STAT3: An Emerging Therapeutic Target for Hepatocellular Carcinoma

1
Department of Surgery, The Chinese University of Hong Kong, Hong Kong, China
2
Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong, China
*
Author to whom correspondence should be addressed.
Cancers 2019, 11(11), 1646; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11111646
Submission received: 8 October 2019
/
Revised: 21 October 2019
/
Accepted: 22 October 2019
/
Published: 25 October 2019
Abstract
:Hepatocellular carcinoma (HCC) is a major global health problem and its treatment options have been limited. Signal transducer and activator of transcription 3 (STAT3) is a transcription factor important for various cellular processes. Overexpression and constitutive activation of STAT3 have been frequently found in HCC and associated with poor prognosis. Ample evidence has shown that STAT3 plays pivotal roles in the initiation, progression, metastasis and immune suppression of HCC. Thus, STAT3 has attracted attention as a novel therapeutic target in HCC. Clinical trials have investigated STAT3-targeted therapeutics either as monotherapy or in combination with chemotherapeutic agents, immune checkpoint inhibitors and alternative targeted drugs. Some of these studies have yielded encouraging results. Particularly, napabucasin—a cancer stemness inhibitor targeting STAT3-driven gene transcription—has stood out with its promising clinical efficacy and safety profile. Nonetheless, clinical investigations of STAT3-targeted therapies in HCC are limited and more efforts are strongly urged to evaluate their clinical performance in HCC. Here, we provide a comprehensive review of the roles of STAT3 in HCC and follow by comprehensive analysis of STAT3 targeted strategies.
1. Introduction
Primary liver cancer is the sixth most prevalent cancer and the second leading cause of cancer mortality worldwide [1]. The most common primary liver cancer is hepatocellular carcinoma (HCC), which accounts for >85% of all cases [2]. HCC predominantly arises in the setting of cirrhosis associated with hepatitis B and C virus infections, alcohol abuse, non-alcoholic steatohepatitis and metabolic diseases [3,4]. Early-stage HCC patients are often subjected to surgical resection and liver transplantation. However, 5-year recurrence rates after resection reach >70% [5] and the efficiency of transplantation is limited by organ shortage and technical issues [6]. Although local ablation and transarterial chemoembolization respectively confer 5-year survival rates of 50–70% and survival benefit of >6 months, they are confined to patients with single tumors or multinodular tumors with good liver reserve [7,8]. Chemotherapy is not routinely used due to the chemoresistant character of HCC [9]. For advanced HCC, sorafenib, a multi-tyrosine kinase inhibitor (MTKI), has been the only approved systemic first-line agent for over a decade, until lenvatinib, another MTKI, recently came into play. Yet, their clinical efficacy was suboptimal, with only ~3 months of prolonged survival [10,11]. Second-line treatments include regorafenib and cabozantinib, also MTKIs [12,13]; nivolumab and pembrolizumab, immune checkpoint inhibitors targeting programmed death-1 (PD-1) [14,15]; and ramucirumab, the first biomarker-driven therapeutics approved for HCC that targets the angiogenic vascular endothelial growth factor receptor (VEGFR) [16], all of which have not been established until the past few years and require further investigations. So far, treatment outcomes for HCC are far from satisfactory, with a 5-year survival rate of only ~18% [17]. Evidently, there is an urgent need to develop more effective therapeutic strategies in HCC.
Signal transducer and activator of transcription 3 (STAT3) has recently emerged as a potential therapeutic target for HCC due to its crucial roles in oncogenesis. STAT3 was initially determined to control acute-phase genes in response to interleukin-6 (IL-6) and epidermal growth factor (EGF) during inflammation [18]. It belongs to the STAT family of cytoplasmic transcription factors that mediate signal transduction from the plasma membrane to the nucleus in various cellular activities [19]. The STAT family comprises seven members: STAT1, 2, 3, 4, 5a, 5b and 6. Each of them consists of (i) an N-terminal domain for oligomerization, (ii) a coiled-coil domain for interaction with regulatory proteins, (iii) a DNA-binding domain for recognition of specific DNA sequences, (iv) a Src homology-2 (SH2) domain that triggers phosphorylation and dimerization after docking to phosphorylated receptors and (iv) a C-terminal transactivation domain with specific tyrosine (Y) (present in all STATs) and serine (S) residues (absent in STAT2 and 6) that are phosphorylated upon transcriptional activation [19,20].
Intensive investigation has been done on STAT3 since its discovery, revealing its physiological roles in early embryonic development, growth and differentiation of various adult tissues [21]. In addition, its pathogenic roles in cancer initiation, progression, metastasis, chemoresistance and immunoevasion have been uncovered [22]. To date, STAT3 is widely recognized as an oncogenic factor in diverse human cancers. Therefore, targeting STAT3 might be an attractive therapeutic strategy for HCC treatment. In this review, we summarize the oncogenic roles of STAT3 in HCC and the current clinical development of STAT3-targeted therapeutics.
2. The STAT3 Signaling Pathway
2.1. Activation and Regulation of STAT3
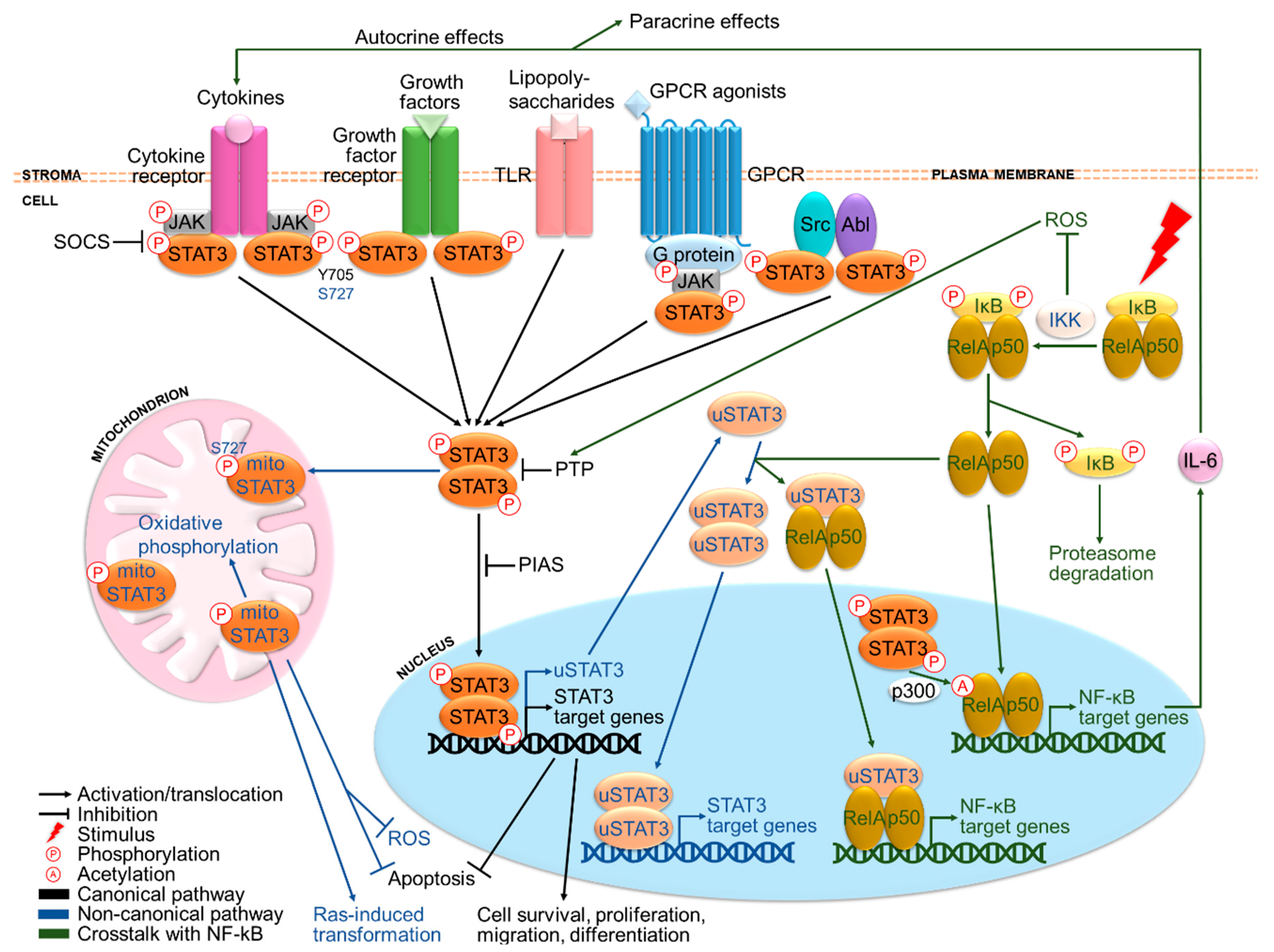
Essentially, STAT3 is a transcription factor that activates survival and proliferation signaling upon cytokine and growth factor stimuli. STAT3 can signal through both canonical and non-canonical pathways (Figure 1). Canonically, the binding of ligands to their cognate receptors leads to the recruitment and phosphorylation of tyrosine kinases, which recruit and phosphorylate STAT3 at Y705 (pY705) [21]. Subsequently, STAT3 proteins dimerize and translocate to the nucleus where they bind to promoter elements of target genes and modulate their transcription [21]. These include cell cycle regulatory genes such as fos, cyclin D, c-Myc, pim1 and anti-apoptotic genes such as B-cell CLL/Lymphoma-2 (Bcl-2), Bcl-xL, survivin and X-linked inhibitor of apoptosis protein (XIAP) [23]. Non-canonically, STAT3 may function independently of pY705 and nuclear localization (Figure 1). pS727 is required for maximal activation although pY705 plays a key activating role [24,25]. pS727 can also stimulate mitochondrial STAT3, where it may trigger oxidative phosphorylation [26], confer stress protection by reducing reactive oxygen species (ROS) accumulation and apoptosis [27,28] and support Ras-induced malignant transformation [29]. STAT3 can also autoregulate its own transcription, which will be discussed further in the next section.
STAT3 is primarily activated by ligand binding to (i) cytokine receptors and (ii) growth factor receptors, (iii) toll-like receptors (TLRs) [30], (iv) G-protein coupled receptors (GPCRs) [31] and (v) cytoplasmic tyrosine kinases such as Src and Abl [32,33] (Figure 1). A notable example is the proinflammatory cytokine IL-6, which binds to IL-6R/gp130 receptors and stimulates Janus kinases (JAKs), leading to STAT3 activation [34]. Other key STAT3-activating molecules include IL-6 family cytokines like IL-22 [35] and growth factors including EGF and vascular endothelial growth factor (VEGF) [36,37]. Except for growth factor receptors with intrinsic receptor tyrosine kinases (RTKs) and cytoplasmic kinases, other receptors rely on JAKs for phosphorylation. Under normal conditions, STAT3 activation is strictly governed by negative regulators, namely suppressor of cytokine signaling (SOCS) proteins [38], protein inhibitor of activated STAT (PIAS) proteins [39] and protein tyrosine phosphatases (PTPs) such as Src-homology-2 protein phosphatase-1 and 2 (SHP1/2) [40].
