Acute Promyelocytic Leukemia: Update on the Mechanisms of Leukemogenesis, Resistance and on Innovative Treatment Strategies

 ,
, 
Abstract
:
1. Introduction
2. APL Pathophysiology
3. Additional Genetic Events
4. Immunophenotypic Characteristics
5. Insight into the Mechanisms of Treatment Resistance in APL
6. Experimental Strategies for the Treatment of Resistant APL
7. Prophylaxis for Incidence of CNS Relapse
8. Conclusions and Future Perspectives
Reference
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Thé, H.; Chomienne, C.; Lanotte, M.; Degos, L.; Dejean, A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor α gene to a novel transcribed locus. Nature 1990, 347, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, M.; Zangrilli, D.; Pandolfi, P.P.; Longo, L.; Mencarelli, A.; Giacomucci, A.; Rocchi, M.; Biondi, A.; Rambaldi, A.; Lo Coco, F.; et al. Translocation breakpoint of acute promyelocytic leukemia lies within the retinoic acid receptor α locus. Proc. Natl. Acad. Sci. USA 1991, 88, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Grignani, F.; Ferrucci, P.F.; Testa, U.; Talamo, G.; Fagioli, M.; Alcalay, M.; Mencarelli, A.; Grignani, F.; Peschle, C.; Nicoletti, I.; et al. The acute promyelocytic leukemia-specific PML-RARα fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell 1993, 74, 423–431. [Google Scholar] [CrossRef]
- Di Masi, A.; Leboffe, L.; De Marinis, E.; Pagano, F.; Cicconi, L.; Rochette-Egly, C.; Lo-Coco, F.; Ascenzi, P.; Nervi, C. Retinoic acid receptors: From molecular mechanisms to cancer therapy. Mol. Aspects Med. 2015. [Google Scholar] [CrossRef]
- Hadjimichael, C.; Chanoumidou, K.; Nikolaou, C.; Klonizakis, A.; Theodosi, G.I.; Makatounakis, T.; Papamatheakis, J.; Kretsovali, A. Promyelocytic leukemia protein is an essential regulator of stem cell pluripotency and somatic cell reprogramming. Stem Cell Rep. 2017, 8, 1366–1378. [Google Scholar] [CrossRef]
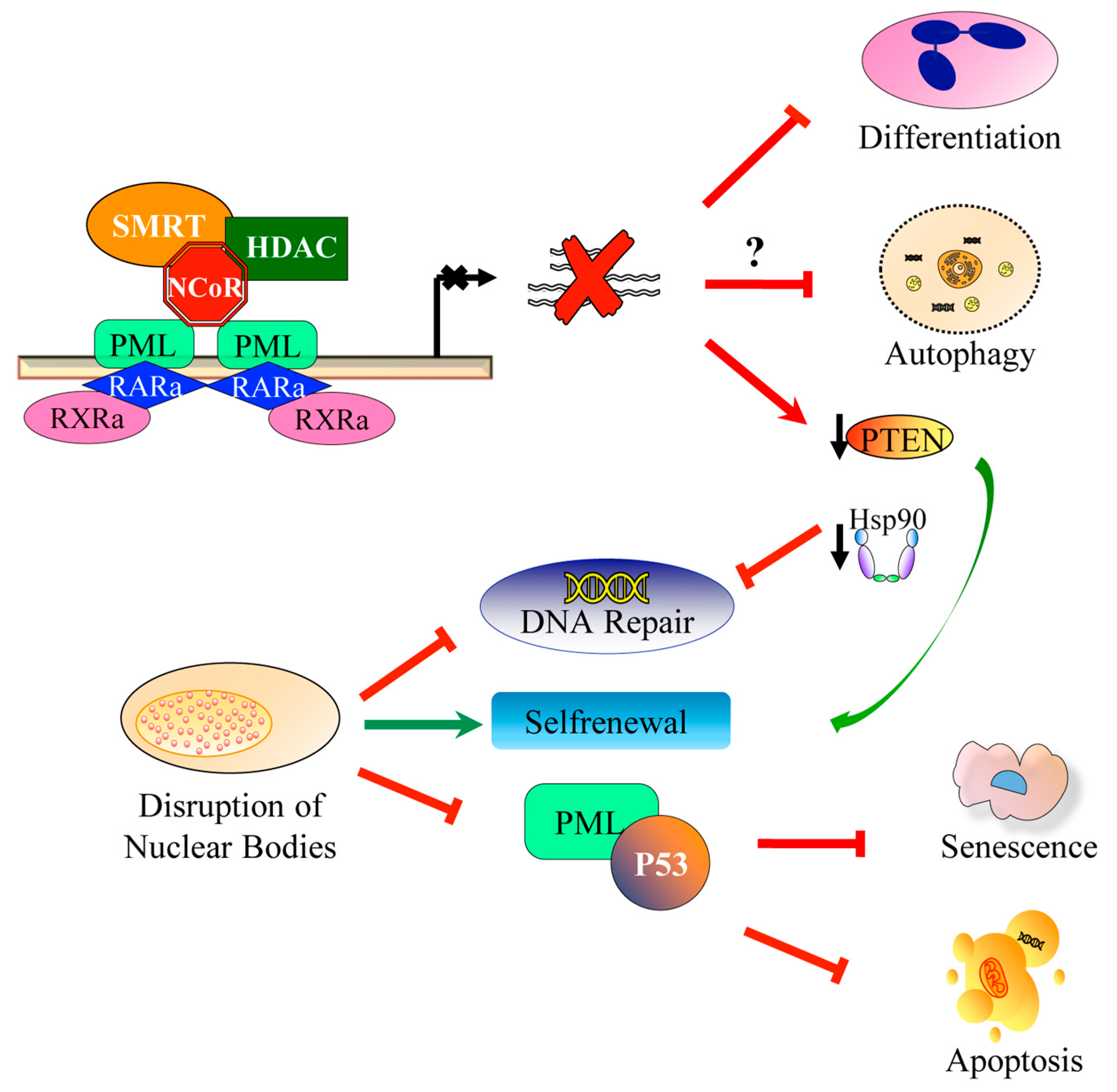
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010. [Google Scholar] [CrossRef]
- Grignani, F.; De Matteis, S.; Nervi, C.; Tomassoni, L.; Gelmetti, V.; Cioce, M.; Fanelli, M.; Ruthardt, M.; Ferrara, F.F.; Zamir, I.; et al. Fusion proteins of the retinoic acid receptor-α recruit histone deacetylase in promyelocytic leukaemia. Nature 1998, 391, 815–818. [Google Scholar] [CrossRef]
- Wang, K.; Wang, P.; Shi, J.; Zhu, X.; He, M.; Jia, X.; Yang, X.; Qiu, F.; Jin, W.; Qian, M.; et al. PML/RARα targets promoter regions containing PU.1 consensus and RARE half sites in acute promyelocytic leukemia. Cancer Cell 2010, 17, 186–197. [Google Scholar] [CrossRef]
- Martens, J.H.A.; Brinkman, A.B.; Simmer, F.; Francoijs, K.J.; Nebbioso, A.; Ferrara, F.; Altucci, L.; Stunnenberg, H.G. PML-RARα/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell 2010, 17, 173–185. [Google Scholar] [CrossRef]
- Fabiani, E.; Falconi, G.; Noguera, N.I.; Saulle, E.; Cicconi, L.; Divona, M.; Banella, C.; Picardi, A.; Cerio, A.M.; Boe, L.; et al. The forkhead box C1 (FOXC1) transcription factor is downregulated in acute promyelocytic leukemia. Oncotarget 2017, 8, 84074–84085. [Google Scholar] [CrossRef] [Green Version]
- Koken, M.H.; Puvion-Dutilleul, F.; Guillemin, M.C.; Viron, A.; Linares-Cruz, G.; Stuurman, N.; de Jong, L.; Szostecki, C.; Calvo, F.; Chomienne, C. The t(15;17) translocation alters a nuclear body in a retinoic acid-reversible fashion. EMBO J. 1994, 13, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Kogan, S.; Lagasse, E.; Weissman, I.; Alcalay, M.; Pelicci, P.G.; Atwater, S.; Bishop, J.M. A PMLRARα transgene initiates murine acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 1997, 94, 2551–2556. [Google Scholar] [CrossRef] [PubMed]
- Bischof, O.; Kirsh, O.; Pearson, M.; Itahana, K.; Pelicci, P.G.; Dejean, A. Deconstructing PML-induced premature senescence. EMBO J. 2002, 21, 3358–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottifredi, V.; Prives, C. P53 and PML: New partners in tumor suppression. Trends Cell Biol. 2001, 11, 184–187. [Google Scholar] [CrossRef]
- De Stanchina, E.; Querido, E.; Narita, M.; Davuluri, R.V.; Pandolfi, P.P.; Ferbeyre, G.; Lowe, S.W. PML is a direct p53 target that modulates p53 effector functions. Mol. Cell 2004, 13, 523–535. [Google Scholar] [CrossRef]
- Zhu, J.; Lallemand-Breitenbach, V.; De Thé, H. Pathways of retinoic acid- or arsenic- trioxide-induced PML/RARα catabolism, role of oncogene degradation in disease remission. Oncogene 2001, 20, 7257–7265. [Google Scholar] [CrossRef]
- Lo-Coco, F.; Hasan, S.K. Understanding the molecular pathogenesis of acute promyelocytic leukemia. Best Pract. Res. Clin. Haematol. 2014, 27, 3–9. [Google Scholar] [CrossRef]
