Targeting Glycolysis with Epigallocatechin-3-Gallate Enhances the Efficacy of Chemotherapeutics in Pancreatic Cancer Cells and Xenografts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
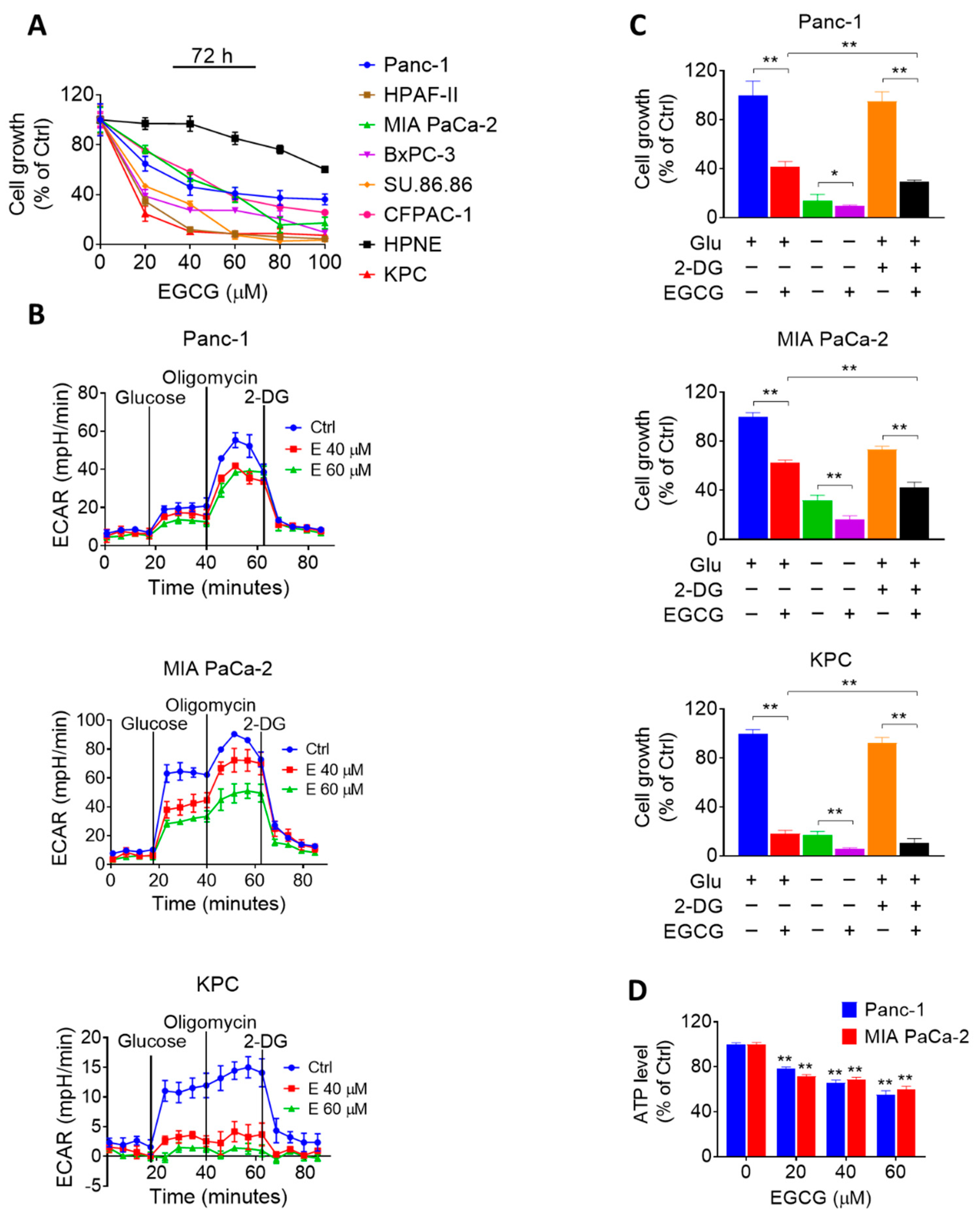
2.1. EGCG Affects Glycolysis in Pancreatic Cancer Cells
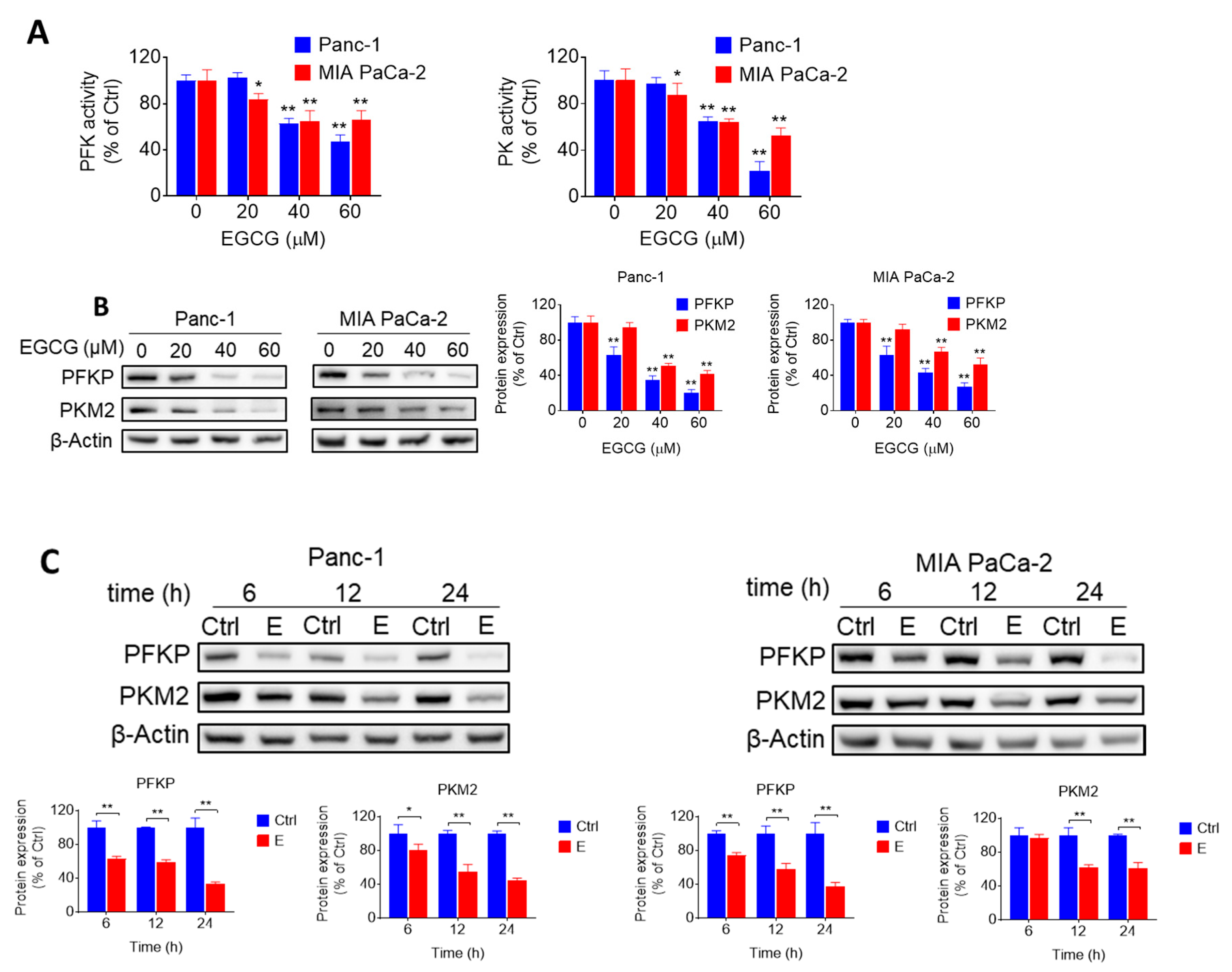
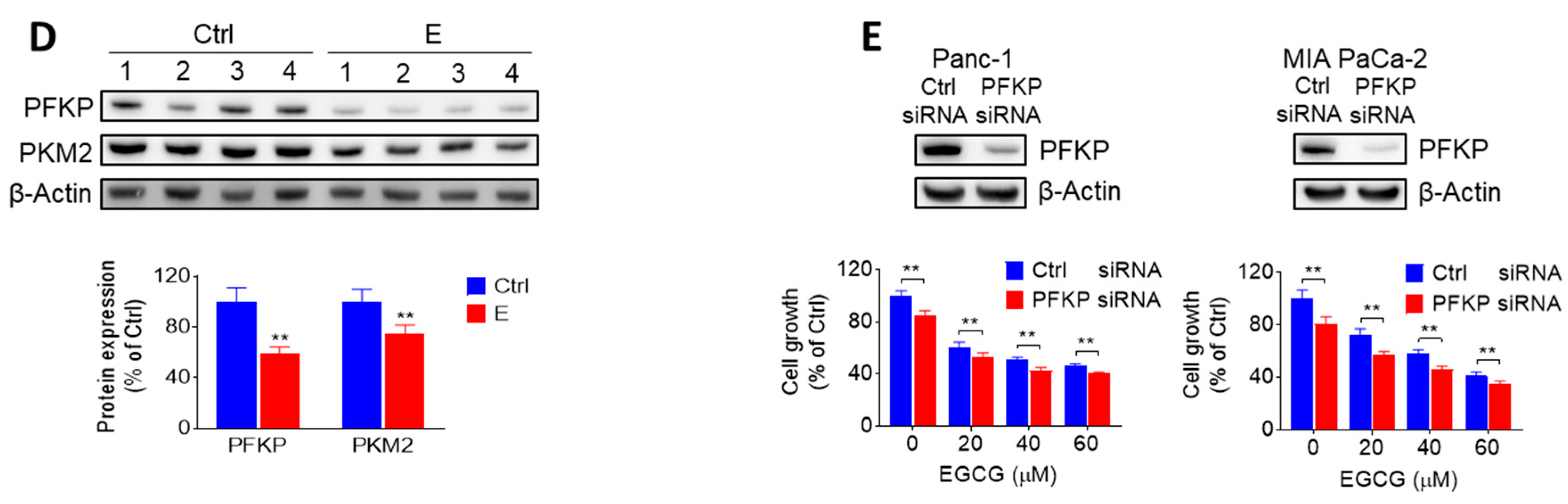
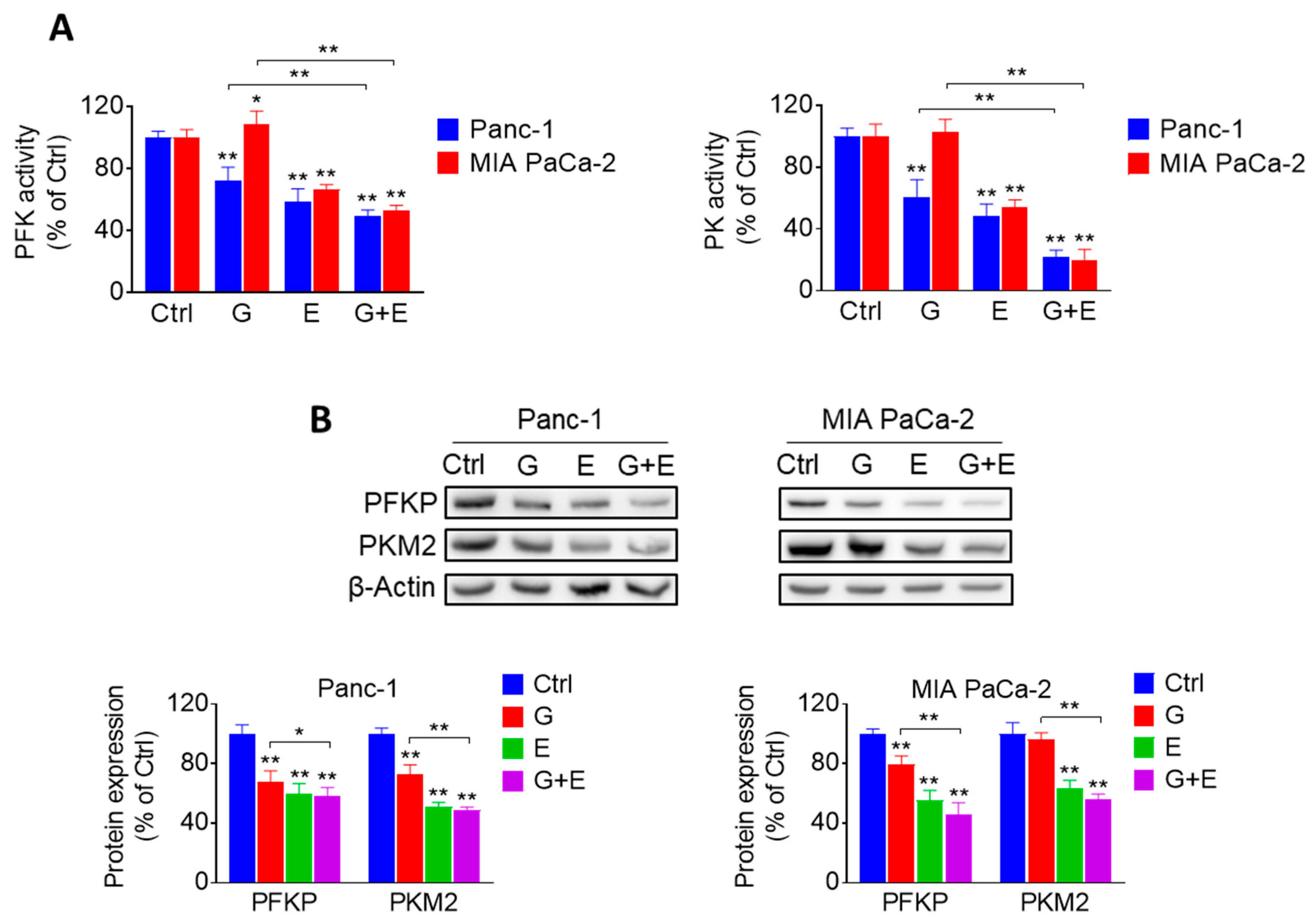
2.2. EGCG Inhibits Glycolysis through Suppressing Rate-Limiting Enzymes
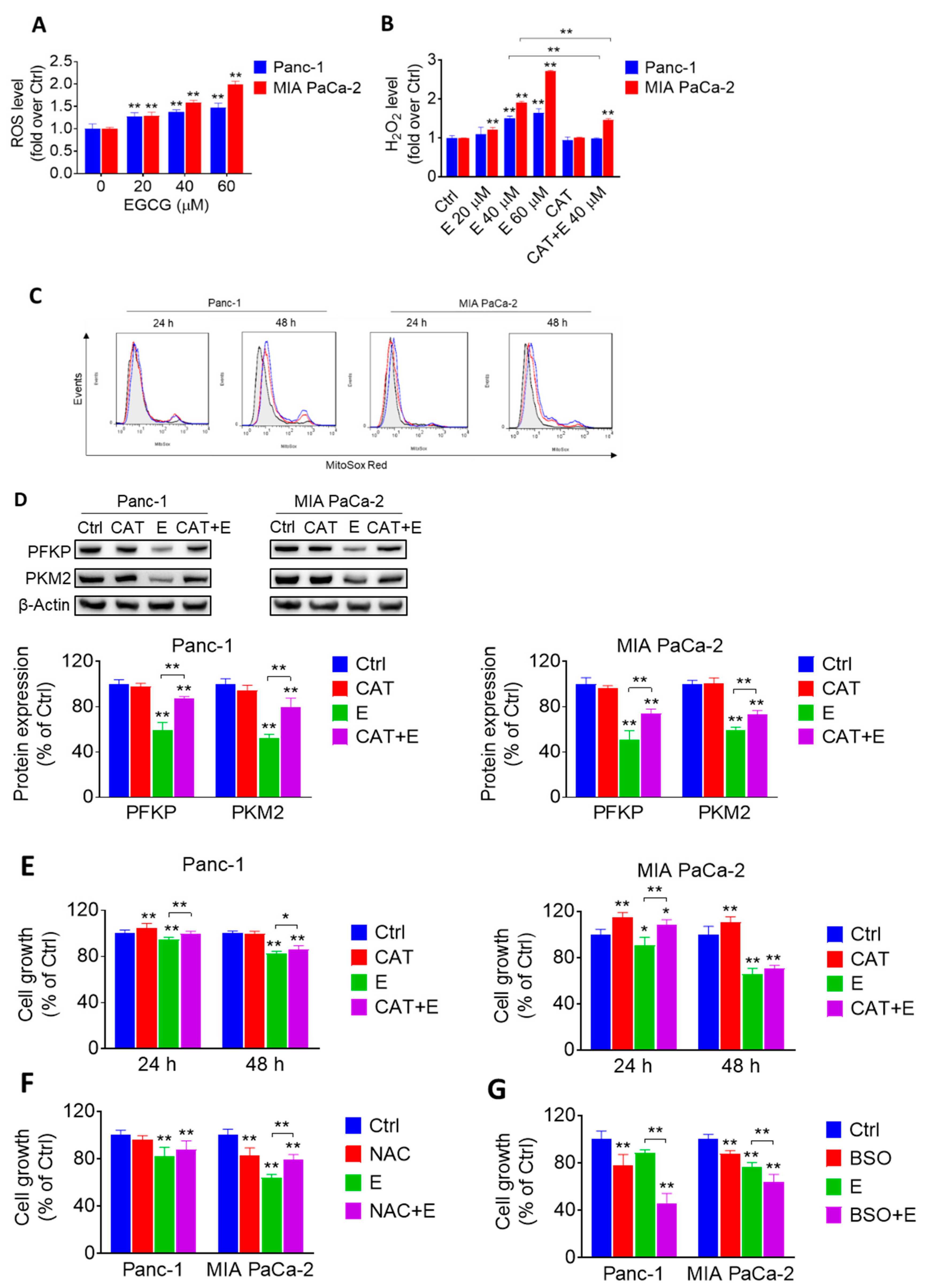
2.3. EGCG Affects the Glycolytic Pathway in Pancreatic Cancer Cells through a ROS-Dependent Manner
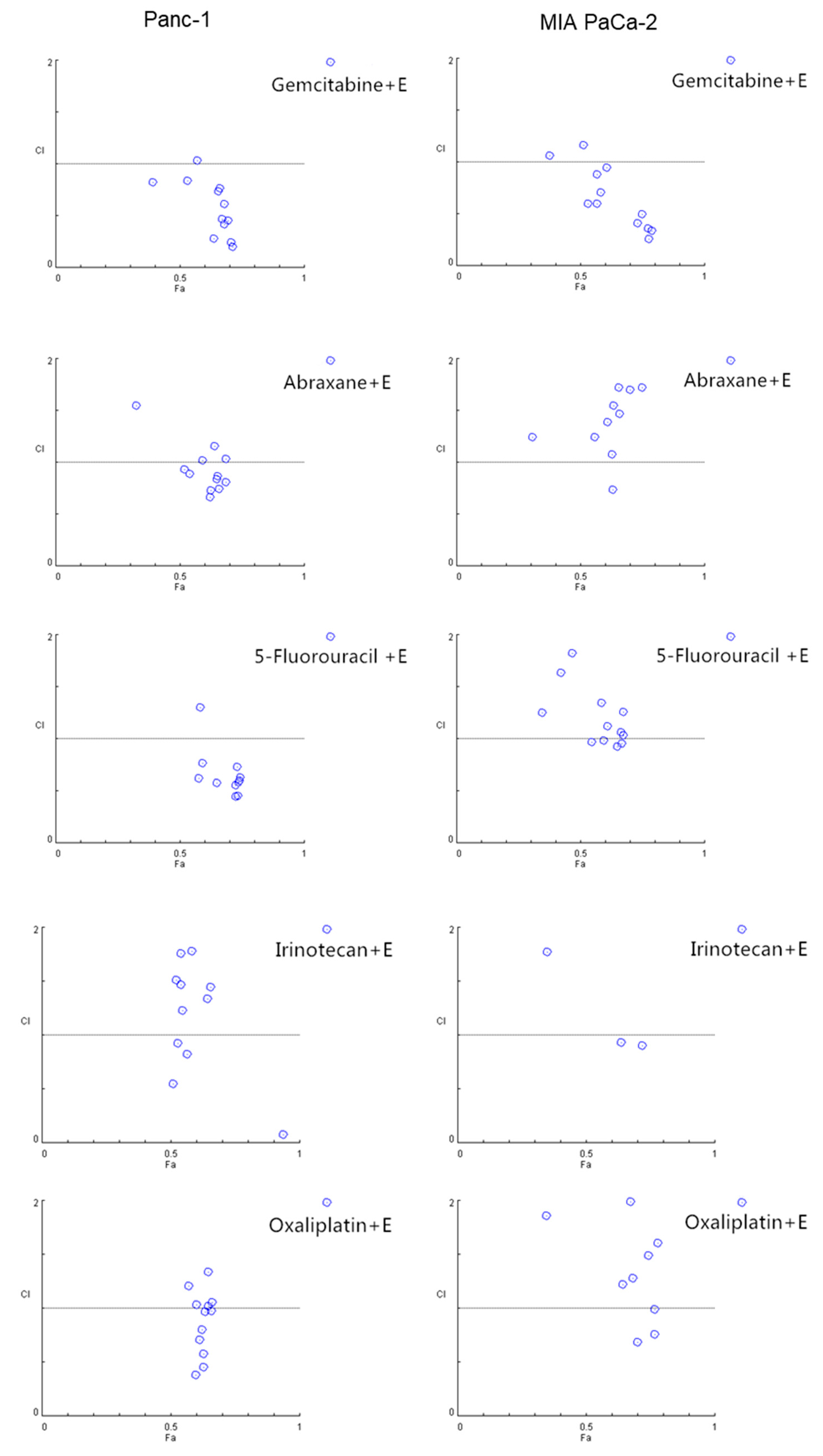
2.4. EGCG Sensitizes Pancreatic Cancer Cells to Gemcitabine In Vitro
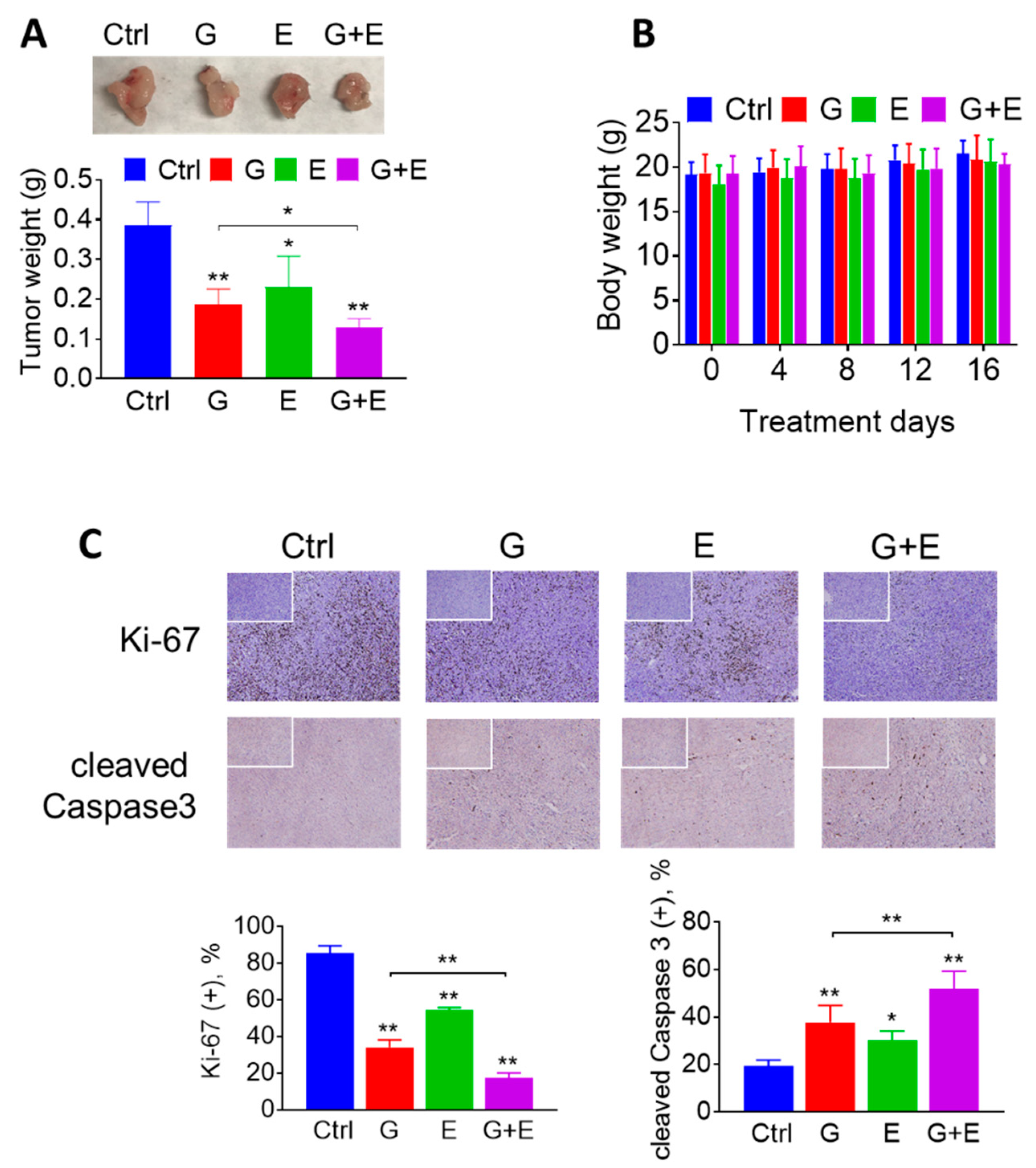
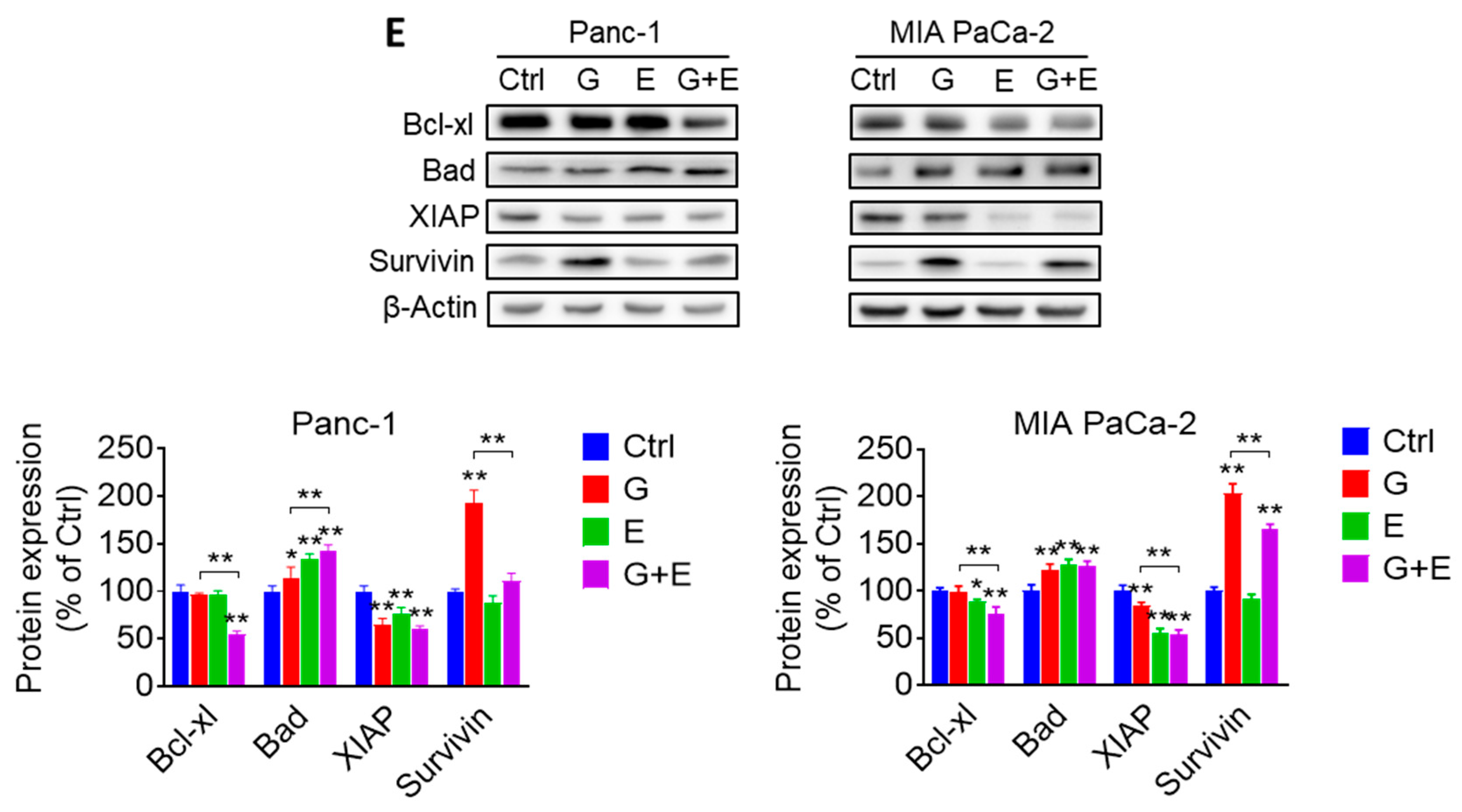
2.5. EGCG Enhances the Anticancer Effect of Gemcitabine in Pancreatic Cancer Xenografts through a Strong Cytokinetic Effect
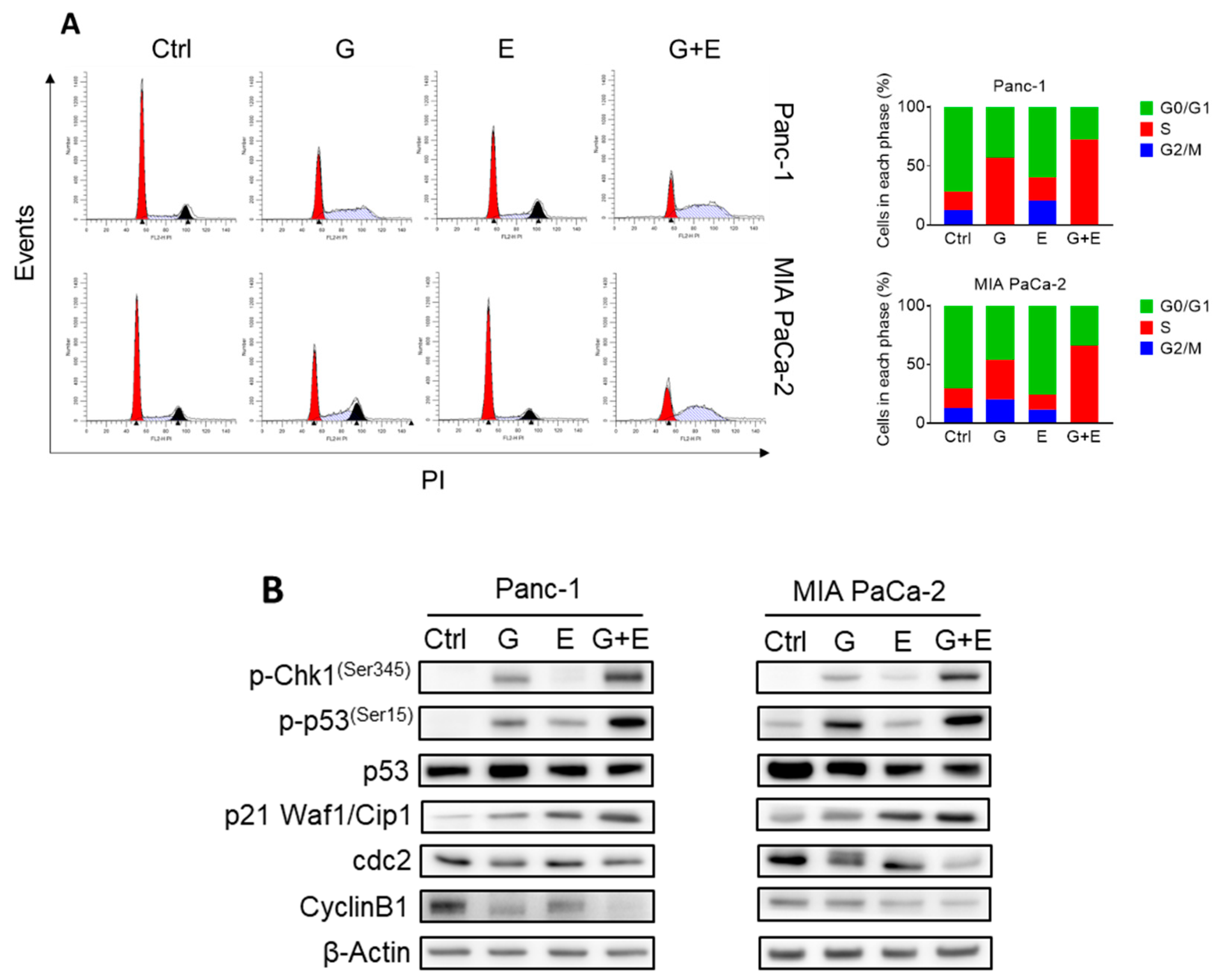
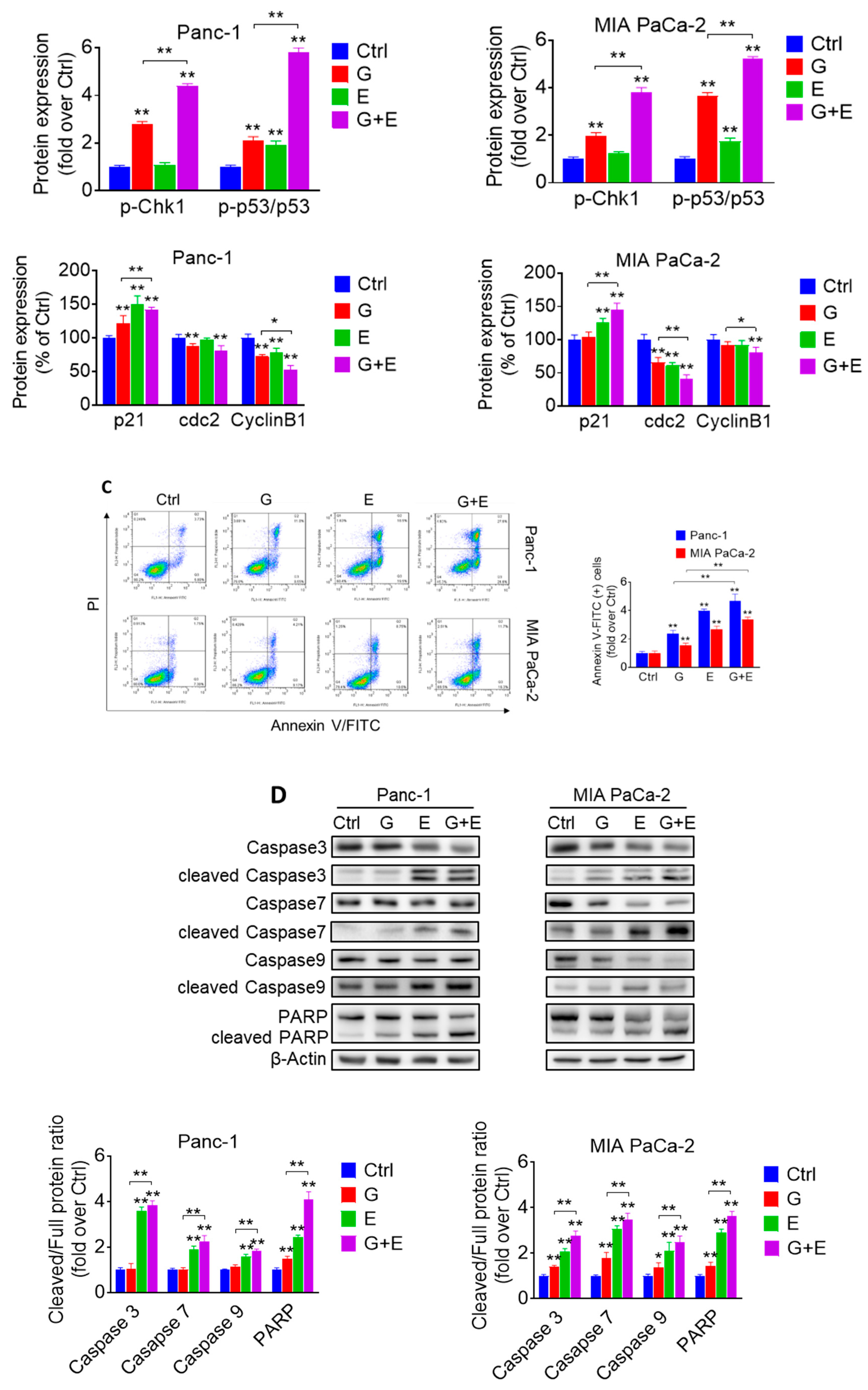
2.6. EGCG Plus Gemcitabine Further Inhibits Glycolysis
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Cell Viability