2.2. Crosstalk between STAT3 and NF-κB
Particularly, activation of STAT3 has been found to be associated with nuclear factor-kappa B (NF-kB), a family of inflammatory transcription factors consisting of NF-κB1 (p50), NF-κB2 (p52), Rel-A (p65), Rel-B and c-Rel subunits [41]. In response to proinflammatory stimuli, NF-kB translocates from the cytoplasm to the nucleus after being released from inhibitor of NF-κB (IκB), which is then phosphorylated by IκB kinase (IKK) and degraded (Figure 1). Activated NF-kB increases secretion of a panel of cytokines including IL-6 at the sites of inflammation [34], which activates STAT3 signaling. In fact, the NF-kB/IL-6/STAT3 pathway plays an important oncogenic role in cancers that arise from chronic inflammation such as HCC [42]. By contrast, NF-κB activation has also been shown to inhibit STAT3 signaling by preventing ROS accumulation responsible for oxidizing PTPs [43,44,45]. On the other hand, activated STAT3 can directly bind to the major NF-κB subunit Rel-A and promote its p300-dependent acetylation, causing prolonged nuclear localization and constitutive activation of NF-κB [34]. Moreover, STAT3 autoregulation produces unphosphorylated STAT3 (u-STAT3) molecules that bind to and facilitate nuclear translocation of unphosphorylated NF-κB (u-NF-κB), coregulating another set of target genes such as IL-6, IL-8, mesenchymal-epithelial transition factor (MET) and muscle RAS (MRAS) [46].
3. The Roles of STAT3 in HCC
3.1. Hepatic STAT3 Functions
Under physiological conditions, STAT3 is only transiently activated under the tight control of negative regulators and exerts various functions in the liver. In hepatocytes, the IL-6/STAT3 pathway is involved in hepatoprotection upon liver damage [35,47] and glucose homeostasis by inhibiting gluconeogenesis upon increase in plasma insulin [48,49]. In non-parenchymal hepatic cells, STAT3 activation by different stimuli also offers protective effects against cell injury, including cholangiocytes [50], stellate cells [51], endothelial cells [52] and liver-specific immune cells [53,54].
3.2. Clinical Implication of STAT3 in HCC
In contrast to transient activation in physiological states, STAT3 becomes persistently activated in the majority of malignancies [55]. Importantly, overexpression and constitutive activation of STAT3 has been found to be closely associated with pathogenesis and survival outcomes of HCC. He et al. has reported that ~60% of HCC cases exhibit nuclear STAT3 pY705 in tumoral but not surrounding non-tumoral tissues [44]. In addition, overexpression of STAT3 pY705 and/or pS727 in tumoral tissues has been found to be correlated with poor prognosis and clinicopathological features including larger tumor size, vascular invasion, advanced disease stage and cirrhosis in HCC patients [56,57,58,59]. Moreover, a significant association has been reported between STAT3 activity in stromal monocytes and poor prognosis in HCC, indicating the role of STAT3 in regulating the tumor microenvironment [60]. These data clearly substantiate the clinical significance of STAT3 in HCC.
Constitutive STAT3 activation in HCC could be explained by several causes. The foremost reason would be the elevated levels of STAT3-inducing signals, particularly, IL-6 and IL-22, which exert oncogenic functions via STAT3 activation in HCC [57,61,62]. Second, disruption of negative regulators of STAT3, such as SOCS3 and SHP1/2, may enhance STAT3 activation and promote HCC development [44,63]. Third, activating mutations in the gene encoding the gp130 subunit of IL-6R in benign hepatic adenomas have been found to cause STAT3 activation and HCC development when accompanied by β-catenin mutations, albeit at low frequency [64]. Intriguingly, no oncogenic mutations of STAT3 or JAKs have yet been detected in HCC [64].
3.3. Functions of STAT3 in HCC
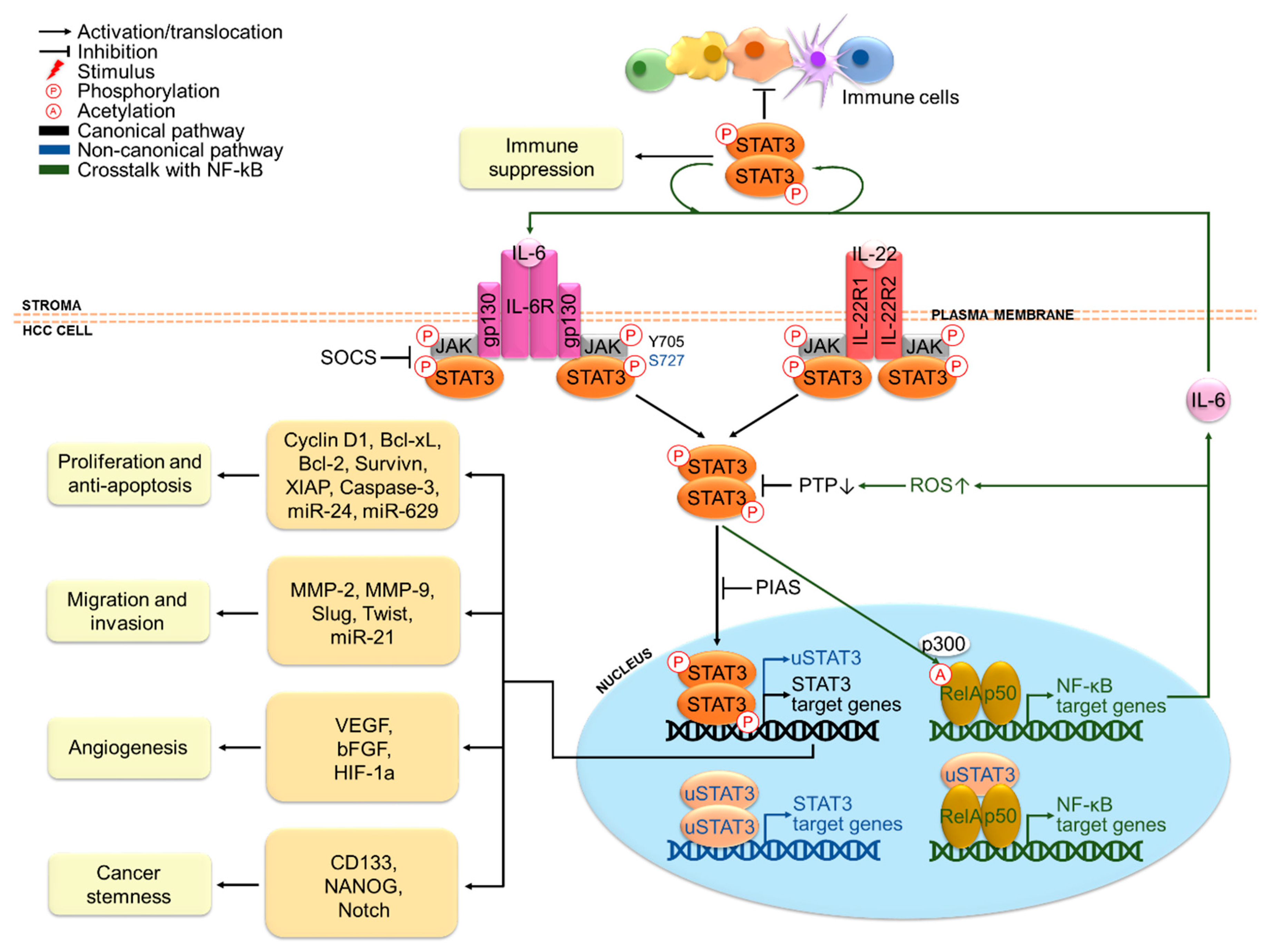
Indeed, the oncogenic functions of STAT3 in HCC have been extensively reported with relation to cancer cell proliferation, anti-apoptosis, migration, invasion, angiogenesis, stemness properties and immune suppression (Figure 2). These functions are mainly exerted via transcriptional regulation of different oncogenic target genes. The cooperation between STAT3 and NF-kB may also occur, given that around one third of HCC tumors display concomitant activation of STAT3 and NF-kB [44]. In addition, STAT3-mediated microRNA (miRNA) expression is also emerging as an epigenetic mechanism for driving hepatic oncogenesis and in turn, miRNA can also play a role in the regulation of STAT3 signaling [65].
In terms of proliferation and anti-apoptosis, STAT3 antisense oligonucleotides have been reported to inhibit proliferation and survival of several HCC cell lines [66]. In the same study, they have also been shown to impede tumorigenicity of a highly tumorigenic HCC cell line upon transplantation into mice [66]. Likewise, diethylnitrosamine (DEN)-induced HCC cells transduced with STAT3 short hairpin RNA (shRNA) have failed to form subcutaneous HCC tumors when transplanted into mice [44]. Besides, hepatocyte-specific STAT3-deficient mice have shown reduced size and multiplicity upon DEN treatment [44]. These results strongly justify the importance of STAT3 in HCC growth and tumor formation. Moreover, an epigenetic circuit involving multiple miRNAs has been demonstrated to promote HCC formation [67]. A key event in this mechanism is the downregulation of the hepatocyte nuclear factor 4α (HNF4α), a suppressor of hepatic oncogenesis, via IL-6/STAT3-dependent activation of miR-24 and miR-629. In turn, HNF4α-regulated expression of miR-124 is switched off, which releases its negative regulation on IL-6R, forming a positive feedback loop. In fact, the pro-proliferative role of STAT3 is highly related to its anti-apoptotic functions on HCC cells. Abrogation of STAT3 signaling by the Jak2 inhibitor, AG490, has triggered cell cycle arrest at G0/G1 phase in HCC cells via cyclin D1 downregulation and induced apoptosis by downregulating anti-apoptotic proteins Bcl-xL, survivin and XIAP [68]. STAT3 antisense treatment of HCCLM3 cells has also markedly impaired STAT3-dependent transcription of these genes and activated the main apoptosis executor caspase-3, leading to induction of apoptosis [66]. These studies provide support for the role of STAT3 in driving cell cycle progression and blocking apoptosis of HCC cells. By contrast, in mice transplanted with a highly metastatic HCC cell line and IL-22+ tumor-infiltrating lymphocytes from HCC patients, increased expression of pSTAT3 in tumor tissues has been detected along with an upregulation of cyclin D1, Bcl-2 and Bcl-xL, indicating the role of IL-22-mediated STAT3 activation in HCC tumor growth and resistance to apoptosis [57]. Collectively, these findings underpin the significance of STAT3 in HCC initiation and development.
In terms of migration and invasion, it has been shown that STAT3 antisense treatment has reduced the invasiveness of HCC cells via downregulation of matrix metalloproteinases (MMP)-2 and MMP-9, which are engaged in the digestion of extracellular matrices [66]. Conversely, STAT3 activation has enhanced migration and invasion of HCC cells by transcriptionally inducing epithelial-to-mesenchymal transition (EMT) markers including Slug and Twist [69,70], suggesting that STAT3 activation may induce invasion and metastasis via mediation of EMT in HCC. In addition, the role STAT3 in migration and invasion has also been found to involve regulation of miR-21 and its targets [71].