- Di Masi, A.; Cilli, D.; Berardinelli, F.; Talarico, A.; Pallavicini, I.; Pennisi, R.; Leone, S.; Antoccia, A.; Noguera, N.I.; Lo-Coco, F.; et al. PML nuclear body disruption impairs DNA double-strand break sensing and repair in APL. Cell Death Dis. 2016. [Google Scholar] [CrossRef]
- Voisset, E.; Moravcsik, E.; Stratford, E.W.; Jaye, A.; Palgrave, C.J.; Hills, R.K.; Salomoni, P.; Kogan, S.C.; Solomon, E.; Grimwade, D. PML nuclear body disruption cooperates in APL pathogenesis and impairs DNA damage repair pathways in mice. Blood 2018, 131, 636–648. [Google Scholar] [CrossRef]
- Pennisi, R.; Ascenzi, P.; di Masi, A. Hsp90: A new player in DNA Repair? Biomolecules 2015, 5, 2589–2618. [Google Scholar] [CrossRef]
- Piredda, M.L.; Gaur, G.; Catalano, G.; Divona, M.; Banella, C.; Travaglini, S.; Puzzangara, M.C.; Voso, M.T.; Lo-Coco, F.; Noguera, N.I. PML/RARA inhibits expression of HSP90 and its target AKT. Br. J. Haematol. 2019, 184, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Bernardi, R.; Morotti, A.; Matsuoka, S.; Saglio, G.; Ikeda, Y.; Rosenblatt, J.; Avigan, D.E.; Teruya-Feldstein, J.; Pandolfi, P.P. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 2008, 453, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Regad, T.; Bellodi, C.; Nicotera, P.; Salomoni, P. The tumor suppressor Pml regulates cell fate in the developing neocortex. Nat. Neurosci. 2009, 12, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, Ö.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Grindley, J.C.; Yin, T.; Jayasinghe, S.; He, X.C.; Ross, J.T.; Haug, J.S.; Rupp, D.; Porter-Westpfahl, K.S.; Wiedemann, L.M.; et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006, 441, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Carracedo, A.; Egia, A.; Lo-Coco, F.; Teruya-Feldstein, J.; Pandolfi, P.P. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 2008, 455, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Noguera, N.I.; Song, M.S.; Divona, M.; Catalano, G.; Calvo, K.L.; García, F.; Ottone, T.; Florenzano, F.; Faraoni, I.; Battistini, L.; et al. Nucleophosmin/B26 regulates PTEN through interaction with HAUSP in acute myeloid leukemia. Leukemia 2013, 27, 1037–1043. [Google Scholar] [CrossRef]
- Orfali, N.; O’Donovan, T.R.; Nyhan, M.J.; Britschgi, A.; Tschan, M.P.; Cahill, M.R.; Mongan, N.P.; Gudas, L.J.; McKenna, S.L. Induction of autophagy is a key component of all-trans-retinoic acid-induced differentiation in leukemia cells and a potential target for pharmacologic modulation. Exp. Hematol. 2015, 43, 781–793. [Google Scholar] [CrossRef]
- Brigger, D.; Proikas-Cezanne, T.; Tschan, M.P. WIPI-Dependent autophagy during neutrophil differentiation of NB4 acute promyelocytic leukemia cells. Cell Death Dis. 2014. [Google Scholar] [CrossRef]
- Jin, J.; Britschgi, A.; Schläfli, A.M.; Humbert, M.; Shan-Krauer, D.; Batliner, J.; Federzoni, E.A.; Ernst, M.; Torbett, B.E.; Yousefi, S.; et al. Low autophagy (ATG) gene expression is associated with an immature AML blast cell phenotype and can be restored during AML differentiation therapy. Oxid. Med. Cell. Longev. 2018. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 11, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, M.A.; Djavaheri-Mergny, M. Autophagy: New Insights into mechanisms of action and resistance of treatment in acute promyelocytic leukemia. Int. J. Mol. Sci. 2019, 20, 3559. [Google Scholar] [CrossRef] [PubMed]
- Ronchini, C.; Brozzi, A.; Riva, L.; Luzi, L.; Gruszka, A.M.; Melloni, G.E.M.; Scanziani, E.; Dharmalingam, G.; Mutarelli, M.; Belcastro, V.; et al. PML-RARA-Associated cooperating mutations belong to a transcriptional network that is deregulated in myeloid leukemias. Leukemia 2017, 31, 1975–1986. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hou, J.K.; Chen, T.T.; Zhao, X.Y.; Yan, Z.W.; Zhang, J.; Yang, J.; Kogan, S.C.; Chen, G.Q. PML-RARα enhances constitutive autophagic activity through inhibiting the Akt/mTOR pathway. Autophagy 2011, 7, 1132–1144. [Google Scholar] [CrossRef]
- Yamamoto, J.F.; Goodman, M.T. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997–2002. Cancer Causes Control 2008, 19, 379–390. [Google Scholar] [CrossRef]
- Douer, D. The epidemiology of acute promyelocytic leukaemia. Best Pract. Res. Clin. Haematol. 2003, 16, 357–367. [Google Scholar] [CrossRef]
- Matasar, M.J.; Ritchie, E.K.; Consedine, N.; Magai, C.; Neugut, A.I. Incidence rates of acute promyelocytic leukemia among Hispanics, blacks, Asians, and non-Hispanic whites in the United States. Eur. J. Cancer Prev. 2006, 15, 367–370. [Google Scholar] [CrossRef]
- Kogan, S.C. Mouse models of acute promyelocytic leukemia. Curr. Top. Microbiol. Immunol. 2007, 313, 3–29. [Google Scholar]
