STAT5A and STAT5B—Twins with Different Personalities in Hematopoiesis and Leukemia

Institute of Pharmacology and Toxicology, University of Veterinary Medicine, 1210 Vienna, Austria
*
Author to whom correspondence should be addressed.
†
Equal first author contribution.
Cancers 2019, 11(11), 1726; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11111726
Submission received: 30 September 2019
/
Revised: 25 October 2019
/
Accepted: 1 November 2019
/
Published: 4 November 2019
(This article belongs to the Special Issue JAK-STAT Signalling Pathway in Cancer)
Abstract
:The transcription factors STAT5A and STAT5B have essential roles in survival and proliferation of hematopoietic cells—which have been considered largely redundant. Mutations of upstream kinases, copy number gains, or activating mutations in STAT5A, or more frequently in STAT5B, cause altered hematopoiesis and cancer. Interfering with their activity by pharmacological intervention is an up-and-coming therapeutic avenue. Precision medicine requests detailed knowledge of STAT5A’s and STAT5B’s individual functions. Recent evidence highlights the privileged role for STAT5B over STAT5A in normal and malignant hematopoiesis. Here, we provide an overview on their individual functions within the hematopoietic system.
Keywords:
STAT5A; STAT5B; hematopoietic stem cells; STAT5BN642H; STAT5 mouse models; BCR–ABL; leukemia; hematopoiesis1. Introduction
The transcription factors signal transducer and activator of transcription 5A (STAT5A) and 5B (STAT5B) are part of the highly conserved Janus kinase (JAK)/STAT signaling pathway. They fulfill critical functions in processes like proliferation, differentiation, survival, and senescence. Triggers for JAK activation come from the stimulation of upstream membrane receptors which respond to cytokines or growth factors. Upon activation, JAK family members phosphorylate STAT5A/B on a critical tyrosine residue (pYSTAT5A/B), which induces a conformational change to parallel STAT5A/B dimers, exposing the DNA binding domain. After nuclear import, gene transcription is typically initiated at gamma interferon-activated sequence (GAS) motifs [1,2]. Beside the “classical” canonical signal transduction pathway, STAT5A/B can function via tyrosine phosphorylation-independent mechanisms (unphosphorylated STAT5A/B, uSTAT5A/B). As uSTATs, they seem to have a more global role by interacting with epigenetic and chromatin modifiers [3,4,5].
STAT5A/B signaling is enhanced in diverse hematopoietic cancers and is believed to drive disease. Enhanced STAT5A/B activation is achieved by copy number gains, enhanced protein expression, or gain-of-function (GOF) mutations, leading to higher pYSTAT5A/B levels contributing to tumor cell survival and disease progression. As such, STAT5A and STAT5B are in the focus of current pharmaceutical research [6]. Activating mutations occur much more frequently in STAT5B than in STAT5A, the underlying reason being widely enigmatic. The recent evidence has provoked studies that provide insights into specific roles of STAT5A and STAT5B in hematopoiesis, immune cell functions, and leukemogenesis—knowledge needed for future drug development approaches. This review focuses on the specific roles of STAT5A and STAT5B in different hematopoietic cell types and their impact on hematopoietic malignancies and treatment options.
2. Differences and Similarities of STAT5A and STAT5B
STAT5A and STAT5B arose approximately 310 to 130 million years ago in the course of early eutherian evolution. While birds and several other animals harbor one STAT5 gene, the co-presence of STAT5A and STAT5B became a mammal-specific feature [7,8], along with STAT5A’s master regulatory function in the mammary gland [9]. Interestingly, zebrafish developed an independent duplication of the stat5 gene, consisting of stat5.1 and stat5.2 [10]. The intron–exon structure of the zebrafish and the murine isoforms are highly concordant. In contrast to the mammalian isoforms, zebrafish stat5.1 and stat5.2 are located on different chromosomes. Stat5.1 is highly homologous to mammalian STAT5A and STAT5B, while stat5.2 lacks a mammalian orthologue [11,12]. Mutated stat5.1 zebrafish displayed a reduced body size, in line with reduced growth hormone (gh)1 mRNA levels and Stat5.1 binding to the gh1 promoter, while stat5.2-mutated zebrafish showed no developmental defect [13].
Initially, STAT5A/B was described as a prolactin-responsive DNA binding protein in mammary epithelial cells [14,15]. Soon afterwards, it was found that interleukin (IL)-2, IL-3, and erythropoietin (EPO) signaling activate the protein by tyrosine phosphorylation and that it exists in two flavors—STAT5A and STAT5B [16,17,18,19]. STAT5A and STAT5B are encoded by two juxtaposed genes with the transcriptional start sites within 10.7 kb of each other, mapping to chromosome #17 in humans and to chromosome #11 in mice. They are translated to two more than 90% homologous proteins differing primarily at their C-termini [20] (see Figure S1). Similar to other STAT proteins, STAT5A and STAT5B consist of six functional domains (Figure 1): The N-terminus is important for oligomerization, and the C-terminus contains the phosphorylation sites involved in STAT5A/B activation [21,22,23,24]. Comparing their protein structures, STAT5A has 12 amino acids more on the C-terminus. The last 20 amino acids of STAT5A and the last 8 amino acids of STAT5B are unique to the respective proteins. STAT5A differs in one residue and lacks 5 residues between the Src-homology 2 (SH2) and transactivation domain, the so-called phosphotyrosyl tail [25,26], depicted in Figure 1 and Figure S1. These differences may account for the non-redundant roles of STAT5A and STAT5B by affecting gene regulation or specific protein–protein interactions [27,28]. The DNA binding domain differs by five amino acids which contribute to homodimer-specific DNA binding affinities [25]. These individual DNA binding specificities of pYSTAT5A/B homo- or heterodimers may influence the transcription of target genes [25,29], but the formation of pYSTAT5A/B homo- and heterodimers was suggested to occur randomly [30]. Different STAT5A/B expression levels, cytokine receptor affinities, and oligomerization properties are further factors probably influencing the signaling response in each cell type.
STAT5A/B functions are modified via post-translational modifications at different sites (Figure 1). The critical tyrosine phosphorylation sites for activation are Y694 in STAT5A and Y699 in STAT5B [31]. In addition, serine phosphorylation at S726 and S780 for STAT5A (corresponding mouse serine phosphorylation sites S725 and S779) and at S715 and S731 for STAT5B enables enhanced activation and nuclear translocation [32,33]. STAT5A contains two additional phosphorylation sites: STAT5A S127/S128 involved in ERB4-mediated activation; and STAT5A T682/T683 associated with IL-3 signaling [34,35]. STAT5B comprises additional phosphorylation sites taking part in inducing or inhibiting transcription, e.g., S193 is associated with mTOR kinase activity [36,37,38,39,40]. Known upstream kinases for serine phosphorylation are the MAPK family, ERKs, JNK, p38 MAPK, PAK kinases in a RHO/RAC dependent manner, and CDK8. The latter was associated with enhanced mediator complex occupancy at its target genes [32,41,42]. Additionally, STAT5B tyrosine phosphorylation sites Y725, Y740, and Y743 were described to be highly induced by epidermal growth factor (EGF) stimulation. While Y740 and Y743 were reported as negative regulators of transcription by reducing Y699 phosphorylation, Y725 displayed a much weaker effect with controversial transcriptional contributions [40,43,44]. STAT5B also contains SUMOylation (inhibiting STAT5 phosphorylation) and acetylation (promotes STAT5 phosphorylation) sites—lysine acetylation may even be a prerequisite for efficient STAT5 dimerization, translocation, and activation of transcription [45,46,47]. O-GlcNAcylation of STAT5A’s T92 was described to enhance tyrosine phosphorylation and, consequently, transactivation [48].
A different mode of action of STAT5A/B is added by non-canonical functions of uSTAT5 first shown in Drosophila [49]. In a colon cancer model, uSTAT5A stabilized heterochromatin by binding to heterochromatin protein 1α (HP1α) and suppressing the “cancer expression signature” [3]. In hematopoietic progenitor cells, uSTAT5 prevented megakaryocyte differentiation [5], as discussed below. A very recent study focusing on uSTAT5A and uSTAT5B in acute myeloid leukemia (AML) suggested that uSTAT5B is a key regulator of differentiation of AML cells. Isoform-specific interaction partners were identified in AML cell lines: uSTAT5A interacts with DBC1, while uSTAT5B interacts with ETV6 [50].
Various activating and repressing interactions with transcriptional co-factors and epigenetic modulators have been described for STAT5A/B, which have been recently reviewed [51]. In the following, we focus on our current understanding of STAT5A and STAT5B functions in the differentiation of hematopoietic lineages.
3. STAT5A/B Deficiency in Mice and Men
To understand the roles of STAT5A/B, genetically engineered mice were generated (Table 1). First insights were derived by Stat5a/bΔN mice, which expressed truncated N-termini of STAT5A and STAT5B [52,53,54,55,56]. STAT5A/BΔN proteins still formed dimers and bound DNA, but tetramer formation and complete target gene transcription were significantly impaired [21]. Hematopoiesis in Stat5aΔN, Stat5bΔN, and Stat5a/bΔN mice was affected to a minor degree [56]. Likewise, tetramer formation was blocked in the Stat5a/bDKI (double knock-in) mouse model, in which mutations were introduced into the N-termini of Stat5a or Stat5b. Both mouse models showed reduced numbers of natural killer (NK) cells, while T cell numbers were exclusively reduced in Stat5a/bΔN mice [57,58].
The complete genetic abrogation of STAT5A and STAT5B (Stat5a/b−/−) resulted in perinatal lethality; the few survivors displayed severe microcytic anemia, reduced numbers of CD8+ T cells, and a block in the pre–pro-B cell stage. The anemia was explained by apoptosis of fetal liver cells and reduced expression of iron-regulatory protein 2 (IRP2) and transferrin receptor 1 (TFR1) [52,59,64]. To study tissue-specific STAT5A/B functions or allow conditional deletion, Stat5a/b floxed mice [59] were crossed with Mx1-Cre, vav1-Cre, or Tie2-Cre mice to elucidate STAT5A/B’s function in the hematopoietic system [60,61,62]. As the prenatal lethality of Stat5a/b−/− mice was connected to severe combined immunodeficiency, erythroid defects, and subsequent anemia [52,64,65], it was somehow surprising that hematopoietic-specific STAT5A/B deletion led to anemia and lymphopenia, but did not influence survival [60,61].
Moreover, mouse models either lacking STAT5A [9] or STAT5B [63] gave insight into gene-specific functions. Stat5a−/− females showed defective mammary gland formation and failed to lactate, while Stat5b−/− mice were smaller, showed altered GH signaling, and displayed stronger hematopoietic defects [9,63,66]. “Compensatory” mechanisms due to the absence of STAT5A or STAT5B in the whole organism cannot be ruled out. Murine models with floxed loci of either Stat5a or Stat5b are so far unavailable.
Humans with STAT5B deficiency suffer from a rare autosomal disorder resulting in dwarfism, prominent forehead, eczema, and a high-pitched voice. In line with the role of STAT5B as mediator of IL-2 signaling, these patients undergo recurrent infections due to immunodeficiency caused by impairment in T, regulatory T (Treg), and NK cell differentiation and activation. The homozygous mis- or nonsense mutations in these patients lead to non-detectable STAT5B expression [67,68,69,70,71].
Recently, dominant negative germline mutations of STAT5B were discovered in patients. Here, the wild-type (wt)–mutant heterodimers fail to translocate to the nucleus or bind DNA. This leads to growth failure and hyper-IgE syndrome [72].
Importantly, STAT5B deficiency-associated diseases confirm that STAT5A does not compensate for all functions of STAT5B. Until now, STAT5A deficiency in humans has not been reported. One may speculate that this provokes only a very mild or absent phenotype, or—the opposite—its absence would be fatal.
4. STAT5B as the Dominant Player in Hematopoietic Lineages
STAT5A/B are fundamental for myelopoiesis, lymphoid development, macrophage functions, megakaryopoiesis, basophil, eosinophil, and mast cell functions [73,74,75]. This is explained by STAT5A/B’s function as key signaling molecules downstream of various cytokine and growth factor receptors, e.g., IL-2, -3, -4, -5, -7, -9, -13, -15, -21, EPO, thrombopoietin (TPO), GH, prolactin, stem cell factor (SCF), Flt3, granulocyte-macrophage (GM) colony-stimulating factor (CSF) or GCSF [73,76]. In all differentiated hematopoietic cell types, STAT5B is expressed at higher levels compared to STAT5A (Figure 2) [77].
4.1. STAT5A/B as Regulators of Erythropoiesis
The importance of STAT5A/B in erythroid differentiation downstream of EPO/EPOR/JAK2 signaling has been well established [78,79]. Hematopoietic deletion of STAT5A/B resulted in anemia, defining STAT5A/B as regulator of iron uptake (control of TFR1 expression) and survival genes in erythroid cells [60,61]. Their essential role was demonstrated by Stat5aS710F (cS5F, a hyperactive Stat5a variant) expression in Epor−/− or Jak2−/− fetal liver cells, enabling self-renewal and erythroid differentiation [80]. Overexpression of cS5F in human CD34+ cells induced erythroid differentiation [81]. In an elegant experimental set-up, Villarino and colleagues determined the specific functions of STAT5A and STAT5B in single-allele expressing mice and found no difference in the hematocrit of these mice [66], suggesting a redundant role of both genes in erythroid development. Transgenic expression of cS5F, STAT5B, or hyperactive STAT5BN642H under the hematopoietic vav promoter [82] did not affect hematocrit levels [83,84], which points to a strictly controlled regulation of pYSTAT5A/B signaling in erythrocytes.
4.2. Megakaryopoiesis—Non-Canonical STAT5A/B Prevent Differentiation
TPO activates the megakaryocytic differentiation program via JAK2-dependent pYSTAT5A/B activation, regulating, e.g., Bcl-xl expression and cell survival [85]. Human CD34+ cells differentiated to megakaryocytes upon STAT5A/B downregulation in line with the prevention of megakaryocyte development by activated STAT5A expression (cS5F) [81].
A non-canonical function for nucleus-located uSTAT5 was described in megakaryocyte differentiation: uSTAT5 bound to CTCF binding sites and suppressed differentiation by antagonizing ERG in the absence of TPO. Upon TPO stimulation, STAT5 was tyrosine phosphorylated and redistributed to canonical GAS sites [5]. Based on the knockdown experiments of Park et al., this non-canonical role was mainly assigned to STAT5B—at least in the studied experimental system (HPC-7 cells). Further evidence stems from enforced expression of STAT5BY699F, a mutant incapable of getting phosphorylated: Upon stimulation with TPO, the mutant protected uSTAT5-bound enhancers from deacetylation. This observation underlined the role of uSTAT5 in maintaining regulatory elements [4]. Of note, uSTAT5A was found to participate in chromatin compaction by binding to HP1α [3]—so far not reported in hematopoietic cells. Further studies are needed to determine whether similar functions of nuclear u- and pY-STAT5A or -STAT5B, respectively, are important for the chromatin landscape and consequently lineage determination.