In terms of angiogenesis, mice bearing STAT3 antisense-transfected HCC tumors have displayed decreased microvessel density and reduced circulating proteins of VEGF and basic fibroblast growth factor (bFGF), which are potent inducers of angiogenesis [66]. This clearly suggests the pro-angiogenic role of STAT3 in HCC. Mechanistically, STAT3 upregulates and recruits hypoxia inducible factor (HIF)-1α to form a transcriptional complex that binds to the VEGF promoter, thus inducing VEGF expression under hypoxia [72]. Indeed, increased VEGF expression has been correlated with poor prognosis in HCC patients, which is in accord with the marked vascularity characteristic of advanced HCC [73].
In terms of cancer stemness, IL6/STAT3 signaling has been found to induce expression of the cancer stem cell (CSC) marker CD133 via interaction of STAT3 with NF-κB and HIF-1α in HCC [74]. Besides, CD24-mediated STAT3 activation has been reported to regulate expression of the stem cell-associated protein, NANOG [75]. STAT3 can also promote stemness in HCC cells by activating Notch signaling, which is implicated in self-renewal and proliferation of CSCs [76]. These results reveal an important role of STAT3 in maintaining HCC stem cell phenotypes, which confer chemoresistance and contribute to recurrence upon chemotherapy treatment.
In terms of immune suppression, signaling molecules produced in the tumor microenvironment may establish a reciprocal STAT3 activation loop between tumor and stromal cells in a paracrine fashion, which exerts suppressive effects in various immune cells in HCC. STAT3 activation promotes various protumorigenic effects in stromal cells in HCC, including the production of immunosuppressive molecules from dendritic cells [77], polarization of tumorigenic M2 macrophages to antitumorigenic M1 subtype [78], cancer-associated fibroblast-mediated generation of myeloid-derived suppressor cells, inhibition of T cell proliferation and functions [79] and impairment of natural killer cell-mediated cytotoxicity [80]. These contribute to impaired effectiveness of immune surveillance against HCC.
Interestingly, studies have also revealed antioncogenic roles of STAT3 in RAS-dependent HCC [81] and early-stage HCC developed from carbon tetrachloride (CCl4)-induced liver fibrosis [82], implying that specific genetic context and etiology of the disease may impact the outcome of STAT3-targeted therapeutics. Nonetheless, as appreciated from the abundance of evidence illustrating the oncogenic roles of STAT3 in HCC, STAT3 is recognized as a vital oncogene in HCC and may serve as a potential therapeutic target for HCC therapy.
4. Clinical Trials of STAT3 Targeting Therapies
Given the constitutive activation and critical oncogenic roles of STAT3 in HCC and other cancer types, the STAT3 signaling pathway has emerged as a promising target for pharmacological intervention in cancer treatment. A myriad of STAT3-targeted drugs has been developed, which can be categorized into six major classes: (i) N-terminal domain inhibitors, (ii) DNA-binding domain inhibitors, (iii) SH2 domain inhibitors, (iv) antisense molecules, (v) inhibitors of downstream target genes and (vi) inhibitors of upstream activators or regulators. The first five classes are direct STAT3 inhibitors which respectively prevent interaction with regulatory proteins, DNA binding ability, phosphorylation and dimerization, protein expression and gene transcription. The sixth class acts indirectly by inhibiting upstream receptors such as JAK1/2 or stimulating negative regulators such as SHP1/2, of which a detailed analysis would be beyond the scope of this review due to its relative non-specificity for STAT3. Indeed, many of them have been demonstrated to exert antioncogenic effects in preclinical models of HCC and other cancers [83,84]. The remainder of this review will focus on direct STAT3 inhibitors that have advanced into clinical trials with promising therapeutic potential as monotherapy or in combination with other treatment modalities in HCC and other cancers.
4.1. Napabucasin: Cancer Stemness Inhibitor Targeting STAT3-Driven Gene Transcription
Napabucasin (BBI608) is the most extensively investigated STAT3-targeted agent against cancers thus far. It is a first-in-class cancer stemness inhibitor that inhibits transcription of STAT3 downstream target genes. In preclinical settings, it has shown to reduce the expression of stemness genes β-catenin, NANOG, smoothened and sex-determining region Y-box protein 2 (Sox2), impede self-renewal and survival of various cancer cells including HCC cells in vitro, as well as prevent cancer relapse and metastasis in vivo [85]. Besides, it has also been demonstrated to sensitize stemness-high gastric cancer cells to standard chemotherapeutic agent paclitaxel [86].
Napabucasin is the only agent that has advanced into phase III trials among other STAT3-targeted therapeutics (Table 1). This orally administered drug has been proven safe at 240–480/500 mg twice daily (n = 41) (NCT01775423) [87] and has shown promising antitumor effects and potential to sensitize patients to conventional therapies in various cancers. Unfortunately, no studies have yet been reported on its clinical impact in HCC patients. The antitumor efficacy of napabucasin as a monotherapy has been particularly observed in colorectal cancer (CRC) among others. It has demonstrated a disease control rate (DCR) of 67% in CRC patients and 29% stable disease (SD) in other solid cancer patients in a phase Ib study (n = 24) (NCT01775423) [88]. It has also resulted in improved overall survival (OS) of advanced refractory CRC patients from 3 to 5.1 months after pSTAT3 stratification in another phase III trial (n = 46) (NCT01830621) [89].
In combination with chemotherapeutic drugs, full-dose napabucasin (500 mg, twice daily) and paclitaxel has passed safety and tolerability tests and shown encouraging clinical responses in patients with different advanced solid tumors, including gastric and gastroesophageal junction (GEJ) adenocarcinoma [91], platinum-resistant ovarian cancer (PROC) [92], pancreatic ductal adenocarcinoma (PDAC) [93], triple-negative breast cancer (TNBC) [94] and other cancer types [90] in individual cohorts of a phase Ib/II trial (NCT01325441). Particularly, in the gastric and GEJ adenocarcinoma cohort, this combination has shown promising results in both taxane-naïve (n = 16) and taxane-exposed patients (n = 19), with DCR being 75% versus 68% [91]. When combined with gemcitabine plus nab-paclitaxel, half-dose napabucasin (240 mg, twice daily) has been proven to be safe, with 93% DCR, 80% tumor regression and 47% partial response (PR) in metastatic PDAC patients in a phase Ib/II trial (n = 37) (NCT02231723) [99]. The combination of folinic acid-5-fluorouracil-irinotecan (FOLFIRI) regimen in the presence or absence of bevacizumab with half-dose napabucasin has also been well tolerated in advanced CRC patients. In a phase Ib/II study (NCT02024607) [97], the DCR of FOLFIRI-naïve (n = 34) and FOLFIRI-exposed patients (n = 29) has been 82% versus 72%. Interestingly, patients with pSTAT3high (n = 30) and pSTAT3low (n = 27) statuses have shown DCR of 83% and 89% respectively, suggesting possible synergism between napabucasin and FOLFIRI irrespective of pSTAT3 status. The result of its extension study has also been positive, with DCR of 90% in FOLFIRI-exposed patients (n = 19) [98].
The targeted drug panitumumab, a human anti-EGFR monoclonal antibody, could also be safely combined with full-dose napabucasin in KRAS wild-type metastatic (m) CRC as reported in a phase Ib/II trial (NCT10776307) [95,96]. This combination has shown positive antitumor activity in both anti-EGFR-naïve patients (n = 24) and those who failed anti-EGFR therapy (n = 48), with DCR being 48% versus 59% and PFS being 16.9 weeks versus 9 weeks respectively, suggesting that prior anti-EGFR exposure is not limiting the efficacy of this combination therapy and that napabucasin may sensitize patients to repeat anti-EGFR therapy.
Apart from good antitumor activity, napabucasin has shown a manageable toxicity profile in patients with various cancers. The most common adverse events have been mild gastrointestinal symptoms, such as diarrhea, nausea and vomiting, which could be generally kept under control by antidiarrheal and antiemetic agents. At present, different trials are ongoing to assess the efficacy of combination regimens of napabucasin with (i) chemotherapeutic drugs, including paclitaxel, gemcitabine plus nab-paclitaxel and FOLIFIRI, (ii) the standard systemic targeted drug for advanced HCC, sorafenib and (iii) immune checkpoint inhibitors ipilimumab, nivolumab and pembrolizumab. Besides, the pro-drug of napabucasin, DSP-0337, is now being evaluated for its safety, tolerability, pharmacokinetics and antitumor activity in a phase I trial for advanced solid tumors (NCT03416816) and may serve as an alternative for napabucasin. Although clinical benefits of napabucasin has been demonstrated in different types of solid tumors, an important unmet need exists to gain better understanding of its clinical effects in HCC patients.
4.2. AZD9150: STAT3-Targeted Antisense Oligonucleotide
AZD9150 (ISIS481464), a STAT3-targeted antisense oligonucleotide that reduces STAT3 mRNA expression, has also shown promising antioncogenic effects. Preclinical findings have demonstrated its ability to decrease the expression of STAT3 and its downstream oncogenic target genes in a broad range of cancer cells [100]. In particular, this drug has been effective against leukemia and lymphoma both in vitro and in vivo [100,101], while it has also inhibited primary or secondary tumor growth respectively in xenografts of lung cancer and neuroblastoma [100,102].
Clinically, the only study of AZD9150 in HCC completed thus far is a phase I trial evaluating its safety and antitumor activity in patients with advanced or metastatic HCC (n = 58) (NCT01839604) (Table 2) [103]. It has been well tolerated at doses up to 3 mg/kg, applying 3 infusions in the first week followed by weekly infusions, with only mild and few serious adverse reactions. Of note, another phase I trial in treatment-refractory cancer patients, with half of them suffering from advanced lymphoma and the others having various types of solid tumors, has reported promising therapeutic effects (n = 25) (NCT number not stated) [100]. The DCR has reached 44%, including 3 patients with diffuse large B-cell lymphoma (DLBCL) displaying tumor shrinkage and 2 patients showing durable PR. Besides, 33% of the patients have displayed >30% of post-treatment reductions in circulating concentrations of IL-6. IL-6 is a prime stimulus for STAT3 activation and elevated serum levels of IL-6 have been associated with poor prognosis in various cancers [104]. Thus, such decline in circulating IL-6 may serve as an indicator of STAT3 pathway inhibition. Encouraging results have also been described in a more recent phase I trial in relapsed or refractory lymphoma patients primarily consisting of a DLBCL population (n = 30) (NCT01563302) [105]. The DCR has been 17%, including 2 complete responses with median response duration of 10.7 months, 2 PRs and 1 SD.
In general, AZD9150 is deemed safe and effective in advanced cancer patients especially those with lymphoma. There have been few unacceptable toxicity events, with the most common adverse events being elevated levels of aspartate and alanine aminotransferase and thrombocytopenia. Many phase I/II trials of AZD9150 are now underway in numerous cancer types including solid tumors and hematological malignancies, either as a single agent or in combination with chemotherapeutic, targeted or immunomodulatory agents. Nonetheless, like napabucasin, clinical investigations of AZD9150 in HCC are still in its infancy and further efforts are urgently needed.
