Identification of Circulating Genomic and Metabolic Biomarkers in Intrahepatic Cholangiocarcinoma

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Sequencing of Circulating Tumour DNA from ICC Patients
2.2. Changes in Variants following SIRT Radiotherapy Treatment
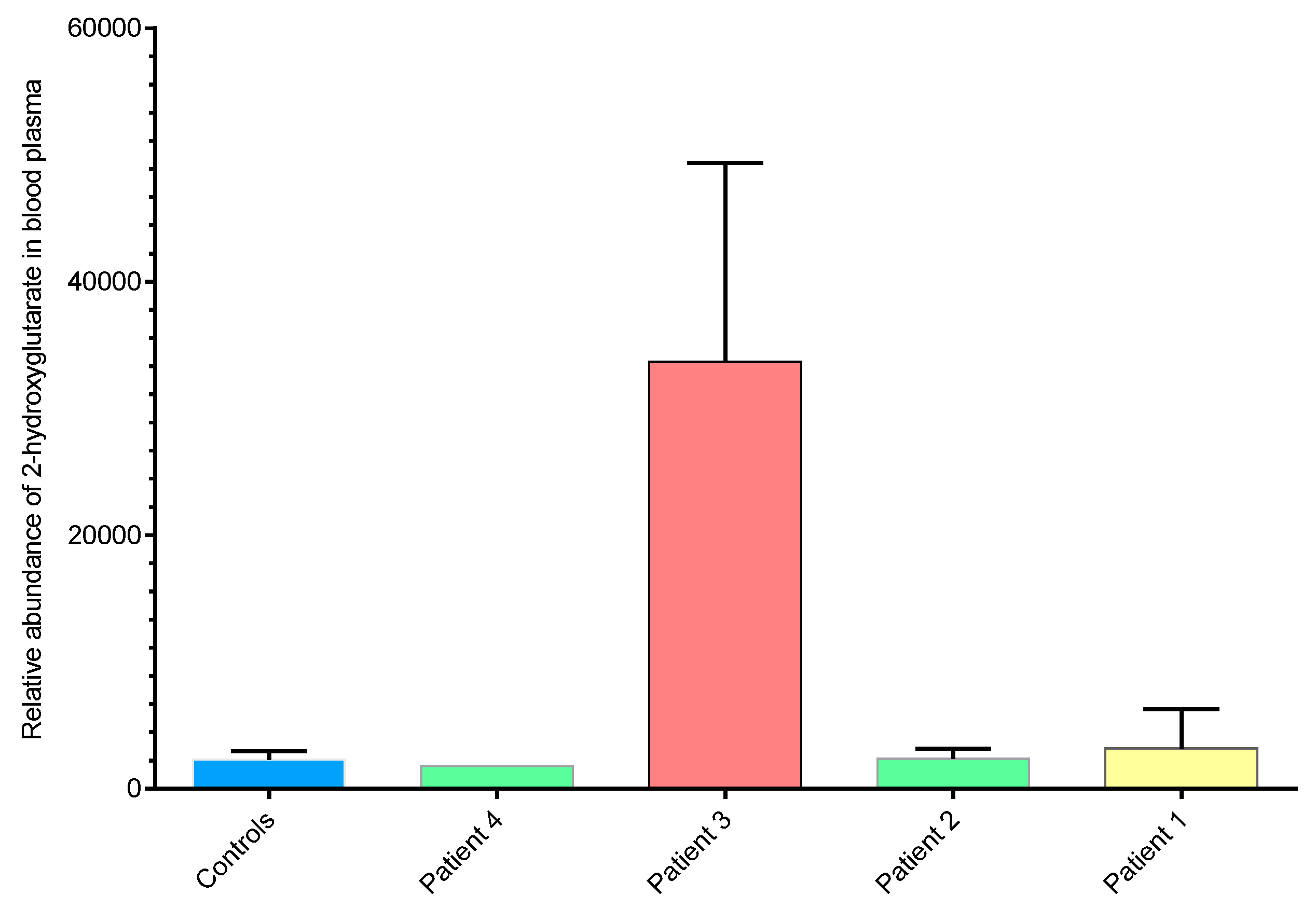
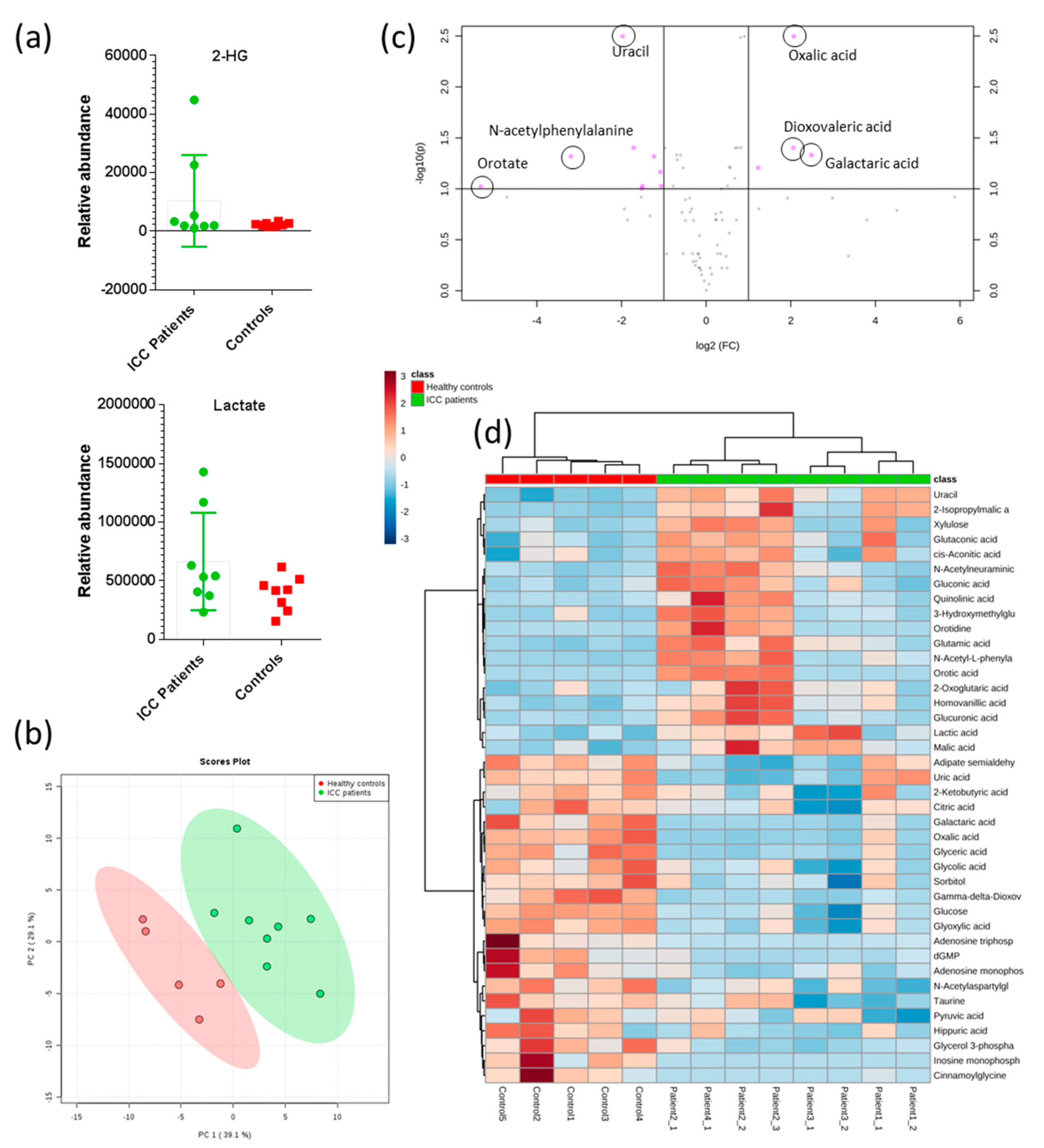
2.3. 2-hydroxyglutarate Abundance Correlates with ctDNA IDH1 Status
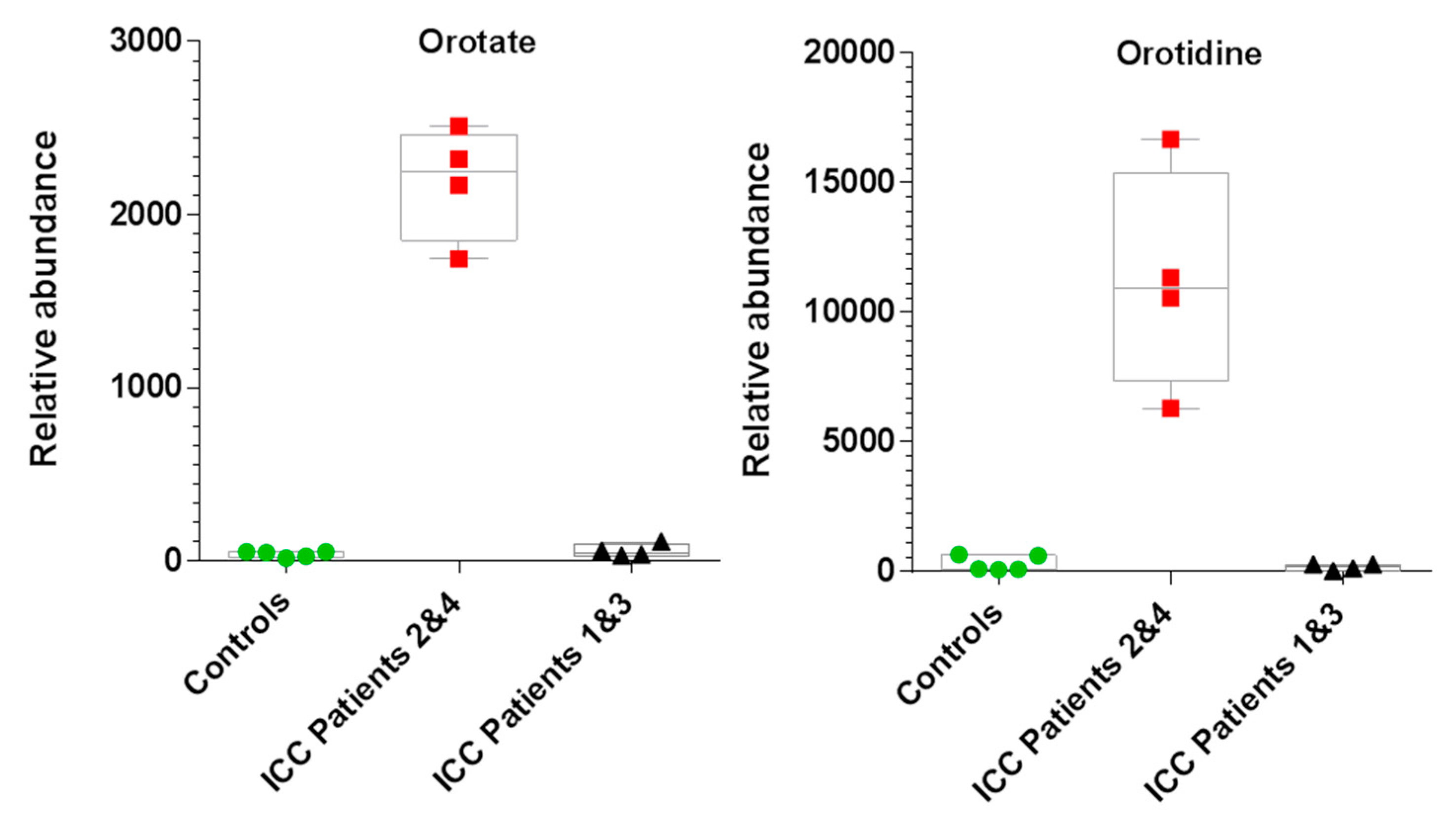
2.4. Untargeted Metabolomics Reveals Altered Metabolite Profiles in ICC Patients
3. Discussion
4. Materials and Methods
4.1. Methods and Patients
4.2. Sample Processing and Extraction of ctDNA and gDNA
4.3. Targeted Sequencing Using 50 Cancer Gene Panel
4.4. Library Preparation for whole Genome Sequencing of Plasma DNA Samples
4.5. Library Preparation for whole Genome Sequencing of Genomic DNA Samples
4.6. Mapping and Variant Calling in whole Genome Sequence (WGS) Data
4.7. Metabolomics by IC-MS/MS
4.7.1. Metabolite Extraction
4.7.2. IC-MS Analysis
4.7.3. Data Processing and Analysis
4.7.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Konstantinidis, I.T.; Groot Koerkamp, B.; Do, R.K.; Gonen, M.; Fong, Y.; Allen, P.J.; D’Angelica, M.I.; Kingham, T.P.; DeMatteo, R.P.; Klimstra, D.S.; et al. Unresectable intrahepatic cholangiocarcinoma: Systemic plus hepatic arterial infusion chemotherapy is associated with longer survival in comparison with systemic chemotherapy alone. Cancer 2016, 122, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, S.H.; Chang, H.M.; Kim, J.S.; Choi, H.J.; Lee, M.A.; Jang, J.S.; Jeung, H.C.; Kang, J.H.; Lee, H.W.; et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012, 13, 181–188. [Google Scholar] [CrossRef]
- Malka, D.; Cervera, P.; Foulon, S.; Trarbach, T.; de la Fouchardiere, C.; Boucher, E.; Fartoux, L.; Faivre, S.; Blanc, J.F.; Viret, F.; et al. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGO): A randomised, open-label, non-comparative phase 2 trial. Lancet Oncol. 2014, 15, 819–828. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef]
- Sahu, S.; Sun, W. Targeted therapy in biliary tract cancers-current limitations and potentials in the future. J. Gastrointest. Oncol. 2017, 8, 324–336. [Google Scholar] [CrossRef]