- Wartman, L.D.; Larson, D.E.; Xiang, Z.; Ding, L.; Chen, K.; Lin, L.; Cahan, P.; Klco, J.M.; Welch, J.S.; Li, C.; et al. Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. J. Clin. Investig. 2011, 121, 1445–1455. [Google Scholar] [CrossRef] [Green Version]
- Agger, K.; Cloos, P.A.C.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef]
- Terashima, M.; Ishimura, A.; Yoshida, M.; Suzuki, Y.; Sugano, S.; Suzuki, T. The tumor suppressor Rb and its related Rbl2 genes are regulated by Utx histone demethylase. Biochem. Biophys. Res. Commun. 2010, 399, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.K.; Tsai, M.C.; Poulin, G.; Adler, A.S.; Chen, S.; Liu, H.; Shi, Y.; Chang, H.Y. The histone demethylase UTX enables RB-dependent cell fate control. Genes Dev. 2010, 24, 327–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herz, H.M.; Madden, L.D.; Chen, Z.; Bolduc, C.; Buff, E.; Gupta, R.; Davuluri, R.; Shilatifard, A.; Hariharan, I.K.; Bergmann, A. The H3K27me3 demethylase dUTX is a suppressor of notch- and rb-dependent tumors in drosophila. Mol. Cell. Biol. 2010, 30, 2485–2497. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Shyamsunder, P.; Han, L.; Mayakonda, A.; Nagata, Y.; Sundaresan, J.; Kanojia, D.; Yoshida, K.; Ganesan, S.; Hattori, N.; et al. Comprehensive mutational analysis of primary and relapse acute promyelocytic leukemia. Leukemia 2016, 30, 2430. [Google Scholar] [CrossRef]
- Yin, J.; Sun, A.N.; Tian, X.P.; Tian, H.; Wang, R.X.; Yang, Z.; Wang, X.L.; Wu, D.P.; Qiu, H.Y.; Pan, J.L.; et al. Clinical significance of common leukemia gene mutations in patients with acute promyelocytic leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2013. [Google Scholar] [CrossRef]
- Iaccarino, L.; Ottone, T.; Alfonso, V.; Cicconi, L.; Divona, M.; Lavorgna, S.; Travaglini, S.; Ferrantini, A.; Falconi, G.; Baer, C.; et al. Mutational landscape of patients with acute promyelocytic leukemia at diagnosis and relapse. Am. J. Hematol. 2019, 94, 1091–1097. [Google Scholar] [CrossRef]
- Calvo, K.L.; Ojeda, M.J.; Ammatuna, E.; Lavorgna, S.; Ottone, T.; Targovnik, H.M.; Lo-Coco, F.; Noguera, N.I. Detection of the nucleophosmin gene mutations in acute myelogenous leukemia through RT-PCR and polyacrylamide gel electrophoresis. Eur. J. Haematol. 2009, 82, 69–72. [Google Scholar] [CrossRef]
- Gaur, G.C.; Ramadan, S.M.; Cicconi, L.; Noguera, N.I.; Luna, I.; Such, E.; Lavorgna, S.; Di Giandomenico, J.; Sanz, M.A.; Lo-Coco, F. Analysis of mutational status, SNP rs16754, and expression levels of Wilms tumor 1 (WT1) gene in acute promyelocytic leukemia. Ann. Hematol. 2012, 91, 1855–1860. [Google Scholar] [CrossRef]
- Noguera, N.I.; Breccia, M.; Divona, M.; Diverio, D.; Costa, V.; De Santis, S.; Avvisati, G.; Pinazzi, M.B.; Petti, M.C.; Mandelli, F.; et al. Alterations of the FLT3 gene in acute promyelocytic leukemia: Association with diagnostic characteristics and analysis of clinical outcome in patients treated with the Italian AIDA protocol. Leukemia 2002, 16, 2185–2189. [Google Scholar] [CrossRef]
- Beitinjaneh, A.; Jang, S.; Roukoz, H.; Majhail, N.S. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations in acute promyelocytic leukemia: A systematic review. Leuk. Res. 2010, 34, 831–836. [Google Scholar] [CrossRef]
- Kelly, L.M.; Kutok, J.L.; Williams, I.R.; Boulton, C.L.; Amaral, S.M.; Curley, D.P.; Ley, T.J.; Gilliland, D.G. PML/RARα and FLT3-ITD induce an APL-like disease in a mouse model. Proc. Natl. Acad. Sci. USA 2002, 99, 8283–8288. [Google Scholar] [CrossRef] [PubMed]
- McCormack, E.; Bruserud, O.; Gjertsen, B.T. Review: Genetic models of acute myeloid leukaemia. Oncogene 2008, 27, 3765–3779. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Rahmé, R.; Rice, K.L.; Berthier, C.; Gaillard, C.; Quentin, S.; Maubert, A.L.; Kogan, S.; de Thé, H. FLT3-ITD impedes retinoic acid, but not arsenic, responses in murine acute promyelocytic leukemias. Blood 2019, 133, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Hecht, A.; Nowak, D.; Nowak, V.; Hanfstein, B.; Büchner, T.; Spiekermann, K.; Weiß, C.; Hofmann, W.K.; Lengfelder, E.; Nolte, F. A molecular risk score integrating BAALC, ERG and WT1 expression levels for risk stratification in acute promyelocytic leukemia. Leuk. Res. 2015. [Google Scholar] [CrossRef]
- Hecht, A.; Doll, S.; Altmann, H.; Nowak, D.; Lengfelder, E.; Röllig, C.; Ehninger, G.; Spiekermann, K.; Hiddemann, W.; Weiß, C.; et al. Validation of a molecular risk score for prognosis of patients with acute promyelocytic leukemia treated with all-trans retinoic acid and chemotherapy-containing regimens. Clin. Lymphoma Myeloma Leuk. 2017, 17, 889–896.e5. [Google Scholar] [CrossRef]
- Lucena-Araujo, A.R.; Coelho-Silva, J.L.; Pereira-Martins, D.A.; Silveira, D.R.; Koury, L.C.; Melo, R.A.M.; Bittencourt, R.; Pagnano, K.; Pasquini, R.; Nunes, E.C.; et al. Combining gene mutation with gene expression analysis improves outcomes prediction in acute promyelocytic leukemia. Blood 2019. [Google Scholar] [CrossRef]
