PAK4-NAMPT Dual Inhibition as a Novel Strategy for Therapy Resistant Pancreatic Neuroendocrine Tumors

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
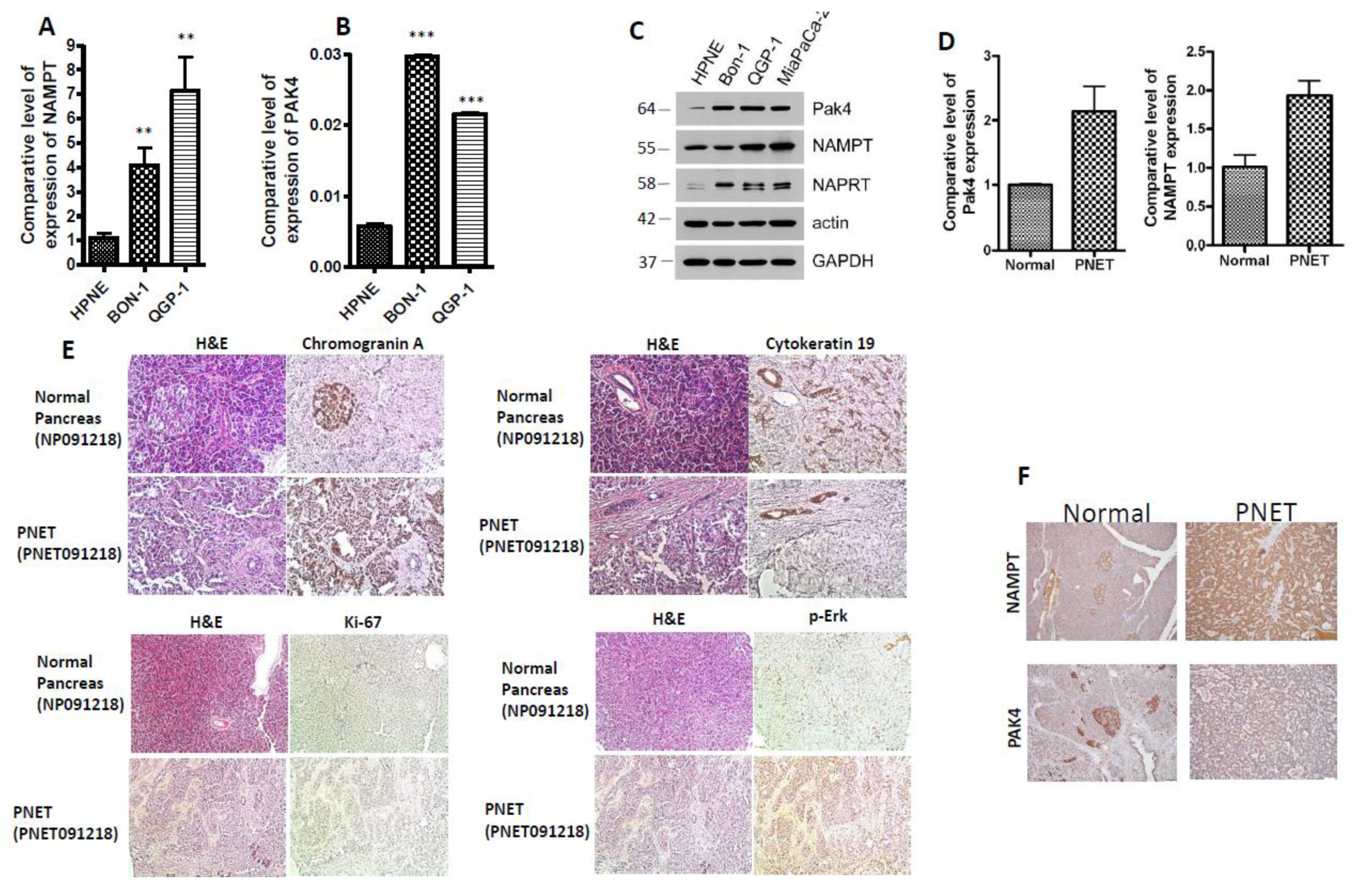
2.1. PAK4 and NAMPT Are Overexpressed in PNET
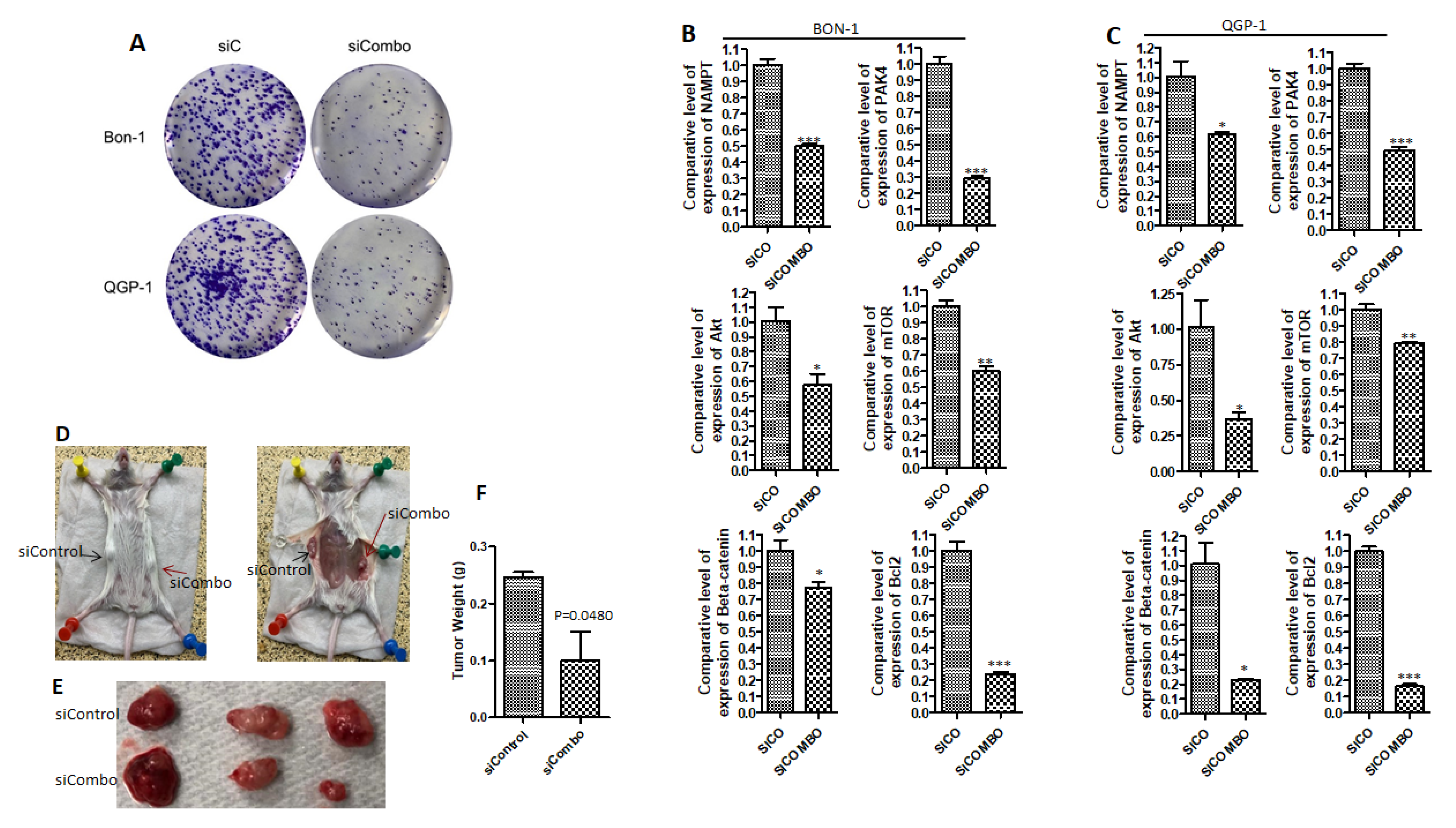
2.2. PAK4 and NAMPT Promote PNET Survival
