Functionalized Tobacco Mosaic Virus Coat Protein Monomers and Oligomers as Nanocarriers for Anti-Cancer Peptides


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
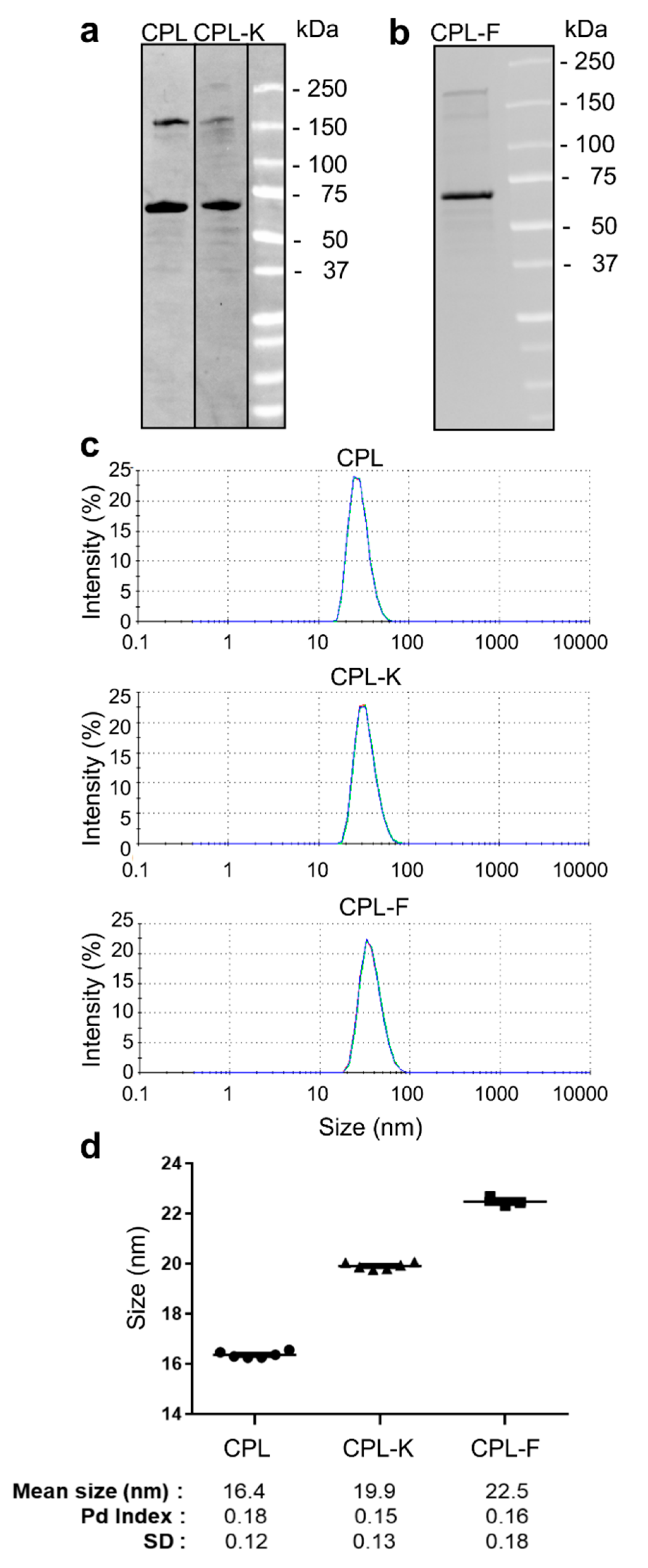
2.1. Characterization of Coat Protein (CP) Fusion Proteins Produced in Bacteria
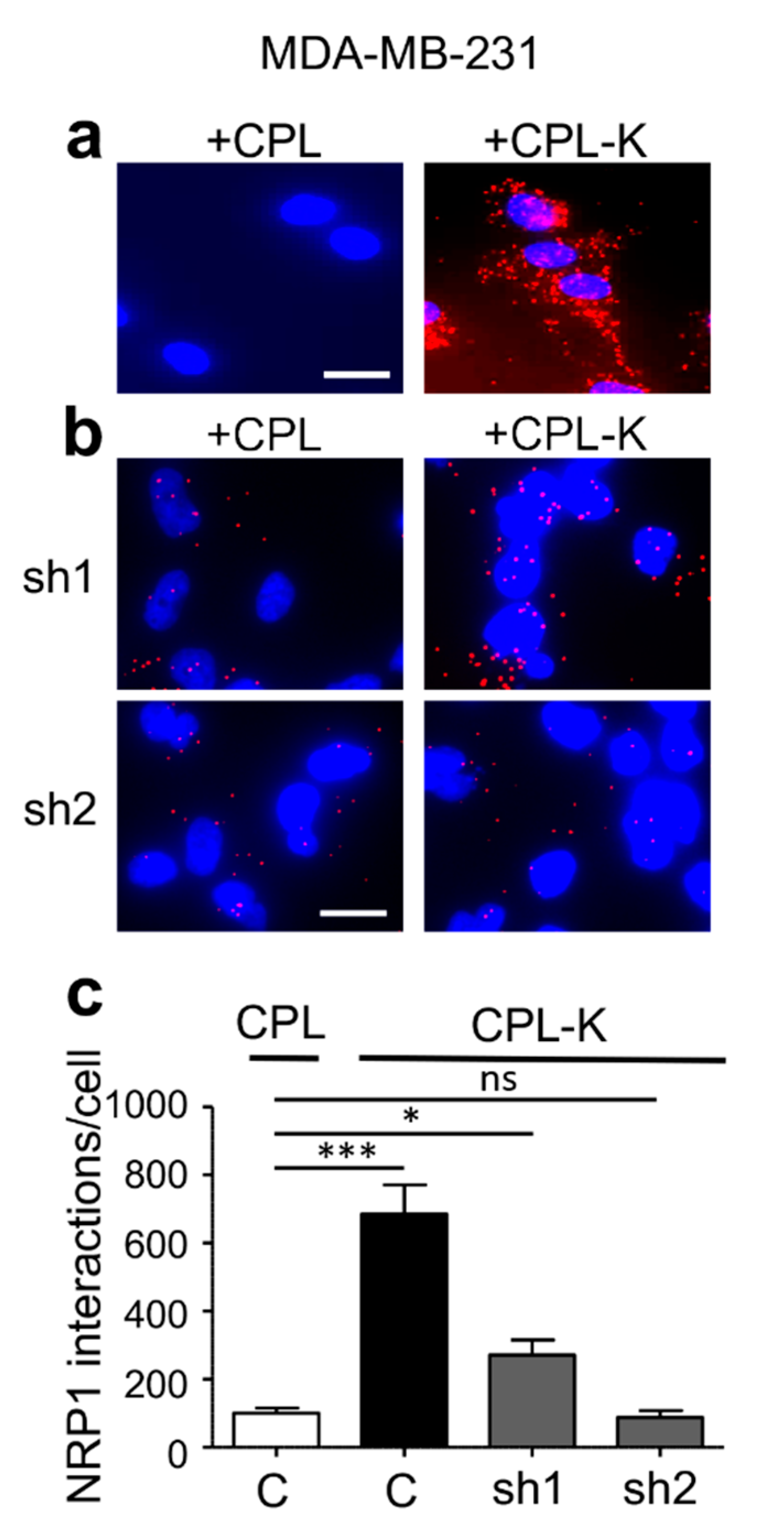
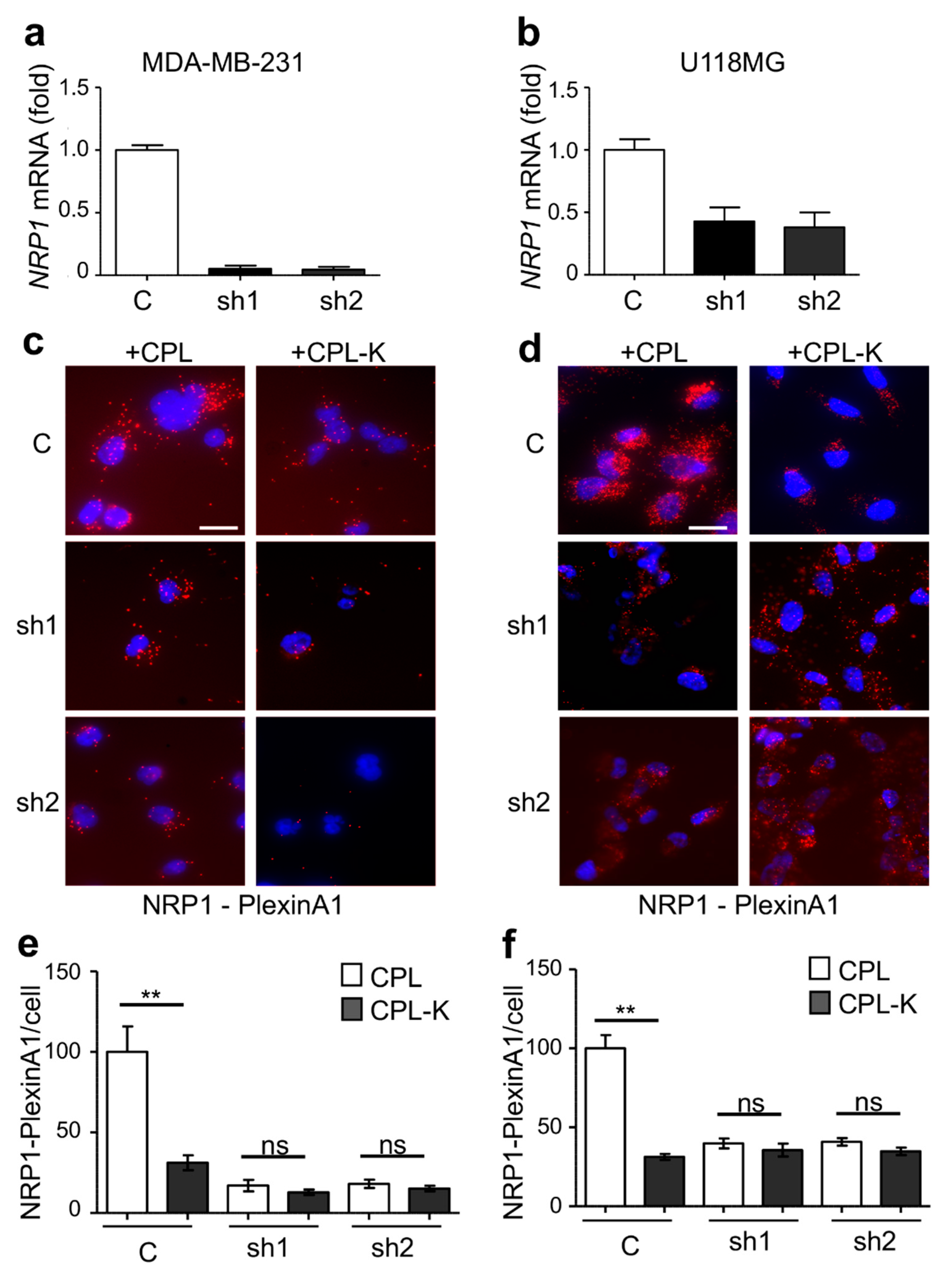
2.2. CPL-K Interacts with NRP1 and Inhibits NRP1 Binding to Plexin A1
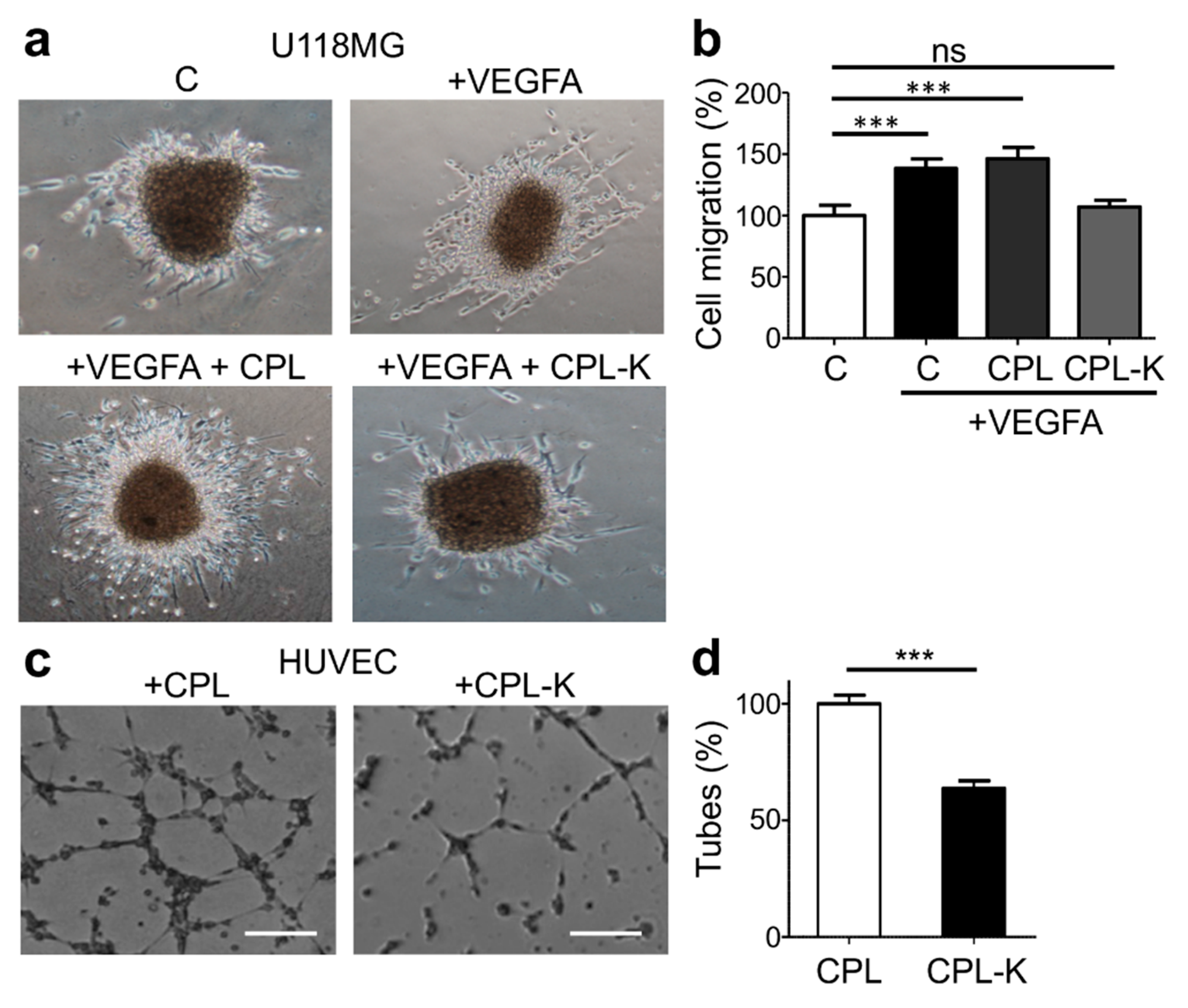
2.3. CPL-K Inhibits VEGFA-Induced Tumor Cell Migration and Human Umbilical Vein Endothelial Cell (HUVEC) Tubulogenesis
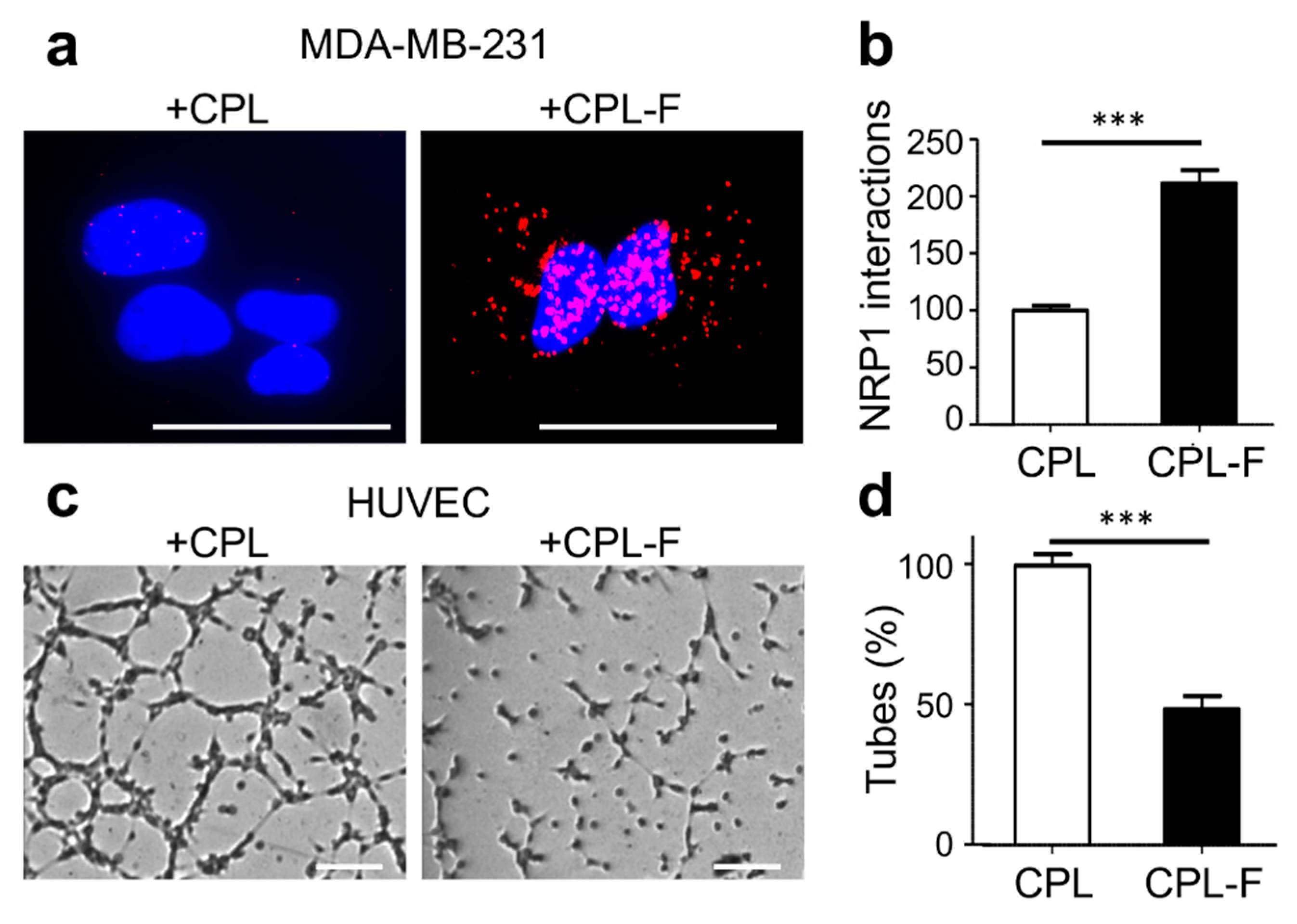
2.4. CPL-F Binds to NRP1 and Inhibits HUVEC Tubulogenesis
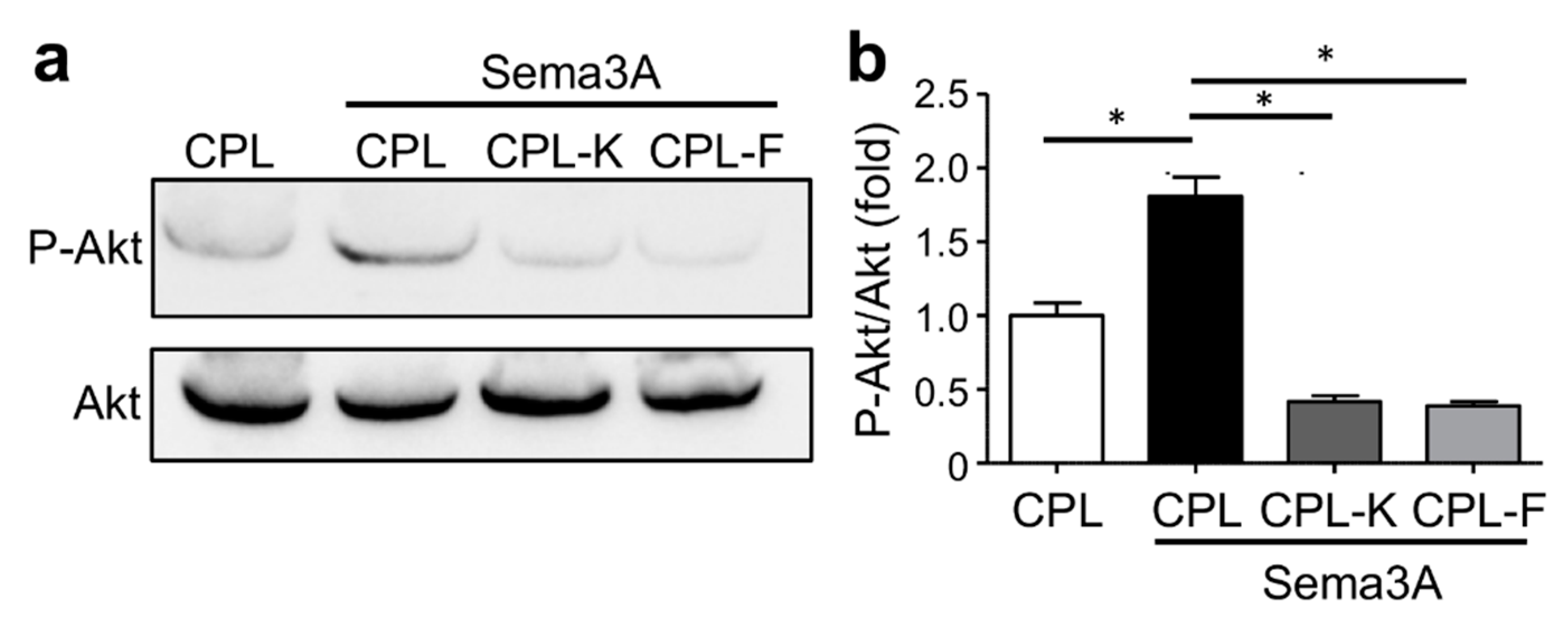
2.5. CPL-K and CPL-F Inhibit NRP1-Dependent Sema3A-Induced Downstream Signaling
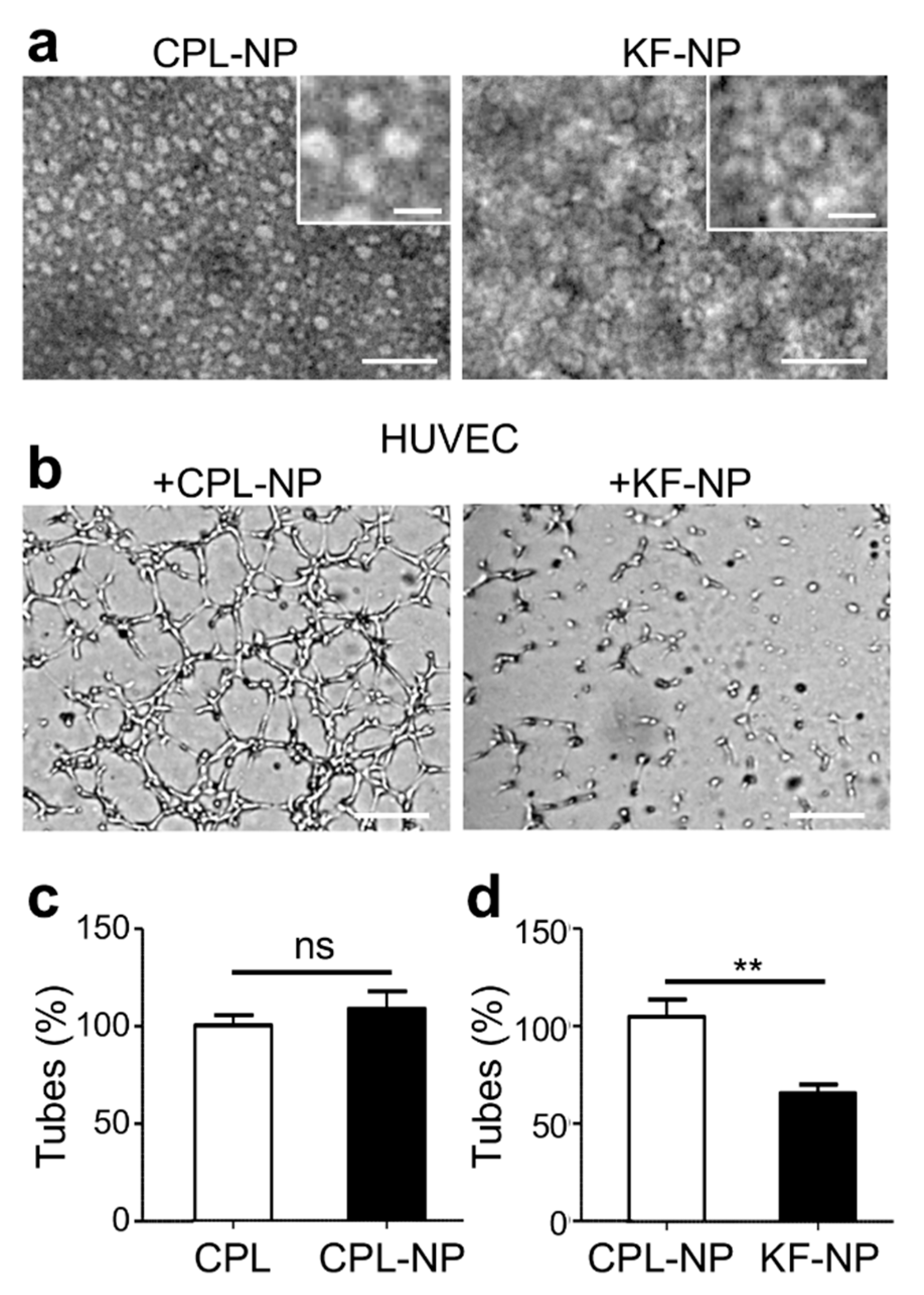
2.6. A Nanoparticle Formulation of CPL/CPL-K/CPL-F Inhibits HUVEC Tubulogenesis
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Plasmids
4.3. Protein Expression and Purification
4.4. Western Blot for Detection of Akt Phosphorylation
4.5. Dynamic Light-Scattering (DLS) Analysis
4.6. Proximity Ligation Assay (PLA)
4.7. Cell Migration Assay
4.8. Angiogenesis Assay
4.9. Disk Assembly
4.10. Transmission Electron Microscopy (TEM) Imaging
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Emerich, D.F.; Thanos, C.G. Targeted nanoparticle-based drug delivery and diagnosis. J. Drug Target. 2007, 15, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Miller, G. Biomedicine. Nanoparticle treatment reverses cerebral palsy in rabbits. Science 2012, 336, 286. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Deng, H.; Peng, H.; Wang, Q. Functional nanoparticles in targeting glioma diagnosis and therapies. J. Nanosci. Nanotechnol. 2014, 14, 415–432. [Google Scholar] [CrossRef] [PubMed]