- Paietta, E.; Goloubeva, O.; Neuberg, D.; Bennett, J.M.; Gallagher, R.; Racevskis, J.; Dewald, G.; Wiernik, P.H.; Tallman, M.S. A surrogate marker profile for PML/RAR? expressing acute promyelocytic leukemia and the association of immunophenotypic markers with morphologic and molecular subtypes. Cytometry 2004, 59, 1–9. [Google Scholar] [CrossRef]
- Paietta, E. Expression of cell-surface antigens in acute promyelocytic leukaemia. Best Pract. Res. Clin. Haematol. 2003, 16, 369–385. [Google Scholar] [CrossRef]
- Albano, F.; Mestice, A.; Pannunzio, A.; Lanza, F.; Martino, B.; Pastore, D.; Ferrara, F.; Carluccio, P.; Nobile, F.; Castoldi, G.; et al. The biological characteristics of CD34+ CD2+ adult acute promyelocytic leukemia and the CD34- CD2- hypergranular (M3) and microgranular (M3v) phenotypes. Haematologica 2006, 91, 311–316. [Google Scholar]
- Montesinos, P.; Rayón, C.; Vellenga, E.; Brunet, S.; González, J.; González, M.; Holowiecka, A.; Esteve, J.; Bergua, J.; González, J.D.; et al. Clinical significance of CD56 expression in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline-based regimens. Blood 2011, 117, 1799–1805. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.Y.; Li, Y.Y.; Li, C.W.; Wang, Y.S.; Liu, Y.; Wang, C.; Ru, K.; Mi, Y.C.; Wang, J.X.; Wang, H.J. Application of immunophenotypic analysis and molecular genetics in the diagnosis of acute promyelocytic leukemia. Zhonghua Xue Ye Xue Za Zhi 2019, 40, 288–293. [Google Scholar] [CrossRef] [PubMed]
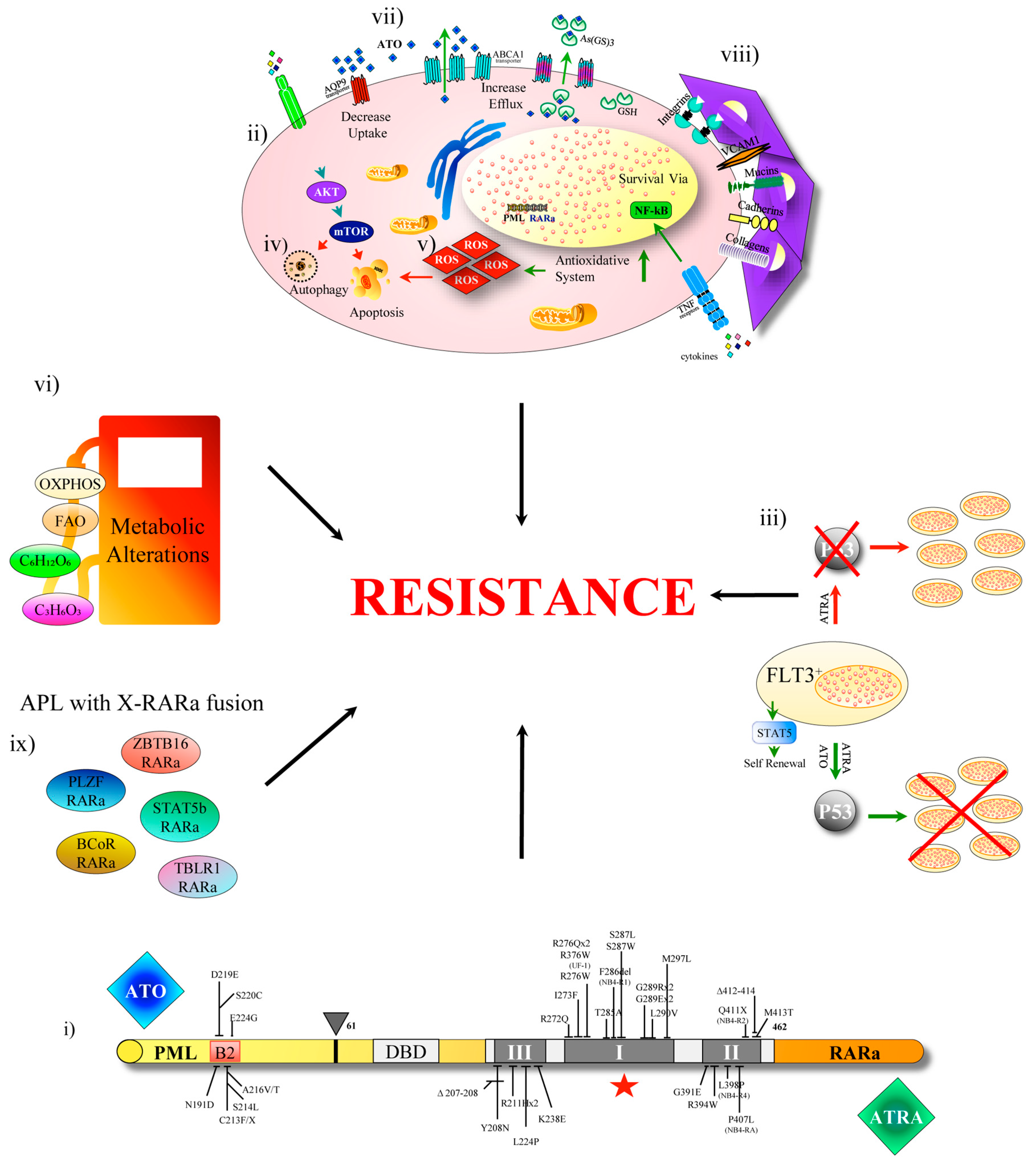
- Shao, W.; Benedetti, L.; Lamph, W.W.; Nervi, C.; Miller, W.H. A retinoid-resistant acute promyelocytic leukemia subclone expresses a dominant negative PML-RARα mutation. Blood 1997, 89, 4282–4289. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, R.E.; Moser, B.K.; Racevskis, J.; Poiré, X.; Bloomfield, C.D.; Carroll, A.J.; Ketterling, R.P.; Roulston, D.; Schachter-Tokarz, E.; Zhou, D.C.; et al. Treatment-influenced associations of PML-RARα mutations, FLT3 mutations, and additional chromosome abnormalities in relapsed acute promyelocytic leukemia. Blood 2012, 120, 2098–2108. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Che, J.; Bally, C.; Letouzé, E.; Berthier, C.; Yuan, H.; Jollivet, F.; Ades, L.; Cassinat, B.; Hirsch, P.; Pigneux, A.; et al. Dual origin of relapses in retinoic-Acid resistant acute promyelocytic leukemia. Nat. Commun. 2018, 9, 2407. [Google Scholar] [CrossRef] [PubMed]
- Jeanne, M.; Lallemand-Breitenbach, V.; Ferhi, O.; Koken, M.; Le Bras, M.; Duffort, S.; Peres, L.; Berthier, C.; Soilihi, H.; Raught, B.; et al. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell 2010, 18, 88–98. [Google Scholar] [CrossRef]
- Van Delft, F.W.; Horsley, S.; Colman, S.; Anderson, K.; Bateman, C.; Kempski, H.; Zuna, J.; Eckert, C.; Saha, V.; Kearney, L.; et al. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 2011, 117, 6247–6254. [Google Scholar] [CrossRef] [Green Version]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Bowen, D.; Kell, J.; Knapper, S.; Morgan, Y.G.; Lok, J.; Grech, A.; Jones, G.; et al. Arsenic trioxide and all-trans retinoic acid treatment for acute promyelocytic leukaemia in all risk groups (AML17): Results of a randomised, controlled, phase 3 trial. Lancet Oncol. 2015, 16, 1295–1305. [Google Scholar] [CrossRef]
- Isakson, P.; Bjørås, M.; Bøe, S.O.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010, 116, 2324–2331. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, L.; Kang, R.; Yang, M.; Liu, L.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARα oncoprotein. Autophagy 2011, 7, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Liu, J.; Jin, J.; Ni, W.; Xu, W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk. Res. 2007, 31, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Alex, A.A.; Chendamarai, E.; Ganesan, S.; Balasundaram, N.; Palani, H.K.; David, S.; Mathews, V. Arsenic trioxide resistance: More to it than mutations in PML-RARalpha. Blood 2014, 124, 3605. [Google Scholar] [CrossRef]
- Balasundaram, N.; Ganesan, S.; Palani, H.K.; Alex, A.A.; David, S.; Korula, A.; George, B.; Chomienne, C.; Balasubramanian, P.; Mathews, V. Metabolic Rewiring Drives Resistance to Arsenic Trioxide in Acute Promyelocytic Leukemia. Blood 2016, 128, 3956. [Google Scholar] [CrossRef]
- Chendamarai, E.; Ganesan, S.; Alex, A.A.; Kamath, V.; Nair, S.C.; Nellickal, A.J.; Janet, N.B.; Srivastava, V.; Lakshmi, K.M.; Viswabandya, A. Comparison of newly diagnosed and relapsed patients with acute promyelocytic leukemia treated with arsenic trioxide: Insight into mechanisms of resistanc. PLoS ONE 2015, 10, e0121912. [Google Scholar] [CrossRef]