- Kim, Y.H.; Hong, E.K.; Kong, S.Y.; Han, S.S.; Kim, S.H.; Rhee, J.K.; Hwang, S.K.; Park, S.J.; Kim, T.M. Two classes of intrahepatic cholangiocarcinoma defined by relative abundance of mutations and copy number alterations. Oncotarget 2016, 7, 23825–23836. [Google Scholar] [CrossRef]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef]
- Xie, D.; Ren, Z.; Fan, J.; Gao, Q. Genetic profiling of intrahepatic cholangiocarcinoma and its clinical implication in targeted therapy. Am. J. Cancer Res. 2016, 6, 577–586. [Google Scholar]
- Zou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef]
- Voss, J.S.; Holtegaard, L.M.; Kerr, S.E.; Fritcher, E.G.; Roberts, L.R.; Gores, G.J.; Zhang, J.; Highsmith, W.E.; Halling, K.C.; Kipp, B.R. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum. Pathol. 2013, 44, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.P.; Barr Fritcher, E.G.; Pestova, E.; Schulz, J.; Sitailo, L.A.; Vasmatzis, G.; Murphy, S.J.; McWilliams, R.R.; Hart, S.N.; Halling, K.C.; et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum. Pathol. 2014, 45, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Boissel, N.; Nibourel, O.; Renneville, A.; Gardin, C.; Reman, O.; Contentin, N.; Bordessoule, D.; Pautas, C.; de Revel, T.; Quesnel, B.; et al. Prognostic impact of isocitrate dehydrogenase enzyme isoforms 1 and 2 mutations in acute myeloid leukemia: A study by the Acute Leukemia French Association group. J. Clin. Oncol. 2010, 28, 3717–3723. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Koszarska, M.; Bors, A.; Feczko, A.; Meggyesi, N.; Batai, A.; Csomor, J.; Adam, E.; Kozma, A.; Orban, T.I.; Lovas, N.; et al. Type and location of isocitrate dehydrogenase mutations influence clinical characteristics and disease outcome of acute myeloid leukemia. Leuk. Lymphoma 2013, 54, 1028–1035. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Fujiwara, H.; Tateishi, K.; Kato, H.; Nakatsuka, T.; Yamamoto, K.; Tanaka, Y.; Ijichi, H.; Takahara, N.; Mizuno, S.; Kogure, H.; et al. Isocitrate dehydrogenase 1 mutation sensitizes intrahepatic cholangiocarcinoma to the BET inhibitor JQ1. Cancer Sci. 2018, 109, 3602–3610. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Emir, U.E.; Larkin, S.J.; de Pennington, N.; Voets, N.; Plaha, P.; Stacey, R.; Al-Qahtani, K.; McCullagh, J.; Schofield, C.J.; Clare, S.; et al. Noninvasive Quantification of 2-Hydroxyglutarate in Human Gliomas with IDH1 and IDH2 Mutations. Cancer Res. 2016, 76, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Voets, N.L.; Larkin, S.J.; de Pennington, N.; Plaha, P.; Stacey, R.; McCullagh, J.S.O.; Schofield, C.J.; Clare, S.; Jezzard, P.; et al. A Noninvasive Comparison Study between Human Gliomas with IDH1 and IDH2 Mutations by MR Spectroscopy. Metabolites 2019, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Tarhonskaya, H.; Rydzik, A.M.; Leung, I.K.; Loik, N.D.; Chan, M.C.; Kawamura, A.; McCullagh, J.S.; Claridge, T.D.; Flashman, E.; Schofield, C.J. Non-enzymatic chemistry enables 2-hydroxyglutarate-mediated activation of 2-oxoglutarate oxygenases. Nat. Commun. 2014, 5, 3423. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Touat, M.; Maher, E.; De La Fuente, M.; Cloughesy, T.F.; Holdhoff, M.; Cote, G.M.; Burris, H.; Janku, F.; Huang, R.; et al. ACTR-46. AG-120, A first-in-class mutant idh1 inhibitor in patients with recurrent or progressive idh1 mutant glioma: updated results from the phase 1 non-enhancing glioma population. Neuro Oncol. 2017, 19, vi10–vi11. [Google Scholar] [CrossRef]
- Saha, S.K.; Gordan, J.D.; Kleinstiver, B.P.; Vu, P.; Najem, M.S.; Yeo, J.C.; Shi, L.; Kato, Y.; Levin, R.S.; Webber, J.T.; et al. Isocitrate Dehydrogenase Mutations Confer Dasatinib Hypersensitivity and SRC Dependence in Intrahepatic Cholangiocarcinoma. Cancer Discov. 2016, 6, 727–739. [Google Scholar] [CrossRef]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef]
- Andersen, R.F.; Jakobsen, A. Screening for circulating RAS/RAF mutations by multiplex digital PCR. Cli. Chim. Acta Int. J. Clin. Chem. 2016, 458, 138–143. [Google Scholar] [CrossRef]
- Mody, K.; Kasi, P.M.; Yang, J.; Surapaneni, P.K.; Bekaii-Saab, T.; Ahn, D.H.; Mahipal, A.; Sonbol, M.B.; Starr, J.S.; Roberts, A.; et al. Circulating Tumor DNA Profiling of Advanced Biliary Tract Cancers. JCO Precis. Oncol. 2019, 1–9. [Google Scholar] [CrossRef]