- Saadeh, Y.; Leung, T.; Vyas, A.; Chaturvedi, L.S.; Perumal, O.; Vyas, D. Applications of nanomedicine in breast cancer detection, imaging, and therapy. J. Nanosci. Nanotechnol. 2014, 14, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Nan, A. Combination drug delivery approaches in metastatic breast cancer. J. Drug Deliv. 2012, 2012, 915375. [Google Scholar] [CrossRef]
- Czapar, A.E.; Steinmetz, N.F. Plant viruses and bacteriophages for drug delivery in medicine and biotechnology. Curr. Opin. Chem. Biol. 2017, 38, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Wen, A.M.; Steinmetz, N.F. Design of virus-based nanomaterials for medicine, biotechnology, and energy. Chem. Soc. Rev. 2016, 45, 4074–4126. [Google Scholar] [CrossRef] [Green Version]
- Steinmetz, N.F. Viral nanoparticles as platforms for next-generation therapeutics and imaging devices. Nanomedicine 2010, 6, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Steinmetz, N.F.; Calder, G.; Lomonossoff, G.P.; Evans, D.J. Plant viral capsids as nanobuilding blocks: Construction of arrays on solid supports. Langmuir 2006, 22, 10032–10037. [Google Scholar] [CrossRef]
- Young, M.; Willits, D.; Uchida, M.; Douglas, T. Plant viruses as biotemplates for materials and their use in nanotechnology. Annu. Rev. Phytopathol. 2008, 46, 361–384. [Google Scholar] [CrossRef]
- Lewis, J.D.; Destito, G.; Zijlstra, A.; Gonzalez, M.J.; Quigley, J.P.; Manchester, M.; Stuhlmann, H. Viral nanoparticles as tools for intravital vascular imaging. Nat. Med. 2006, 12, 354–360. [Google Scholar] [CrossRef] [Green Version]
- Pitek, A.S.; Jameson, S.A.; Veliz, F.A.; Shukla, S.; Steinmetz, N.F. Serum albumin ‘camouflage’ of plant virus based nanoparticles prevents their antibody recognition and enhances pharmacokinetics. Biomaterials 2016, 89, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, N.F.; Evans, D.J. Utilisation of plant viruses in bionanotechnology. Org. Biomol. Chem 2007, 5, 2891–2902. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef]
- Cho, C.F.; Ablack, A.; Leong, H.S.; Zijlstra, A.; Lewis, J. Evaluation of nanoparticle uptake in tumors in real time using intravital imaging. J. Vis. Exp. 2011, 52, e2808. [Google Scholar] [CrossRef]