- Takeshita, A.; Shinjo, K.; Naito, K.; Matsui, H.; Shigeno, K.; Nakamura, S.; Horii, T.; Maekawa, M.; Kitamura, K.; Naoe, T.; et al. P-glycoprotein (P-gp) and multidrug resistance-associated protein 1 (MRP1) are induced by arsenic trioxide (As2O3), but are not the main mechanism of As2O3-resistance in acute promyelocytic leukemia cells. Leukemia 2003. [Google Scholar] [CrossRef]
- Conserva, M.R.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. The pleiotropic role of retinoic acid/retinoic acid receptors signaling: from vitamin a metabolism to gene rearrangements in acute promyelocytic leukemia. Int. J. Mol. Sci. 2019, 20, 2921. [Google Scholar] [CrossRef]
- Hisada, K.; Hata, K.; Ichida, F.; Matsubara, T.; Orimo, H.; Nakano, T.; Yatani, H.; Nishimura, R.; Yoneda, T. Retinoic acid regulates commitment of undifferentiated mesenchymal stem cells into osteoblasts and adipocytes. J. Bone Miner. Metab. 2013, 31, 53–63. [Google Scholar] [CrossRef]
- Cao, J.; Ma, Y.; Yao, W.; Zhang, X.; Wu, D. Retinoids regulate adipogenesis involving the TGFβ/SMAD and Wnt/β-catenin pathways in human bone marrow mesenchymal stem cells. Int. J. Mol. Sci. 2017, 18, 842. [Google Scholar] [CrossRef]
- Su, M.; Alonso, S.; Jones, J.W.; Yu, J.; Kane, M.A.; Jones, R.J.; Ghiaur, G. All-trans retinoic acid activity in acute myeloid leukemia: Role of cytochrome P450 enzyme expression by the microenvironment. PLoS ONE 2015, 10, e0127790. [Google Scholar] [CrossRef]
- Wang, Y.T.; Chen, J.; Chang, C.W.; Jen, J.; Huang, T.Y.; Chen, C.M.; Shen, R.; Liang, S.Y.; Cheng, I.C.; Yang, S.C.; et al. Ubiquitination of tumor suppressor PML regulates prometastatic and immunosuppressive tumor microenvironment. J. Clin. Investig. 2017, 127, 2982–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesan, S.; Alex, A.A.; Chendamarai, E.; Balasundaram, N.; Palani, H.K.; David, S.; Kulkarni, U.; Aiyaz, M.; Mugasimangalam, R.; Korula, A.; et al. Rationale and efficacy of proteasome inhibitor combined with arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia 2016, 30, 2169–2178. [Google Scholar] [CrossRef] [PubMed]
- Sainty, D.; Liso, V.; Cantu-Rajnoldi, A.; Head, D.; Mozziconacci, M.J.; Arnoulet, C.; Benattar, L.; Fenu, S.; Mancini, M.; Duchayne, E.; et al. A new morphologic classification system for acute promyelocytic leukemia distinguishes cases with underlying PLZF/RARA gene rearrangements. Blood 2000, 96, 1287–1296. [Google Scholar] [PubMed]
- Wells, R.A.; Catzavelos, C.; Kamel-Reid, S. Fusion of retinoic acid receptor α to NuMA, the nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat. Genet. 1997, 17, 109–113. [Google Scholar] [CrossRef]
- Arnould, C.; Philippe, C.; Bourdon, V.; Grégoire, M.J.; Berger, R.; Jonveaux, P. The signal transducer and activator of transcription STAT5b gene is a new partner of retinoic acid receptor α in acute promyelocytic-like leukaemia. Hum. Mol. Genet. 1999, 8, 1741–1749. [Google Scholar] [CrossRef]
- Kondo, T.; Mori, A.; Darmanin, S.; Hashino, S.; Tanaka, J.; Asaka, M. The seventh pathogenic fusion gene FIP1L1-RARA was isolated from a t(4;17)-positive acute promyelocytic leukemia. Haematologica 2008, 93, 1414–1416. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.J.; Lu, X.; Zeisig, B.B.; Ma, Z.; Cai, X.; Chen, S.; Gronemeyer, H.; Tweardy, D.J.; So, C.W.E.; Dong, S. Leukemic transformation by the APL fusion protein PRKAR1A-RARα critically depends on recruitment of RXRα. Blood 2010, 115, 643–652. [Google Scholar] [CrossRef]
- Adams, J.; Nassiri, M. Acute promyelocytic leukemia a review and discussion of variant translocations. Arch. Pathol. Lab. Med. 2015, 139, 1308–1313. [Google Scholar] [CrossRef]
- Ichikawa, S.; Ichikawa, S.; Ishikawa, I.; Takahashi, T.; Fujiwara, T.; Harigae, H. Successful treatment of acute promyelocytic leukemia with a t(X;17)(p11.4;q21) and BCOR-RARA fusion gene. Cancer Genet. 2015, 208, 162–163. [Google Scholar] [CrossRef]
- Won, D.; Shin, S.Y.; Park, C.J.; Jang, S.; Chi, H.S.; Lee, K.H.; Lee, J.O.; Seo, E.J. OBFC2A/RARA: A novel fusion gene in variant acute promyelocytic leukemia. Blood 2013, 121, 1432–1435. [Google Scholar] [CrossRef]