- Wasenang, W.; Chaiyarit, P.; Proungvitaya, S.; Limpaiboon, T. Serum cell-free DNA methylation of OPCML and HOXD9 as a biomarker that may aid in differential diagnosis between cholangiocarcinoma and other biliary diseases. Clin. Epigenetics 2019, 11, 39. [Google Scholar] [CrossRef] [Green Version]
- Hamblin, A.; Wordsworth, S.; Fermont, J.M.; Page, S.; Kaur, K.; Camps, C.; Kaisaki, P.; Gupta, A.; Talbot, D.; Middleton, M.; et al. Clinical applicability and cost of a 46-gene panel for genomic analysis of solid tumours: Retrospective validation and prospective audit in the UK National Health Service. PLoS Med. 2017, 14, e1002230. [Google Scholar] [CrossRef] [PubMed]
- Misumi, K.; Hayashi, A.; Shibahara, J.; Arita, J.; Sakamoto, Y.; Hasegawa, K.; Kokudo, N.; Fukayama, M. Intrahepatic cholangiocarcinoma frequently shows loss of BAP1 and PBRM1 expression, and demonstrates specific clinicopathological and genetic characteristics with BAP1 loss. Histopathology 2017, 70, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.; Bahra, M.; Lenze, D.; Bradtmoller, M.; Guse, K.; Gehlhaar, C.; Blaker, H.; Heppner, F.L.; Koch, A. Genome wide DNA copy number analysis in cholangiocarcinoma using high resolution molecular inversion probe single nucleotide polymorphism assay. Exp. Mol. Pathol. 2015, 99, 344–353. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The meaning of p16(ink4a) expression in tumors: Functional significance, clinical associations and future developments. Cell cycle (Georgetown, Tex.) 2011, 10, 2497–2503. [Google Scholar] [CrossRef] [Green Version]
- Akhavantabasi, S.; Akman, H.B.; Sapmaz, A.; Keller, J.; Petty, E.M.; Erson, A.E. USP32 is an active, membrane-bound ubiquitin protease overexpressed in breast cancers. Mamm. Genome 2010, 21, 388–397. [Google Scholar] [CrossRef]
- Monni, O.; Barlund, M.; Mousses, S.; Kononen, J.; Sauter, G.; Heiskanen, M.; Paavola, P.; Avela, K.; Chen, Y.; Bittner, M.L.; et al. Comprehensive copy number and gene expression profiling of the 17q23 amplicon in human breast cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 5711–5716. [Google Scholar] [CrossRef] [Green Version]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 2017, 47, 466–480. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, G.; Corona, G.; Bellu, L.; Della Puppa, A.; Pambuku, A.; Fiduccia, P.; Bertorelle, R.; Gardiman, M.P.; D’Avella, D.; Toffoli, G.; et al. Diagnostic value of plasma and urinary 2-hydroxyglutarate to identify patients with isocitrate dehydrogenase-mutated glioma. Oncologist 2015, 20, 562–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ramos, A.; Velazquez-Campoy, A.; Grande-Garcia, A.; Moreno-Morcillo, M.; Ramon-Maiques, S. Structure and Functional Characterization of Human Aspartate Transcarbamoylase, the Target of the Anti-tumoral Drug PALA. Structure 2016, 24, 1081–1094. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; Nickerson, D.A.; et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010, 42, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Rainger, J.; Bengani, H.; Campbell, L.; Anderson, E.; Sokhi, K.; Lam, W.; Riess, A.; Ansari, M.; Smithson, S.; Lees, M.; et al. Miller (Genee-Wiedemann) syndrome represents a clinically and biochemically distinct subgroup of postaxial acrofacial dysostosis associated with partial deficiency of DHODH. Hum. Mol. Genet. 2012, 21, 3969–3983. [Google Scholar] [CrossRef] [Green Version]
- Columbano, A.; Ledda, G.M.; Rao, P.M.; Rajalakshmi, S.; Sarma, D.S. Dietary orotic acid, a new selective growth stimulus for carcinogen altered hepatocytes in rat. Cancer Lett. 1982, 16, 191–196. [Google Scholar] [CrossRef]
- Laurier, C.; Tatematsu, M.; Rao, P.M.; Rajalakshmi, S.; Sarma, D.S. Promotion by orotic acid of liver carcinogenesis in rats initiated by 1,2-dimethylhydrazine. Cancer Res. 1984, 44, 2186–2191. [Google Scholar]
- Rao, P.M.; Rajalakshmi, S.; Alam, A.; Sarma, D.S.; Pala, M.; Parodi, S. Orotic acid, a promoter of liver carcinogenesis induces DNA damage in rat liver. Carcinogenesis 1985, 6, 765–768. [Google Scholar] [CrossRef]
- Sidransky, H.; Verney, E. Influence of orotic acid on liver tumorigenesis in rats ingesting ethionine, N-2-fluorenylacetamide, and 3′-methyl-dimethylaminoazobenzene. J. Natl. Cancer Inst. 1970, 44, 1201–1215. [Google Scholar]
- Rogers, S. Inhibitory influence of a normally occurring pyrimidine precursor upon methylcholanthrene carcinogenesis. Proc. Soc. Exp. Biol. Med. 1957, 96, 464–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, E.W.; Malmgren, R.A. Effect of orotic acid on the metastasis of mammary tumors in mice. Cancer Res. 1964, 24, 671–673. [Google Scholar] [PubMed]