- Craeger, A.N.; Scholthof, K.B.; Citovsky, V.; Scholthof, H.B. Tobacco mosaic virus: Pioneering research for a century. Plant Cell 1999, 11, 301–308. [Google Scholar] [CrossRef]
- Scholthof, K.B. Tobacco mosaic virus: A model system for plant biology. Ann. Rev. Phytopathol. 2004, 42, 13–34. [Google Scholar] [CrossRef]
- Scholthof, K.B.; Adkins, S.; Czosnek, H.; Palukaitis, P.; Jacquot, E.; Hohn, T.; Hohn, B.; Saunders, K.; Candresse, T.; Ahlquist, P.; et al. Top 10 plant viruses in molecular plant pathology. Mol. Plant Pathol. 2011, 12, 938–954. [Google Scholar] [CrossRef]
- Namba, K.; Stubbs, G. Structure of tobacco mosaic virus at 3.6 a resolution: Implications for assembly. Science 1986, 231, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Namba, K.; Pattanayek, R.; Stubbs, G. Visualization of protein-nucleic acid interactions in a virus. Refined structure of intact tobacco mosaic virus at 2.9 a resolution by X-ray fiber diffraction. J. Mol. Biol. 1989, 208, 307–325. [Google Scholar] [CrossRef]
- Sachse, C.; Chen, J.Z.; Coureux, P.D.; Stroupe, M.E.; Fandrich, M.; Grigorieff, N. High-resolution electron microscopy of helical specimens: A fresh look at tobacco mosaic virus. J. Mol. Biol. 2007, 371, 812–835. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Zhou, Z.H. Hydrogen-bonding networks and RNA bases revealed by cryo electron microscopy suggest a triggering mechanism for calcium switches. Proc. Natl. Acad. Sci. USA 2011, 108, 9637–9642. [Google Scholar] [CrossRef] [Green Version]
- Fromm, S.A.; Bharat, T.A.; Jakobi, A.J.; Hagen, W.J.; Sachse, C. Seeing tobacco mosaic virus through direct electron detectors. J. Struct. Biol. 2015, 189, 87–97. [Google Scholar] [CrossRef]
- Fraenkel-Conrat, H.; Williams, R.C. Reconstitution of active tobacco mosaic virus from its inactive protein and nucleic acid components. Proc. Natl. Acad. Sci. USA 1955, 41, 690–698. [Google Scholar] [CrossRef]
- Okada, Y. Molecular assembly of tobacco mosaic virus in vitro. Adv. Biophys. 1986, 22, 95–149. [Google Scholar] [CrossRef]