- Chen, Y.; Li, S.; Zhou, C.; Li, C.; Ru, K.; Rao, Q.; Xing, H.; Tian, Z.; Tang, K.; Mi, Y.; et al. TBLR1 fuses to retinoid acid receptor α in a variant t(3;17)(q26;q21) translocation of acute promyelocytic leukemia. Blood 2014, 124, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhong, H.Y.; Zhang, Y.; Xiao, L.; Bai, L.H.; Liu, S.F.; Zhou, G.B.; Zhang, G. Sen GTF2I-RARA is a novel fusion transcript in a t(7;17) variant of acute promyelocytic leukaemia with clinical resistance to retinoic acid. Br. J. Haematol. 2015, 168, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.C.; Jain, N.; Mehrotra, M.; Zhang, J.; Protopopov, A.; Zuo, Z.; Pemmaraju, N.; DiNardo, C.; Hirsch-Ginsberg, C.; Wang, S.A.; et al. Identification of a novel fusion gene, IRF2BP2-RARA, in acute promyelocytic leukemia. J. Natl. Compr. Cancer Netw. 2015, 13, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Wang, A.Z.; Wong, T.H.Y.; Wan, T.S.K.; Cheung, J.S.; Raghupathy, R.; Chan, N.P.H.; Ng, M.H.L. To the editor: FNDC3B is another novel partner fused to RARA in the t(3;17)(q26;q21) variant of acute promyelocytic leukemia. Blood 2017, 129, 2705–2709. [Google Scholar] [CrossRef] [PubMed]
- Guidez, F.; Parks, S.; Wong, H.; Jovanovic, J.V.; Mays, A.; Gilkes, A.F.; Mills, K.I.; Guillemin, M.C.; Hobbs, R.M.; Pandolfi, P.P.; et al. RARα-PLZF overcomes PLZF-mediated repression of CRABPI, contributing to retinoid resistance in t(11;17) acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2007, 104, 18694–18699. [Google Scholar] [CrossRef]
- Breccia, M.; Diverio, D.; Noguera, N.I.; Visani, G.; Santoro, A.; Locatelli, F.; Damiani, D.; Marmont, F.; Vignetti, M.; Petti, M.C.; et al. Clinico-biological features and outcome of acute promyelocytic leukemia patients with persistent polymerase chain reaction-detectable disease after AIDA front-line induction and consolidation therapy. Haematologica 2004, 89, 29–33. [Google Scholar] [PubMed]
- Sanz, M.A.; Lo-Coco, F. Modern approaches to treating acute promyelocytic leukemia. J. Clin. Oncol. 2011, 29, 495–503. [Google Scholar] [CrossRef]
- Montesinos, P.; González, J.D.; González, J.; Rayón, C.; De Lisa, E.; Amigo, M.L.; Ossenkoppele, G.J.; Peñarrubia, M.J.; Pérez-Encinas, M.; Bergua, J.; et al. Therapy-related myeloid neoplasms in patients with acute promyelocytic leukemia treated with all-trans-retinoic acid and anthracycline-based chemotherapy. J. Clin. Oncol. 2010, 28, 3872–3879. [Google Scholar] [CrossRef]
- Fenaux, P.; Wang, Z.Z.; Degos, L. Treatment of acute promyelocytic leukemia by retinoids. Curr. Top. Microbiol. Immunol. 2007, 313, 101–128. [Google Scholar]
- De Thé, H.; Pandolfi, P.P.; Chen, Z. Acute promyelocytic leukemia: A paradigm for oncoprotein-targeted cure. Cancer Cell 2017, 32, 552–560. [Google Scholar] [CrossRef]
- Watts, J.M.; Tallman, M.S. Acute promyelocytic leukemia: What is the new standard of care? Blood Rev. 2014, 28, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, A.; Asou, N.; Atsuta, Y.; Sakura, T.; Ueda, Y.; Sawa, M.; Dobashi, N.; Taniguchi, Y.; Suzuki, R.; Nakagawa, M.; et al. Tamibarotene maintenance improved relapse-free survival of acute promyelocytic leukemia: A final result of prospective, randomized, JALSG-APL204 study. Leukemia 2019, 33, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.C.; Kim, S.H.; Ding, W.; Schultz, C.; Warrell, R.P.; Gallagher, R.E. Frequent mutations in the ligand-binding domain of PML-RARα after multiple relapses of acute promyelocytic leukemia: Analysis for functional relationship to response to all-trans retinoic acid and histone deacetylase inhibitors in vitro and in vivo. Blood 2002, 99, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Lo-Coco, F.; Cimino, G.; Breccia, M.; Noguera, N.I.; Diverio, D.; Finolezzi, E.; Pogliani, E.M.; Di Bona, E.; Micalizzi, C.; Kropp, M.; et al. Gemtuzumab ozogamicin (Mylotarg) as a single agent for molecularly relapsed acute promyelocytic leukemia. Blood 2004, 104, 1995–1999. [Google Scholar] [CrossRef] [PubMed]
- Gale, R.E.; Hills, R.; Pizzey, A.R.; Kottaridis, P.D.; Swirsky, D.; Gilkes, A.F.; Nugent, E.; Mills, K.I.; Wheatley, K.; Solomon, E.; et al. Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood 2005, 106, 3768–3776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Coco, F.; Ammatuna, E.; Noguera, N. Treatment of acute promyelocytic leukemia with gemtuzumab ozogamicin. Clin. Adv. Hematol. Oncol. 2006, 4, 57–62, 76–77. [Google Scholar]
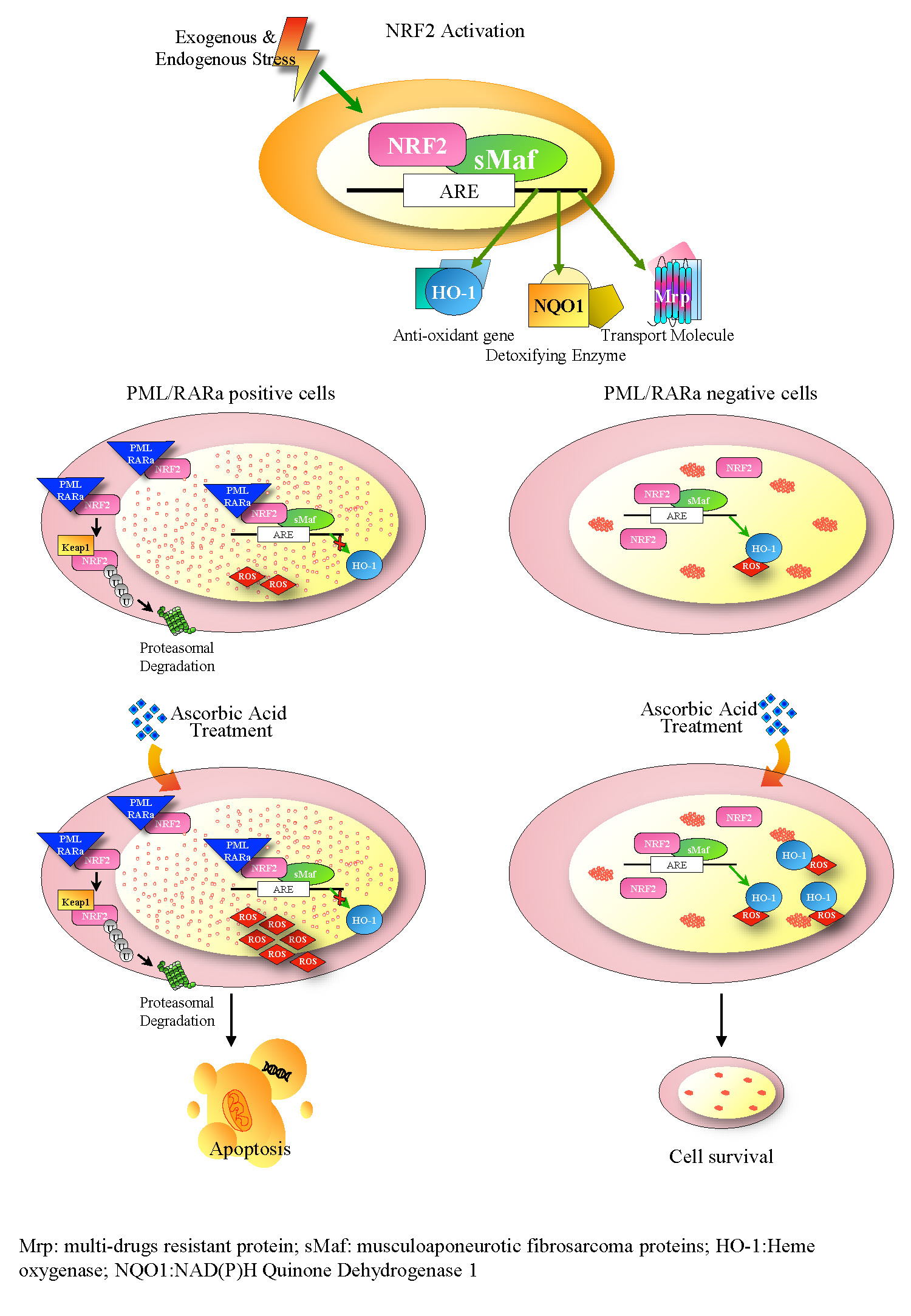
- Noguera, N.I.; Pelosi, E.; Angelini, D.F.; Piredda, M.L.; Guerrera, G.; Piras, E.; Battistini, L.; Massai, L.; Berardi, A.; Catalano, G.; et al. High-dose ascorbate and arsenic trioxide selectively kill acute myeloid leukemia and acute promyelocytic leukemia blasts in vitro. Oncotarget 2017, 8, 32550–32565. [Google Scholar] [CrossRef]
- Mastrangelo, D.; Massai, L.; Lo Coco, F.; Noguera, N.I.; Borgia, L.; Fioritoni, G.; Berardi, A.; Iacone, A.; Muscettola, M.; Pelosi, E.; et al. Cytotoxic effects of high concentrations of sodium ascorbate on human myeloid cell lines. Ann. Hematol. 2015, 94, 1807–1816. [Google Scholar] [CrossRef]
- Bernardini, S.; Nuccetelli, M.; Noguera, N.I.; Bellincampi, L.; Lunghi, P.; Bonati, A.; Mann, K.; Miller, W.H., Jr.; Federici, G.; Lo Coco, F. Role of GSTP1-1 in mediating the effect of As2O3 in the acute promyelocytic leukemia cell line NB4. Ann. Hematol. 2006, 85, 681–687. [Google Scholar] [CrossRef]