4.3. STAT5A/B Promote Survival and Differentiation of B Cells
The IL-2 family cytokines, characterized by signaling through the common gamma chain, regulate STAT5A/B activation and play important parts in the immune system [2]. In the few surviving Stat5a/b−/− mice, B cell development was blocked in the pre–pro-B cell stage [52]. This block was explained by the absence of Mcl-1, a direct STAT5 target gene promoting survival during early B cell stages [86]. IL-7/STAT5A/B signaling is essential for B cell development—IL-7R signaling-deficient mice were blocked at the earliest stages of B cell development and lacked mature B cells in the periphery. A constitutive active (ca) Stat5b rescued B cell development by upregulating pro-survival genes [54,86,87], and ca STAT5B (Stat5b-CA-tg) transgenic mice had an increased number of pro-B cells [88]. In Stat5a+/−; Stat5b−/− mice, B cell numbers were increased and auto-antibodies were enriched, a phenotype more modest in Stat5a−/−; Stat5b+/− mice [66].
In Stat5b-CA-tg mice, STAT5B bound to genes involved in normal B cell development like pre-B cell receptor (BCR) genes (Syk, Blk, Blnk, Carma1, Irf4, Irf8, or Ikaros) and blocked B cell differentiation. This was considered to contribute to transformation in B cell acute lymphocytic leukemia (B-ALL) [89]. Whether this is an exclusive function of STAT5B or whether it is shared by STAT5A remains to be determined.
4.4. STAT5B is the Major Player in NK Cells
NK cells represent an important part of the innate immune system and function as immediate effector cells against viral infections, pathogens, and malignant cells. They also depend on IL-2 family cytokines—especially IL-2 and IL-15 signaling are essential in NK cells. STAT5A/B is a master regulator of NK cell proliferation, survival, and cytotoxicity [2,90,91,92]. Accordingly, NK cells were grossly absent in Stat5a/b−/− [65], as well as NK cell-specific Stat5a/b−/− [93] and Jak1−/− mice [94]. NK cell survival was rescued by overexpression of BCL-2 [95]. STAT5A/B dimers were sufficient for NK cell development, whereas tetramers were needed for maturation [96].
Murine and human data pinpoint to a key role of STAT5B in NK cells: STAT5B deficiency in mice resulted in a more pronounced reduction of NK cell numbers, activity after IL-2 and IL-15 stimulation, and cytolytic function compared to STAT5A [97,98]. This may be explained by the higher expression levels of STAT5B compared to STAT5A in NK cells (Figure 2).
Loss-of-function (LOF) mutations in STAT5B led to human primary immune deficiencies affecting NK cells [99]—so far not reported for STAT5A [91]. Interestingly, a chemical-induced mutation in the linker domain of murine Stat5a led to reduced STAT5A levels and negatively influenced NK cell development, maturation and activation [100]. These results argue for the need of correct stochiometric ratios of STAT5A and STAT5B to generate complete NK cell functionality,
4.5. CD8+ T Cells are Sensitive to Elevated pYSTAT5A/B Levels
STAT5A and STAT5B proteins are essential downstream mediators of IL-2R and IL-7R signaling to regulate T cell differentiation [101]. The reduced number of thymocytes in Stat5a/b−/− mice resulted in a severely decreased number of CD8+ T cells, loss of γδ T cells [52] and a higher proportion of CD4−CD8− thymocytes [65]. Deletion of STAT5A/B at the CD4+CD8+ T cell differentiation stage also resulted in a massive reduction of CD8+ T cells [52]. Differentiation of CD8+ T cells is regulated by STAT5A/B in a dose-dependent manner [102,103] and STAT5A/B-tetramers are required for expansion of antigen-specific activated CD8+ T cells [57]. Interestingly, IL-7R-mediated STAT5A/B signaling upregulated Runx3, Bcl-2, as well as Mcl1 mRNA expression, which allowed to bypass T cell receptor signaling to induce CD8+ T cell differentiation [104].
Stat5b deficiency led to a more pronounced CD8+ T cell reduction compared to Stat5a loss [66]. Vice versa, overexpression of STAT5B led to an increase in CD8+ and γδ T cells [84,105]. Expression of cS5F in CD8+ T cells enhanced effector and memory CD8+ T cell survival [106,107] and its broad hematopoietic expression induced CD8+ T cell leukemia [83].
4.6. CD4+ T Cell Development—Quantities Matter
CD4+ T cells can be further subdivided into T helper (Th) 1 cells, Th2 cells, Tregs, follicular helper T cells, Th9, Th17, and T helper type GM-CSF cells whose differentiation is induced by specific cytokines. For all of these cell types, STAT5A/B signaling contributes to differentiation, function, or survival, which has been recently reviewed [108].
STAT5B deficiency in mice had a greater impact on CD4+ T cell numbers compared to STAT5A deficiency. This difference was rather explained by higher expression levels of STAT5B in CD4+ T cells than by differences in DNA binding site occupancy [66]. These data generated from knockout mouse models do not completely reflect the insights gained from human cells: Knockdown of STAT5B in primary human CD4+ T cells reduced expression of IL-2Rα and FOXP3—the main markers for Tregs—to a greater extent than deletion of STAT5A. In contrast, BCL-X expression was affected primarily by STAT5A knockdown [109]. In a global ChIP-Seq approach in human CD4+ T cells, Kanai et al. confirmed the preferential occupation of IL2RA and FOXP3 by STAT5B after IL-2 stimulation, and further showed STAT5B binding to DOCK8 and SNX9, both functioning in T cell immune responses. Exclusive STAT5A binding sites were described to take part in neural development and function. Common STAT5A and STAT5B binding sites were found at proliferation and survival genes implicating redundant functions in these processes [27].
Despite many redundant roles of STAT5A and STAT5B in T and NK cells, the higher expression levels of STAT5B define it as the prominent isoform in immune cells.
5. STAT5A/B are Required for Hematopoietic Stem Cell Maintenance and Self-Renewal
Hematopoietic stem cells (HSCs) are defined by their ability to reconstitute the hematopoietic tree with blood cells of all lineages, while maintaining the ability to produce a multipotent HSC by self-renewal. To test the role of STAT5A/B in the repopulation capacity of stem cells, competitive and non-competitive transplantations of wt, Stat5a/b∆N and STAT5A/B-deficient bone marrow (BM) or fetal liver cells were performed. Stat5a/b∆N cells exert a drastic reduction in the ability to reconstitute the hematopoietic system [110,111,112]. Upon conditional or hematopoietic-specific Stat5a/b deletion, this defect was recapitulated by a depletion of the long term (LT)-HSC pool [60,62]. In line with these results, RNAi-mediated downregulation of STAT5A/B resulted in decreased long-term expansion capacity of human progenitor cells [113,114]. Collectively, these data demonstrate a role for STAT5A/B in HSC maintenance and self-renewal.
Bunting and colleagues linked the defect in LT-HSC maintenance of Stat5a/b−/− cells to increased apoptosis and loss of quiescence. Quiescence genes like Tie2, Mpl, Slamf1, or Cited2 were downregulated in HSCs derived from STAT5A/B-deficient BM transplants. They also showed that the Slamf1 locus is directly bound by STAT5A/B. In addition, the TPO-induced HSC-related genes Tie-2 and p57 were downregulated in cells lacking STAT5A/B [60,62]. A recent study employed single-cell qPCR to study the deregulation of several quiescence- and HSC-associated genes in STAT5A/B-deficient LSKs and LT-HSCs. Downregulation of quiescence genes like Mpl, Tie2, or Cited2 in HSCs derived from STAT5A/B-deficient mice was confirmed. Loss of STAT5A/B in LT-HSCs induced myeloid and lymphoid–myeloid multi-lineage priming based on mRNA expression profiles [115], assigning STAT5A/B as a keeper of HSC quiescence.
Using an inducible system in CD34+ human cord blood cells, the induction of STAT5A/B activity provided an advantage in long-term proliferation of HSCs, but not in multi-lineage progenitors [116]. This effect was even enhanced upon down-modulation of GATA1 and allowed the identification of GATA1 (erythroid committed)-independent STAT5A/B target genes [117]. Besides well-known target genes like Pim1 or Osm, STAT5A/B directly bound the HIF2α promoter and induced gene transcription. As HIF2α is critical for glucose uptake, this observation suggests a major role for STAT5A/B in maintaining self-renewal under hypoxic conditions [116].
Moreover, STAT5A/B regulates the expression of miR-193b, which controls expansion of HSCs by reducing c-KIT expression. This c-KIT reduction inhibits cytokine-induced STAT5A/B and AKT signaling and prevents uncontrolled HSC expansion [118]. Of interest, mice lacking the typical STAT5 target gene Pim1 failed to reconstitute lethally irradiated recipient mice. Pim1 activity regulates CXCR4 expression, suggesting an important role for STAT5A/B in homing and migration of HSCs [119].
Despite the profound role of STAT5A/B in HSC renewal, quiescence, and lineage differentiation, none of these studies focused on the individual roles of STAT5A and STAT5B in HSC biology. In contrast to differentiated hematopoietic cells, mRNA levels of STAT5A and STAT5B are comparable in HSCs (Figure 2).
As mentioned, numbers of LT-HSCs were reduced in STAT5A/B-deficient mouse models [62], while they were drastically increased in transgenic STAT5BN642H and STAT5B wild-type mice [84]. Further evidence stems from the expression of recombinant oncogenic STAT5A or STAT5B variants in HSCs and progenitors. Here, STAT5A S779 phosphorylation fine-tuned proliferation and transformation of HSCs and progenitors [120]. Given the comparable expression levels of STAT5A and STAT5B in HSCs, it remains to be determined whether and how they induce the same set of target genes, which is currently enigmatic.
A further layer of complexity is provided by the interferon (IFN) signaling pathway that mediates cell cycle induction in LT-HSCs [121,122]. IFNs first activate dormant LT-HSCs, and later on cause them to re-enter their quiescent state to avoid apoptosis and DNA damage [123]. LT-HSCs which have once experienced IFNs show a reduced potential of reconstitution even if they have regained their quiescent state. These observations are in line with those of STAT5A/B deficiency [124].
One potential mechanism, how STAT5A/B interferes with the decreased repopulation capacity upon IFN signaling, might be via SOCS1 upregulation. SOCS1 negatively regulates the levels of pYSTAT5A/B. It impairs TPO signaling, which finally ends up in lower HSC self-renewal and reconstitution [125,126,127]. Vice versa, activated STAT5B may repress IFN-α/β and IFN-γ signaling in HSCs, as has been recently demonstrated in transformed pro-B cells [128].
6. STAT5A/B as Oncogenes in Hematopoietic Cancer
STAT5A/B are deregulated in a variety of hematopoietic and non-hematopoietic tumors. Amongst others, ALL, myeloproliferative neoplasms, AML, chronic myeloid leukemia (CML), B-ALL, and peripheral T cell leukemia/lymphoma (PTCL) show enhanced STAT5A/B signaling [129,130]. STAT5A and STAT5B act as proto-oncogenes by regulating proliferation and survival [131,132]. They directly promote transcription of anti-apoptotic genes like Mcl-1, Bcl-2, Bcl-xL, miR15/16 or C-Myc, D-type cyclins D1, D2 and D3, cytokines/cytokine receptor chain expression exemplified by OSM, IL-2Rα, IL-4Rα or IL-7Rα, and are associated with growth factor receptor signaling, contributing to many essential functions in cancer [110,132,133,134,135,136]. STAT5A/B activation is, in most cases, induced by hyperactive upstream tyrosine kinases (TK) (e.g., JAK2V617F, BCR-ABL, FLT3-ITD, KITD816V). Recurrent somatic point mutations in STAT5B in mature NK/T cell neoplasms recently concentrated research on STAT5B mutations and how they drive disease [84,137].
6.1. STAT5A and STAT5B Mutations as Disease Drivers
Although redundant functions have been assigned to STAT5A and STAT5B in T cells [66,84], the STAT5B gene is predominantly affected in NK/T cell neoplasia by point mutations localized mainly in the SH2 domain. These GOF mutations lead to enhanced parallel dimerization, nuclear translocation, gene regulation, and persistence against dephosphorylation. Examples for STAT5B mutations are N642H, G596V, Y665F, T648S, or T628S [138], of which some have been analyzed in vitro [84,137,139,140,141,142].
The most recurrent mutation, STAT5BN642H, was detected across many forms of PTCL [139,140,141,143,144,145,146,147,148] and has also been reported in myeloid neoplasia with eosinophilia [149], as well as neutrophilic leukemia [150].
The STAT5BN642H mutation stabilizes dimer formation and leads to increased phosphotyrosine levels [139,140,141,143,144,145,146,147,148]. Confirmation stems from the recently published crystal structure of STAT5BN642H: Hyperactivation is explained by an “open” SH2 domain state, which allows facilitated access to the peptide binding pocket [137]. However, upstream cytokine signaling is still a prerequisite for the activation of STAT5B [84,143,151,152].
Transgenic mouse models expressing either wt Stat5b, or ca Stat5a or ca Stat5b in hematopoietic lineages (summarized in Table 2) were used to study the role of STAT5A/B in leukemia. When expressing high levels of cS5F under the hematopoietic vav promoter, mice developed CD8+ T cell leukemia/lymphoma [83]. The first transgenic mouse model expressing STAT5BN642H in the hematopoietic system (vav promoter) developed an aggressive CD8+ T cell leukemia with organ infiltrations by CD8+, CD4+, and γδ T cells [84,137]. Transplantation models derived from this transgenic mouse verified the oncogenic role for STAT5BN642H in NKT [152] and γδ T cells [137].
Despite the phenotypic similarities of the cS5F vav mouse model compared to the STAT5BN642H transgenic mice, the latter disease model is far more drastic and aggressive. This pinpoints to a greater oncogenic potential of STAT5B compared to STAT5A. The higher number of deregulated genes in STAT5BN642H- compared to STAT5AS710F-mutated CD8+ T cells supports this concept. Interestingly, both mutations resulted in exclusive sets of deregulated genes pinpointing to specific functions [83]. In addition, certain γδ T cell subsets reacted differently—STAT5BN642H supported IFN-γ-producing CD27+ γδ T cells, whereas expression of STAT5AS710F led to expansion of IL-17-producing γδ T cells—explained by an inverse regulation of Tbet [156]. In summary, both transgenic mouse models verified the privileged role of STAT5A/B signaling in CD8+ T cells and the high sensitivity to altered pYSTAT5A/B levels [83,84].