4.3. OPB Compounds, Pyrimethamine and TTI-101: STAT3 SH2 Domain Inhibitor
In the realm of SH2 domain inhibitors, OPB-31121 is one of the earliest oral drugs developed by Otsuka Pharmaceuticals. It prevents STAT3 dimerization upon phosphorylation by binding with high affinity to the SH2 domain of STAT3 [106]. Preclinically, OPB-31121 has demonstrated good antitumor activity in leukemic [107] and gastric cancer cells [108] and even elicited synergistic effects against gastric cancer when combined with chemotherapeutic agents [108]. However, a phase I clinical trial in patients with advanced solid tumors has not yielded any objective responses, accompanied with a highly varied pharmacokinetic profile (n = 14) (NCT00955812) [109] (Table 3). As such, further development of the compound has been discontinued.
In the same phase I study, OPB-111077, the primary metabolite of OPB-31121, has been found to be accumulated at higher tissue levels [109]. In vitro studies have shown that OPB-111077 profoundly inhibits the growth of various cancer cell types [109]. Subsequently, two phase I trials have been conducted in patients with advanced HCC and other solid tumors respectively (Table 3). Although no objective responses have been achieved in the HCC trial (n = 33) (NCT01942083) [111], antitumor responses have been more encouraging in the other study, with one PR in a DLBCL patient and 39% patients showing SD or minor responses (n = 145) (NCT01711034) [110]. In both studies, the drug has been well-tolerated and the recommended phase II dose has been determined to be 250 mg once daily. These indicate the feasibility of STAT3 inhibition with OPB-111077 but more trials are essential for assessing its safety, pharmacokinetics and therapeutic efficacy in larger cohorts of patients with HCC or other cancer types. Results of two recently completed studies in advanced solid tumors (NCT02250170) and relapsed/refractory acute myeloid leukemia (AML) (NCT03197714) are eagerly awaited. Three other ongoing studies are evaluating OPB-111077 either as a single agent or in combination with chemo- and targeted therapy.
On the other hand, OPB-51602 has also demonstrated inhibitory effects against different cancer models [115] but intolerability issues of this drug have been revealed in the clinic (Table 3). A phase I study in refractory solid tumors has demonstrated poorer tolerability for continuous dosing, compared with intermittent dosing at 4 mg daily with two weeks of treatment followed by one week of rest (n = 51) (NCT01184807) [112]. Modest antitumor responses have been reported, with two patients showing PR at 5 mg intermittently and 4 mg continuously, both of whom suffer from EGFR mutation-positive non-small-cell lung cancer with prior EGFR-TKI therapy. The recommended phase II dose has also been determined to be 4 mg in another study in relapsed/refractory hematological malignancies (n = 20) (NCT02058017) [113]. However, because of poor tolerability and lack of responses in long-term daily administration at higher doses, further clinical development of the drug with daily dosing in hematological malignancies has been halted. Another study in advanced nasopharyngeal carcinoma (NPC) has also been terminated due to patient intolerability to lactic and metabolic acidosis elicited by the drug (n = 9) (NCT02058017) [114]. There has not been published record for two other completed phase I trials, including a safety and tolerability study in advanced cancers (NCT01423903) and a pharmacodynamic and pharmacogenetic biomarker study in advanced solid tumors (NCT01867073).
While OPB-111077 is probably the most promising OPB candidate by far, pyrimethamine and TTI-101 are two other STAT3 SH2 domain inhibitors currently undergoing early phase clinical trials in relapsed chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL) (NCT01066663) and other advanced cancers including HCC (NCT03195699) respectively (Table 3). Both of them have shown significant inhibitory effects in preclinical breast cancer models [116,117].
4.4. STAT3 Decoy: STAT3-DNA Binding Inhibitor
STAT3 decoy is a 15-bp double-stranded oligonucleotide that competitively inhibits STAT3 binding to the response element within the c-fos promoter. It has been shown to effectively induce apoptosis, suppress growth and downregulate STAT3 target genes in in vitro and in vivo cancer models, including liver [118], lung [119,120], head and neck [121,122] and ovarian cancer [123]. Notably, Sen et al. has conducted the first phase 0 clinical trial in head and neck tumors from patients undergoing surgical resection (n = 32) and reported that intratumoral injection of STAT3 decoy abrogates expression of STAT3 target genes including cyclin D1 and Bcl-xL without toxicities [122]. Although preliminary, this study may serve as a basis for future endeavors in the application of STAT3 decoy in biopsies of HCC or other solid tumors and in more advanced clinical settings.
4.5. Current Status and Future Perspectives of STAT3-Targeted Therapies in HCC
The existing preclinical and clinical evidence strongly justifies the use of STAT3-targeted drugs as a promising therapeutic approach against HCC. To date, napabucasin is considered the most potent STAT3 inhibitor with an acceptable safety profile in the clinic. Although the STAT3-targeted antisense oligonucleotide, AZD91150, have demonstrated antitumor activities, its efficient delivery and stability in vivo remain technically challenging. For SH2 domain inhibitors, the development of OPB compounds is somewhat limited by tolerability issues, whereas others are still under early clinical investigation. Proof of concept for DNA-binding domain inhibition by STAT3 decoy has sufficed to warrant further clinical studies but it shares the same concerns as antisense oligonucleotides. Despite these limitations, direct STAT3 inhibitors are better options than indirect inhibitors targeting upstream regulators of STAT3, as the latter lack specificity to the STAT3 pathway and may produce a range of undesired pleiotropic effects. While many direct STAT3 inhibitors have not been clinically well explored in HCC, current clinical data in various other cancers suggest their potential benefit in HCC patients.
Importantly, a combinational approach for STAT3-targeted agents may be more effective than STAT3-targeted monotherapy. It has been elucidated that STAT3 is involved in extensive crosstalk with other signaling pathways and that single activating mutation of STAT3 is rare in HCC. Combination of STAT3-targeted drugs with other anticancer therapeutics may address these issues by simultaneously targeting different mechanisms of action, thus eliciting more powerful antitumor responses. While targeted agents specifically block molecular pathways that promote oncogenesis, chemotherapeutic and immunotherapeutic agents respectively inhibit growth of all dividing cells and stimulate immune responses to attack tumor cells. So far, napabucasin is the only STAT3-targeted drug that has been clinically studied in combination with other therapies. As discussed before, early-phase trials have revealed promising antitumor efficacy when combining napabucasin with standard chemodrugs including paclitaxel [90,91,92,93,94], gemcitabine plus nab-paclitaxel [99] and FOLFIRI with or without bevacizumab [97,98], as well as another targeted drug panitumumab [95,96] in several solid malignancies. Preclinical results have also suggested that napabucasin may synergize with paclitaxel to overcome drug resistance [86] and sensitize CRC to immune checkpoint inhibitors in syngeneic tumor models [124]. These findings support the notion that napabucasin may sensitize refractory cancer patients to chemotherapy, other targeted therapy and immunotherapy. Given that therapeutic resistance is a common phenomenon in advanced HCC, a combinational approach for STAT3-targeted drugs may be more effective than monotherapy. Notably, blockade of the immune checkpoint, programmed death receptor-1 (PD-1), with the fully human monoclonal antibody nivolumab has been shown to safely induce durable objective responses in patients with cancer types including HCC [14,125]. Thus, strategies to combine STAT3-targeted drugs with immune checkpoint inhibitors, which reactivate immune responses from suppression, may be of great value. Nevertheless, it must be emphasized that current clinical studies of STAT3-targeted agents have been chiefly based on cancer types other than HCC and more efforts to evaluate their clinical performance in HCC are strongly urged.
Apart from combinational strategies, novel STAT3 inhibitors and better biomarker strategies may improve the therapeutic efficacy of STAT3-targeted agents. It is expected that advancement in technology, for instance, high-throughput screening platforms of protein-protein interaction inhibitors, would boost the discovery of novel STAT3-targeted drugs. As STAT3 is a pivotal regulator of cellular metabolism under physiological conditions, an ideal STAT3-targeted agent should minimize its toxicities in normal cells while preserving its specificity and efficacy against tumor-associated components. Besides, a more robust biomarker strategy should be established for patient stratification. Although overexpression of STAT3 pY705 is the typical definition of STAT3 activation, it may not be broadly representative. Further investigations on biomarkers of STAT3 activation may be helpful in identifying cancer patients for STAT3-targeted therapies and thus improve clinical outcomes.
5. Conclusions
HCC is an extremely deadly tumor and the search for innovative treatment strategies is never ending. Overexpression and constitutive activation of STAT3 in HCC tumors has been found to associate with disease development and patient prognosis. The oncogenic functions of STAT3 have also been well established in numerous HCC models. These preclinical and clinical findings provide rationale for the use of STAT3 as a novel therapeutic target in HCC. Various types of STAT3 inhibitors have since been developed. Currently, clinical trials evaluating different STAT3-targeted strategies as monotherapy or combination therapy are ongoing. Although encouraging results have been shown in various cancer types, clinical trials on STAT3-targeted therapies in HCC are still limited. This stresses the need for more assessments of STAT3 inhibitors in HCC patients, especially in combination with other anticancer therapeutics. Future development of novel STAT3 inhibitors with lower toxicities and higher efficacy is anticipated. Besides, more efforts are required to delineate a comprehensive STAT3-associated biomarker profile, which helps define a subset of HCC patients that are more susceptible to STAT3 inhibition.
Author Contributions
Conceptualization, C.L. and S.T.C.; writing–original draft preparation, C.L.; writing–review and editing, C.L. and S.T.C.; supervision, S.T.C.; funding acquisition, S.T.C.