- De Thé, H.; Le Bras, M.; Lallemand-Breitenbach, V. Acute promyelocytic leukemia, arsenic, and PML bodies. J. Cell Biol. 2012, 198, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Mastrangelo, D.; Massai, L.; Fioritoni, G.; Coco, F.L.; Noguera, N.; Testa, U. High doses of vitamin C and leukemia: In vitro update. In Myeloid Leukemia; Lasfar, A., Ed.; IntechOpen: London, UK, 2018. [Google Scholar]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [PubMed]
- Masciarelli, S.; Capuano, E.; Ottone, T.; Divona, M.; De Panfilis, S.; Banella, C.; Noguera, N.I.; Picardi, A.; Fontemaggi, G.; Blandino, G.; et al. Retinoic acid and arsenic trioxide sensitize acute promyelocytic leukemia cells to ER stress. Leukemia 2018, 32, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.M.; Wu, Y.L.; Zhou, M.Y.; Liu, C.X.; Xu, H.Z.; Yan, H.; Zhao, Y.; Huang, Y.; Sun, H.D.; Chen, G.Q. Pharicin B stabilizes retinoic acid receptor-α and presents synergistic differentiation induction with ATRA in myeloid leukemic cells. Blood 2010, 116, 5289–5297. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, Q.; Lv, F.; Liu, N.; Xu, Y.; Liu, M.; Chen, Y.; Yi, Z. LG-362B targets PML-RARα and blocks ATRA resistance of acute promyelocytic leukemia. Leukemia 2016, 30, 1465–1474. [Google Scholar] [CrossRef]
- Calvo, K.L.; Ronco, M.T.; Noguera, N.I.; García, F. Benznidazole modulates cell proliferation in acute leukemia cells. Immunopharmacol. Immunotoxicol. 2013, 35, 478–486. [Google Scholar] [CrossRef]
- Ying, L.; Jin-Song, Y.; Li, X.; Kang Qin, Q.-Q.Y.; Hong-Tao, X.; Meng-Qing, G.; Xiao-Ning, Q.; Yu-Ting, S.; Guo-Qiang, C. 2-Bromopalmitate targets retinoic acid receptor alpha and overcomes all-trans retinoic acid resistance of acute promyelocytic leukemia. Haematologica 2019, 104, 102–112. [Google Scholar] [CrossRef]
- Hussain, L.; Maimaitiyiming, Y.; Su, L.D.; Wang, Q.Q.; Naranmandura, H. Phenylarsine oxide can induce degradation of PLZF-RARα variant fusion protein of acute promyelocytic leukemia. Chem. Res. Toxicol. 2019, 32, 548–550. [Google Scholar] [CrossRef]
- Furuya, A.; Kawahara, M.; Kumode, M.; Ohira, Y.; Usui, A.; Nagai, S.; Hosoba, S.; Minamiguchi, H.; Kito, K.; Andoh, A. Central nervous system involvement of acute promyelocytic leukemia, three case reports. Clin. Case Rep. 2017, 5, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Montesinos, P.; Díaz-Mediavilla, J.; Debén, G.; Prates, V.; Tormo, M.; Rubio, V.; Pérez, I.; Fernández, I.; Viguria, M.; Rayón, C.; et al. Central nervous system involvement at first relapse in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline monochemotherapy without intrathecal prophylaxis. Haematologica 2009. [Google Scholar] [CrossRef]
- Sanz, M.A.; Grimwade, D.; Tallman, M.S.; Lowenberg, B.; Fenaux, P.; Estey, E.H.; Naoe, T.; Lengfelder, E.; Büchner, T.; Döhner, H.; et al. Management of acute promyelocytic leukemia: Recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009, 113, 1875–1891. [Google Scholar] [CrossRef] [PubMed]
- Specchia, G.; Lo Coco, F.; Vignetti, M.; Avvisati, G.; Fazi, P.; Albano, F.; Di Raimondo, F.; Martino, B.; Ferrara, F.; Selleri, C.; et al. Extramedullary involvement at relapse in acute promyelocytic leukemia patients treated or not with all-trans retinoic acid: A report by the Gruppo Italiano Malattie Ematologiche dell’Adulto. J. Clin. Oncol. 2001, 19, 4023–4028. [Google Scholar] [CrossRef] [PubMed]
- Abaza, Y.; Kantarjian, H.; Garcia-Manero, G.; Estey, E.; Borthakur, G.; Jabbour, E.; Faderl, S.; O’Brien, S.; Wierda, W.; Pierce, S.; et al. Long-term outcome of acute promyelocytic leukemia treated with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab. Blood 2017, 129, 1275–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iland, H.J.; Collins, M.; Bradstock, K.; Supple, S.G.; Catalano, A.; Hertzberg, M.; Browett, P.; Grigg, A.; Firkin, F.; Campbell, L.J.; et al. Use of arsenic trioxide in remission induction and consolidation therapy for acute promyelocytic leukaemia in the Australasian Leukaemia and Lymphoma Group (ALLG) APML4 study: A non-randomised phase 2 trial. Lancet Haematol. 2015, 2, e357–e366. [Google Scholar] [CrossRef]
- Osman, A.E.G.; Anderson, J.; Churpek, J.E.; Christ, T.N.; Curran, E.; Godley, L.A.; Liu, H.; Thirman, M.J.; Odenike, T.; Stock, W.; et al. Treatment of acute promyelocytic leukemia in adults. J. Oncol. Pract. 2018, 14, 649–657. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Author | Source | Number of Samples | Method | Molecular Alterations | Ref | ||
|---|---|---|---|---|---|---|---|
| Dx | Relapse | Dx | Relapse | ||||