- Ser, Z.; Gao, X.; Johnson, C.; Mehrmohamadi, M.; Liu, X.; Li, S.; Locasale, J.W. Targeting One Carbon Metabolism with an Antimetabolite Disrupts Pyrimidine Homeostasis and Induces Nucleotide Overflow. Cell Rep 2016, 15, 2367–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochiai, T.; Umeki, M.; Miyake, H.; Iida, T.; Okumura, M.; Ohno, K.; Sakamoto, M.; Miyoshi, N.; Takahashi, M.; Tsumura, H.; et al. Impact of 5-fluorouracil metabolizing enzymes on chemotherapy in patients with resectable colorectal cancer. Oncol. Rep. 2014, 32, 887–892. [Google Scholar] [CrossRef] [Green Version]
- Reyes, P.; Guganig, M.E. Studies on a pyrimidine phosphoribosyltransferase from murine leukemia P1534J. Partial purification, substrate specificity, and evidence for its existence as a bifunctional complex with orotidine 5-phosphate decarboxylase. J. Biol. Chem. 1975, 250, 5097–5108. [Google Scholar]
- Villa, E.; Ali, E.S.; Sahu, U.; Ben-Sahra, I. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers 2019, 11, 688. [Google Scholar] [CrossRef] [Green Version]
- Bardeleben, C.; Sharma, S.; Reeve, J.R.; Bassilian, S.; Frost, P.; Hoang, B.; Shi, Y.; Lichtenstein, A. Metabolomics identifies pyrimidine starvation as the mechanism of 5-aminoimidazole-4-carboxamide-1-beta-riboside-induced apoptosis in multiple myeloma cells. Mol. Cancer Ther. 2013, 12, 1310–1321. [Google Scholar] [CrossRef] [Green Version]
- Akahoshi, K.; Ban, D.; Kuboki, R.; Oba, A.; Ono, H.; Mitsunori, Y.; Kudo, A.; Tanaka, S.; Tanabe, M. Orotate phosphoribosyltransferase as a predictor of benefit from S-1 adjuvant chemotherapy for cholangiocarcinoma patients. J. Gastroenterol. Hepatol. 2019, 34, 1108–1115. [Google Scholar] [CrossRef]
- Page, K.; Guttery, D.S.; Zahra, N.; Primrose, L.; Elshaw, S.R.; Pringle, J.H.; Blighe, K.; Marchese, S.D.; Hills, A.; Woodley, L.; et al. Influence of plasma processing on recovery and analysis of circulating nucleic acids. PLoS ONE 2013, 8, e77963. [Google Scholar] [CrossRef] [Green Version]