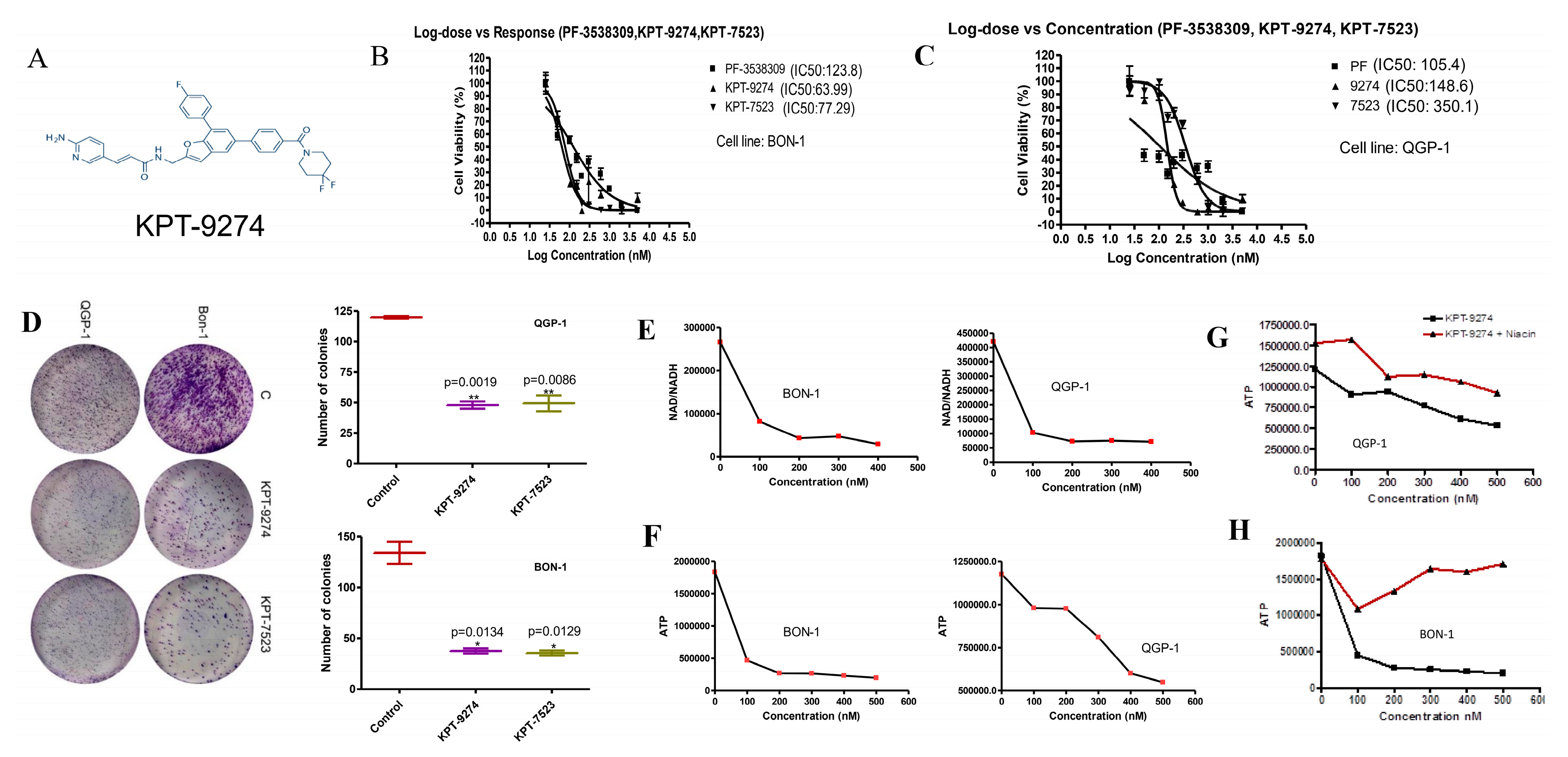
2.3. KPT-9274 and Analog KPT-7523 Decrease PNET Cell Survival and Growth
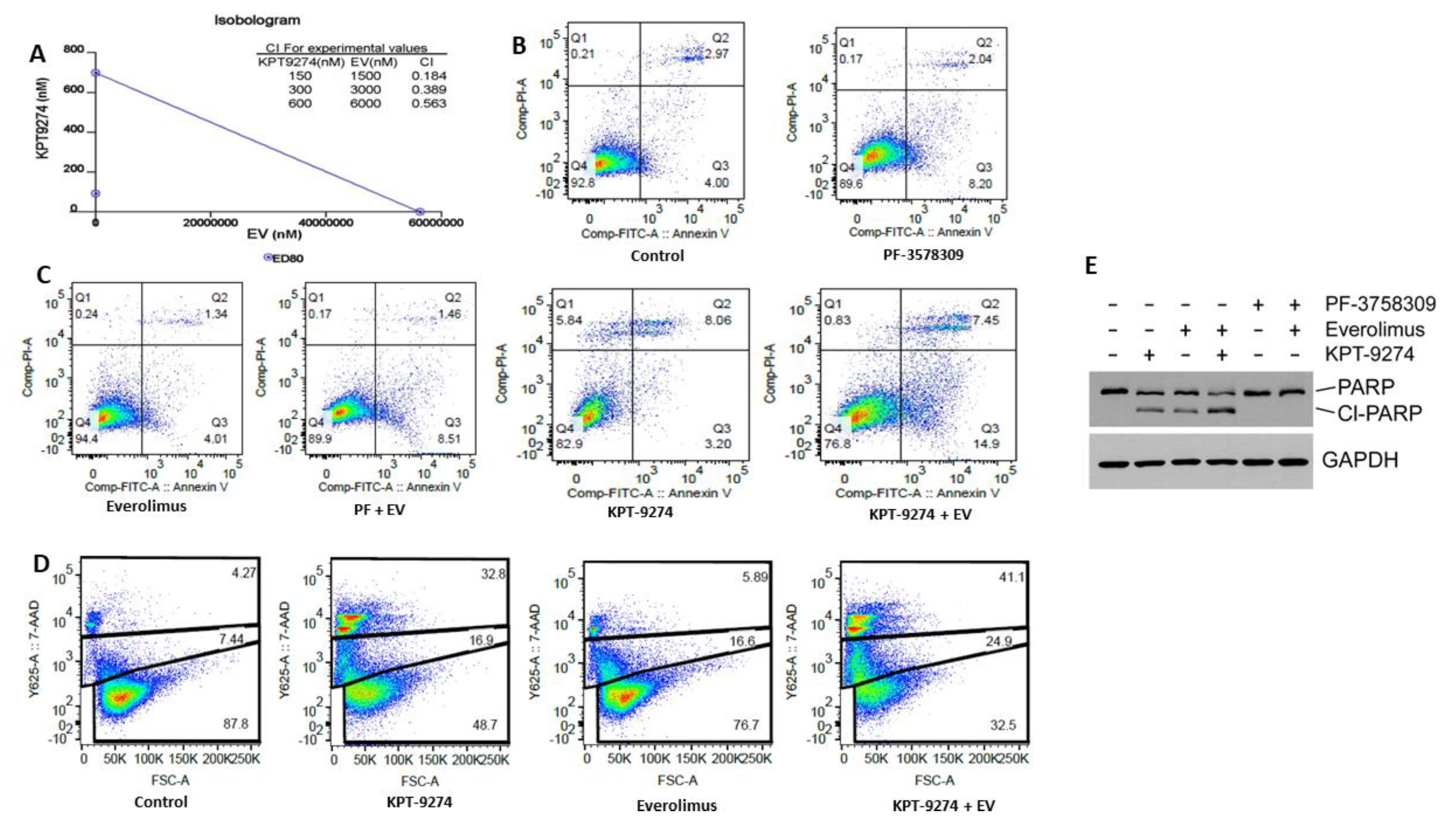