- Butler, P.J. Self-assembly of Tobacco mosaic virus: The role of an intermediate aggregate in generating both specificity and speed. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 537–550. [Google Scholar] [CrossRef]
- Sleat, D.E.; Turner, P.C.; Finch, J.T.; Butler, P.J.; Wilson, T.M. Packaging of recombinant RNA molecules into pseudovirus particles directed by the origin-of-assembly sequence from tobacco mosaic virus RNA. Virology 1986, 155, 299–308. [Google Scholar] [CrossRef]
- Klug, A. The tobacco mosaic virus particle: Structure and assembly. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 531–535. [Google Scholar] [CrossRef]
- Durham, A.C.; Finch, J.T.; Klug, A. States of aggregation of tobacco mosaic virus protein. Nat. New Biol. 1971, 229, 37–42. [Google Scholar] [CrossRef]
- Bruckman, M.A.; Soto, C.M.; McDowell, H.; Liu, J.L.; Ratna, B.R.; Korpany, K.V.; Zahr, O.K.; Blum, A.S. Role of hexahistidine in directed nanoassemblies of tobacco mosaic virus coat protein. ACS Nano 2011, 5, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.D.; Naves, L.; Wang, X.; Ghodssi, R.; Culver, J.N. Carboxylate-directed in vivo assembly of virus-like nanorods and tubes for the display of functional peptides and residues. Biomacromolecules 2013, 14, 3123–3129. [Google Scholar] [CrossRef]
- Shire, S.J.; McKay, P.; Leung, D.W.; Cachianes, G.J.; Jackson, E.; Wood, W.I.; Raghavendra, K.; Khairallah, L.; Schuster, T.M. Preparation and properties of recombinant DNA derived tobacco mosaic virus coat protein. Biochemistry 1990, 29, 5119–5126. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Eiben, S.; Wang, Q. Coassembly of tobacco mosaic virus coat proteins into nanotubes with uniform length and improved physical stability. ACS Appl. Mater. Interfaces 2016, 8, 13192–13196. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.J.; Roberts, I.M.; Wilson, T.M. Assembly of tobacco mosaic virus and TMV-like pseudovirus particles in Escherichia coli. Arch. Virol. Suppl. 1994, 9, 543–558. [Google Scholar] [PubMed]