So far, novel DNA binding sites or interaction partners of STAT5BN642H have not been thoroughly analyzed. In a T-ALL model, co-operative HOXA9/STAT5BN642H transcription enhanced the STAT5 transcriptional signature [157]. Decreased methylation of potential polycomb repressor complex 2 (PCR2) binding sites in STAT5BN642H-expressing CD8+ T cells and consequent upregulation of aurora kinases, known PRC2 target genes, suggest combinatorial treatments [84]. Understanding the consequences of STAT5BN642H is a prerequisite to establish targeted treatments.
6.2. STAT5B—The Major Player Downstream of BCR–ABL
BCR–ABL+ leukemia is one of the best studied experimental model systems of a STAT5-dependent disease. BCR–ABL is a fusion protein with a potent and constitutive kinase activity. Almost 100% of all CML and ~30% of B-ALL cases are associated with the t(9;22)(q34;q11) reciprocal translocation resulting in the Philadelphia chromosome. Imatinib, a TK inhibitor targeting BCR–ABL, and its follow-up inhibitors improved the prognosis of CML patients incredibly, but treatment-resistant leukemic stem cells (LSCs) remain [158].
STAT5A/B acts as critical node in the signaling network downstream of BCR–ABL [52] and is indispensable for initiation and maintenance of BCR–ABL+ leukemia [159,160]. BCR–ABL is capable of directly or indirectly phosphorylating and activating STAT5A/B [161]. Even LSCs require STAT5A/B signaling—lowering STAT5A/B levels in an already established leukemia blocks the disease and disables BCR–ABL+ LSCs. Elevated levels of STAT5A/B contribute to a higher resistance rate to TK inhibitors in BCR–ABL+ leukemia [162]. The STAT5 target gene PIM2 contributes to imatinib resistance and its inhibition sensitized LSCs towards pharmacological treatment [163]. Suppression of the STAT5 target genes PIM1 and BCL2 (PIM kinase inhibitor AZD1208 and BCL2 antagonist Sabutoclax) induced apoptosis in BCR–ABL+ ALL cells [164].
Until recently, we lacked an understanding of whether and how STAT5A and STAT5B contribute individually to BCR–ABL-driven diseases. STAT5A-specific knockdown in human cells revealed no effect on survival, while STAT5B-diminished cells displayed increased levels of apoptosis and lost their self-renewal potential [35,165,166]. BCR–ABL directly activates STAT5B to a higher extent than STAT5A, as STAT5A remains partially in the cytoplasm [35]. Imatinib-resistant cell lines upregulate STAT5A; vice versa, cells are increasingly sensitive towards TK inhibitor-treatment upon knockdown of STAT5A [165].
Using BM of Stat5a−/− and Stat5b−/− mice, STAT5B was identified as the dominant isoform downstream of BCR–ABL, as it facilitates transformation via suppressing IFN-α/β and IFN-γ signaling. The relevance of this finding is supported by data from human patients suffering from STAT5B-GOF mutant PTCL. There, the picture was inverse: IFN signaling was downregulated upon STAT5B hyperactivation [128].
Disrupting the STAT5(B)–BCR–ABL interaction in STAT5-dependent hematopoietic diseases is of therapeutic relevance. Whether this defined role of STAT5B as predominant onco-protein extends also to other TK-driven malignancies (such as JAK2V617F or FLT3-ITD) remains to be elucidated.
6.3. STAT5A/B as Potential Opponents in NPM–ALK+ Lymphoma
The oncogenic fusion protein of anaplastic lymphoma kinase (ALK) with nucleophosmin 1 (NPM1) in anaplastic large cell lymphoma leads to the activation of multiple intracellular signal transduction pathways including PI3K–AKT, MAPK/ERK, mTOR, STAT3, and STAT5B [167,168]. NPM–ALK+ cells predominately express STAT5B, which controls proliferation and survival. Downregulation of STAT5A was explained by epigenetic silencing via methylation of its promoter by the NPM–ALK/STAT3 signaling axis. Forced expression of STAT5A led to downregulation of NPM–ALK through direct transcriptional inhibition [169]. This might indicate opposing roles, namely tumor-suppressive STAT5A and oncogenic STAT5B, in NPM–ALK-driven ALCL. Currently, NPM–ALK is the only oncogene for which an antagonistic function of STAT5A and STAT5B has been described.
7. Direct STAT5A/B Inhibition Remains Challenging
STAT5A/B hyperactivation is a common feature of hematopoietic malignancies, with point mutations being primarily reported for STAT5B. Due to its disease-driving role in various forms of myeloid and lymphoid leukemia/lymphomas, it represents a potential therapeutic target [170]. The lack of an enzymatic activity in the transcription factors STAT5A and STAT5B makes the development of specific inhibitors difficult. The structural similarity of STAT5A and STAT5B to each other, as well as to other STAT proteins, adds a further level of complexity. Here, we focus on direct STAT5 inhibitors, since inhibitors of upstream JAKs have been rigorously reviewed [171,172].
Although promiscuous with respect to their peptide binding motifs, a promising option is to target the SH2 domain of STAT5A/B to prevent tyrosine phosphorylation, activation, and nuclear translocation. The lead compound AC-3-19, whose structure is based on salicylic acid, turned out to not be potent enough for clinical translation [83,173,174]. Its further optimization led to AC-4-130, which was used successfully for in vitro and in vivo treatment of FLT3–ITD+ AML cell lines, as well as on primary AML cells. Its cytotoxic potential was enhanced by combinatorial treatment with the JAK1/2 inhibitor ruxolitinib or the p300/pCAF inhibitor garcinol [175]. The sterical confirmation of STAT5BN642H hinders AC-4-130 binding [175], making it not applicable for targeted treatment of patients who carry STAT5BN642H. Another example for a STAT5A/B SH2 domain inhibitor, IST5-002, blocked phosphorylation and nuclear translocation of STAT5A/B in BCR–ABL+ in vitro and in vivo systems [164,176]. Via a virtual compound library screening approach and further structural adaptions, the first STAT5A SH2 domain inhibitor was identified, showing a modest reduction of pYSTAT5A levels in BCR–ABL+ cells [177]. All compounds require further modifications to improve the bioavailability, stability, and potency.
A high selectivity for STAT5B-tyrosine phosphorylation inhibition over STAT5A was attributed to the catechol bisphosphate derivatives Capstafin [178] and Stafib-1 [179], which has been further modified to Stafib-2 [180]. STAT5B selectivity was assigned to a STAT5B-specific amino acid in the linker domain, which might represent a novel design approach [181]. So far, in vivo data are not available.
Nucleic acid-based approaches aiming to interfere with STAT5A/B DNA binding (e.g., dominant negative constructs, G-quartet oligonucleotides, decoy oligonucleotides, metal-based inhibitors) or STAT5A/B expression (antisense or siRNA) have been successful in vitro and in vivo [19,182,183,184]. What remains problematic in the clinic is delivery of these constructs to their preferred site [185]. Furthermore, cell-permeable peptides or mimetics have been identified, which bind to the protein or even the STAT5A/B DNA binding domain itself [186]. Permeability and stability of the peptides still represent a hurdle for clinical usage [187]. Recently, ATP was found to bind to and inhibit the STAT5B SH2 domain [188], which needs to be validated in cellular systems. Until now, none of these defined inhibitors met the requirements for entrance into clinical assessment.
Insights into mutation-specific changes in the transcriptome or methylome of STAT5B-GOF mutated cancer cells may open novel therapeutic avenues. Combinatorial treatments will be helpful to prevent resistance development and reduce side effects. In the transgenic STAT5BN642H mouse model, RNA-seq analysis revealed that STAT5BN642H CD8+ T cells upregulated cell cycle-driving and EZH2 target genes like Top2A and Aurkb [84]. Importantly, STAT5A/B and EZH2 have been shown to interact [189]. In STAT5BN642H CD8+ T cells, DNA methylation was reduced at EZH2 and SUZ12 binding sites, both components of the chromatin remodeling complex PRC2 [190]. These observations indicate a competition of EZH2 and STAT5BN642H for binding sites, leading to an altered transcriptome, including aurora kinases. Aurora kinases are important for cell division and represent promising targets in leukemia treatment. Specific inhibitors are currently under investigation in clinical trials [191,192,193]. Combining JAK and aurora kinase inhibitors resulted in the selective cell death of STAT5BN642H-expressing CD8+ T cells and NKTCL cell lines, which offers a targeted treatment option in STAT5BN642H+ PTCL patients [84,146]. Whether JAK/aurora kinase inhibition can effectively eradicate or ameliorate the STAT5BN642H-driven T cell disease in vivo remains to be shown.
Selective and effective low dosage STAT5A, STAT5B, or STAT5A/B inhibitors are not clinically available. Suppression of the immune system may be the downside of STAT5A/B inhibition comparable to the side effects of JAK inhibitors [194,195,196]. Similarly, anemia, thrombocytopenia, diarrhea, or neurotoxicity have to be explored in pre-clinical trials before new drug treatment options can be concluded. As the mode of action of STAT proteins is distinct from JAK action in many circumstances, side effects cannot be anticipated. Finally, combination therapies using epigenetic inhibitors targeting STAT5 cofactors, such as bromodomain and extra terminal inhibitors (BETi) and histone deacetylase inhibitors (HDACi), might offer alternative therapeutic approaches [197,198,199,200,201,202].
8. Conclusions
Until now, many studies focused on the collective roles of STAT5A and STAT5B in healthy and malignant hematopoiesis. Establishment of tools distinguishing between STAT5A and STAT5B will help to clarify redundant and non-redundant contributions in hematopoiesis and leukemogenesis. Only recently is a privileged role for STAT5B unveiling. The discoveries of STAT5B mutations in NK and T cell leukemia/lymphoma and STAT5B-deficient patients unambiguously indicate STAT5B’s particular importance. Its distinct role may stem from specific or preferential DNA binding sites and target gene expression, distinct protein–protein interactions, or non-canonical signaling (Figure 3). Exclusive targeting of STAT5B—while sparing STAT5A—is a great challenge. The most promising avenue to date encompasses the combined blockage of JAK with specific downstream targets of mutated STAT5B—as exemplified with aurora kinase inhibition. These downstream targets may be specific for the upstream TK mutation, the STAT5B-GOF mutation, and the affected cell type, which complicates clinical approaches. A more detailed understanding of STAT5A’s and STAT5B’s physiological roles will facilitate future clinical interventions in hematopoietic malignancies.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2072-6694/11/11/1726/s1: Figure S1. Protein alignment of human or murine STAT5A and STAT5B.
Author Contributions
All authors have made substantial, direct, and intellectual contribution to the work. B.M., S.K., and J.P. reviewed the literature and wrote the manuscript. A.H.-K. corrected the review and V.S. corrected, edited, and approved the final version.
Funding
This work is supported by the Austrian Science Fund FWF SFB-F6107.