2.4. KPT-9274 Synergizes with Everolimus in PNET Cellular Models
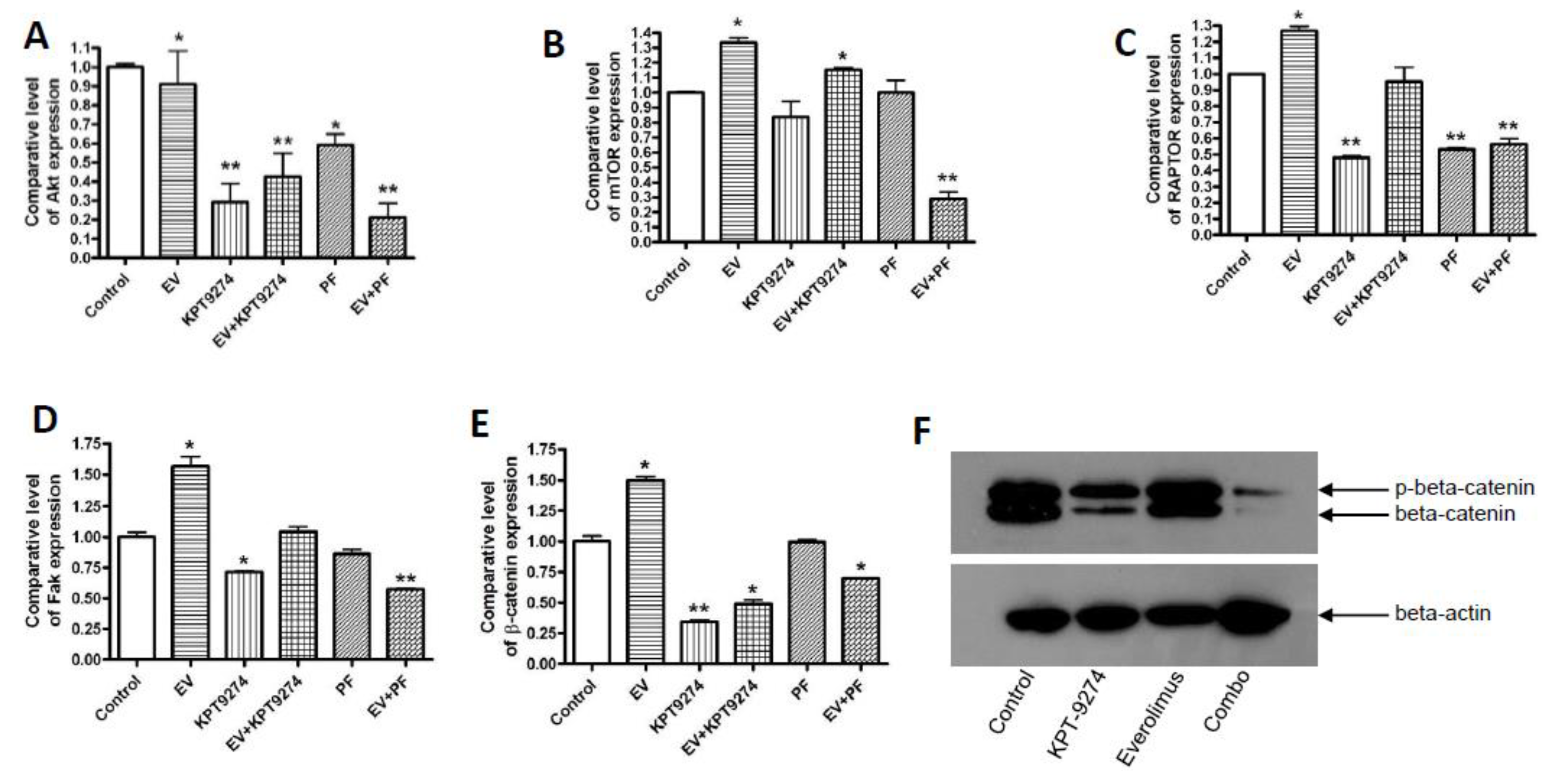
2.5. Molecular Analysis of KPT-9274-Everolimus Combination
