholar] [CrossRef]
- Merlot, S.; Firtel, R.A. Leading the way: Directional sensing through phosphatidylinositol 3-kinase and other signaling pathways. J. Cell Sci. 2003, 116, 3471–3478. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buffa, F.M.; Harris, A.L.; West, C.M.; Miller, C.J. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer 2010, 102, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Chibon, F.; Lagarde, P.; Salas, S.; Pérot, G.; Brouste, V.; Tirode, F.; Lucchesi, C.; de Reynies, A.; Kauffmann, A.; Bui, B.; et al. Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nat. Med. 2010, 16, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Scheck, A.C.; Cloughesy, T.F.; Lai, A.; Dong, J.; Farooqi, H.K.; Liau, L.M.; Horvath, S.; Mischel, P.S.; Nelson, S.F. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med. Genom. 2008, 1, 52. [Google Scholar] [CrossRef] [PubMed]
- Van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martínez-Ledesma, E.; Martínez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Peña, J.G.; Treviño, V. SurvExpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, D.; Lucien, F.; Dubois, C.M. Hypoxia enhances cancer cell invasion through relocalization of the proprotein convertase furin from the trans-Golgi network to the cell surface. J. Cell. Physiol. 2012, 227, 789–800. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harper, K.; Brochu-Gaudreau, K.; Saucier, C.; Dubois, C.M. Hypoxia Downregulates LPP3 and Promotes the Spatial Segregation of ATX and LPP1 During Cancer Cell Invasion. Cancers 2019, 11, 1403. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11091403
Harper K, Brochu-Gaudreau K, Saucier C, Dubois CM. Hypoxia Downregulates LPP3 and Promotes the Spatial Segregation of ATX and LPP1 During Cancer Cell Invasion. Cancers. 2019; 11(9):1403. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11091403
Chicago/Turabian StyleHarper, Kelly, Karine Brochu-Gaudreau, Caroline Saucier, and Claire M. Dubois. 2019. "Hypoxia Downregulates LPP3 and Promotes the Spatial Segregation of ATX and LPP1 During Cancer Cell Invasion" Cancers 11, no. 9: 1403. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11091403