- Dedeo, M.T.; Duderstadt, K.E.; Berger, J.M.; Francis, M.B. Nanoscale protein assemblies from a circular permutant of the tobacco mosaic virus. Nano Lett. 2010, 10, 181–186. [Google Scholar] [CrossRef]
- Smith, M.L.; Fitzmaurice, W.P.; Turpen, T.H.; Palmer, K.E. Display of peptides on the surface of tobacco mosaic virus particles. Curr. Top. Microbiol. Immunol. 2009, 332, 13–31. [Google Scholar]
- Liu, X.; Yang, T.; Han, Y.; Zou, L.; Yang, H.; Jiang, J.; Liu, S.; Zhao, Q.; Huang, W. In situ growth of CuS/SiO2-Based multifunctional nanotherapeutic agents for combined photodynamic/photothermal cancer therapy. ACS Appl. Mater. Interfaces 2018, 10, 31008–31018. [Google Scholar] [CrossRef]
- Klimpel, A.; Lutzenburg, T.; Neundorf, I. Recent advances of anti-cancer therapies including the use of cell-penetrating peptides. Curr. Opin. Pharmacol. 2019, 47, 8–13. [Google Scholar] [CrossRef]
- Baig, M.H.; Ahmad, K.; Saeed, M.; Alharbi, A.M.; Barreto, G.E.; Ashraf, G.M.; Choi, I. Peptide based therapeutics and their use for the treatment of neurodegenerative and other diseases. Biomed. Pharmacother. 2018, 103, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Bielenberg, D.R.; Pettaway, C.A.; Takashima, S.; Klagsbrun, M. Neuropilins in neoplasms: Expression, regulation, and function. Exp. Cell Res. 2006, 312, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Grandclement, C.; Borg, C. Neuropilins: A new target for cancer therapy. Cancers 2011, 3, 1899–1928. [Google Scholar] [CrossRef] [PubMed]
- Zachary, I. Neuropilins: Role in signalling, angiogenesis and disease. Chem. Immunol. Allergy 2014, 99, 37–70. [Google Scholar]
- Miao, H.Q.; Klagsbrun, M. Neuropilin is a mediator of angiogenesis. Cancer Metastasis Rev. 2000, 19, 29–37. [Google Scholar] [CrossRef]
- Roth, L.; Nasarre, C.; Dirrig-Grosch, S.; Aunis, D.; Cremel, G.; Hubert, P.; Bagnard, D. Transmembrane domain interactions control biological functions of neuropilin-1. Mol. Biol. Cell 2008, 19, 646–654. [Google Scholar] [CrossRef]