- Merriman, B.; Ion Torrent Team, R.D.; Rothberg, J.M. Progress in ion torrent semiconductor chip based sequencing. Electrophoresis 2012, 33, 3397–3417. [Google Scholar] [CrossRef]
- Lamble, S.; Batty, E.; Attar, M.; Buck, D.; Bowden, R.; Lunter, G.; Crook, D.; El-Fahmawi, B.; Piazza, P. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 2013, 13, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunter, G.; Goodson, M. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011, 21, 936–939. [Google Scholar] [CrossRef] [Green Version]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.F.; Wilkie, A.O.M.; McVean, G.; Lunter, G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Kallberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [Green Version]
- De Cima, S.; Polo, L.M.; Diez-Fernandez, C.; Martinez, A.I.; Cervera, J.; Fita, I.; Rubio, V. Structure of human carbamoyl phosphate synthetase: Deciphering the on/off switch of human ureagenesis. Sci. Rep. 2015, 5, 16950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande-Garcia, A.; Lallous, N.; Diaz-Tejada, C.; Ramon-Maiques, S. Structure, functional characterization, and evolution of the dihydroorotase domain of human CAD. Structure 2014, 22, 185–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walse, B.; Dufe, V.T.; Svensson, B.; Fritzson, I.; Dahlberg, L.; Khairoullina, A.; Wellmar, U.; Al-Karadaghi, S. The structures of human dihydroorotate dehydrogenase with and without inhibitor reveal conformational flexibility in the inhibitor and substrate binding sites. Biochemistry 2008, 47, 8929–8936. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient | Baseline Quantity ctDNA (ng/µL) | Mutation (Single Nucleotide Variants (SNV)) | VAF (%) Baseline | VAF (%) 4 Weeks Post-SIRT | VAF (%) 10 Weeks Post-SIRT |
|---|---|---|---|---|---|
| 1 | 1.71 | NRAS, NM_002524.5: c.182A > G; p.Q61R | 0.6 (T = 5168, C = 31) | na | nd |
| 2 | 18 | NRAS, NM_002524.5: c.182A > G; p.Q61R | 46 (T = 4408, C = 3773) | 28 (T = 6375, C = 2518) | 32 (T = 4254, C = 2000) |
| 3 | 2.98 | NRAS, NM_002524.5: c.182A > G; p.Q61R | 21 (T = 7509, C = 2014) | 8 (T = 7819, C = 680) | na |
| IDH1, NM_005896.3: c.394C > T; p.R132C | 12 (G = 8490, A = 1109) | 5 (G = 9369, A = 480) | na | ||