Acknowledgments
The authors thank the group of Richard Moriggl for continuous discussion.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Villarino, A.V.; Kanno, Y.; Ferdinand, J.R.; O’Shea, J.J. Mechanisms of Jak/STAT Signaling in Immunity and Disease. J. Immunol. 2015, 194, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signalling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dutta, P.; Tsurumi, A.; Li, J.; Wang, J.; Land, H.; Li, W.X. Unphosphorylated STAT5A stabilizes heterochromatin and suppresses tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 10213–10218. [Google Scholar] [CrossRef] [Green Version]
- Comoglio, F.; Park, H.J.; Schoenfelder, S.; Barozzi, I.; Bode, D.; Fraser, P.; Green, A.R. Thrombopoietin signaling to chromatin elicits rapid and pervasive epigenome remodeling within poised chromatin architectures. Genome Res. 2018, 28, 295–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.J.; Li, J.; Hannah, R.; Biddie, S.; Leal-Cervantes, A.I.; Kirschner, K.; Flores Santa Cruz, D.; Sexl, V.; Göttgens, B.; Green, A.R. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J. 2016, 35, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Groner, B.; von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef]
- Wang, Y.; Levy, D.E. Comparative evolutionary genomics of the STAT family of transcription factors. JAK-STAT 2012, 1, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Liongue, C.; Ward, A.C. Evolution of the JAK-STAT pathway. JAK-STAT 2013, 2, e22756. [Google Scholar] [CrossRef]
- Liu, X.; Robinson, G.W.; Wagner, K.U.; Garrett, L.; Wynshaw-Boris, A.; Hennighausen, L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997, 11, 179–186. [Google Scholar] [CrossRef]
- Liongue, C.; O’Sullivan, L.A.; Trengove, M.C.; Ward, A.C. Evolution of JAK-STAT Pathway Components: Mechanisms and Role in Immune System Development. PLoS ONE 2012, 7, e32777. [Google Scholar] [CrossRef]
- Lewis, R.S.; Ward, A.C. Conservation, duplication and divergence of the zebrafish stat5 genes. Gene 2004, 338, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Mei, J.; Huang, P.; Jing, J.; Li, Z.; Kang, J.; Gui, J.F. Essential roles of stat5.1/stat5b in controlling fish somatic growth. J. Genet. Genom. 2017, 44, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Paffett-Lugassy, N.; Hsia, N.; Fraenkel, P.G.; Paw, B.; Leshinsky, I.; Barut, B.; Bahary, N.; Caro, J.; Handin, R.; Zon, L.I. Functional conservation of erythropoietin signaling in zebrafish. Blood 2007, 110, 2718–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakao, H.; Gouilleux, F.; Groner, B. Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J. 1994, 13, 2182–2191. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Ney, M.; Doppler, W.; Ball, R.K.; Groner, B. Beta-casein gene promoter activity is regulated by the hormone-mediated relief of transcriptional repression and a mammary-gland-specific nuclear factor. Mol. Cell. Biol. 1991, 11, 3745–3755. [Google Scholar] [CrossRef]
- Wakao, H.; Harada, N.; Kitamura, T.; Mui, A.L.; Miyajima, A. Interleukin 2 and erythropoietin activate STAT5/MGF via distinct pathways. EMBO J. 1995, 14, 2527–2535. [Google Scholar] [CrossRef]
- Mui, A.L.; Wakao, H.; O’Farrell, A.M.; Harada, N.; Miyajima, A. Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO J. 1995, 14, 1166–1175. [Google Scholar] [CrossRef]
- Gouilleux, F.; Wakao, H.; Mundt, M.; Groner, B. Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO J. 1994, 13, 4361–4369. [Google Scholar] [CrossRef]
- Liu, K.D.; Lai, S.Y.; Goldsmith, M.A.; Greene, W.C. Identification of a Variable Region within the Cytoplasmic Tail of the IL-2 Receptor β Chain That Is Required for Growth Signal Transduction. J. Biol. Chem. 1995, 270, 22176–22181. [Google Scholar] [CrossRef]
- Miyoshi, K.; Cui, Y.; Riedlinger, G.; Robinson, P.; Lehoczky, J.; Zon, L.; Oka, T.; Dewar, K.; Hennighausen, L. Structure of the Mouse Stat 3/5 Locus: Evolution from Drosophila to Zebrafish to Mouse. Genomics 2001, 71, 150–155. [Google Scholar] [CrossRef]
- Moriggl, R.; Sexl, V.; Kenner, L.; Duntsch, C.; Stangl, K.; Gingras, S.; Hoffmeyer, A.; Bauer, A.; Piekorz, R.; Wang, D.; et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell 2005, 7, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar] [CrossRef]
- Kornfeld, J.W.; Grebien, F.; Kerenyi, M.A.; Friedbichler, K.; Kovacic, B.; Zankl, B.; Hoelbl, A.; Nivarti, H.; Beug, H.; Sexl, V.; et al. The different functions of Stat5 and chromatin alteration through Stat5 proteins. Front. Biosci. 2008, 13, 6237–6254. [Google Scholar] [CrossRef] [PubMed]
- Grimley, P.M.; Dong, F.; Rui, H. Stat5a and Stat5b: Fraternal twins of signal transduction and transcriptional activation. Cytokine Growth Factor Rev. 1999, 10, 131–157. [Google Scholar] [CrossRef]
- Boucheron, C.; Dumon, S.; Santos, S.C.R.; Moriggl, R.; Hennighausen, L.; Gisselbrecht, S.; Gouilleux, F. A Single Amino Acid in the DNA Binding Regions of STAT5A and STAT5B Confers Distinct DNA Binding Specificities. J. Biol. Chem. 1998, 273, 33936–33941. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.X.; Mietz, J.; Modi, W.S.; John, S.; Leonard, W.J. Cloning of Human Stat5B: RECONSTITUTION OF INTERLEUKIN-2-INDUCED Stat5A AND Stat5B DNA BINDING ACTIVITY IN COS-7 CELLS. J. Biol. Chem. 1996, 271, 10738–10744. [Google Scholar] [CrossRef]
- Kanai, T.; Seki, S.; Jenks, J.; Kohli, A.; Kawli, T.; Martin, D.; Snyder, M.; Baccetta, R.; Nadeau, K. Identification of STATA and STAT5B target genes in human T cells. PLoS ONE 2014, 9, e86790. [Google Scholar] [CrossRef]
- Basham, B.; Sathe, M.; Grein, J.; McClanahan, T.; D‘Andrea, A.; Lees, E.; Rascle, A. In vivo identification of novel STAT5 target genes. Nucleic Acids Res. 2008, 36, 3802–3818. [Google Scholar] [CrossRef] [Green Version]
- Meinke, A.; Barahmand-Pour, F.; Wöhrl, S.; Stoiber, D.; Decker, T. Activation of different Stat5 isoforms contributes to cell-type-restricted signaling in response to interferons. Mol. Cell. Biol. 1996, 16, 6937–6944. [Google Scholar] [CrossRef] [Green Version]
- Boehm, M.E.; Adlung, L.; Schilling, M.; Roth, S.; Klingmüller, U.; Lehmann, W.D. Identification of Isoform-Specific Dynamics in Phosphorylation-Dependent STAT5 Dimerization by Quantitative Mass Spectrometry and Mathematical Modeling. J. Proteome Res. 2014, 13, 5685–5694. [Google Scholar] [CrossRef]
- Imada, K.; Leonard, W.J. The Jak-STAT pathway. Mol. Immunol. 2000, 37, 1–11. [Google Scholar] [CrossRef]
- Berger, A.; Hoelbl-Kovacic, A.; Bourgeais, J.; Hoefling, L.; Warsch, W.; Grundschober, E.; Uras, I.Z.; Menzl, I.; Putz, E.M.; Hoermann, G.; et al. PAK-dependent STAT5 serine phosphorylation is required for BCR-ABL-induced leukemogenesis. Leukemia 2014, 28, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Friedbichler, K.; Kerenyi, M.A.; Kovacic, B.; Li, G.; Hoelbl, A.; Yahiaoui, S.; Sexl, V.; Mullner, E.W.; Fajmann, S.; Cerny-Reiterer, S.; et al. Stat5a serine 725 and 779 phosphorylation is a prerequisite for hematopoietic transformation. Blood 2010, 116, 1548–1558. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E.; Williams, C.C.; Duplessis, T.T.; Moring, K.L.; Notwick, A.R.; Long, W.; Lane, W.S.; Beuvink, I.; Hynes, N.E.; Jones, F.E. ERBB4/HER4 potentiates STAT5A transcriptional activity by regulating novel STAT5A serine phosphorylation events. J. Biol. Chem. 2005, 280, 24175–24180. [Google Scholar] [CrossRef] [PubMed]
- Schaller-Schonitz, M.; Barzan, D.; Williamson, A.J.; Griffiths, J.R.; Dallmann, I.; Battmer, K.; Ganser, A.; Whetton, A.D.; Scherr, M.; Eder, M. BCR-ABL affects STAT5A and STAT5B differentially. PLoS ONE 2014, 9, e97243. [Google Scholar] [CrossRef]
- Mitra, A.; Ross, J.A.; Rodriguez, G.; Nagy, Z.S.; Wilson, H.L.; Kirken, R.A. Signal transducer and activator of transcription 5b (Stat5b) serine 193 is a novel cytokine-induced phospho-regulatory site that is constitutively activated in primary hematopoietic malignancies. J. Biol. Chem. 2012, 287, 16596–16608. [Google Scholar] [CrossRef]
- Bartalucci, N.; Calabresi, L.; Balliu, M.; Martinelli, S.; Rossi, M.C.; Villeval, J.L.; Annunziato, F.; Guglielmelli, P.; Vannucchi, A.M. Inhibitors of the PI3K/mTOR pathway prevent STAT5 phosphorylation in JAK2V617F mutated cells through PP2A/CIP2A axis. Oncotarget 2017, 8, 96710–96724. [Google Scholar] [CrossRef] [Green Version]
- Kabotyanski, E.B.; Rosen, J.M. Signal transduction pathways regulated by prolactin and Src result in different conformations of activated Stat5b. J. Biol. Chem. 2003, 278, 17218–17227. [Google Scholar] [CrossRef]
- Fox, E.M.; Bernaciak, T.M.; Wen, J.; Weaver, A.M.; Shupnik, M.A.; Silva, C.M. Signal transducer and activator of transcription 5b, c-Src, and epidermal growth factor receptor signaling play integral roles in estrogen-stimulated proliferation of estrogen receptor-positive breast cancer cells. Mol. Endocrinol. 2008, 22, 1781–1796. [Google Scholar] [CrossRef]
- Weaver, A.M.; Silva, C.M. S731 in the transactivation domain modulates STAT5b activity. Biochem. Biophys. Res. Commun. 2007, 362, 1026–1030. [Google Scholar] [CrossRef] [Green Version]
- Bancerek, J.; Poss, Z.C.; Steinparzer, I.; Sedlyarov, V.; Pfaffenwimmer, T.; Mikulic, I.; Dölken, L.; Strobl, B.; Müller, M.; Taatjes, D.J.; et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 2013, 38, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Beuvink, I.; Hess, D.; Flotow, H.; Hofsteenge, J.; Groner, B.; Hynes, N.E. Stat5a Serine Phosphorylation: SERINE 779 IS CONSTITUTIVELY PHOSPHORYLATED IN THE MAMMARY GLAND, AND SERINE 725 PHOSPHORYLATION INFLUENCES PROLACTIN-STIMULATEDIN VITRO DNA BINDING ACTIVITY. J. Biol. Chem. 2000, 275, 10247–10255. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.M.; Silva, C.M. Modulation of Signal Transducer and Activator of Transcription 5b Activity in Breast Cancer Cells by Mutation of Tyrosines within the Transactivation Domain. Mol. Endocrinol. 2006, 20, 2392–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloth, M.T.; Catling, A.D.; Silva, C.M. Novel Activation of STAT5b in Response to Epidermal Growth Factor. J. Biol. Chem. 2002, 277, 8693–8701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nguyen, T.; Angkasekwinai, P.; Dou, H.; Lin, F.M.; Lu, L.S.; Cheng, J.; Chin, Y.E.; Dong, C.; Yeh, E.T. SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol. Cell 2012, 45, 210–221. [Google Scholar] [CrossRef]
- Yamamoto, M.; Iguchi, G.; Fukuoka, H.; Suda, K.; Bando, H.; Takahashi, M.; Nishizawa, H.; Seino, S.; Takahashi, Y. SIRT1 regulates adaptive response of the growth hormone--insulin-like growth factor-I axis under fasting conditions in liver. Proc. Natl. Acad. Sci. USA 2013, 110, 14948–14953. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, T.; Kasai, H.; Kondo, M. Acetylation Modulates IL-2 Receptor Signaling in T Cells. J. Immunol. 2016, 197, 4334–4343. [Google Scholar] [CrossRef] [Green Version]
- Freund, P.; Kerenyi, M.A.; Hager, M.; Wagner, T.; Wingelhofer, B.; Pham, H.T.T.; Elabd, M.; Han, X.; Valent, P.; Gouilleux, F.; et al. O-GlcNAcylation of STAT5 controls tyrosine phosphorylation and oncogenic transcription in STAT5-dependent malignancies. Leukemia 2017, 31, 2132. [Google Scholar] [CrossRef]
- Shi, S.; Larson, K.; Guo, D.; Lim, S.J.; Dutta, P.; Yan, S.J.; Li, W.X. Drosophila STAT is required for directly maintaining HP1 localization and heterochromatin stability. Nat. Cell Biol. 2008, 10, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Szybinski, J. Role of Unphosphorylated STAT5 in Maintenance of Acute Myeloid Leukemia Cells; Johannes-Gutenberg-Universität Mainz: Mainz, Germany, 2019. [Google Scholar]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef] [Green Version]
- Hoelbl, A.; Kovacic, B.; Kerenyi, M.A.; Simma, O.; Warsch, W.; Cui, Y.; Beug, H.; Hennighausen, L.; Moriggl, R.; Sexl, V. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood 2006, 107, 4898–4906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sexl, V.; Piekorz, R.; Moriggl, R.; Rohrer, J.; Brown, M.P.; Bunting, K.D.; Rothammer, K.; Roussel, M.F.; Ihle, J.N. Stat5a/b contribute to interleukin 7–induced B-cell precursor expansion, but abl-andbcr/abl-induced transformation are independent of Stat5. Blood 2000, 96, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.A.; Harmon, I.R.; O’Neil, J.J.; Burchill, M.A.; Farrar, M.A. STAT5 Activation Underlies IL7 Receptor-Dependent B Cell Development. J. Immunol. 2004, 172, 4770–4778. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; DiBenedetto, B.; Narayan, K.; Zhao, H.; Der, S.D.; Chambers, C.A. STAT5 Is Required for Thymopoiesis in a Development Stage-Specific Manner. J. Immunol. 2004, 173, 2307–2314. [Google Scholar] [CrossRef]