- Nasarre, C.; Roth, M.; Jacob, L.; Roth, L.; Koncina, E.; Thien, A.; Labourdette, G.; Poulet, P.; Hubert, P.; Cremel, G.; et al. Peptide-based interference of the transmembrane domain of neuropilin-1 inhibits glioma growth in vivo. Oncogene 2010, 29, 2381–2392. [Google Scholar] [CrossRef] [Green Version]
- Arpel, A.; Gamper, C.; Spenle, C.; Fernandez, A.; Jacob, L.; Baumlin, N.; Laquerriere, P.; Orend, G.; Cremel, G.; Bagnard, D. Inhibition of primary breast tumor growth and metastasis using a neuropilin-1 transmembrane domain interfering peptide. Oncotarget 2016, 7, 54723–54732. [Google Scholar] [CrossRef] [Green Version]
- Prud’homme, G.J.; Glinka, Y. Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget 2012, 3, 921–939. [Google Scholar] [CrossRef] [Green Version]
- Meyer, L.A.; Fritz, J.; Pierdant-Mancera, M.; Bagnard, D. Current drug design to target the Semaphorin/Neuropilin/Plexin complexes. Cell Adh. Migr. 2016, 10, 700–708. [Google Scholar] [CrossRef] [Green Version]
- Tirand, L.; Frochot, C.; Vanderesse, R.; Thomas, N.; Trinquet, E.; Pinel, S.; Viriot, M.L.; Guillemin, F.; Barberi-Heyob, M. A peptide competing with VEGF165 binding on neuropilin-1 mediates targeting of a chlorin-type photosensitizer and potentiates its photodynamic activity in human endothelial cells. J. Control. Release 2006, 111, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.; Bechet, D.; Becuwe, P.; Tirand, L.; Vanderesse, R.; Frochot, C.; Guillemin, F.; Barberi-Heyob, M. Peptide-conjugated chlorin-type photosensitizer binds neuropilin-1 in vitro and in vivo. J. Photochem. Photobiol. B Biol. 2009, 96, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Bechet, D.; Tirand, L.; Faivre, B.; Plenat, F.; Bonnet, C.; Bastogne, T.; Frochot, C.; Guillemin, F.; Barberi-Heyob, M. Neuropilin-1 targeting photosensitization-induced early stages of thrombosis via tissue factor release. Pharm. Res. 2010, 27, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Jacob, L.; Sawma, P.; Garnier, N.; Meyer, L.A.; Fritz, J.; Hussenet, T.; Spenle, C.; Goetz, J.; Vermot, J.; Fernandez, A.; et al. Inhibition of PlexA1-mediated brain tumor growth and tumor-associated angiogenesis using a transmembrane domain targeting peptide. Oncotarget 2016, 7, 57851–57865. [Google Scholar] [CrossRef]