Funding
This review was supported in part by the Health and Medical Research Fund (05160556) and the Terry Fox Foundation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012 v1.0; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- McGlynn, K.A.; Tsao, L.; Hsing, A.W.; Devesa, S.S.; Fraumeni, J.F., Jr. International trends and patterns of primary liver cancer. Int. J. Cancer 2001, 94, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Aravalli, R.N.; Cressman, E.N.K.; Steer, C.J. Cellular and molecular mechanisms of hepatocellular carcinoma: An update. Arch. Toxicol. 2013, 87, 227–247. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Matsuyama, Y.; Tanaka, E.; Ohkubo, T.; Hasegawa, K.; Miyagawa, S.; Sugawara, Y.; Minagawa, M.; Takayama, T.; Kawasaki, S.; et al. Risk factors contributing to early and late phase intrahepatic recurrence of hepatocellular carcinoma after hepatectomy. J. Hepatol. 2003, 38, 200–207. [Google Scholar] [CrossRef]
- Jadlowiec, C.C.; Taner, T. Liver transplantation: Current status and challenges. World J. Gastroenterol. 2016, 22, 4438–4445. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver; European Organisation for Research and Treatment of Cancer. EASL–EORTC Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [Google Scholar] [CrossRef]
- Arizumi, T.; Ueshima, K.; Iwanishi, M.; Minami, T.; Chishina, H.; Kono, M.; Takita, M.; Yada, N.; Hagiwara, S.; Minami, Y.; et al. The Overall Survival of Patients with Hepatocellular Carcinoma Correlates with the Newly Defined Time to Progression after Transarterial Chemoembolization. Liver Cancer 2017, 6, 227–235. [Google Scholar] [CrossRef]
- Kim, D.W.; Talati, C.; Kim, R. Hepatocellular carcinoma (HCC): Beyond sorafenib—chemotherapy. J. Gastrointest. Oncol. 2017, 8, 256. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.H.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab (pembro) in patients with advanced hepatocellular carcinoma (HCC): KEYNOTE-224 update. J. Clin. Oncol. 2018, 36, 4020. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- American Cancer Society Inc. Cancer Facts & Figures 2018; American Cancer Society: Atlanta, GA, USA, 2018. [Google Scholar]
- Zhong, Z.; Wen, Z.; Darnell, J.E. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef]
- Quesnelle, K.M.; Boehm, A.L.; Grandis, J.R. STAT-mediated EGFR signaling in cancer. J. Cell. Biochem. 2007, 102, 311–319. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.E.; Lee, C.-k. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Carpenter, R.; Lo, H.-W. STAT3 target genes relevant to human cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.A. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007, 251, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Serge, H.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegrzyn, J.; Potla, R.; Chwae, Y.-J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Szczepanek, K.; Chen, Q.; Larner, A.C.; Lesnefsky, E.J. Cytoprotection by the modulation of mitochondrial electron transport chain: The emerging role of mitochondrial STAT3. Mitochondrion 2012, 12, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Peuckert, C.; Wölfl, S. Essential role of mitochondrial Stat3 in p38 MAPK mediated apoptosis under oxidative stress. Sci. Rep. 2017, 7, 15388. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Kortylewski, M.; Kujawski, M.; Herrmann, A.; Yang, C.; Wang, L.; Liu, Y.; Salcedo, R.; Yu, H. Toll-like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist-based immunotherapy. Cancer Res. 2009, 69, 2497–2505. [Google Scholar] [CrossRef]
- Xin, H.; Lu, R.; Lee, H.; Zhang, W.; Zhang, C.; Deng, J.; Liu, Y.; Shen, S.; Wagner, K.-U.; Forman, S. G-protein-coupled receptor agonist BV8/prokineticin-2 and STAT3 protein form a feed-forward loop in both normal and malignant myeloid cells. J. Biol. Chem. 2013, 288, 13842–13849. [Google Scholar] [CrossRef]
- Byers, L.A.; Sen, B.; Saigal, B.; Diao, L.; Wang, J.; Nanjundan, M.; Cascone, T.; Mills, G.B.; Heymach, J.V.; Johnson, F.M. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin. Cancer Res. 2009, 15, 6852–6861. [Google Scholar] [CrossRef]
- Danial, N.N.; Pernis, A.; Rothman, P.B. Jak-STAT signaling induced by the v-abl oncogene. Science 1995, 269, 1875–1877. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Herrmann, A.; Deng, J.-H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-κB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Mair, M.; Blaas, L.; Osterreicher, C.; Casanova, E.; Eferl, R. JAK-STAT signaling in hepatic fibrosis. Front. Biosci. (Landmark Ed) 2011, 16, 2794–2811. [Google Scholar] [CrossRef] [PubMed]
- Park, O.K.; Schaefer, T.S.; Nathans, D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 13704–13708. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Chen, H.; Yel, F.; Wang, F.; Xie, X. VEGF induces phosphorylation of STAT3 through binding VEGFR2 in ovarian carcinoma cells in vitro. Eur. J. Gynaecol. Oncol. 2006, 27, 363–369. [Google Scholar]
- Croker, B.A.; Kiu, H.; Nicholson, S.E. SOCS regulation of the JAK/STAT signalling pathway. Semin. Cell Developmental Biol. 2018, 19, 414–422. [Google Scholar] [CrossRef]
- Shuai, K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 2006, 16, 196. [Google Scholar] [CrossRef]
- Kim, M.; Morales, L.; Jang, I.-S.; Cho, Y.-Y.; Kim, D. Protein tyrosine phosphatases as potential regulators of STAT3 signaling. Int. J. Mol. Sci. 2018, 19, 2708. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584. [Google Scholar] [CrossRef]
- Kamata, H.; Honda, S.-i.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Yu, G.-Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKβ/NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C. Ferritin heavy chain upregulation by NF-κB inhibits TNFα-induced apoptosis by suppressing reactive oxygen species. Cell 2004, 119, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.G.; Albrecht, U.; Häussinger, D.; Heinrich, P.C.; Schaper, F. Hepatic acute phase proteins–regulation by IL-6-and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. Eur. J. Cell Biol. 2012, 91, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Ogawa, W.; Asakawa, A.; Okamoto, Y.; Nishizawa, A.; Matsumoto, M.; Teshigawara, K.; Matsuki, Y.; Watanabe, E.; Hiramatsu, R. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab. 2006, 3, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Inoue, H.; Ogawa, W.; Ozaki, M.; Haga, S.; Matsumoto, M.; Furukawa, K.; Hashimoto, N.; Kido, Y.; Mori, T.; Sakaue, H. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat. Med. 2004, 10, 168. [Google Scholar] [CrossRef]
- Nozaki, I.; Lunz, J.G.; Specht, S.; Stolz, D.B.; Taguchi, K.; Subbotin, V.M.; Murase, N.; Demetris, A.J. Small proline-rich proteins 2 are noncoordinately upregulated by IL-6/STAT3 signaling after bile duct ligation. Lab. Investig. 2005, 85, 109. [Google Scholar] [CrossRef]
- Saxena, N.K.; Ikeda, K.; Rockey, D.C.; Friedman, S.L.; Anania, F.A. Leptin in hepatic fibrosis: Evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology 2002, 35, 762–771. [Google Scholar] [CrossRef]
- Miller, A.M.; Wang, H.; Park, O.; Horiguchi, N.; Lafdil, F.; Mukhopadhyay, P.; Moh, A.; Fu, X.Y.; Kunos, G.; Pacher, P. Anti-inflammatory and anti-apoptotic roles of endothelial cell STAT3 in alcoholic liver injury. Alcohol. Clin. Exp. Res. 2010, 34, 719–725. [Google Scholar] [CrossRef]
- Lafdil, F.; Wang, H.; Park, O.; Zhang, W.; Moritoki, Y.; Yin, S.; Fu, X.Y.; Gershwin, M.E.; Lian, Z.X.; Gao, B. Myeloid STAT3 inhibits T cell-mediated hepatitis by regulating T helper 1 cytokine and interleukin-17 production. Gastroenterology 2009, 137, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, N.; Wang, L.; Mukhopadhyay, P.; Park, O.; Jeong, W.I.; Lafdil, F.; Osei–Hyiaman, D.; Moh, A.; Fu, X.Y.; Pacher, P. Cell type–dependent pro-and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology 2008, 134, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.A.; Grandis, J.R. STAT3 signaling: Anticancer strategies and challenges. Mol. Interv. 2011, 11, 18–26. [Google Scholar] [CrossRef]
- Liang, C.; Xu, Y.; Ge, H.; Li, G.; Wu, J. Clinicopathological significance and prognostic role of p-sTaT3 in patients with hepatocellular carcinoma. Oncotargets Ther. 2018, 11, 1203. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Tan, Z.; Deng, L.; Chen, Y.; Xia, Y.; Gao, Y.; Wang, X.; Sun, B. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology 2011, 54, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- De Yun Feng, H.Z.; Tan, Y.; Cheng, R.X. Effect of phosphorylation of MAPK and Stat3 and expression of c-fos and c-jun proteins on hepatocarcinogenesis and their clinical significance. World J. Gastroenterol. 2001, 7, 33. [Google Scholar] [CrossRef]
- Wu, W.-Y.; Li, J.; Wu, Z.-S.; Zhang, C.-L.; Meng, X.-L. STAT3 activation in monocytes accelerates liver cancer progression. BMC Cancer 2011, 11, 506. [Google Scholar] [CrossRef]
- Van Hees, S.; Michielsen, P.; Vanwolleghem, T. Circulating predictive and diagnostic biomarkers for hepatitis B virus-associated hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 8271. [Google Scholar] [CrossRef]
- Kao, J.-T.; Feng, C.-L.; Yu, C.-J.; Tsai, S.-M.; Hsu, P.-N.; Chen, Y.-L.; Wu, Y.-Y. IL-6, through p-STAT3 rather than p-STAT1, activates hepatocarcinogenesis and affects survival of hepatocellular carcinoma patients: A cohort study. BMC Gastroenterol. 2015, 15, 50. [Google Scholar] [CrossRef]
- Riehle, K.J.; Campbell, J.S.; McMahan, R.S.; Johnson, M.M.; Beyer, R.P.; Bammler, T.K.; Fausto, N. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J. Exp. Med. 2008, 205, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Rebouissou, S.; Amessou, M.; Couchy, G.; Poussin, K.; Imbeaud, S.; Pilati, C.; Izard, T.; Balabaud, C.; Bioulac-Sage, P.; Zucman-Rossi, J. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 2009, 457, 200. [Google Scholar] [CrossRef] [PubMed]