- Teglund, S.; McKay, C.; Schuetz, E.; van Deursen, J.M.; Stravopodis, D.; Wang, D.; Brown, M.; Bodner, S.; Grosveld, G.; Ihle, J.N. Stat5a and Stat5b Proteins Have Essential and Nonessential, or Redundant, Roles in Cytokine Responses. Cell 1998, 93, 841–850. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.X.; Li, P.; Liu, D.; Jin, H.T.; He, J.; Ata Ur Rasheed, M.; Rochman, Y.; Wang, L.; Cui, K.; Liu, C.; et al. Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity 2012, 36, 586–599. [Google Scholar] [CrossRef]
- Moriggl, R.; Topham, D.J.; Teglund, S.; Sexl, V.; McKay, C.; Wang, D.; Hoffmeyer, A.; van Deursen, J.; Sangster, M.Y.; Bunting, K.D.; et al. Stat5 Is Required for IL-2-Induced Cell Cycle Progression of Peripheral T Cells. Immunity 1999, 10, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Riedlinger, G.; Miyoshi, K.; Tang, W.; Li, C.; Deng, C.X.; Robinson, G.W.; Hennighausen, L. Inactivation of Stat5 in Mouse Mammary Epithelium during Pregnancy Reveals Distinct Functions in Cell Proliferation, Survival, and Differentiation. Mol. Cell. Biol. 2004, 24, 8037–8047. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Medrzycki, M.; Bunting, S.T.; Bunting, K.D. Stat5-deficient hematopoiesis is permissive for Myc-induced B-cell leukemogenesis. Oncotarget 2015, 6, 28961–28972. [Google Scholar] [CrossRef]
- Zhu, B.M.; McLaughlin, S.K.; Na, R.; Liu, J.; Cui, Y.; Martin, C.; Kimura, A.; Robinson, G.W.; Andrews, N.C.; Hennighausen, L. Hematopoietic-specific Stat5-null mice display microcytic hypochromic anemia associated with reduced transferrin receptor gene expression. Blood 2008, 112, 2071–2080. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, G.; Tse, W.; Bunting, K.D. Conditional deletion of STAT5 in adult mouse hematopoietic stem cells causes loss of quiescence and permits efficient nonablative stem cell replacement. Blood 2009, 113, 4856–4865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udy, G.B.; Towers, R.P.; Snell, R.G.; Wilkins, R.J.; Park, S.H.; Ram, P.A.; Waxman, D.J.; Davey, H.W. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 7239–7244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerenyi, M.A.; Grebien, F.; Gehart, H.; Schifrer, M.; Artaker, M.; Kovacic, B.; Beug, H.; Moriggl, R.; Müllner, E.W. Stat5 regulates cellular iron uptake of erythroid cells via IRP-2 and TfR-1. Blood 2008, 112, 3878–3888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Cui, Y.; Watford, W.T.; Bream, J.H.; Yamaoka, K.; Hissong, B.D.; Li, D.; Durum, S.K.; Jiang, Q.; Bhandoola, A.; et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 1000–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarino, A.; Laurence, A.; Robinson, G.W.; Bonelli, M.; Dema, B.; Afzali, B.; Shih, H.Y.; Sun, H.W.; Brooks, S.R.; Hennighausen, L.; et al. Signal transducer and activator of transcription 5 (STAT5) paralog dose governs T cell effector and regulatory functions. eLife 2016, 5, e08384. [Google Scholar] [CrossRef]
- Bernasconi, A.; Marino, R.; Ribas, A.; Rossi, J.; Ciaccio, M.; Oleastro, M.; Ornani, A.; Paz, R.; Rivarola, M.A.; Zelazko, M.; et al. Characterization of Immunodeficiency in a Patient With Growth Hormone Insensitivity Secondary to a Novel STAT5b Gene Mutation. Pediatrics 2006, 118, e1584–e1592. [Google Scholar] [CrossRef]
- Cohen, A.C.; Nadeau, K.C.; Tu, W.; Hwa, V.; Dionis, K.; Bezrodnik, L.; Teper, A.; Gaillard, M.; Heinrich, J.; Krensky, A.M.; et al. Cutting Edge: Decreased Accumulation and Regulatory Function of CD4 + CD25high T Cells in Human STAT5b Deficiency. J. Immunol. 2006, 177, 2770–2774. [Google Scholar] [CrossRef]
- Kofoed, E.M.; Hwa, V.; Little, B.; Woods, K.A.; Buckway, C.K.; Tsubaki, J.; Pratt, K.L.; Bezrodnik, L.; Jasper, H.; Tepper, A.; et al. Growth Hormone Insensitivity Associated with a STAT5b Mutation. N. Engl. J. Med. 2003, 349, 1139–1147. [Google Scholar] [CrossRef]
- Bezrodnik, L.; Di Giovanni, D.; Caldirola, M.S.; Azcoiti, M.E.; Torgerson, T.; Gaillard, M.I. Long-Term Follow-up of STAT5B Deficiency in Three Argentinian Patients: Clinical and Immunological Features. J. Clin. Immunol. 2015, 35, 264–272. [Google Scholar] [CrossRef]
- Hwa, V. STAT5B deficiency: Impacts on human growth and immunity. Growth Horm. IGF Res. 2016, 28, 16–20. [Google Scholar] [CrossRef]
- Klammt, J.; Neumann, D.; Gevers, E.F.; Andrew, S.F.; Schwartz, I.D.; Rockstroh, D.; Colombo, R.; Sanchez, M.A.; Vokurkova, D.; Kowalczyk, J.; et al. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat. Commun. 2018, 9, 2105. [Google Scholar] [CrossRef] [PubMed]
- Nivarthi, H.; Friedbichler, K.; Moriggl, R. Stat5 as a Hematopoietic Master Regulator for Differentiation and Neoplasia Development. In Jak-Stat Signaling: From Basics to Disease; Decker, T., Mueller, M., Eds.; Springer: Vienna, Austria, 2012; pp. 153–167. [Google Scholar]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak–STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Murphy, J.J. STAT5 in Cancer and Immunity. J. Interferon Cytokine Res. 2015, 36, 226–237. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2012, 32, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- Bagger, F.O.; Sasivarevic, D.; Sohi, S.H.; Laursen, L.G.; Pundhir, S.; Sønderby, C.K.; Winther, O.; Rapin, N.; Porse, B.T. BloodSpot: A database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res. 2015, 44, D917–D924. [Google Scholar] [CrossRef]
- Gillinder, K.R.; Tuckey, H.; Bell, C.C.; Magor, G.W.; Huang, S.; Ilsley, M.D.; Perkins, A.C. Direct targets of pSTAT5 signalling in erythropoiesis. PLoS ONE 2017, 12, e0180922. [Google Scholar] [CrossRef]
- Perreault, A.A.; Venters, B.J. Integrative view on how erythropoietin signaling controls transcription patterns in erythroid cells. Curr. Opin. Hematol. 2018, 25, 189–195. [Google Scholar] [CrossRef]
- Grebien, F.; Kerenyi, M.A.; Kovacic, B.; Kolbe, T.; Becker, V.; Dolznig, H.; Pfeffer, K.; Klingmüller, U.; Müller, M.; Beug, H.; et al. Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2. Blood 2008, 111, 4511–4522. [Google Scholar] [CrossRef] [Green Version]
- Olthof, S.G.; Fatrai, S.; Drayer, A.L.; Tyl, M.R.; Vellenga, E.; Schuringa, J.J. Downregulation of Signal Transducer and Activator of Transcription 5 (STAT5) in CD34+ Cells Promotes Megakaryocytic Development, Whereas Activation of STAT5 Drives Erythropoiesis. Stem Cells 2008, 26, 1732–1742. [Google Scholar] [CrossRef]
- Ogilvy, S.; Metcalf, D.; Gibson, L.; Bath, M.L.; Harris, A.W.; Adams, J.M. Promoter Elements of vav Drive Transgene Expression In Vivo Throughout the Hematopoietic Compartment. Blood 1999, 94, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.; Nivarthi, H.; Wingelhofer, B.; Pham, H.T.T.; Schlederer, M.; Suske, T.; Grausenburger, R.; Schiefer, A.I.; Prchal-Murphy, M.; Chen, D.; et al. High activation of STAT5A drives peripheral T-cell lymphoma and leukemia. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.T.T.; Maurer, B.; Prchal-Murphy, M.; Grausenburger, R.; Grundschober, E.; Javaheri, T.; Nivarthi, H.; Boersma, A.; Kolbe, T.; Elabd, M.; et al. STAT5BN642H is a driver mutation for T cell neoplasia. J. Clin. Investig. 2018, 128, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Kirito, K.; Watanabe, T.; Sawada, K.I.; Endo, H.; Ozawa, K.; Komatsu, N. Thrombopoietin Regulates Bcl-xL Gene Expression through Stat5 and Phosphatidylinositol 3-Kinase Activation Pathways. J. Biol. Chem. 2002, 277, 8329–8337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malin, S.; McManus, S.; Cobaleda, C.; Novatchkova, M.; Delogu, A.; Bouillet, P.; Strasser, A.; Busslinger, M. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat. Immunol. 2010, 11, 171–179. [Google Scholar] [CrossRef]
- Malin, S.; McManus, S.; Busslinger, M. STAT5 in B cell development and leukemia. Curr. Opin. Immunol. 2010, 22, 168–176. [Google Scholar] [CrossRef]
- Burchill, M.A.; Goetz, C.A.; Prlic, M.; O’Neil, J.J.; Harmon, I.R.; Bensinger, S.J.; Turka, L.A.; Brennan, P.; Jameson, S.C.; Farrar, M.A. Distinct Effects of STAT5 Activation on CD4+ and CD8+ T Cell Homeostasis: Development of CD4+CD25+ Regulatory T Cells versus CD8+ Memory T Cells. J. Immunol. 2003, 171, 5853–5864. [Google Scholar] [CrossRef]
- Katerndahl, C.D.S.; Heltemes-Harris, L.M.; Willette, M.J.L.; Henzler, C.M.; Frietze, S.; Yang, R.; Schjerven, H.; Silverstein, K.A.T.; Ramsey, L.B.; Hubbard, G.; et al. Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat. Immunol. 2017, 18, 694–704. [Google Scholar] [CrossRef] [Green Version]
- Gotthardt, D.; Sexl, V. STATs in NK-Cells: The Good, the Bad, and the Ugly. Front. Immunol. 2017, 7, 694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas-Hernández, A.; Forbes, L.R. JAK/STAT proteins and their biological impact on NK cell development and function. Mol. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Hernández, A.; Witalisz-Siepracka, A.; Prchal-Murphy, M.; Klein, K.; Mahapatra, S.; Al-Herz, W.; Mace, E.M.; Carisey, A.F.; Orange, J.S.; Sexl, V.; et al. Human STAT5b mutation causes dysregulated human natural killer cell maturation and impaired lytic function. J. Allergy Clin. Immunol. 2019, S0091674919312527. [Google Scholar] [CrossRef]
- Eckelhart, E.; Warsch, W.; Zebedin, E.; Simma, O.; Stoiber, D.; Kolbe, T.; Rülicke, T.; Mueller, M.; Casanova, E.; Sexl, V. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood 2011, 117, 1565–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witalisz-Siepracka, A.; Klein, K.; Prinz, D.; Leidenfrost, N.; Schabbauer, G.; Dohnal, A.; Sexl, V. Loss of JAK1 Drives Innate Immune Deficiency. Front. Immunol. 2019, 9, 3108. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, D.; Putz, E.M.; Grundschober, E.; Prchal-Murphy, M.; Straka, E.; Kudweis, P.; Heller, G.; Bago-Horvath, Z.; Witalisz-Siepracka, A.; Cumaraswamy, A.A.; et al. STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discov. 2016, 6, 414–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.X.; Du, N.; Li, P.; Kazemian, M.; Gebregiorgis, T.; Spolski, R.; Leonard, W.J. Critical functions for STAT5 tetramers in the maturation and survival of natural killer cells. Nat. Commun. 2017, 8, 1320. [Google Scholar] [CrossRef]
- Villarino, A.V.; Sciumè, G.; Davis, F.P.; Iwata, S.; Zitti, B.; Robinson, G.W.; Hennighausen, L.; Kanno, Y.; O’Shea, J.J. Subset- and tissue-defined STAT5 thresholds control homeostasis and function of innate lymphoid cells. J. Exp. Med. 2017, 214, 2999–3014. [Google Scholar] [CrossRef]
- Imada, K.; Bloom, E.T.; Nakajima, H.; Horvath-Arcidiacono, J.A.; Udy, G.B.; Davey, H.W.; Leonard, W.J. Stat5b Is Essential for Natural Killer Cell–mediated Proliferation and Cytolytic Activity. J. Exp. Med. 1998, 188, 2067–2074. [Google Scholar] [CrossRef]
- Lorenzini, T.; Dotta, L.; Giacomelli, M.; Vairo, D.; Badolato, R. STAT mutations as program switchers: Turning primary immunodeficiencies into autoimmune diseases. J. Leukoc. Biol. 2017, 101, 29–38. [Google Scholar] [CrossRef]
- Chehboun, S.; Leiva-Torres, G.A.; Charbonneau, B.; Eveleigh, R.; Bourque, G.; Vidal, S.M. A point mutation in the linker domain of mouse STAT5A is associated with impaired NK-cell regulation. Genes Immun. 2019. [CrossRef]
- Heltemes-Harris, L.M.; Farrar, M.A. The role of STAT5 in lymphocyte development and transformation. Curr. Opin. Immunol. 2012, 24, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Ermakova, O.; Piszczek, L.; Luciani, L.; Cavalli, F.M.; Ferreira, T.; Farley, D.; Rizzo, S.; Paolicelli, R.C.; Al-Banchaabouchi, M.; Nerlov, C.; et al. Sensitized phenotypic screening identifies gene dosage sensitive region on chromosome 11 that predisposes to disease in mice. EMBO Mol. Med. 2011, 3, 50–66. [Google Scholar] [CrossRef]
- Nivarthi, H.; Prchal-Murphy, M.; Swoboda, A.; Hager, M.; Schlederer, M.; Kenner, L.; Tuckermann, J.; Sexl, V.; Moriggl, R.; Ermakova, O. Stat5 gene dosage in T cells modulates CD8+ T-cell homeostasis and attenuates contact hypersensitivity response in mice. Allergy 2015, 70, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Adoro, S.; Guinter, T.; Erman, B.; Alag, A.S.; Catalfamo, M.; Kimura, M.Y.; Cui, Y.; Lucas, P.J.; Gress, R.E.; et al. Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxic-lineage T cells. Nat. Immunol. 2010, 11, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, J.; Spolski, R.; Imada, K.; Bollenbacher, J.; Lee, S.; Leonard, W.J. A Role for Stat5 in CD8+ T Cell Homeostasis. J. Immunol. 2003, 170, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Hand, T.W.; Cui, W.; Jung, Y.W.; Sefik, E.; Joshi, N.S.; Chandele, A.; Liu, Y.; Kaech, S.M. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc. Natl. Acad. Sci. USA 2010, 107, 16601–16606. [Google Scholar] [CrossRef] [Green Version]
- Grange, M.; Buferne, M.; Verdeil, G.; Leserman, L.; Schmitt-Verhulst, A.; Auphan-Anezin, N. Activated STAT5 Promotes Long-Lived Cytotoxic CD8+ T Cells That Induce Regression of Autochthonous Melanoma. Cancer Res. 2012, 72, 76–87. [Google Scholar] [CrossRef]