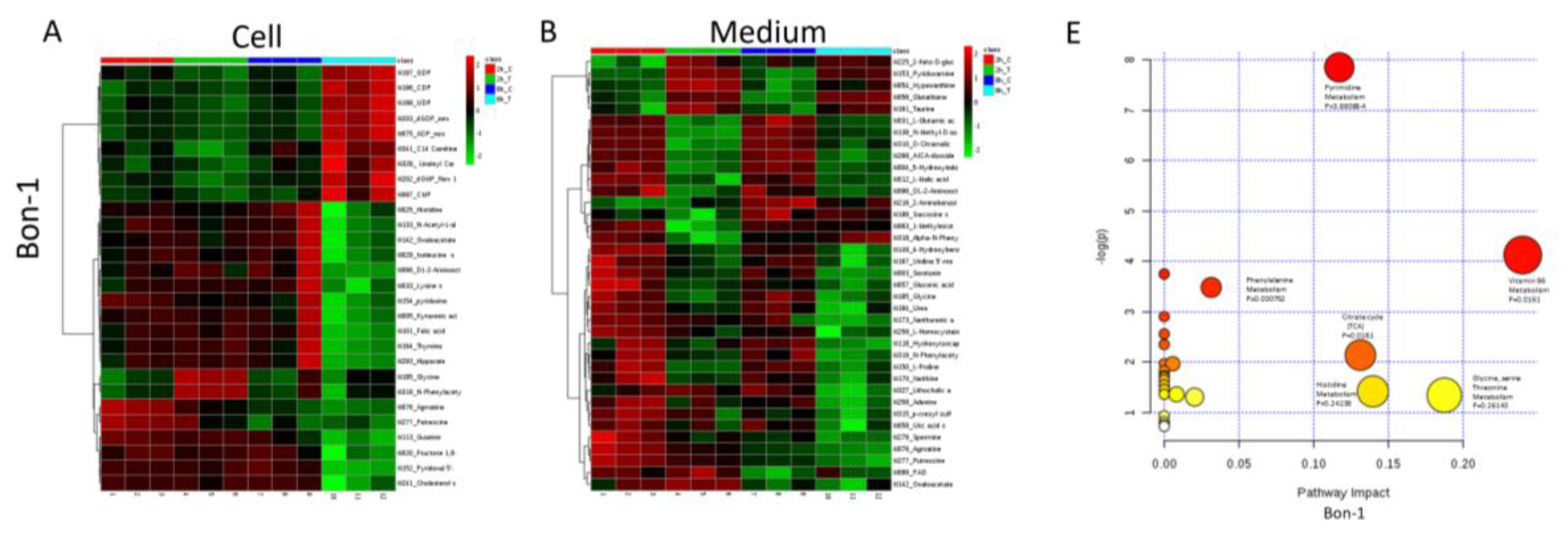
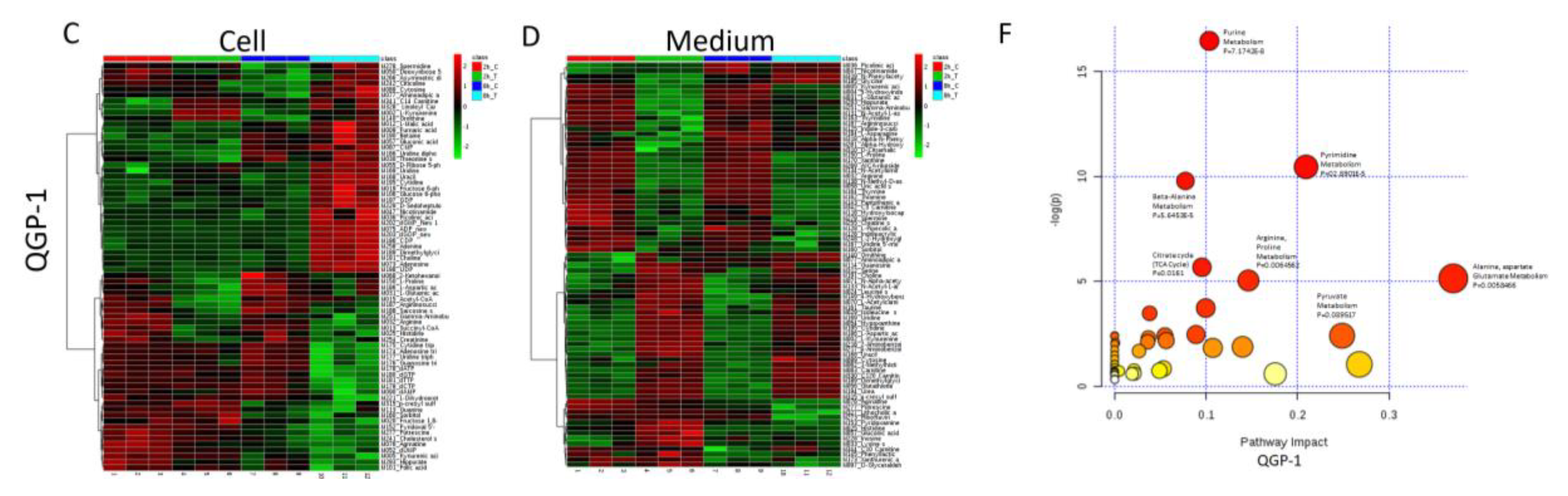
2.6. Metabolomic Analysis of PNET Cells Treated with KPT-9274
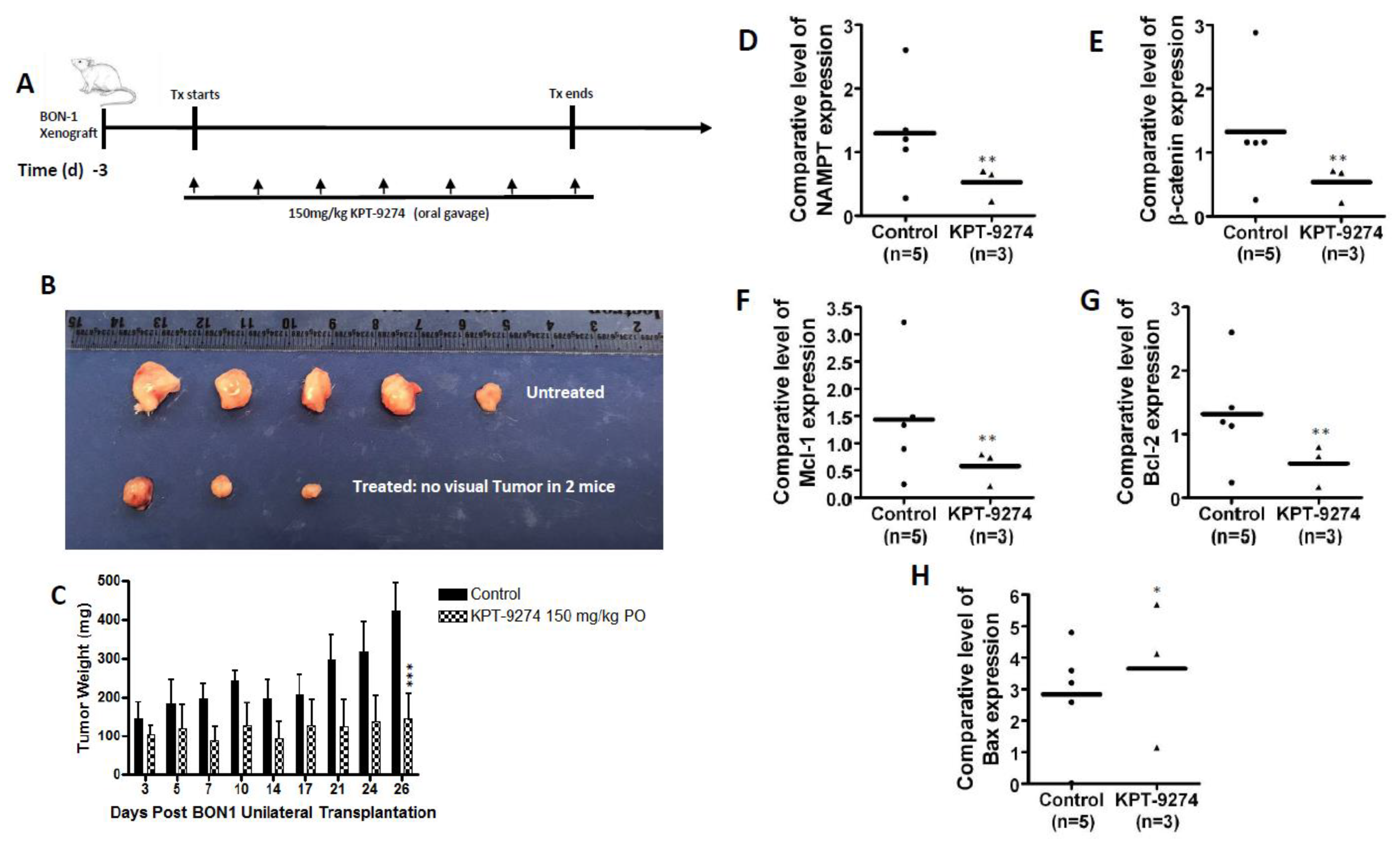
2.7. KPT-9274 Shows Single Agent Anti-Tumor Activity in PNET
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Small Interference RNA and Transfection