4.4. Cellular Glycolytic Rate Measurements
4.5. Cellular ATP Levels
4.6. Glycolysis-Related Enzymes Activity
4.7. Gene Silencing
4.8. Determination of ROS Levels
4.9. Mitochondrial Superoxide Level Analysis
4.10. Cell Apoptosis
4.11. Cell Cycle Analysis
4.12. Western Blot
4.13. Animal Studies
4.14. Immunohistochemistry
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Aier, I.; Semwal, R.; Sharma, A.; Varadwaj, P.K. A systematic assessment of statistics, risk factors, and underlying features involved in pancreatic cancer. Cancer Epidemiol. 2019, 58, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Dhup, S.; Dadhich, R.K.; Porporato, P.E.; Sonveaux, P. Multiple biological activities of lactic acid in cancer: Influences on tumor growth, angiogenesis and metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, B.; Mohd Omar, M.F.; Soong, R. The warburg effect and drug resistance. Br. J. Pharmacol. 2016, 173, 970–979. [Google Scholar] [CrossRef]
- Lewis, K.A.; Jordan, H.R.; Tollefsbol, T.O. Effects of SAHA and EGCG on growth potentiation of triple-negative breast cancer cells. Cancers 2018, 11, 23. [Google Scholar] [CrossRef]
- Du, G.J.; Zhang, Z.Y.; Wen, X.D.; Yu, C.H.; Calway, T.; Yuan, C.S.; Wang, C.Z. Epigallocatechin Gallate (EGCG) Is the most effective cancer chemopreventive polyphenol in green tea. Nutrients 2012, 4, 1679–1691. [Google Scholar] [CrossRef]
- Wang, Y.K.; Chen, W.C.; Lai, Y.H.; Chen, Y.H.; Wu, M.T.; Kuo, C.T.; Wang, Y.Y.; Yuan, S.S.F.; Liu, Y.P.; Wu, I.C. Influence of tea consumption on the development of second esophageal neoplasm in patients with head and neck cancer. Cancers 2019, 11, 387. [Google Scholar] [CrossRef]
- Wei, R.; Mao, L.; Xu, P.; Zheng, X.; Hackman, R.M.; Mackenzie, G.G.; Wang, Y. Suppressing glucose metabolism with epigallocatechin-3-gallate (EGCG) reduces breast cancer cell growth in preclinical models. Food Funct. 2018, 9, 5682–5696. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, L.; Feng, J.; Li, J.; Liu, T.; Zhang, R.; Xu, S.; Cheng, K.; Zhou, Y.; Zhou, S.; et al. In vitro and in vivo study of epigallocatechin-3-gallate-induced apoptosis in aerobic glycolytic hepatocellular carcinoma cells involving inhibition of phosphofructokinase activity. Sci. Rep. 2016, 6, e28479. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Li, M.; Liu, W.B.; Zhou, Z.S.; Zhang, R.; Li, J.L.; Zhou, K.C. Epigallocatechin gallate inhibits human tongue carcinoma cells via HK2mediated glycolysis. Oncol. Rep. 2015, 33, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Long, L.H.; Clement, M.V.; Halliwell, B. Artifacts in cell culture: Rapid generation of hydrogen peroxide on addition of (-)-epigallocatechin, (-)-epigallocatechin gallate, (+)-catechin, and quercetin to commonly used cell culture media. Biochem. Biophys. Res. Commun. 2000, 273, 50–53. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, e17. [Google Scholar] [CrossRef]
- Villa, E.; Ricci, J.E. How does metabolism affect cell death in cancer? Febs. J. 2016, 283, 2653–2660. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, P.; Ling, T.; Wang, Y.; Dong, R.; Zhang, C.; Zhang, L.; Han, M.; Wang, D.; Wan, X.; et al. Certain (-)-epigallocatechin-3-gallate (EGCG) auto-oxidation products (EAOPs) retain the cytotoxic activities of EGCG. Food Chem. 2016, 204, 218–226. [Google Scholar] [CrossRef]
- Lambert, J.D.; Kennett, M.J.; Sang, S.; Reuhl, K.R.; Ju, J.; Yang, C.S. Hepatotoxicity of high oral dose (-)-epigallocatechin-3-gallate in mice. Food Chem. Toxicol. 2010, 48, 409–416. [Google Scholar] [CrossRef]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17, 7–12. [Google Scholar] [CrossRef]
- Yu, L.; Chen, X.; Wang, L.; Chen, S. The sweet trap in tumors: Aerobic glycolysis and potential targets for therapy. Oncotarget 2016, 7, 38908–38926. [Google Scholar] [CrossRef]
- Lee, J.H.; Liu, R.; Li, J.; Zhang, C.; Wang, Y.; Cai, Q.; Qian, X.; Xia, Y.; Zheng, Y.; Piao, Y.; et al. Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat. Commun. 2017, 8, e949. [Google Scholar] [CrossRef] [PubMed]
- Lang, L.; Chemmalakuzhy, R.; Shay, C.; Teng, Y. PFKP signaling at a glance: An emerging mediator of cancer cell metabolism. Adv. Exp. Med. Biol. 2019, 1134, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Azoitei, N.; Becher, A.; Steinestel, K.; Rouhi, A.; Diepold, K.; Genze, F.; Simmet, T.; Seufferlein, T. PKM2 promotes tumor angiogenesis by regulating HIF-1alpha through NF-kappaB activation. Mol. Cancer 2016, 15, e3. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huang, L.Q.; Mackenzie, G.G.; Rigas, B. Oxidative stress mediates through apoptosis the anticancer effect of phospho-nonsteroidal anti-Inflammatory drugs: Implications for the role of oxidative stress in the action of anticancer agents. J. Pharmacol. Exp. Ther. 2011, 338, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, C.; Kovacs, K.; Polgar, Z.; Regdon, Z.; Szabo, E.; Robaszkiewicz, A.; Forman, H.J.; Martner, A.; Virag, L. Redox control of cancer cell destruction. Redox. Biol. 2018, 16, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Qanungo, S.; Das, M.; Haldar, S.; Basu, A. Epigallocatechin-3-gallate induces mitochondrial membrane depolarization and caspase-dependent apoptosis in pancreatic cancer cells. Carcinogenesis 2005, 26, 958–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, R.; Penso, N.E.C.; Hackman, R.M.; Wang, Y.; Mackenzie, G.G. Epigallocatechin-3-gallate (EGCG) suppresses pancreatic cancer cell growth, invasion, and migration partly through the inhibition of Akt pathway and epithelial-mesenchymal transition: Enhanced efficacy when combined with gemcitabine. Nutrients 2019, 11, 1856. [Google Scholar] [CrossRef]
- Frese, K.K.; Neesse, A.; Cook, N.; Bapiro, T.E.; Lolkema, M.P.; Jodrell, D.I.; Tuveson, D.A. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012, 2, 260–269. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Shi, Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 2002, 9, 459–470. [Google Scholar] [CrossRef]
- Mallangada, N.A.; Vargas, J.M.; Thomas, S.; DiGiovanni, M.G.; Vaeth, B.M.; Nemesure, M.D.; Wang, R.X.; LaComb, J.F.; Williams, J.L.; Golub, L.M.; et al. A novel tricarbonylmethane agent (CMC2.24) reduces human pancreatic tumor growth in mice by targeting Ras. Mol. Carcinogen. 2018, 57, 1130–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, G.G.; Huang, L.; Alston, N.; Ouyang, N.; Vrankova, K.; Mattheolabakis, G.; Constantinides, P.P.; Rigas, B. Targeting mitochondrial STAT3 with the novel phospho-valproic acid (MDC-1112) inhibits pancreatic cancer growth in mice. PLoS ONE 2013, 8, e61532. [Google Scholar] [CrossRef] [PubMed]