- Owen, D.L.; Farrar, M.A. STAT5 and CD4 (+) T Cell Immunity. F1000Research 2017, 6, 32. [Google Scholar] [CrossRef]
- Jenks, J.A.; Seki, S.; Kanai, T.; Huang, J.; Morgan, A.A.; Scalco, R.C.; Nath, R.; Bucayu, R.; Wit, J.M.; Al-Herz, W.; et al. Differentiating the roles of STAT5B and STAT5A in human CD4+ T cells. Clin. Immunol. 2013, 148, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Miskimen, K.L.; Wang, Z.; Xie, X.Y.; Brenzovich, J.; Ryan, J.J.; Tse, W.; Moriggl, R.; Bunting, K.D. STAT5 requires the N-domain for suppression of miR15/16, induction of bcl-2, and survival signaling in myeloproliferative disease. Blood 2010, 115, 1416–1424. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Wang, Z.; Zhang, Y.; Kang, Z.; Haviernikova, E.; Cui, Y.; Hennighausen, L.; Moriggl, R.; Wang, D.; Tse, W.; et al. STAT5 requires the N-domain to maintain hematopoietic stem cell repopulating function and appropriate lymphoid-myeloid lineage output. Exp. Hematol. 2007, 35, 1684–1694. [Google Scholar] [CrossRef] [Green Version]
- Snow, J.W.; Abraham, N.; Ma, M.C.; Abbey, N.W.; Herndier, B.; Goldsmith, M.A. STAT5 promotes multilineage hematolymphoid development in vivo through effects on early hematopoietic progenitor cells. Blood 2002, 99, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Schepers, H.; van Gosliga, D.; Wierenga, A.T.J.; Eggen, B.J.L.; Schuringa, J.J.; Vellenga, E. STAT5 is required for long-term maintenance of normal and leukemic human stem/progenitor cells. Blood 2007, 110, 2880–2888. [Google Scholar] [CrossRef] [Green Version]
- Scherr, M.; Chaturvedi, A.; Battmer, K.; Dallmann, I.; Schultheis, B.; Ganser, A.; Eder, M. Enhanced sensitivity to inhibition of SHP2, STAT5, and Gab2 expression in chronic myeloid leukemia (CML). Blood 2006, 107, 3279–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Bunting, K.D. Stat5 deficiency decreases transcriptional heterogeneity and supports emergence of hematopoietic sub-populations. Oncotarget 2017, 8, 22477–22482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatrai, S.; Wierenga, A.T.J.; Daenen, S.M.G.J.; Vellenga, E.; Schuringa, J.J. Identification of HIF2α as an important STAT5 target gene in human hematopoietic stem cells. Blood 2011, 117, 3320–3330. [Google Scholar] [CrossRef]
- Wierenga, A.T.J.; Vellenga, E.; Schuringa, J.J. Down-regulation of GATA1 uncouples STAT5-induced erythroid differentiation from stem/progenitor cell proliferation. Blood 2010, 115, 4367–4376. [Google Scholar] [CrossRef] [Green Version]
- Haetscher, N.; Feuermann, Y.; Wingert, S.; Rehage, M.; Thalheimer, F.B.; Weiser, C.; Bohnenberger, H.; Jung, K.; Schroeder, T.; Serve, H.; et al. STAT5-regulated microRNA-193b controls haematopoietic stem and progenitor cell expansion by modulating cytokine receptor signalling. Nat. Commun. 2015, 6, 8928. [Google Scholar] [CrossRef]
- Grundler, R.; Brault, L.; Gasser, C.; Bullock, A.N.; Dechow, T.; Woetzel, S.; Pogacic, V.; Villa, A.; Ehret, S.; Berridge, G.; et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J. Exp. Med. 2009, 206, 1957–1970. [Google Scholar] [CrossRef]
- Ghanem, S.; Friedbichler, K.; Boudot, C.; Bourgeais, J.; Gouilleux-Gruart, V.; Régnier, A.; Herault, O.; Moriggl, R.; Gouilleux, F. STAT5A/5B-specific expansion and transformation of hematopoietic stem cells. Blood Cancer J. 2017, 7, e514. [Google Scholar] [CrossRef]
- Essers, M.A.G.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNα activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904. [Google Scholar] [CrossRef]
- Baldridge, M.T.; King, K.Y.; Boles, N.C.; Weksberg, D.C.; Goodell, M.A. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature 2010, 465, 793. [Google Scholar] [CrossRef]
- Morales-Mantilla, D.E.; King, K.Y. The Role of Interferon-Gamma in Hematopoietic Stem Cell Development, Homeostasis, and Disease. Curr. Stem Cell Rep. 2018, 4, 264–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirche, C.; Frenz, T.; Haas, S.F.; Döring, M.; Borst, K.; Tegtmeyer, P.K.; Brizic, I.; Jordan, S.; Keyser, K.; Chhatbar, C.; et al. Systemic Virus Infections Differentially Modulate Cell Cycle State and Functionality of Long-Term Hematopoietic Stem Cells In Vivo. Cell Rep. 2017, 19, 2345–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Iwama, A.; Tadokoro, Y.; Shimoda, K.; Minoguchi, M.; Akira, S.; Tanaka, M.; Miyajima, A.; Kitamura, T.; Nakauchi, H. Selective activation of STAT5 unveils its role in stem cell self-renewal in normal and leukemic hematopoiesis. J. Exp. Med. 2005, 202, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bruin, A.M.; Demirel, Ö.; Hooibrink, B.; Brandts, C.H.; Nolte, M.A. Interferon-γ impairs proliferation of hematopoietic stem cells in mice. Blood 2013, 121, 3578–3585. [Google Scholar] [CrossRef]
- Yoshihara, H.; Arai, F.; Hosokawa, K.; Hagiwara, T.; Takubo, K.; Nakamura, Y.; Gomei, Y.; Iwasaki, H.; Matsuoka, S.; Miyamoto, K.; et al. Thrombopoietin/MPL Signaling Regulates Hematopoietic Stem Cell Quiescence and Interaction with the Osteoblastic Niche. Cell Stem Cell 2007, 1, 685–697. [Google Scholar] [CrossRef] [Green Version]
- Kollmann, S.; Grundschober, E.; Maurer, B.; Warsch, W.; Grausenburger, R.; Edlinger, L.; Huuhtanen, J.; Lagger, S.; Hennighausen, L.; Valent, P.; et al. Twins with different personalities: STAT5B—but not STAT5A—has a key role in BCR/ABL-induced leukemia. Leukemia 2019. [Google Scholar] [CrossRef]
- Joliot, V.; Cormier, F.; Medyouf, H.; Alcalde, H.; Ghysdael, J. Constitutive STAT5 activation specifically cooperates with the loss of p53 function in B-cell lymphomagenesis. Oncogene 2006, 25, 4573–4584. [Google Scholar] [CrossRef]
- Orlova, A.; Wingelhofer, B.; Neubauer, H.A.; Maurer, B.; Berger-Becvar, A.; Keserű, G.M.; Gunning, P.T.; Valent, P.; Moriggl, R. Emerging therapeutic targets in myeloproliferative neoplasms and peripheral T-cell leukemia and lymphomas. Expert Opin. Ther. Targets 2018, 22, 45–57. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97. [Google Scholar] [CrossRef]
- Ferbeyre, G.; Moriggl, R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim. Biophys. Acta 2011, 1815, 104–114. [Google Scholar] [CrossRef]
- Dagvadorj, A.; Kirken, R.A.; Leiby, B.; Karras, J.; Nevalainen, M.T. Transcription Factor Signal Transducer and Activator of Transcription 5 Promotes Growth of Human Prostate Cancer Cells In vivo. Clin. Cancer Res. 2008, 14, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Wong, L.; Flavell, R.; Thompson, S.A.; Wells, A.; Larner, A.C.; Johnson, G.R. STAT Activation by Epidermal Growth Factor (EGF) and Amphiregulin: REQUIREMENT FOR THE EGF RECEPTOR KINASE BUT NOT FOR TYROSINE PHOSPHORYLATION SITES OR JAK1. J. Biol. Chem. 1996, 271, 9185–9188. [Google Scholar] [CrossRef] [PubMed]
- Blaas, L.; Kornfeld, J.W.; Schramek, D.; Musteanu, M.; Zollner, G.; Gumhold, J.; van Zijl, F.; Schneller, D.; Esterbauer, H.; Egger, G.; et al. Disruption of the growth hormone--signal transducer and activator of transcription 5--insulinlike growth factor 1 axis severely aggravates liver fibrosis in a mouse model of cholestasis. Hepatology 2010, 51, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Sonkin, D.; Regnier, C.; Rong, X.; Fanton, C.; Palmer, M.; Holash, J.; Squires, M.; Sirulnik, L.A.; Radimerski, T.; Schlegel, R.; et al. Identification of pSTAT5 gene signature in hematologic malignancy. J. Clin. Oncol. 2013, 31, 7111. [Google Scholar] [CrossRef]
- De Araujo, E.D.; Erdogan, F.; Neubauer, H.A.; Meneksedag-Erol, D.; Manaswiyoungkul, P.; Eram, M.S.; Seo, H.S.; Qadree, A.K.; Israelian, J.; Orlova, A.; et al. Structural and functional consequences of the STAT5B(N642H) driver mutation. Nat. Commun. 2019, 10, 2517. [Google Scholar] [CrossRef]
- COSMIC. STAT5B Mutations Gene View. Available online: https://cancer.sanger.ac.uk/cosmic/gene/analysis?all_data=&coords=AA%3AAA&dr=&end=788&gd=&id=5369&ln=STAT5B&seqlen=788&sn=haematopoietic_and_lymphoid_tissue&start=1#ts (accessed on 19 September 2019).
- Bandapalli, O.R.; Schuessele, S.; Kunz, J.B.; Rausch, T.; Stütz, A.M.; Tal, N.; Geron, I.; Gershman, N.; Izraeli, S.; Eilers, J.; et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica 2014, 99, e188–e192. [Google Scholar] [CrossRef]
- Kontro, M.; Kuusanmaki, H.; Eldfors, S.; Burmeister, T.; Andersson, E.I.; Bruserud, O.; Brummendorf, T.H.; Edgren, H.; Gjertsen, B.T.; Itala-Remes, M.; et al. Novel activating STAT5B mutations as putative drivers of T-cell acute lymphoblastic leukemia. Leukemia 2014, 28, 1738–1742. [Google Scholar] [CrossRef]
- Schrader, A.; Crispatzu, G.; Oberbeck, S.; Mayer, P.; Pützer, S.; von Jan, J.; Vasyutina, E.; Warner, K.; Weit, N.; Pflug, N.; et al. Actionable perturbations of damage responses by TCL1/ATM and epigenetic lesions form the basis of T-PLL. Nat. Commun. 2018, 9, 697. [Google Scholar] [CrossRef]
- McKinney, M.; Moffitt, A.B.; Gaulard, P.; Travert, M.; De Leval, L.; Nicolae, A.; Raffeld, M.; Jaffe, E.S.; Pittaluga, S.; Xi, L.; et al. The Genetic Basis of Hepatosplenic T Cell Lymphoma. Cancer Discov. 2017. [Google Scholar] [CrossRef]
- Küçük, C.; Jiang, B.; Hu, X.; Zhang, W.; Chan, J.K.C.; Xiao, W.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, K.N.; et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Rajala, H.L.M.; Eldfors, S.; Kuusanmäki, H.; van Adrichem, A.J.; Olson, T.; Lagström, S.; Andersson, E.I.; Jerez, A.; Clemente, M.J.; Yan, Y.; et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 2013, 121, 4541–4550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiel, M.J.; Velusamy, T.; Rolland, D.; Sahasrabuddhe, A.A.; Chung, F.; Bailey, N.G.; Schrader, A.; Li, B.; Li, J.Z.; Ozel, A.B.; et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood 2014, 124, 1460–1472. [Google Scholar] [CrossRef] [Green Version]
- Dufva, O.; Kankainen, M.; Kelkka, T.; Sekiguchi, N.; Awad, S.A.; Eldfors, S.; Yadav, B.; Kuusanmäki, H.; Malani, D.; Andersson, E.I.; et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat. Commun. 2018, 9, 1567. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.T.T.; Hengstschläger, M.; Moriggl, R. A haunted beast: Targeting STAT5B(N642H) in T-Cell Neoplasia. Mol. Cell. Oncol. 2018, 5, e1435181. [Google Scholar] [CrossRef]
- Nicolae, A.; Xi, L.; Pittaluga, S.; Abdullaev, Z.; Pack, S.D.; Chen, J.; Waldmann, T.A.; Jaffe, E.S.; Raffeld, M. Frequent STAT5B mutations in [gamma][delta] hepatosplenic T-cell lymphomas. Leukemia 2014, 28, 2244–2248. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.C.P.; Hoade, Y.; Tapper, W.J.; Carreno-Tarragona, G.; Fanelli, T.; Jawhar, M.; Naumann, N.; Pieniak, I.; Lübke, J.; Ali, S.; et al. Recurrent activating STAT5B N642H mutation in myeloid neoplasms with eosinophilia. Leukemia 2018. [Google Scholar] [CrossRef]
- Luo, Q.; Shen, J.; Yang, Y.; Tang, H.; Shi, M.; Liu, J.; Liu, Z.; Shi, X.; Yi, Y. CSF3R T618I, ASXL1 G942 fs and STAT5B N642H trimutation co-contribute to a rare chronic neutrophilic leukaemia manifested by rapidly progressive leucocytosis, severe infections, persistent fever and deep venous thrombosis. Br. J. Haematol. 2018, 180, 892–894. [Google Scholar] [CrossRef]
- Waldmann, T.A.; Chen, J. Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Immunotherapy. Annu. Rev. Immunol. 2017, 35, 533–550. [Google Scholar] [CrossRef]
- Klein, K.; Witalisz-Siepracka, A.; Maurer, B.; Prinz, D.; Heller, G.; Leidenfrost, N.; Prchal-Murphy, M.; Suske, T.; Moriggl, R.; Sexl, V. STAT5BN642H drives transformation of NKT cells: A novel mouse model for CD56+ T-LGL leukemia. Leukemia 2019. [Google Scholar] [CrossRef]
- Kelly, J.A.; Spolski, R.; Kovanen, P.E.; Suzuki, T.; Bollenbacher, J.; Pise-Masison, C.A.; Radonovich, M.F.; Lee, S.; Jenkins, N.A.; Copeland, N.G.; et al. Stat5 Synergizes with T Cell Receptor/Antigen Stimulation in the Development of Lymphoblastic Lymphoma. J. Exp. Med. 2003, 198, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Yi, B.; Mao, R.; Liu, H.; Wang, J.; Sharma, A.; Peiper, S.; Leonard, W.J.; She, J.X. Enhanced T Cell Lymphoma in NOD.Stat5b Transgenic Mice Is Caused by Hyperactivation of Stat5b in CD8(+) Thymocytes. PLoS ONE 2013, 8, e56600. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Schmidt, J.W.; Creamer, B.A.; Triplett, A.A.; Wagner, K.U. Gain-of-Function of Stat5 Leads to Excessive Granulopoiesis and Lethal Extravasation of Granulocytes to the Lung. PLoS ONE 2013, 8, e60902. [Google Scholar] [CrossRef] [PubMed]
- Kadekar, D.; Agerholm, R.; Rizk, J.; Neubauer, H.; Suske, T.; Maurer, B.; Viñals, M.T.; Comelli, E.; Taibi, A.; Moriggl, R.; et al. The neonatal microenvironment programs conventional and intestinal Tbet+ γδT17 cells through the transcription factor STAT5. bioRxiv 2019, 658542. [Google Scholar] [CrossRef]