4.3. MTT Assay
4.4. Colonogenic Assay
4.5. Annexin V and 7AAD Apoptosis Analysis
4.6. Immunohistochemistry Analysis
4.7. Western Blot Analysis
4.8. Quantitative Real-Time PCR
4.9. Metabolomic Analysis.
4.10. Animal Studies
4.11. Statistical Consideration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Modlin, I.M.; Oberg, K.; Chung, D.C.; Jensen, R.T.; de Herder, W.W.; Thakker, R.V.; Caplin, M.; Delle, F.G.; Kaltsas, G.A.; Krenning, E.P.; et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008, 9, 61–72. [Google Scholar] [CrossRef]
- Modlin, I.M.; Lye, K.D.; Kidd, M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer 2003, 97, 934–959. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; de Braud, F.; Festinese, F.; Bregant, C.; Lorenzoni, A.; Maccauro, M.; Milione, M.; Concas, L.; Formisano, B.; Leuzzi, L.; et al. Evolution in the treatment of gastroenteropancreatic-neuroendocrine neoplasms, focus on systemic therapeutic options: A systematic review. Future Oncol. 2015, 11, 1947–1959. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, M.; Caterina, I.; de Divitiis, C.; von Arx, C.; Maiolino, P.; Tatangelo, F.; Cavalcanti, E.; di Girolamo, E.; Iaffaioli, R.V.; Scala, S.; et al. Everolimus and pancreatic neuroendocrine tumors (PNETs): Activity, resistance and how to overcome it. Int. J. Surg. 2015, 21 (Suppl. 1), S89–S94. [Google Scholar] [CrossRef] [PubMed]
- Djukom, C.; Porro, L.J.; Mrazek, A.; Townsend, C.M., Jr.; Hellmich, M.R.; Chao, C. Dual inhibition of PI3K and mTOR signaling pathways decreases human pancreatic neuroendocrine tumor metastatic progression. Pancreas 2014, 43, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Francois, R.A.; Maeng, K.; Nawab, A.; Kaye, F.J.; Hochwald, S.N.; Zajac-Kaye, M. Targeting Focal Adhesion Kinase and Resistance to mTOR Inhibition in Pancreatic Neuroendocrine Tumors. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Aristizabal, P.E.T.; Auernhammer, C.J. Targeted therapy of gastroenteropancreatic neuroendocrine tumours: Preclinical strategies and future targets. Endocr. Connect. 2018, 7, R1–R25. [Google Scholar] [CrossRef]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef]
- Dirckx, N.; Tower, R.J.; Mercken, E.M.; Vangoitsenhoven, R.; Moreau-Triby, C.; Breugelmans, T.; Nefyodova, E.; Cardoen, R.; Mathieu, C.; van der Schueren, B.; et al. Vhl deletion in osteoblasts boosts cellular glycolysis and improves global glucose metabolism. J. Clin. Investig. 2018, 128, 1087–1105. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Dart, A.E.; Box, G.M.; Court, W.; Gale, M.E.; Brown, J.P.; Pinder, S.E.; Eccles, S.A.; Wells, C.M. PAK4 promotes kinase-independent stabilization of RhoU to modulate cell adhesion. J. Cell Biol. 2015, 211, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Hakoshima, T.; Shimizu, T.; Maesaki, R. Structural basis of the Rho GTPase signaling. J. Biochem. 2003, 134, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.P.; Crossley, L.; Zhan, Q.; Huang, R.; Robinson, D.; Badwey, J.A. The P21-activated protein kinases (Paks) receive and integrate messages from a variety of signaling pathways. Adv. Exp. Med. Biol. 2002, 507, 497–502. [Google Scholar] [PubMed]
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 2014, 14, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C.; Hezel, A.F.; Aguirre, A.J.; Zheng, H.; Paik, J.H.; Ying, H.; Chu, G.C.; Zhang, J.X.; Sahin, E.; Yeo, G.; et al. Genomic alterations link Rho family of GTPases to the highly invasive phenotype of pancreas cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 19372–19377. [Google Scholar] [CrossRef] [PubMed]
- Mahlamaki, E.H.; Kauraniemi, P.; Monni, O.; Wolf, M.; Hautaniemi, S.; Kallioniemi, A. High-resolution genomic and expression profiling reveals 105 putative amplification target genes in pancreatic cancer. Neoplasia 2004, 6, 432–439. [Google Scholar] [CrossRef]
- Beauchamp, R.L.; James, M.F.; DeSouza, P.A.; Wagh, V.; Zhao, W.N.; Jordan, J.T.; Stemmer-Rachamimov, A.; Plotkin, S.R.; Gusella, J.F.; Haggarty, S.J.; et al. A high-throughput kinome screen reveals serum/glucocorticoid-regulated kinase 1 as a therapeutic target for NF2-deficient meningiomas. Oncotarget 2015, 6, 16981–16997. [Google Scholar] [CrossRef]
- Yun, C.Y.; You, S.T.; Kim, J.H.; Chung, J.H.; Han, S.B.; Shin, E.Y.; Kim, E.G. p21-activated kinase 4 critically regulates melanogenesis via activation of the CREB/MITF and beta-catenin/MITF pathways. J. Investig. Dermatol. 2015, 135, 1385–1394. [Google Scholar] [CrossRef]