- Arpel, A.; Sawma, P.; Spenle, C.; Fritz, J.; Meyer, L.; Garnier, N.; Velazquez-Quesada, I.; Hussenet, T.; Aci-Seche, S.; Baumlin, N.; et al. Transmembrane domain targeting peptide antagonizing ErbB2/Neu inhibits breast tumor growth and metastasis. Cell Rep. 2014, 8, 1714–1721. [Google Scholar] [CrossRef]
- Geretti, E.; Klagsbrun, M. Neuropilins: Novel targets for anti-angiogenesis therapies. Cell Adh. Migr. 2007, 1, 56–61. [Google Scholar] [CrossRef]
- Jubb, A.M.; Strickland, L.A.; Liu, S.D.; Mak, J.; Schmidt, M.; Koeppen, H. Neuropilin-1 expression in cancer and development. J. Pathol. 2012, 226, 50–60. [Google Scholar] [CrossRef]
- Thomas, N.; Tirand, L.; Chatelut, E.; Plenat, F.; Frochot, C.; Dodeller, M.; Guillemin, F.; Barberi-Heyob, M. Tissue distribution and pharmacokinetics of an ATWLPPR-conjugated chlorin-type photosensitizer targeting neuropilin-1 in glioma-bearing nude mice. Photochem. Photobiol. Sci. 2008, 7, 433–441. [Google Scholar] [CrossRef]
- Folwarczna, J.; Moravec, T.; Plchova, H.; Hoffmeisterova, H.; Cerovska, N. Efficient expression of Human papillomavirus 16 E7 oncoprotein fused to C-terminus of Tobacco mosaic virus (TMV) coat protein using molecular chaperones in Escherichia coli. Protein Expr. Purif. 2012, 85, 152–157. [Google Scholar] [CrossRef]
- Lomonossoff, G.P.; Wege, C. TMV particles: The journey from fundamental studies to bionanotechnology applications. Adv. Virus Res. 2018, 102, 149–176. [Google Scholar]
- He, Z.; Tessier-Lavigne, M. Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell 1997, 90, 739–751. [Google Scholar] [CrossRef]
- Gu, C.; Limberg, B.J.; Whitaker, G.B.; Perman, B.; Leahy, D.J.; Rosenbaum, J.S.; Ginty, D.D.; Kolodkin, A.L. Characterization of neuropilin-1 structural features that confer binding to semaphorin 3A and vascular endothelial growth factor 165. J. Biol. Chem. 2002, 277, 18069–18076. [Google Scholar] [CrossRef] [PubMed]