- De Bock, C.E.; Demeyer, S.; Degryse, S.; Verbeke, D.; Sweron, B.; Gielen, O.; Vandepoel, R.; Vicente, C.; Vanden Bempt, M.; Dagklis, A.; et al. HOXA9 Cooperates with Activated JAK/STAT Signaling to Drive Leukemia Development. Cancer Discov. 2018, 8, 616–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol. Med. 2010, 2, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Walz, C.; Ahmed, W.; Lazarides, K.; Betancur, M.; Patel, N.; Hennighausen, L.; Zaleskas, V.M.; Van Etten, R.A. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2V617F in mice. Blood 2012, 119, 3550–3560. [Google Scholar] [CrossRef]
- Hantschel, O.; Warsch, W.; Eckelhart, E.; Kaupe, I.; Grebien, F.; Wagner, K.U.; Superti-Furga, G.; Sexl, V. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat. Chem. Biol. 2012, 8, 285. [Google Scholar] [CrossRef]
- Warsch, W.; Kollmann, K.; Eckelhart, E.; Fajmann, S.; Cerny-Reiterer, S.; Hölbl, A.; Gleixner, K.V.; Dworzak, M.; Mayerhofer, M.; Hoermann, G.; et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood 2011, 117, 3409–3420. [Google Scholar] [CrossRef]
- Ma, L.; Pak, M.L.; Ou, J.; Yu, J.; St. Louis, P.; Shan, Y.; Hutchinson, L.; Li, S.; Brehm, M.A.; Zhu, L.J.; et al. Prosurvival kinase PIM2 is a therapeutic target for eradication of chronic myeloid leukemia stem cells. Proc. Natl. Acad. Sci. USA 2019, 116, 10482–10487. [Google Scholar] [CrossRef] [Green Version]
- Minieri, V.; De Dominici, M.; Porazzi, P.; Mariani, S.A.; Spinelli, O.; Rambaldi, A.; Peterson, L.F.; Porcu, P.; Nevalainen, M.T.; Calabretta, B. Targeting STAT5 or STAT5-regulated pathways suppresses leukemogenesis of Ph+ acute lymphoblastic leukemia. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Najjar, I.; Grimont, A.; Martin, N.; Martin-Lanneree, S.; Bonnet, M.L.; Guilhot, F.; Chomel, J.C.; Lauret, E.; Turhan, A.G.; Dusanter-Fourt, I. Distinct Functions of Stat5A and Stat5B in Chronic Myeloid Leukemia (CML): Stat5B Is Implicated in Survival and Self-Renewal and Stat5A in Imatinib Resistance. Blood 2010, 116, 1214. [Google Scholar] [CrossRef]
- Casetti, L.; Martin-Lannerée, S.; Najjar, I.; Plo, I.; Augé, S.; Roy, L.; Chomel, J.C.; Lauret, E.; Turhan, A.G.; Dusanter-Fourt, I. Differential Contributions of STAT5A and STAT5B to Stress Protection and Tyrosine Kinase Inhibitor Resistance of Chronic Myeloid Leukemia Stem/Progenitor Cells. Cancer Res. 2013, 73, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The Transcriptional Roles of ALK Fusion Proteins in Tumorigenesis. Cancers 2019, 11, 1074. [Google Scholar] [CrossRef]
- Werner, M.T.; Zhao, C.; Zhang, Q.; Wasik, M.A. Nucleophosmin-anaplastic lymphoma kinase: The ultimate oncogene and therapeutic target. Blood 2017, 129, 823–831. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.Y.; Liu, X.; Wasik, M.A. STAT5A is epigenetically silenced by the tyrosine kinase NPM1-ALK and acts as a tumor suppressor by reciprocally inhibiting NPM1-ALK expression. Nat. Med. 2007, 13, 1341. [Google Scholar] [CrossRef]
- Recio, C.; Guerra, B.; Guerra-Rodríguez, M.; Aranda-Tavío, H.; Martín-Rodríguez, P.; de Mirecki-Garrido, M.; Brito-Casillas, Y.; García-Castellano, J.M.; Estévez-Braun, A.; Fernández-Pérez, L. Signal transducer and activator of transcription (STAT)-5: An opportunity for drug development in oncohematology. Oncogene 2019, 38, 4657–4668. [Google Scholar] [CrossRef]
- Gadina, M.; Johnson, C.; Schwartz, D.; Bonelli, M.; Hasni, S.; Kanno, Y.; Changelian, P.; Laurence, A.; O’Shea, J.J. Translational and clinical advances in JAK-STAT biology: The present and future of jakinibs. J. Leukoc. Biol. 2018, 104, 499–514. [Google Scholar] [CrossRef]
- Senkevitch, E.; Durum, S. The promise of Janus kinase inhibitors in the treatment of hematological malignancies. Cytokine 2017, 98, 33–41. [Google Scholar] [CrossRef]
- Cumaraswamy, A.A.; Lewis, A.M.; Geletu, M.; Todic, A.; Diaz, D.B.; Cheng, X.R.; Brown, C.E.; Laister, R.C.; Muench, D.; Kerman, K.; et al. Nanomolar-Potency Small Molecule Inhibitor of STAT5 Protein. ACS Med. Chem. Lett. 2014, 5, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Gleixner, K.V.; Schneeweiss, M.; Eisenwort, G.; Berger, D.; Herrmann, H.; Blatt, K.; Greiner, G.; Byrgazov, K.; Hoermann, G.; Konopleva, M.; et al. Combined targeting of STAT3 and STAT5: A novel approach to overcome drug resistance in chronic myeloid leukemia. Haematologica 2017, 102, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.A.; Berger-Becvar, A.; de Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia 2018, 32, 1135–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Z.; Gu, L.; Vergalli, J.; Mariani, S.A.; De Dominici, M.; Lokareddy, R.K.; Dagvadorj, A.; Purushottamachar, P.; McCue, P.A.; Trabulsi, E.; et al. Structure-Based Screen Identifies a Potent Small Molecule Inhibitor of Stat5a/b with Therapeutic Potential for Prostate Cancer and Chronic Myeloid Leukemia. Mol. Cancer Ther. 2015, 14, 1777–1793. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Müller-Klieser, D.; Rubner, S.; Berg, T. Stafia-1: A STAT5a-selective inhibitor developed via docking-based screening of in silico O-phosphorylated fragments. Chem. A Eur. J. 2019. [Google Scholar] [CrossRef]
- Elumalai, N.; Berg, A.; Rubner, S.; Berg, T. Phosphorylation of Capsaicinoid Derivatives Provides Highly Potent and Selective Inhibitors of the Transcription Factor STAT5b. ACS Chem. Biol. 2015, 10, 2884–2890. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, N.; Berg, A.; Natarajan, K.; Scharow, A.; Berg, T. Nanomolar inhibitors of the transcription factor STAT5b with high selectivity over STAT5a. Angew. Chem. Int. Ed. 2015, 54, 4758–4763. [Google Scholar] [CrossRef]
- Elumalai, N.; Berg, A.; Rubner, S.; Blechschmidt, L.; Song, C.; Natarajan, K.; Matysik, J.; Berg, T. Rational development of Stafib-2: A selective, nanomolar inhibitor of the transcription factor STAT5b. Sci. Rep. 2017, 7, 819. [Google Scholar] [CrossRef]
- Gräb, J.; Berg, A.; Blechschmidt, L.; Klüver, B.; Rubner, S.; Fu, D.Y.; Meiler, J.; Gräber, M.; Berg, T. The STAT5b Linker Domain Mediates the Selectivity of Catechol Bisphosphates for STAT5b over STAT5a. ACS Chem. Biol. 2019, 14, 796–805. [Google Scholar] [CrossRef]
- Behbod, F.; Nagy, Z.S.; Stepkowski, S.M.; Karras, J.; Johnson, C.R.; Jarvis, W.D.; Kirken, R.A. Specific Inhibition of Stat5a/b Promotes Apoptosis of IL-2-Responsive Primary and Tumor-Derived Lymphoid Cells. J. Immunol. 2003, 171, 3919–3927. [Google Scholar] [CrossRef]
- Sen, M.; Grandis, J.R. Nucleic acid-based approaches to STAT inhibition. JAK-STAT 2012, 1, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.J.; Wang, W.; Kang, T.S.; Liang, J.X.; Liu, C.; Kwong, D.W.J.; Wong, V.K.W.; Ma, D.L.; Leung, C.H. Antagonizing STAT5B dimerization with an osmium complex. Sci. Rep. 2016, 6, 36044. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Kim, J.H.; Park, M.; Yeom, J.H.; Go, H.; Kim, S.; Han, M.S.; Lee, K.; Bae, J. Modulation of biological processes in the nucleus by delivery of DNA oligonucleotides conjugated with gold nanoparticles. Biomaterials 2011, 32, 2593–2604. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Borghouts, C.; Brendel, C.; Moriggl, R.; Delis, N.; Brill, B.; Vafaizadeh, V.; Groner, B. The inhibition of stat5 by a Peptide aptamer ligand specific for the DNA binding domain prevents target gene transactivation and the growth of breast and prostate tumor cells. Pharmaceuticals 2013, 6, 960–987. [Google Scholar] [CrossRef]
- Mullard, A. Protein–protein interaction inhibitors get into the groove. Nat. Rev. Drug Discov. 2012, 11, 173. [Google Scholar] [CrossRef]
- Berg, A.; Sperl, B.; Berg, T. ATP Inhibits the Transcription Factor STAT5b. ChemBioChem 2019. [Google Scholar] [CrossRef]
- Mandal, M.; Powers, S.E.; Maienschein-Cline, M.; Bartom, E.T.; Hamel, K.M.; Kee, B.L.; Dinner, A.R.; Clark, M.R. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat. Immunol. 2011, 12, 1212. [Google Scholar] [CrossRef]
- Gall Trošelj, K.; Novak Kujundzic, R.; Ugarkovic, D. Polycomb repressive complex’s evolutionary conserved function: The role of EZH2 status and cellular background. Clin. Epigenetics 2016, 8, 55. [Google Scholar] [CrossRef]
- Goldenson, B.; Crispino, J.D. The aurora kinases in cell cycle and leukemia. Oncogene 2015, 34, 537–545. [Google Scholar] [CrossRef]
- Tang, A.; Gao, K.; Chu, L.; Zhang, R.; Yang, J.; Zheng, J. Aurora kinases: Novel therapy targets in cancers. Oncotarget 2017, 8, 23937–23954. [Google Scholar] [CrossRef]
- Bavetsias, V.; Linardopoulos, S. Aurora Kinase Inhibitors: Current Status and Outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef]
- Porpaczy, E.; Tripolt, S.; Hoelbl-Kovacic, A.; Gisslinger, B.; Bago-Horvath, Z.; Casanova-Hevia, E.; Clappier, E.; Decker, T.; Fajmann, S.; Fux, D.A.; et al. Aggressive B-cell lymphomas in patients with myelofibrosis receiving JAK1/2 inhibitor therapy. Blood 2018, 132, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Bottos, A.; Gotthardt, D.; Gill, J.W.; Gattelli, A.; Frei, A.; Tzankov, A.; Sexl, V.; Wodnar-Filipowicz, A.; Hynes, N.E. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat. Commun. 2016, 7, 12258. [Google Scholar] [CrossRef] [Green Version]
- Gadina, M.; Le, M.T.; Schwartz, D.M.; Silvennoinen, O.; Nakayamada, S.; Yamaoka, K.; O’Shea, J.J. Janus kinases to jakinibs: From basic insights to clinical practice. Rheumatology 2019, 58, i4–i16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Okhovat, J.P.; Hong, E.K.; Kim, Y.H.; Wood, G.S. Preclinical studies support combined inhibition of BET family proteins and histone deacetylases as epigentic therapy for cutaneous T-cell lymphoma. Neoplasia 2019, 21, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Pinz, S.; Unser, S.; Buob, D.; Fischer, P.; Jobst, B.; Rascle, A. Deacetylase inhibitors repress STAT5-mediated transcription by interfering with bromodomain and extra-terminal (BET) protein function. Nucleic Acids Res. 2015, 43, 3524–3545. [Google Scholar] [CrossRef]
- Liu, S.; Walker, S.R.; Nelson, E.A.; Cerulli, R.; Xiang, M.; Toniolo, P.A.; Qi, J.; Stone, R.M.; Wadleigh, M.; Bradner, J.E.; et al. Targeting STAT5 in hematologic maligancies through inhibition of the bromodomain and extra-terminal (BET) bromodomain BRD2. Mol. Cancer Ther. 2014, 13, 1194–1205. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Qi, J.; Shah, B.; Devaraj, S.G.T.; Leveque, C.; Portier, B.P.; Iyer, S.; Bradner, J.E.; Bhalla, K.N. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (KI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol. Cancer Ther. 2014, 13, 2315–2327. [Google Scholar] [CrossRef]
- Nguyen, T.; Dai, Y.; Attkisson, E.; Kramer, L.; Jordan, N.; Nguyen, N.; Kolluri, N.; Muschen, M.; Grant, S. HDAC inhibitors potentiate the activity of the BCAR/ABL kinase inhibitor KW-2449 in imatinib-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clin. Cancer Res. 2011, 17, 3219–3232. [Google Scholar] [CrossRef]
- Rascle, A.; Johnston, J.A.; Amati, B. Deacetylase activity is required for recruitment of the basal transcription machinery and transactivation by STAT5. Mol. Cell Biol. 2003, 23, 4162–4173. [Google Scholar] [CrossRef]
Figure 1.
Differences in the domain structure and post-translational modifications of STAT5A and STAT5B. The protein structure of human STAT5A and STAT5B, including the most prominent Serine (S) and Tyrosine (Y) phosphorylation, Arginine methylation (R-me), and Lysine acetylation (K-ac), as well as O-GlcNAc sites, are shown.
Figure 1.
Differences in the domain structure and post-translational modifications of STAT5A and STAT5B. The protein structure of human STAT5A and STAT5B, including the most prominent Serine (S) and Tyrosine (Y) phosphorylation, Arginine methylation (R-me), and Lysine acetylation (K-ac), as well as O-GlcNAc sites, are shown.

Figure 2.
STAT5A and STAT5B mRNA expression levels in hematopoietic cells. Log2 fold change (fc) of STAT5B/STAT5A mRNA expression ratio of human (left) or murine (right) hematopoietic cells [77] using the human probes #212550_at, 212549_at, and 203010_at, and the murine probes #1421469_a_at and 1422103_a_at. (n.d., not determined).
Figure 2.
STAT5A and STAT5B mRNA expression levels in hematopoietic cells. Log2 fold change (fc) of STAT5B/STAT5A mRNA expression ratio of human (left) or murine (right) hematopoietic cells [77] using the human probes #212550_at, 212549_at, and 203010_at, and the murine probes #1421469_a_at and 1422103_a_at. (n.d., not determined).

Figure 3.
STAT5A and STAT5B hetero- and homodimers may induce different transcriptional programs. STAT5A and STAT5B may have unique DNA binding sites and induce different sets of target genes by interacting with distinct partners.
Figure 3.
STAT5A and STAT5B hetero- and homodimers may induce different transcriptional programs. STAT5A and STAT5B may have unique DNA binding sites and induce different sets of target genes by interacting with distinct partners.