- Vasuri, F.; Visani, M.; Acquaviva, G.; Brand, T.; Fiorentino, M.; Pession, A.; Tallini, G.; D’Errico, A.; de Biase, D. Role of microRNAs in the main molecular pathways of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 2647–2660. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-C.; Ye, S.-L.; Sun, R.-X.; Liu, Y.-K.; Tang, Z.-Y.; Kim, Y.; Karras, J.G.; Zhang, H. Inhibition of growth and metastasis of human hepatocellular carcinoma by antisense oligonucleotide targeting signal transducer and activator of transcription 3. Clin. Cancer Res. 2006, 12, 7140–7148. [Google Scholar] [CrossRef]
- Hatziapostolou, M.; Polytarchou, C.; Aggelidou, E.; Drakaki, A.; Poultsides, G.A.; Jaeger, S.A.; Ogata, H.; Karin, M.; Struhl, K.; Hadzopoulou-Cladaras, M. An HNF4α-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 2011, 147, 1233–1247. [Google Scholar] [CrossRef]
- Kusaba, M.; Nakao, K.; Goto, T.; Nishimura, D.; Kawashimo, H.; Shibata, H.; Motoyoshi, Y.; Taura, N.; Ichikawa, T.; Hamasaki, K. Abrogation of constitutive STAT3 activity sensitizes human hepatoma cells to TRAIL-mediated apoptosis. J. Hepatol. 2007, 47, 546–555. [Google Scholar] [CrossRef]
- Kang, F.B.; Wang, L.; Jia, H.C.; Li, D.; Li, H.J.; Zhang, Y.G.; Sun, D.X. B7-H3 promotes aggression and invasion of hepatocellular carcinoma by targeting epithelial-to-mesenchymal transition via JAK2/STAT3/Slug signaling pathway. Cancer Cell Int. 2015, 15, 45. [Google Scholar] [CrossRef]
- Zhang, C.; Guo, F.; Xu, G.; Jia, W.; Ge, Y. STAT3 activation mediates epithelial-to-mesenchymal transition in human hepatocellular carcinoma cells. Hepato-Gastroenterol. 2014, 61, 1082–1089. [Google Scholar]
- Zhang, N.; Duan, W.D.; Leng, J.J.; Zhou, L.; Wang, X.; Xu, Y.Z.; Wang, X.D.; Zhang, A.Q.; Dong, J.H. STAT3 regulates the migration and invasion of a stem‑like subpopulation through microRNA‑21 and multiple targets in hepatocellular carcinoma. Oncol. Rep. 2015, 33, 1493–1498. [Google Scholar] [CrossRef]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552. [Google Scholar] [CrossRef]
- Tseng, P.L.; Tai, M.H.; Huang, C.C.; Wang, C.C.; Lin, J.W.; Hung, C.H.; Chen, C.H.; Wang, J.H.; Lu, S.N.; Lee, C.M. Overexpression of VEGF is associated with positive p53 immunostaining in hepatocellular carcinoma (HCC) and adverse outcome of HCC patients. J. Surg. Oncol. 2008, 98, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Won, C.; Kim, B.H.; Yi, E.H.; Choi, K.J.; Kim, E.K.; Jeong, J.M.; Lee, J.H.; Jang, J.J.; Yoon, J.H.; Jeong, W.I. Signal transducer and activator of transcription 3-mediated CD133 up-regulation contributes to promotion of hepatocellular carcinoma. Hepatology 2015, 62, 1160–1173. [Google Scholar] [CrossRef]
- Lee, T.K.W.; Castilho, A.; Cheung, V.C.H.; Tang, K.H.; Ma, S.; Ng, I.O.L. CD24+ Liver Tumor-Initiating Cells Drive Self-Renewal and Tumor Initiation through STAT3-Mediated NANOG Regulation. Cell Stem Cell 2011, 9, 50–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, S.; Wang, R.; Chen, Q.; Luo, J.; Wang, J.; Zhao, Z.; Li, Y.; Wang, Y.; Wang, X.; Cheng, B. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am. J. Cancer Res. 2018, 8, 302–316. [Google Scholar] [PubMed]
- Cheng, J.; Deng, Y.; Yi, H.; Wang, G.; Fu, B.; Chen, W.; Liu, W.; Tai, Y.; Peng, Y.; Zhang, Q. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis 2016, 5, e198. [Google Scholar] [CrossRef]
- Yin, Z.; Ma, T.; Lin, Y.; Lu, X.; Zhang, C.; Chen, S.; Jian, Z. IL-6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J. Cell. Biochem. 2018, 119, 9419–9432. [Google Scholar] [CrossRef]
- Deng, Y.; Cheng, J.; Fu, B.; Liu, W.; Chen, G.; Zhang, Q.; Yang, Y. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene 2017, 36, 1090. [Google Scholar] [CrossRef]
- Sun, X.; Sui, Q.; Zhang, C.; Tian, Z.; Zhang, J. Targeting Blockage of STAT3 in Hepatocellular Carcinoma Cells Augments NK Cell Functions via Reverse Hepatocellular Carcinoma–Induced Immune Suppression. Mol. Cancer Ther. 2013, 12, 2885–2896. [Google Scholar] [CrossRef]
- Schneller, D.; Machat, G.; Sousek, A.; Proell, V.; van Zijl, F.; Zulehner, G.; Huber, H.; Mair, M.; Muellner, M.K.; Nijman, S.M. p19ARF/p14ARF controls oncogenic functions of signal transducer and activator of transcription 3 in hepatocellular carcinoma. Hepatology 2011, 54, 164–172. [Google Scholar] [CrossRef]
- Wang, H.; Lafdil, F.; Wang, L.; Park, O.; Yin, S.; Niu, J.; Miller, A.M.; Sun, Z.; Gao, B. Hepatoprotective versus oncogenic functions of STAT3 in liver tumorigenesis. Am. J. Pathol. 2011, 179, 714–724. [Google Scholar] [CrossRef]
- Subramaniam, A.; Shanmugam, M.K.; Perumal, E.; Li, F.; Nachiyappan, A.; Dai, X.; Swamy, S.N.; Ahn, K.S.; Kumar, A.P.; Tan, B.K. Potential role of signal transducer and activator of transcription (STAT) 3 signaling pathway in inflammation, survival, proliferation and invasion of hepatocellular carcinoma. Biochim. Biophys. Acta (BBA) Rev. Cancer 2013, 1835, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Grandis, J.R.; Johnson, D.E. Chapter 7—STAT3 as a Major Contributor to Chemoresistance. In Targeting Cell Survival Pathways to Enhance Response to Chemotherapy; Johnson, D.E., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 3, pp. 145–167. [Google Scholar]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogoff, H.A.; Li, J.; Li, C. Cancer stemness and resistance: Napabucasin (BBI-608) sensitizes stemness-high cancer cells to Paclitaxel by inhibiting the STAT3-MUC1 pathway. In Proceedings of the American Association for Cancer Research Annual Meeting 2017, Washington, DC, USA, 1–5 April 2017. Cancer Res2017, 77 (13 Suppl.) Abstract nr 4777. [Google Scholar] [CrossRef]
- Langleben, A.; Supko, J.G.; Hotte, S.J.; Batist, G.; Hirte, H.W.; Rogoff, H.; Li, Y.; Li, W.; Kerstein, D.; Leggett, D.; et al. A dose-escalation phase I study of a first-in-class cancer stemness inhibitor in patients with advanced malignancies. J. Clin. Oncol. 2013, 31, 2542. [Google Scholar] [CrossRef]
- Jonker, D.J.; Stephenson, J.; Edenfield, W.J.; Supko, J.G.; Li, Y.; Li, W.; Hitron, M.; Leggett, D.; Kerstein, D.; Li, C. A phase I extension study of BBI608, a first-in-class cancer stem cell (CSC) inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 2546. [Google Scholar] [CrossRef]
- Jonker, D.J.; Nott, L.; Yoshino, T.; Gill, S.; Shapiro, J.; Ohtsu, A.; Zalcberg, J.; Vickers, M.M.; Wei, A.C.; Gao, Y.; et al. Napabucasin versus placebo in refractory advanced colorectal cancer: A randomised phase 3 trial. Lancet Gastroenterol. Hepatol. 2018, 3, 263–270. [Google Scholar] [CrossRef]
- Hitron, M.; Stephenson, J.; Chi, K.N.; Edenfield, W.J.; Leggett, D.; Li, Y.; Li, W.; Gada, K.; Li, C. A phase 1b study of the cancer stem cell inhibitor BBI608 administered with paclitaxel in patients with advanced malignancies. J. Clin. Oncol. 2014, 32, 2530. [Google Scholar] [CrossRef]
- Becerra, C.; Stephenson, J.; Jonker, D.J.; Cohn, A.L.; Asmis, T.R.; Bekaii-Saab, T.S.; Conkling, P.R.; Garbo, L.E.; Lenz, H.-J.; Richards, D.A.; et al. Phase Ib/II study of cancer stem cell (CSC) inhibitor BBI608 combined with paclitaxel in advanced gastric and gastroesophageal junction (GEJ) adenocarcinoma. J. Clin. Oncol. 2015, 33, 4069. [Google Scholar] [CrossRef]
- Becerra, C.; Garcia, A.A.; Hays, J.L.; Method, M.W.; Richey, S.L.; Langleben, A.; Richards, D.A.; Cote, G.M.; Kossler, K.; Li, W.; et al. A phase 1b/2 study of napabucasin with weekly paclitaxel in advanced, previously treated platinum resistant ovarian cancer. J. Clin. Oncol. 2017, 35, 5548. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.S.; Mikhail, S.; Langleben, A.; Becerra, C.; Jonker, D.J.; Asmis, T.R.; Cote, G.M.; Wu, C.S.-Y.; Kwak, E.L.; Spira, A.I.; et al. A phase Ib/II study of BBI608 combined with weekly paclitaxel in advanced pancreatic cancer. J. Clin. Oncol. 2016, 34, 196. [Google Scholar] [CrossRef]
- Becerra, C.; Braiteh, F.S.; Spira, A.I.; Langleben, A.; Panasci, L.C.; Vukelja, S.J.; Hinshaw, I.M.; Goodwin, R.A.; Panella, T.J.; Edenfield, W.J.; et al. A Phase Ib/II Study of Cancer Stemness Inhibitor Napabucasin (BB608) Combined with Weekly Paclitaxel in Advanced Triple Negative Breast Cancer. J. Clin. Oncol. 2016, 34, 1094. [Google Scholar] [CrossRef]
- Larson, T.; Ortuzar, W.F.; Bekaii-Saab, T.S.; Becerra, C.; Ciombor, K.K.; Hubbard, J.M.; Edenfield, W.J.; Shao, S.H.; Grothey, A.; Borodyansky, L.; et al. BBI608-224: A phase Ib/II study of cancer stemness inhibitor napabucasin (BBI-608) administered with panitumumab in KRAS wild-type patients with metastatic colorectal cancer. J. Clin. Oncol. 2017, 35, 677. [Google Scholar] [CrossRef]
- Ciombor, K.K.; Edenfield, W.J.; Hubbard, J.M.; O’Dwyer, P.J.; Becerra, C.; Larson, T.; Spira, A.I.; Grothey, A.; Manoharan, D.; Li, W.; et al. A phase Ib/II study of cancer stem cell inhibitor BBI608 administered with panitumumab in KRAS wild-type (wt) patients (pts) with metastatic colorectal cancer (mCRC) following progression on anti-EGFR therapy. J. Clin. Oncol. 2015, 33, 3617. [Google Scholar] [CrossRef]
- Bendell, J.C.; O’Neil, B.H.; Starodub, A.; Jonker, D.J.; Halfdanarson, T.R.; Edenfield, W.J.; El-Rayes, B.F.; Hubbard, J.M.; Pitot, H.C.; Hobday, T.J.; et al. Cancer stemness inhibition and chemosensitization: Phase 1b/II study of cancer stemness inhibitor napabucasin (BBI-608) with FOLFIRI +/− bevacizumab (Bev) administered to colorectal cancer (CRC) patients (pts). J. Clin. Oncol. 2017, 35, 593. [Google Scholar] [CrossRef]
- O’Neil, B.H.; Hubbard, J.M.; Starodub, A.; Jonker, D.J.; Edenfield, W.J.; El-Rayes, B.F.; Halfdanarson, T.R.; Ramanathan, R.K.; Pitot, H.C.; Britten, C.D.; et al. Phase 1b extension study of cancer stemness inhibitor BB608 (napabucasin) administered in combination with FOLFIRI +/− bevacizumab (Bev) in patients (pts) with advanced colorectal cancer (CRC). J. Clin. Oncol. 2016, 34, 3564. [Google Scholar] [CrossRef]