| 4 | 1.19 | None | nd | na | na |
| Gene Symbol | Genomic Location (GRCh37/hg19) | Patient 1 | Patient 2 | Patient 3 | Patient 4 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CNVs in Region of Gene | SNVs | CNVs in Region of Gene | SNVs | Supporting SNV Reads at Baseline (Mutant/Total Reads) | CNVs in Region of Gene | SNVs | CNVs in Region of Gene | SNVs | Supporting SNV Reads at Baseline (Mutant/Total Reads) | ||
| CAD | chr2: 27,440,258-27,466,811 | none | none | chr2 CN gain (incl CAD) | CAD, NM_004341: c.G6343C; p.V2115L (germline) | 14/27 | none | none | none | CAD, NM_004341: c.T274C; p.C92R | 3/24 |
| CAD, NM_004341: c.6139_6140del; p.D2047fs | 4/36 | ||||||||||
| DHODH | chr16: 72,042,487-72,058,955 | chr16 CN loss (incl DHODH) | none | none | none | na | none | none | none | DHODH, NM_001361: c.G1123T; p.A375S | 2/26 |
| UMPS | chr3: 124,449,213-124,468,120 | chr3 CN loss (incl UMPS) | none | chr3 CN loss (incl UMPS) | none | na | none | none | none | none | na |
| TYMS | chr18: 657,604-673,578 | none | none | none | none | na | chr18 LOH (incl TYMS) | none | none | TYMS, NM_001071:c.G13A; p.G5S (germline) | 9/24 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winter, H.; Kaisaki, P.J.; Harvey, J.; Giacopuzzi, E.; Ferla, M.P.; Pentony, M.M.; Knight, S.J.L.; Sharma, R.A.; Taylor, J.C.; McCullagh, J.S.O. Identification of Circulating Genomic and Metabolic Biomarkers in Intrahepatic Cholangiocarcinoma. Cancers 2019, 11, 1895. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11121895
Winter H, Kaisaki PJ, Harvey J, Giacopuzzi E, Ferla MP, Pentony MM, Knight SJL, Sharma RA, Taylor JC, McCullagh JSO. Identification of Circulating Genomic and Metabolic Biomarkers in Intrahepatic Cholangiocarcinoma. Cancers. 2019; 11(12):1895. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11121895
Chicago/Turabian StyleWinter, Helen, Pamela J. Kaisaki, Joe Harvey, Edoardo Giacopuzzi, Matteo P. Ferla, Melissa M. Pentony, Samantha J.L. Knight, Ricky A. Sharma, Jenny C. Taylor, and James S.O. McCullagh. 2019. "Identification of Circulating Genomic and Metabolic Biomarkers in Intrahepatic Cholangiocarcinoma" Cancers 11, no. 12: 1895. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11121895