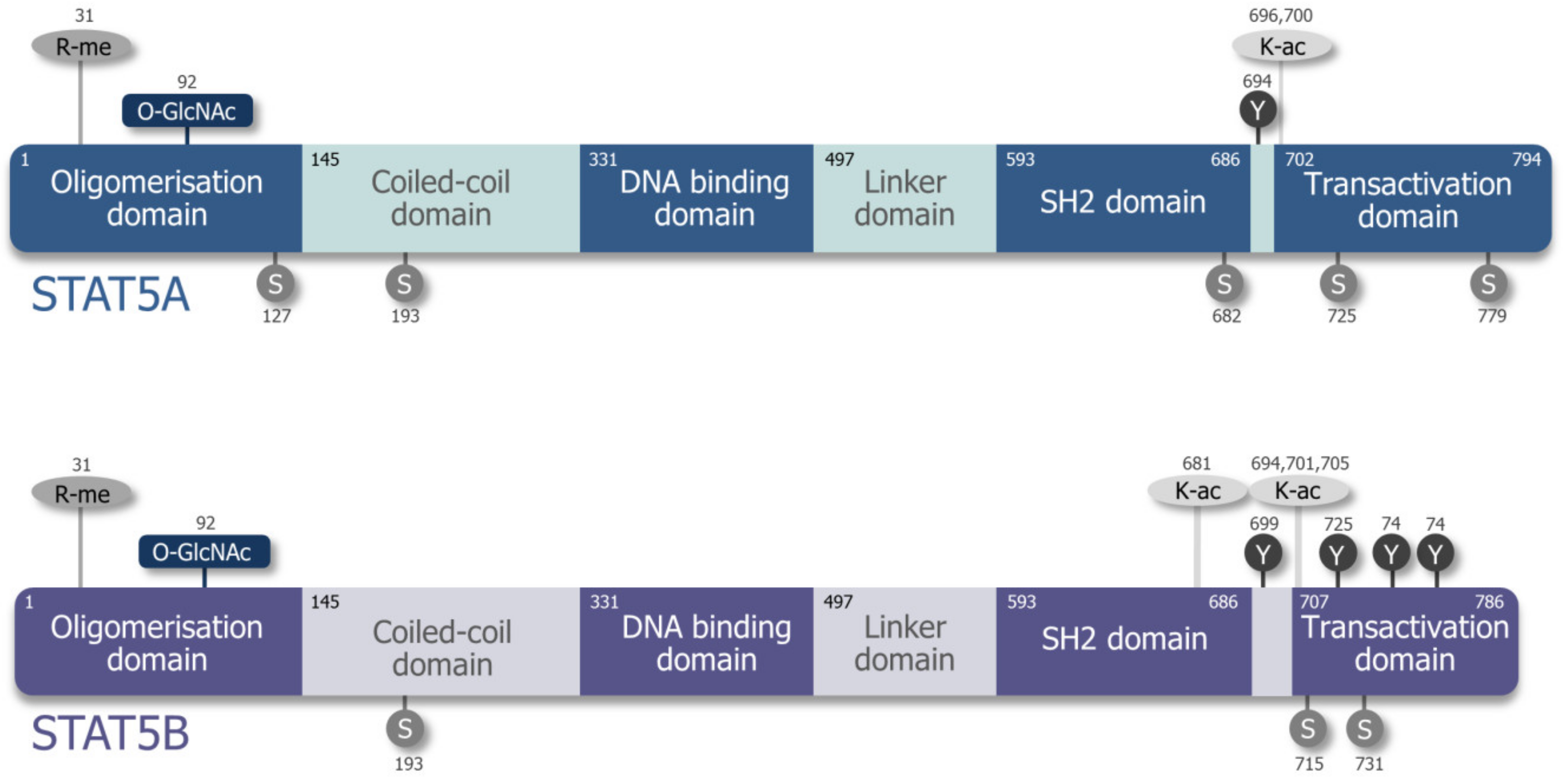{kind=link}
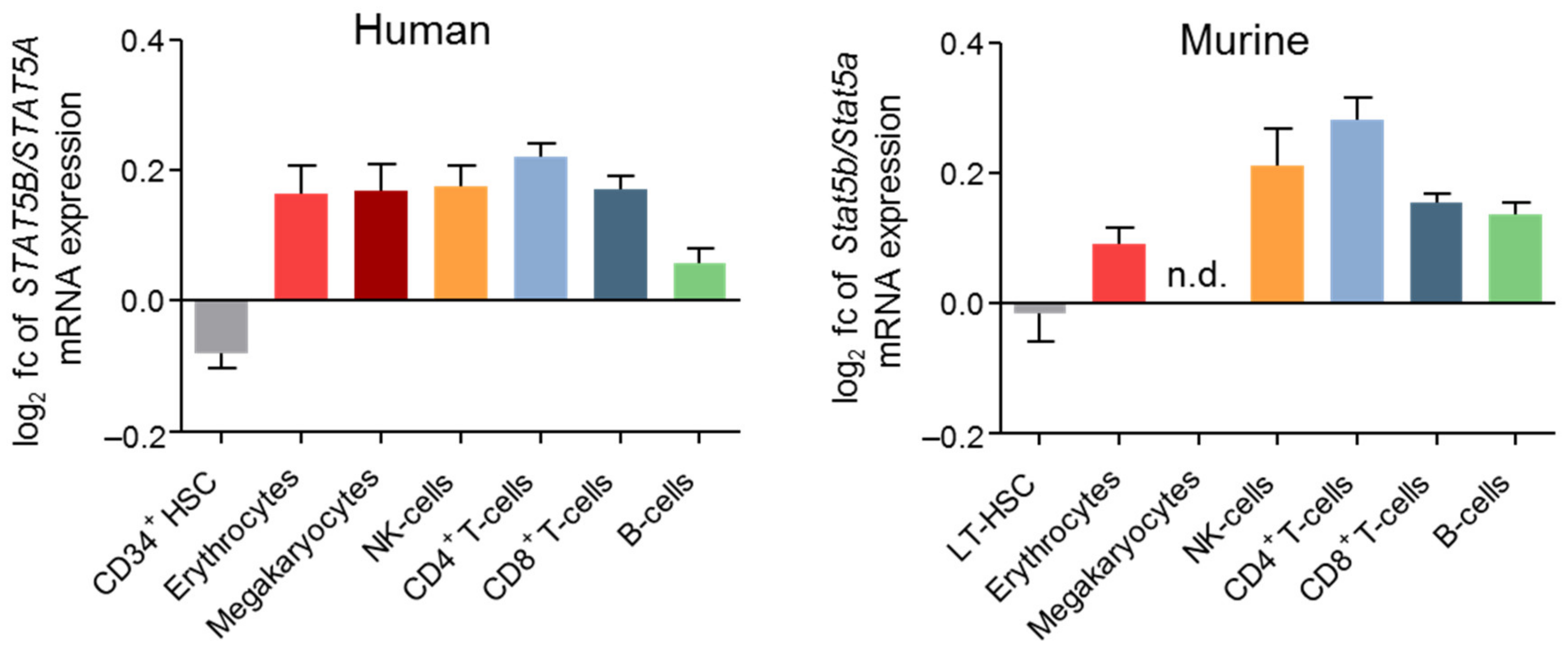{kind=link}
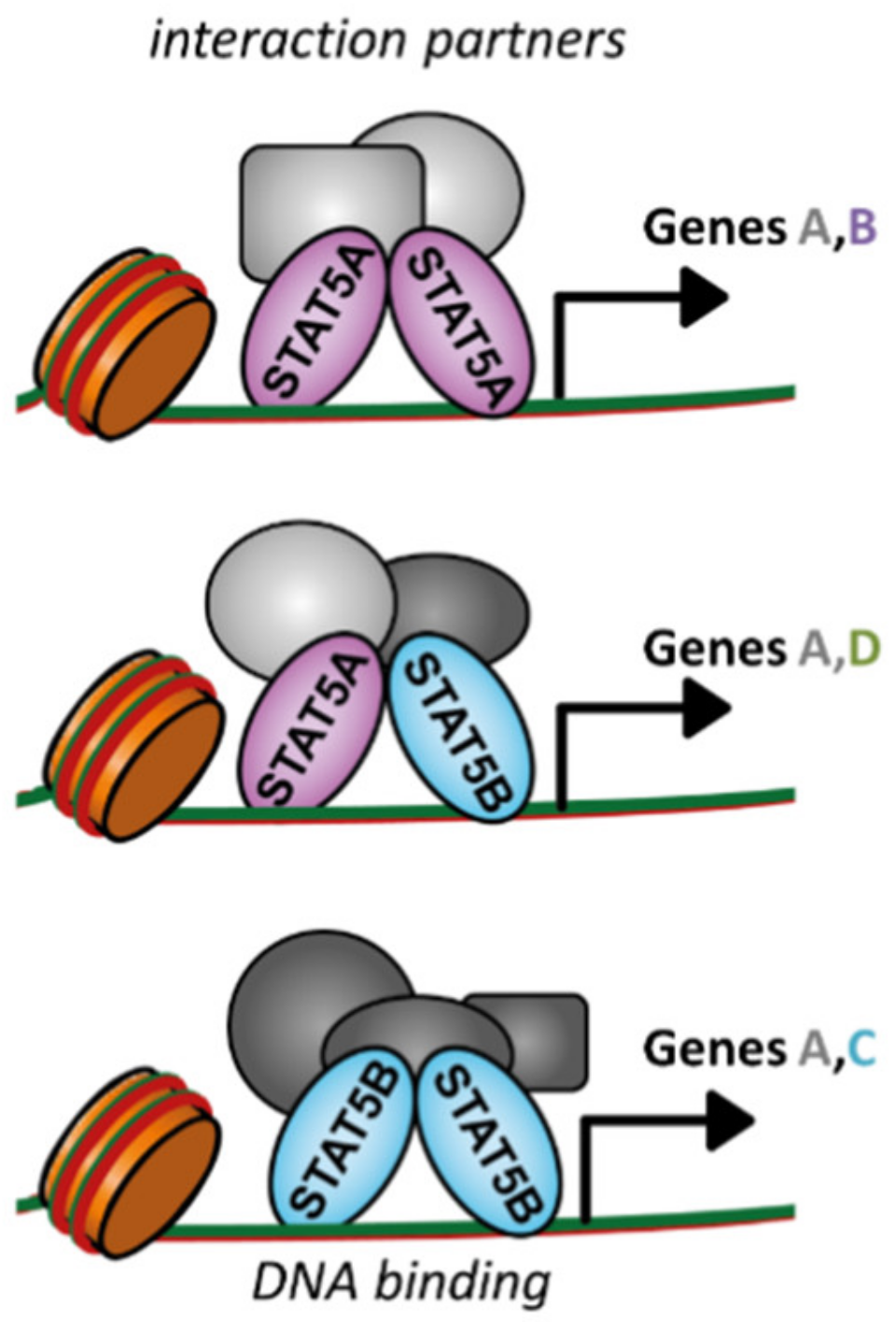{kind=link}
Table 1.
Stat5a/b-deficient mouse models.
| Deficiency | Phenotype | Reference | |
|---|---|---|---|
| STAT5A/B | Stat5a/bΔN | truncated N-termini of Stat5a and Stat5b forms dimers but no tetramers smaller, infertile, less CD25+CD4+ T cells | Teglund et al. 1998 [56] |
| Stat5a/b-/- | total body knockout perinatal lethal, 1–2% survivors dwarfism, anemia, reduced T- and NK cell numbers, block in pre–pro-B cell stage | Cui et al. 2004 [59] | |
| Stat5a/bDKI | N-termini mutations in Stat5a and Stat5b no tetramer formation less CD25+CD4+ and CD8+ T cells, less NK cells | Lin et al. 2012 [57] | |
| Stat5a/bfl/fl | loxP-sites spanning Stat5a and Stat5b for tissue-specific or inducible deletion—e.g., in the hematopoietic system: | Cui et al. 2004 [59] | |
| vav-Cre: Anemia, lymphopenia, reduced repopulation capacity (upon BM transplants) | Wang et al. 2015 [60] | ||
| Tie2-Cre: Anemia, lymphopenia, reduced repopulation capacity (upon BM transplants) | Zhu et al. 2008 [61] | ||
| Mx1-Cre: Reduced repopulation capacity (upon BM transplants) | Wang et al. 2009 [62] | ||
| STAT5A | Stat5aΔN | truncated N-termini of Stat5a forms dimers but no tetramers reduced prolactin signaling (mammary gland) | Teglund et al. 1998 [56] |
| Stat5a−/− | total body knockout of Stat5a failure in mammary gland formation and function | Liu et al. 1997 [9] | |
| Stat5aKI | N-termini mutation of Stat5a no STAT5A tetramers slight reduction of IL-2R expression on T cells | Lin et al. 2012 [57] | |
| STAT5B | Stat5bΔN | truncated N-termini of Stat5b forms dimers but no tetramers dwarfism, reduced IGF-1 levels | Teglund et al. 1998 [56] |
| Stat5b−/− | total body knockout of Stat5b dwarfism, failure in GH signaling, reduced numbers of NK and T cells | Udy et al. 1997 [63] | |
| Stat5bKI | N-termini mutations of Stat5b no STAT5B tetramers same, but more pronounced as in Stat5aKI | Lin et al. 2012 [57] | |
Table 2.
Hematopoietic STAT5A/B transgenic mouse models.
| Transgene | Promoter | Phenotype | Reference |
|---|---|---|---|
| Stat5b-tg | H-2Kb promoter and IgM enhancer (T, B, NK cells) | more CD8+ T cells ~12% thymic T cell lymphoblastic lymphoma (CD8+CD4+ or CD8+) | Kelly et al. 2003 [105,153] |
| NOD background | 75% CD8+ T cell lymphoblastic lymphoma | Chen et al. 2013 [154] | |
| Stat5b-CA-tg | Lck promoter and IgM enhancer | expansion of CD8+ and γδ T cells late emergence of clonal B cell lymphoma/leukemia (low incidence) | Burchill et al. 2003 [88] |
| pre-BCR pathway defects | increased B-ALL incidence | Katerndahl et al. 2017 [89] | |
| cS5F | Eµ enhancer (lymphoid specific) | increase of CD8+ T cells, late emergence of clonal B cell lymphoma/leukemia (low incidence) | Joliot et al. 2006 [129] |
| MMTV-tTA TetO-cS5F | cS5F-Tet-OFF under MMTV-LTR promoter | hematopoiesis unaffected cross to Stat5a/bΔN → expansion of granulocytes | Lin et al. 2013 [155] |
| hSTAT5B | vav promoter | mild expansion of CD8+ T cells no disease | Pham et al. 2018 [84] |
| hSTAT5BN642H | vav promoter | aggressive CD8+ T cell neoplasia enhanced numbers of HSCs and lymphoid progenitors | Pham et al. 2018 [84] |
| cS5F lo | vav promoter | mild expansion of CD8+ T cells no disease | Maurer et al. 2019 [83] |
| cS5F hi | vav promoter | CD8+ T cell neoplasia correlating to PTCL-NOS enhanced numbers of HSCs and lymphoid progenitors | Maurer et al. 2019 [83] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Maurer, B.; Kollmann, S.; Pickem, J.; Hoelbl-Kovacic, A.; Sexl, V. STAT5A and STAT5B—Twins with Different Personalities in Hematopoiesis and Leukemia. Cancers 2019, 11, 1726. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11111726
AMA Style
Maurer B, Kollmann S, Pickem J, Hoelbl-Kovacic A, Sexl V. STAT5A and STAT5B—Twins with Different Personalities in Hematopoiesis and Leukemia. Cancers. 2019; 11(11):1726. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11111726
Chicago/Turabian StyleMaurer, Barbara, Sebastian Kollmann, Judith Pickem, Andrea Hoelbl-Kovacic, and Veronika Sexl. 2019. "STAT5A and STAT5B—Twins with Different Personalities in Hematopoiesis and Leukemia" Cancers 11, no. 11: 1726. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11111726
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.