- Herzog, B.; Pellet-Many, C.; Britton, G.; Hartzoulakis, B.; Zachary, I.C. VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signaling via FAK Tyr407 phosphorylation. Mol. Biol. Cell 2011, 22, 2766–2776. [Google Scholar] [CrossRef] [PubMed]
- Holt, C.A.; Beachy, R.N. In vivo complementation of infectious transcripts from mutant Tobacco mosaic virus cDNAs in transgenic plants. Virology 1991, 181, 109–117. [Google Scholar] [CrossRef]
- Nallamsetty, S.; Austin, B.P.; Penrose, K.J.; Waugh, D.S. Gateway vectors for the production of combinatorially-tagged His6-MBP fusion proteins in the cytoplasm and periplasm of Escherichia coli. Protein Sci. 2005, 14, 2964–2971. [Google Scholar] [CrossRef]
- Nasarre, C.; Koncina, E.; Labourdette, G.; Cremel, G.; Roussel, G.; Aunis, D.; Bagnard, D. Neuropilin-2 acts as a modulator of Sema3A-dependent glioma cell migration. Cell Adh. Migr. 2009, 3, 383–389. [Google Scholar] [CrossRef] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamper, C.; Spenlé, C.; Boscá, S.; van der Heyden, M.; Erhardt, M.; Orend, G.; Bagnard, D.; Heinlein, M. Functionalized Tobacco Mosaic Virus Coat Protein Monomers and Oligomers as Nanocarriers for Anti-Cancer Peptides. Cancers 2019, 11, 1609. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101609
Gamper C, Spenlé C, Boscá S, van der Heyden M, Erhardt M, Orend G, Bagnard D, Heinlein M. Functionalized Tobacco Mosaic Virus Coat Protein Monomers and Oligomers as Nanocarriers for Anti-Cancer Peptides. Cancers. 2019; 11(10):1609. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101609
Chicago/Turabian StyleGamper, Coralie, Caroline Spenlé, Sonia Boscá, Michael van der Heyden, Mathieu Erhardt, Gertraud Orend, Dominique Bagnard, and Manfred Heinlein. 2019. "Functionalized Tobacco Mosaic Virus Coat Protein Monomers and Oligomers as Nanocarriers for Anti-Cancer Peptides" Cancers 11, no. 10: 1609. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101609