- Bartels, L.E.; Mattheolabakis, G.; Vaeth, B.M.; LaComb, J.F.; Wang, R.; Zhi, J.; Komninou, D.; Rigas, B.; Mackenzie, G.G. The novel agent phospho-glycerol-ibuprofen-amide (MDC-330) inhibits glioblastoma growth in mice: An effect mediated by cyclin D1. Carcinogenesis 2016, 37, 420–429. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, R.; Hackman, R.M.; Wang, Y.; Mackenzie, G.G. Targeting Glycolysis with Epigallocatechin-3-Gallate Enhances the Efficacy of Chemotherapeutics in Pancreatic Cancer Cells and Xenografts. Cancers 2019, 11, 1496. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101496
Wei R, Hackman RM, Wang Y, Mackenzie GG. Targeting Glycolysis with Epigallocatechin-3-Gallate Enhances the Efficacy of Chemotherapeutics in Pancreatic Cancer Cells and Xenografts. Cancers. 2019; 11(10):1496. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101496
Chicago/Turabian StyleWei, Ran, Robert M. Hackman, Yuefei Wang, and Gerardo G. Mackenzie. 2019. "Targeting Glycolysis with Epigallocatechin-3-Gallate Enhances the Efficacy of Chemotherapeutics in Pancreatic Cancer Cells and Xenografts" Cancers 11, no. 10: 1496. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101496