- El-Rayes, B.F.; Shahda, S.; Starodub, A.; O’Neil, B.H.; Hanna, W.T.; Shaib, W.L.; Oh, C.; Li, W.; Li, Y.; Borodyansky, L.; et al. A phase Ib extension study of cancer stemness inhibitor BB608 (napabucasin) in combination with gemcitabine and nab-paclitaxel (nab-PTX) in patients (pts) with metastatic pancreatic cancer. J. Clin. Oncol. 2016, 34, 4128. [Google Scholar] [CrossRef]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef]
- Shastri, A.; Choudhary, G.; Teixeira, M.; Gordon-Mitchell, S.; Ramachandra, N.; Bernard, L.; Bhattacharyya, S.; Lopez, R.; Pradhan, K.; Giricz, O. Antisense STAT3 inhibitor decreases viability of myelodysplastic and leukemic stem cells. J. Clin. Investig. 2018, 128. [Google Scholar] [CrossRef]
- Odate, S.; Veschi, V.; Yan, S.; Lam, N.; Woessner, R.; Thiele, C.J. Inhibition of STAT3 with the generation 2.5 antisense oligonucleotide, AZD9150, decreases neuroblastoma tumorigenicity and increases chemosensitivity. Clin. Cancer Res. 2017, 23, 1771–1784. [Google Scholar] [CrossRef]
- AstraZeneca. A Phase I/Ib Study of AZD9150 (ISIS-STAT3Rx) in Patients with Advanced/Metastatic Hepatocellular Carcinoma; AstraZeneca: London, UK, 2017. [Google Scholar]
- Visconti, L.; Nelissen, K.; Deckx, L.; Akker, M.v.d.; Adriaensen, W.; Daniels, L.; Matheï, C.; Linsen, L.; Hellings, N.; Stinissen, P. Prognostic value of circulating cytokines on overall survival and disease-free survival in cancer patients. Biomark. Med. 2014, 8, 297–306. [Google Scholar] [CrossRef]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, L.; Genini, D.; Laurini, E.; Merulla, J.; Perez, L.; Fermeglia, M.; Carbone, G.M.; Pricl, S.; Catapano, C.V. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3). Mol. Oncol. 2015, 9, 1194–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, F.; Sugimoto, K.; Harada, Y.; Hashimoto, N.; Ohi, N.; Kurahashi, S.; Naoe, T. A novel STAT inhibitor, OPB-31121, has a significant antitumor effect on leukemia with STAT-addictive oncokinases. Blood Cancer J. 2013, 3, e166. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Nam, H.-J.; Kim, H.-P.; Han, S.-W.; Im, S.-A.; Kim, T.-Y.; Oh, D.-Y.; Bang, Y.-J. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 2013, 335, 145–152. [Google Scholar] [CrossRef]
- Bendell, J.C.; Hong, D.S.; Burris, H.A.; Naing, A.; Jones, S.F.; Falchook, G.; Bricmont, P.; Elekes, A.; Rock, E.P.; Kurzrock, R. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 125–130. [Google Scholar] [CrossRef]
- Tolcher, A.; Flaherty, K.; Shapiro, G.I.; Berlin, J.; Witzig, T.; Habermann, T.; Bullock, A.; Rock, E.; Elekes, A.; Lin, C. A First-in-Human Phase I Study of OPB-111077, a Small-Molecule STAT3 and Oxidative Phosphorylation Inhibitor, in Patients with Advanced Cancers. Oncology 2018, 23, e658–e672. [Google Scholar] [CrossRef]
- Yoo, C.; Kang, J.; Kim, K.-P.; Lim, H.Y.; Kim, J.H.; Lee, M.A.; Kim, T.-Y.; Ryoo, B.-Y. Phase I dose-finding study of OPB-111077, a novel STAT3 inhibitor, in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2018, 36, 4078. [Google Scholar] [CrossRef]
- Wong, A.; Soo, R.A.; Tan, D.; Lee, S.C.; Lim, J.; Marban, P.; Kong, L.R.; Lee, Y.; Wang, L.; Thuya, W.L. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. 2015, 26, 998–1005. [Google Scholar] [CrossRef]
- Ogura, M.; Uchida, T.; Terui, Y.; Hayakawa, F.; Kobayashi, Y.; Taniwaki, M.; Takamatsu, Y.; Naoe, T.; Tobinai, K.; Munakata, W. Phase I study of OPB-51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015, 106, 896–901. [Google Scholar] [CrossRef]
- National University Hospital Singapore. OPB-51602 in Locally Advanced Nasopharyngeal Carcinoma Prior to Definitive Chemoradiotherapy; National University Hospital: Singapore, 2015. [Google Scholar]
- Genini, D.; Brambilla, L.; Laurini, E.; Merulla, J.; Civenni, G.; Pandit, S.; D’Antuono, R.; Perez, L.; Levy, D.E.; Pricl, S.; et al. Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E4924–E4933. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.W.; Saadalla, A.; Ewida, A.H.; Al-Katranji, K.; Al-Saoudi, G.; Giaccone, Z.T.; Gounari, F.; Zhang, M.; Frank, D.A.; Khazaie, K. The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer. Cancer Immunol. Immunother. 2018, 67, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Kettner, N.M.; Vijayaraghavan, S.; Durak, M.G.; Bui, T.; Kohansal, M.; Ha, M.J.; Liu, B.; Rao, X.; Wang, J.; Yi, M. Combined inhibition of STAT3 and DNA repair in palbociclib-resistant ER-positive breast cancer. Clin. Cancer Res. 2019, 25, 3996–4013. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, J.; Wang, L.; Tian, Z. Growth inhibition of human hepatocellular carcinoma cells by blocking STAT3 activation with decoy-ODN. Cancer Lett. 2008, 262, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Wang, L.; Wei, H.; Tian, Z. Therapeutic effects of STAT3 decoy oligodeoxynucleotide on human lung cancer in xenograft mice. BMC Cancer 2007, 7, 149. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Wei, H.; Tian, Z. STAT3-decoy oligodeoxynucleotide inhibits the growth of human lung cancer via down-regulating its target genes. Oncol. Rep. 2007, 17, 1377–1382. [Google Scholar] [CrossRef]
- Leong, P.L.; Andrews, G.A.; Johnson, D.E.; Dyer, K.F.; Xi, S.; Mai, J.C.; Robbins, P.D.; Gadiparthi, S.; Burke, N.A.; Watkins, S.F. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc. Natl. Acad. Sci. USA 2003, 100, 4138–4143. [Google Scholar] [CrossRef] [Green Version]
- Sen, M.; Thomas, S.M.; Kim, S.; Yeh, J.I.; Ferris, R.L.; Johnson, J.T.; Duvvuri, U.; Lee, J.; Sahu, N.; Joyce, S. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: Implications for cancer therapy. Cancer Discov. 2012, 2, 694–705. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, P.; Zhang, B.; Wang, A.; Yang, M. Role of STAT3 decoy oligodeoxynucleotides on cell invasion and chemosensitivity in human epithelial ovarian cancer cells. Cancer Genet. Cytogenet. 2010, 197, 46–53. [Google Scholar] [CrossRef]
- Gao, Y.; Li, Y.; Hsu, E.; Wang, Y.; Huang, J.; Brooks, E.; Li, C.J. Abstract LB-140: Inhibition of cancer stemness sensitizes colorectal cancer to immune checkpoint inhibitors. Cancer Res. 2017, 77, LB-140. [Google Scholar] [CrossRef]
- Mamdani, H.; Wu, H.; O’Neil, B.H.; Sehdev, A. Excellent Response to Anti-PD1 therapy in a Patient with Hepatocellular Carcinoma: Case Report and Review of Literature. Discov. Med. 2017, 23, 331–336. [Google Scholar]
Figure 1.
The STAT3 signaling pathway and its crosstalk with NF-kB. STAT3 is activated primarily by cytokines and growth factors, in addition to other signaling molecules. Canonically, ligand binding to receptors triggers phosphorylation of tyrosine kinases and subsequently STAT3 at Y705, followed by STAT3 dimerization and translocation to the nucleus where it drives transcription of target genes involved in cell survival and proliferation. Non-canonically, STAT3 can also be phosphorylated at S727, translocate to the mitochondrion, as well as autoregulate its own transcription to produce u-STAT3. Under normal conditions, STAT3 activation is under tight negative regulation by SOCS, PTP and PIAS members. Remarkably, STAT3 is involved in extensive crosstalk with the inflammatory NF-kB pathway. Activated NF-kB has been reported to either activate or inhibit STAT3 signaling, respectively by producing various cytokines including the major STAT3-inducing cytokine IL-6 and preventing reactive oxygen species (ROS) accumulation responsible for oxidizing negative regulators of STAT3. In return, STAT3 may sustain NF-kB activation via p300-mediated acetylation. Moreover, u-STAT3 and u-NF-kB can work in concert to coregulate another set of genes.
Figure 1.
The STAT3 signaling pathway and its crosstalk with NF-kB. STAT3 is activated primarily by cytokines and growth factors, in addition to other signaling molecules. Canonically, ligand binding to receptors triggers phosphorylation of tyrosine kinases and subsequently STAT3 at Y705, followed by STAT3 dimerization and translocation to the nucleus where it drives transcription of target genes involved in cell survival and proliferation. Non-canonically, STAT3 can also be phosphorylated at S727, translocate to the mitochondrion, as well as autoregulate its own transcription to produce u-STAT3. Under normal conditions, STAT3 activation is under tight negative regulation by SOCS, PTP and PIAS members. Remarkably, STAT3 is involved in extensive crosstalk with the inflammatory NF-kB pathway. Activated NF-kB has been reported to either activate or inhibit STAT3 signaling, respectively by producing various cytokines including the major STAT3-inducing cytokine IL-6 and preventing reactive oxygen species (ROS) accumulation responsible for oxidizing negative regulators of STAT3. In return, STAT3 may sustain NF-kB activation via p300-mediated acetylation. Moreover, u-STAT3 and u-NF-kB can work in concert to coregulate another set of genes.

Figure 2.
STAT3-mediated oncogenesis in hepatocellular carcinoma (HCC). IL-6 and IL-22 are major stimuli that activate STAT3 signaling in HCC cells. Activated STAT3 promotes HCC cell proliferation, anti-apoptosis, migration, invasion, angiogenesis and stemness properties via transcriptional regulation of target genes, cooperation with NF-kB and epigenetic regulation involving miRNA. A paracrine STAT3 activation loop between tumor and stromal cells also exerts suppressive effects in various immune cells in the HCC tumor microenvironment.
Figure 2.
STAT3-mediated oncogenesis in hepatocellular carcinoma (HCC). IL-6 and IL-22 are major stimuli that activate STAT3 signaling in HCC cells. Activated STAT3 promotes HCC cell proliferation, anti-apoptosis, migration, invasion, angiogenesis and stemness properties via transcriptional regulation of target genes, cooperation with NF-kB and epigenetic regulation involving miRNA. A paracrine STAT3 activation loop between tumor and stromal cells also exerts suppressive effects in various immune cells in the HCC tumor microenvironment.


{kind=link}
{kind=link}
Table 1.
Clinical trials of napabucasin: cancer stemness inhibitor targeting STAT3-driven gene transcription.
Table 1.