| Madan, et al | Human | 163 | 69 | WGS | FLT3 (43%), WT1 (14%), NRAS (10%) and KRAS (4%), ARID1A (5%), ARID1B (3%), LRP1 (3%) | PML(17%), RARA (10), FLT3-ITD (25%), WT1 (18%), ARID 1B (12%) RUNX1 (5%), FLT3 (5%), NRAS (5%), ARID1B(5%), NRAS (5%), ETV6 (4%), FANCA (3%), TP53 (3%), LRP1 (3%), KMT2C (3%) | [44] |
| Yin J, et al | Human | 84 | - | Genomic DNA-PCR | FLT3-ITD (27%), WT1 (14%), FLT3-TKD (10%), TET2 (8%), N-RAS (6%), ASXL1 (5%), EZH2 (2%), MLL-PTD (1%), IDH1 (1%) and CBL (1%) | - | [45] |
| Iaccarino, et al | Human | 33 | 31 | NGS (31-gene panel) | FLT3-ITD (34%), WT1 (20%), NRAS (7%), RUNX1 (5%), FLT3-TKD (9%), DNMT3A (5%), ETV6 (2%), MYC (2%), SETBP1 (2%), SF3B1 (5%), TET2 (%) | WT1 (13%), FLT3-ITD (10%), DNMT3A (10%), ETV6 (10%), FLT3-TKD (6%), TET2 (6%), ASXL1 (3%), JAK2 (3%), RUNX1 (3%) SRSSF2 (3%), TP53 (3%), U2AF1 (3%), PML (19%), RARa (10%) | [46] |
| Gaur, et al | Human | 103 | - | DNA Sequencing (Ex 7-8) | WT1 (4%) | - | [48] |
| Wartman, et al | Mouse model | - | - | NGS | Jak1 V657F or V658F and Kdm6a | - | [39] |
| Author | Drug | Function | Study | Source | Result | Follow up | p | Ref |
|---|---|---|---|---|---|---|---|---|
| Takeshita, et al. | Tamibarotene (TAM) | RAR α agonist | Clinical trial | 270 Patients | RFS %: TAM 94; ATRA 84 | 7- Year | 0.027 | [102] |
| Lo Coco, et al | Gentuzumab Ozogamicin | Anti CD33 + Calicamicin | Prospective Study | 16 Patients Relapse | RFS % 43 ± 15% | 31 month | - | [104] |
| Gale, et al | CEP-701 (Lestaurtinib) | FLT3 inhibitor | In vitro | Primary APL blast (n = 6) | Greater effect on cell survival/proliferation in FLT3/ITD cells, but this inhibition was reduced in the presence of ATRA | - | - | [105] |
| Mastrangelo, et al | Ascorbate Megadose | Pro-oxidant, | In vitro | Cells Lines (n = 6) | Highly sensitive, with an average 50 % lethal concentration (LC50) of 3 mM Normal CD34+ not sensitive | - | - | [108] |
| Noguera, et al | Ascorbate Megadose | Pro-oxidant, | In vitro | Primary APL (n = 9) and AML (n = 33) Blast; Cells Lines (n = 5) | Higer sensitivity ASC induce PML/RARa and PML degradation ASC potenciate the effect of ATO Normal CD34+ not sensitive | - | < 0.001 | [107] |
| Masciarelli, et al | Tunicamycin | Endoplasmic reticulum (ER) stress-inducing drug | In vitro | Primary APL Blast; ATRA sensitive and resistant APL cell lines | ER stress + ATO induced apoptosis in RA-sensitive an RA-resistant APL cell lines | - | < 0.005 | [114] |
| Gu, et al | pharicin B, | stabilizes RARα protein | In vitro | Primary APL Blast; ATRA sensitive and resistant APL cell lines | Induced apoptosis in RA-sensitive and RA-resistant APL cell lines | - | < 0.001 | [115] |
| Wang, et al | LG-362B, | caspases-mediated degradation of PML-RARα | In vitro e in vivo | Primary APL Blast; ATRA sensitive and resistant APL cell lines Murin models | Inhibits the proliferation of APL in vitro and in vivo Synergistic or additive differentiation effect with ATRA Overcom ATRA resistance | - | RTW: < 0.01 | [116] |
| Ying, et al | 2-bromopalmitate (2-Br) | inhibitor of fatty acid oxidation | In vitro | Primary APL Blast; ATRA sensitive and resistant APL cell lines Murin models | ATRA + 2Br to overcoming ATRA resistance | - | Blast: < 0.05 to < 0.001 (n = 7); > 0.05 (n = 4) RTW: < 0.05 | [118] |
| Ganesan et al | ATO plus Bortezomib | downregulation of the NFĸB pathway, PML-RARa degradation inhibition of the proteasome by bortezomib | In vitro e in vivo | ATO sensitive and resistant APL cell lines Murin models | Synergistic effect in both ATO sensitive and ATO resistant APL cell lines Reduce leukemic burden and induce long-term survival in an APL mouse model | - | OS mouse: 0.0001 | [82] |
| Hussain et al | phenylarsine oxide (PAO) | organic arsenic derivatives | In vitro | Cells Lines transfected with PLZF-RARa | PLZF-RARa degradation | - | - | [119] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noguera, N.I.; Catalano, G.; Banella, C.; Divona, M.; Faraoni, I.; Ottone, T.; Arcese, W.; Voso, M.T. Acute Promyelocytic Leukemia: Update on the Mechanisms of Leukemogenesis, Resistance and on Innovative Treatment Strategies. Cancers 2019, 11, 1591. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101591
Noguera NI, Catalano G, Banella C, Divona M, Faraoni I, Ottone T, Arcese W, Voso MT. Acute Promyelocytic Leukemia: Update on the Mechanisms of Leukemogenesis, Resistance and on Innovative Treatment Strategies. Cancers. 2019; 11(10):1591. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101591
Chicago/Turabian StyleNoguera, N. I., G. Catalano, C. Banella, M. Divona, I. Faraoni, T. Ottone, W. Arcese, and M. T. Voso. 2019. "Acute Promyelocytic Leukemia: Update on the Mechanisms of Leukemogenesis, Resistance and on Innovative Treatment Strategies" Cancers 11, no. 10: 1591. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101591