Clinical trials of napabucasin: cancer stemness inhibitor targeting STAT3-driven gene transcription.
| NCT No. | Phase | Therapy | Cancer Types | Brief Description | Time | Ref |
|---|---|---|---|---|---|---|
| NCT 01775423 | Ib | Monotherapy | Advanced solid cancers (n = 41) | Dose escalation achieved | 2009–2019 | [87] |
| Ib | Advanced solid cancers (n = 24) | Safety and antitumor activity demonstrated in higher strength capsule | [88] | |||
| NCT 01830621 | III | Monotherapy | Advanced CRC (n = 280) | Prolonged OS demonstrated after pSTAT3 stratification | 2013–2016 | [89] |
| NCT 03416816 | I | Monotherapy | Advanced solid tumors (n = 90) | To evaluate the safety, tolerability, pharmacokinetic profile and antitumor activity of pro-drug of napabucasin, DSP-0337 | 2018–2020 | / |
| NCT 01325441 | Ib/II | Combination | Advanced solid cancers (n = 24) | Safety and antitumor activity demonstrated with paclitaxel, particularly in gastric and GEJ adenocarcinoma | 2011–2020 | [90] |
| Ib/II | Advanced gastric and GEJ adenocarcinoma (n = 46) | Safety and antitumor activity demonstrated with paclitaxel, regardless of prior taxane exposure | [91] | |||
| Ib/II | Advanced PROC (n = 98) | Safety and antitumor activity demonstrated with paclitaxel, including 3 completes responses | [92] | |||
| Ib/II | Advanced PDAC (n = 41) | Safety and antitumor activity demonstrated with paclitaxel, particularly in taxane-naïve group | [93] | |||
| Ib/II | Advanced TNBC (n = 35) | Safety and antitumor activity demonstrated with paclitaxel, particularly in taxane-exposed therapy | [94] | |||
| NCT 01776307 | Ib/II | Combination | KRAS-wt mCRC (n = 24) | Safety and antitumor activity demonstrated with panitumumab, regardless of prior anti-EGFR exposure | 2012–2019 | [95] |
| II | KRAS-wt mCRC (n = 72) | [96] | ||||
| NCT 02024607 | Ib/II | Combination | Advanced CRC (n = 63) | Safety and antitumor activity demonstrated with FOLFIRI ± bevacizumab | 2014–2019 | [97] |
| III | Advanced CRC (n = 46) | Safety and antitumor activity demonstrated with FOLFIRI ± bevacizumab, regardless of prior FOLFIRI ± bevacizumab exposure and pSTAT3 status | [98] | |||
| NCT 02231723 | III | Combination | mPDAC (n = 37) | Safety and antitumor activity demonstrated with gemcitabine and nab-paclitaxel | 2014–2020 | [99] |
| NCT 02178956 | III | Combination | Advanced gastric and GEJ adenocarcinoma (n = 680) | To determine if napabucasin in combination with paclitaxel prolongs OS than paclitaxel alone | 2014–2019 | / |
| NCT 02279719 | Ib/II | Combination | Advanced HCC (n = 99) | To evaluate the safety, tolerability, pharmacokinetic profile and antitumor activity in combination with sorafenib in comparison with sorafenib alone | 2014–2019 | / |
| NCT 02358395 | I | Combination | Advanced HCC (n = 12) | To evaluate the safety, tolerability, pharmacokinetic profile and antitumor activity in combination with sorafenib | 2015–2019 | / |
| NCT 02467361 | III | Combination | Advanced cancers (n = 104) | To evaluate the safety, tolerability, pharmacokinetic profile and antitumor activity in combination with immune checkpoint inhibitors | 2015–2019 | / |
| NCT 02753127 | III | Combination | mCRC (n = 1253) | To determine if napabucasin in combination with FOLFIRI prolongs OS than FOLFIRI alone | 2016–2020 | / |
| NCT 02993731 | III | Combination | mPDAC (n = 1134) | To determine if napabucasin in combination with nab-paclitaxel and gemcitabine prolongs OS than nab-paclitaxel and gemcitabine alone | 2016–2020 | / |
| NCT 03416816 | I | Monotherapy | Advanced solid tumors (n = 90) | To evaluate the safety, tolerability, pharmacokinetic profile and antitumor activity | 2018–2020 | / |
Table 2.
Clinical trials of AZD9150: STAT3-targetd antisense oligonucleotide.
| NCT No. | Phase | Therapy | Cancer Types | Brief Description | Time | Ref. |
|---|---|---|---|---|---|---|
| NCT 01563302 | I/II | Monotherapy | Predominantly refractory DLBCL (n = 30) | Safety and antitumor activity demonstrated | 2012–2016 | [105] |
| NCT 01839604 | II | Monotherapy | Advanced HCC (n = 58) | Safety, tolerability and pharmacokinetics evaluated but with limited antitumor activity | 2013–2015 | [103] |
| Not stated | I | Monotherapy | Advanced lymphoma and solid tumors (n = 25) | Safety and antitumor activity demonstrated | 2015 | [100] |
| NCT 02499328 | I & II | Combination | Advanced solid tumors (n = 465) | To evaluate the safety, tolerability and antitumor activity in combination with durvalumab, tremelimumab and AZD5069 | 2015–2020 | / |
| NCT 02549651 | 1 | Combination | Relapsed/refractory DLBCL (n = 32) | To evaluate the safety, tolerability and antitumor activity in combination with durvalumab and tremelimumab compared with durvalumab alone | 2016–2019 | / |
| NCT 02546661 | I | Combination | MIBC (n = 156) | To evaluate the safety, tolerability and antitumor activity of durvalumab in combination with AZD9150 or other novel anticancer agents compared with durvalumab alone | 2016–2020 | / |
| NCT 02983578 | II | Combination | Advanced pancreatic, lung, colorectal cancer (n = 75) | To evaluate the antitumor activity and tumor-based biomarkers in combination with durvalumab | 2017–2021 | / |
| NCT 03334617 | II | Combination | Advanced non-small cell lung cancer (NSCLC) (n = 260) | To evaluate the safety, tolerability and antitumor activity of different combinations of anticancer agents | 2017–2021 | / |
| NCT 03394144 | I | Combination | Advanced solid tumors (n = 110) | To evaluate the safety, tolerability and antitumor activity in combination with durvalumab compared with AZD9150 alone | 2018–2019 | / |
| NCT 03421353 | I & II | Combination | Advanced solid tumors (n = 110) | To evaluate the safety, tolerability and antitumor activity in combination with durvalumab and chemodrugs compared with AZD9150 alone; to compare its bioavailability of subcutaneous and intravenous formulations | 2018–2020 | / |
| NCT 03742102 | Ib/II | Combination | mTNBC (n = 110) | To evaluate the safety, tolerability and antitumor activity of durvalumab and paclitaxel in combination with AZD9150 or other novel anticancer agents | 2018–2020 | / |
| NCT 03819465 | Ib | Combination | Advanced NSCLC (n = 200) | To evaluate the safety, tolerability and antitumor activity of durvalumab in combination with AZD9150 or other novel anticancer agents +/- chemotherapy | 2018–2020 | / |
| NCT 03527147 | I | Combination | Relapsed/refractory non-Hodgkin lymphoma (n = 88) | To evaluate the safety, tolerability and antitumor activity of different combinations of targeted agents | 2018–2021 | / |
Table 3.
Clinical trials of OPB compounds, pyrimethamine, TTI-101: STAT3 SH2 domain inhibitors.
| NCT No. | Phase | Therapy | Cancer Types | Brief Description | Time | Ref. |
|---|---|---|---|---|---|---|
| OPB-31121 | ||||||
| NCT 00955812 | I | Monotherapy | Advanced solid tumors (n = 14) | Unfavorable pharmacokinetic profile and antitumor activity demonstrated, leading to discontinuation of compound development | 2009–2012 | [109] |
| OPB-111077 | ||||||
| NCT 01711034 | I | Monotherapy | Advanced solid tumors (n = 145) | Safety, tolerability and antitumor activity demonstrated, including one PR in DLBCL | 2012–2015 | [110] |
| NCT 01942083 | I | Monotherapy | Advanced HCC (n = 33) | Safety and tolerability demonstrated but no antitumor response was observed | 2013–2017 | [111] |
| NCT 02250170 | I | Monotherapy | Advanced solid tumors (n = 47) | To evaluate the safety, tolerability and antitumor activity | 2014–2019 | / |
| NCT 03197714 | Ib | Monotherapy | Relapsed/refractory AML (n = 15) | To evaluate the safety, tolerability and antitumor activity | 2017–2018 | / |
| NCT 03158324 | IIa | Monotherapy | NPC/refractory tumors (n = 52) | To evaluate the safety, tolerability and antitumor activity | 2017–2020 | / |
| NCT 03063944 | I | Combination | AML (n = 12) | To evaluate the safety, tolerability and antitumor activity in combination with decitabine | 2017–2023 | / |
| NCT 04049825 | I | Combination | Relapsed/refractory DLBCL (n = 65) | To evaluate the safety, tolerability and antitumor activity in combination with bendamustine and rituximab | 2019–2021 | / |
| OPB-51602 | ||||||
| NCT 01184807 | I | Monotherapy | Refractory solid tumors (n = 51) | Better tolerability for intermittent than continuous dosing; antitumor activity demonstrated, including 2 PRs in EGFR mutation-positive NSCLC prior anti-EGFR exposure | 2009–2013 | [112] |
| NCT 01423903 | I | Monotherapy | Advanced cancers (n = 45) | To evaluate the safety, tolerability, pharmacokinetic profile and antitumor activity | 2010–2013 | / |
| NCT 01344876 | I | Monotherapy | Relapsed/refractory hematological malignancies (n = 20) | Safety and tolerability demonstrated but long-term administration at higher doses was difficult with daily dosing and no antitumor response was observed, leading to termination of study | 2011–2014 | [113] |
| NCT 01867073 | I | Monotherapy | Advanced solid tumors (n = 20) | To evaluate the pharmacokinetic profile in relation to pSTAT3 expression in peripheral mononuclear blood cells and single nucleotide polymorphisms in patient tissues | 2011–2015 | / |
| NCT 02058017 | I | Monotherapy | Advanced NPC (n = 9) | Poor tolerability, leading to termination of study | 2013–2015 | [114] |
| Pyrimethamine | ||||||
| NCT 01066663 | I & II | Monotherapy | Relapsed CLL/SLL (n = 26) | To evaluate the safety, tolerability, pharmacokinetic profile and antitumor activity | 2010–2024 | / |
| TTI-101 | ||||||
| NCT 03195699 | I | Monotherapy | Advanced cancers (n = 30) | To evaluate the safety, tolerability and pharmacokinetic profile | 2017–2020 | / |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, C.; Cheung, S.T. STAT3: An Emerging Therapeutic Target for Hepatocellular Carcinoma. Cancers 2019, 11, 1646. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11111646
AMA Style
Lee C, Cheung ST. STAT3: An Emerging Therapeutic Target for Hepatocellular Carcinoma. Cancers. 2019; 11(11):1646. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11111646
Chicago/Turabian StyleLee, Carol, and Siu Tim Cheung. 2019. "STAT3: An Emerging Therapeutic Target for Hepatocellular Carcinoma" Cancers 11, no. 11: 1646. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11111646
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.