- Noda, K.; Nakajima, S.; Godo, S.; Saito, H.; Ikeda, S.; Shimizu, T.; Enkhjargal, B.; Fukumoto, Y.; Tsukita, S.; Yamada, T.; et al. Rho-kinase inhibition ameliorates metabolic disorders through activation of AMPK pathway in mice. PLoS ONE 2014, 9, e110446. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Li, Y.; Muqbil, I.; Aboukameel, A.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Philip, P.A.; Azmi, A.S. Targeting Rho GTPase effector p21 activated kinase 4 (PAK4) suppresses p-Bad-microRNA drug resistance axis leading to inhibition of pancreatic ductal adenocarcinoma proliferation. Small GTPase 2019, 5, 367–377. [Google Scholar] [CrossRef]
- Chini, C.C.; Guerrica, A.M.; Nin, V.; Camacho-Pereira, J.; Escande, C.; Barbosa, M.T.; Chini, E.N. Targeting of NAD metabolism in pancreatic cancer celss: Potential novel therapy for pancreatic tumors. Clin. Cancer Res. 2014, 20, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Moratta, V.; Zatelli, M.C.; Sciammarella, C.; Ambrosio, M.R.; Bondanelli, M.; Colao, A.; Faggiano, A. Chromogranin A as circulating marker for diagnosis and management of neuroendocrine neoplasms: More flaws than fame. Emdocr. Relat. Cancer 2018, 1, R11–R29. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Yang, Z.; Liu, Z.; Miao, X.; Yang, L.; Li, D.; Zou, Q.; Yuan, Y. Glypican-3 and KRT19 are markers associating with metastasis and poor prognosis of pancreatic ductal adenocarcinoma. Cancer Biomark. 2016, 4, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Abu, A.O.; Chen, C.H.; Senapedis, W.; Baloglu, E.; Argueta, C.; Weiss, R.H. Dual and Specific Inhibition of NAMPT and PAK4 By KPT-9274 Decreases Kidney Cancer Growth. Mol. Cancer Ther. 2016, 9, 2119–2129. [Google Scholar]
- Mitchell, S.R.; Larkin, K.; Grieselhuber, N.R.; Lai, T.H.; Cannon, M.; Orwick, S.; Sharma, P.; Asemelash, Y.; Zhang, P.; Goettl, V.M. Selective targeting of NAMPT by KPT-9274 in acute myeloid leukemia. Blood Adv. 2019, 3, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lopez, M.A.; Linares, M.; Kumar, S.; Oliva, S.; Martinez-Lopez, J.; Xu, L.; Xu, Y.; Perini, T.; Senapedis, W.; et al. Dual PAK4-NAMPT Inhibition Impacts Growth and Survival, and Increases Sensitivity to DNA-Damaging Agents in Waldenstrom Macroglobulinemia. Clin. Cancer Res. 2019, 25, 369–377. [Google Scholar] [CrossRef]
- Takao, S.; Chien, W.; Madan, V.; Lin, D.C.; Ding, L.W.; Sun, Q.Y.; Mayakonda, A.; Sudo, M.; Xu, L.; Chen, Y.; et al. Targeting the vulnerability to NAD(+) depletion in B-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 616–625. [Google Scholar] [CrossRef]
- Rane, C.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Crochiere, M.; Das-Gupta, S.; Minden, A. A novel orally bioavailable compound KPT-9274 inhibits PAK4, and blocks triple-negative breast cancer tumor growth. Sci. Rep. 2017, 7, 42555. [Google Scholar] [CrossRef]
- Aboukameel, A.; Muqbil, I.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Shacham, S.; Kauffman, M.; Philip, P.A.; Mohammad, R.M.; Azmi, A.S. Novel p21-Activated Kinase 4 (PAK4) Allosteric Modulators Overcome Drug Resistance and Stemness in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 76–87. [Google Scholar] [CrossRef]
- Yao, J.C.; Phan, A.T.; Jehl, V.; Shah, G.; Meric-Bernstam, F. Everolimus in advanced pancreatic neuroendocrine tumors: The clinical experience. Cancer Res. 2013, 73, 1449–1453. [Google Scholar] [CrossRef]
- Won, S.Y.; Park, J.J.; Shin, E.Y.; Kim, E.G. PAK4 signaling in health and disease: Defining the PAK4-CREB axis. Exp. Mol. Med. 2019, 51, 11. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Lim, S.M.; Yoo, Y.I.; Jung, J.; Park, J.W.; Kim, G.J. Microenvironmental Interaction Between Hypoxia and Endothelial Cells Controls the Migration Ability of Placenta-Derived Mesenchymal Stem Cells via alpha4 Integrin and Rho Signaling. J. Cell Biochem. 2016, 117, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.T.; Hideshima, T.; Chauhan, D.; Roccaro, A.; Sacco, A.; Calimeri, T.; Cottini, F.; et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 2012, 120, 3519–3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, S.; Penke, M.; Gorski, T.; Gebhardt, R.; Weiss, T.S.; Kiess, W.; Garten, A. FK866-induced NAMPT inhibition activates AMPK and downregulates mTOR signaling in hepatocarcinoma cells. Biochem. Biophys. Res. Commun. 2015, 458, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.J.; Subramaniam, P.S.; Tang, L.H.; Grunn, A.; Aburi, M.; Rieckhof, G.; Komissarova, E.V.; Hagan, E.A.; Bodei, L.; Clemons, P.A. A precision oncology approach to pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 2018, 50, 979–989. [Google Scholar] [CrossRef] [PubMed]
- de Figueiredo, L.F.; Gossmann, T.I.; Ziegler, M.; Schuster, S. Pathway analysis of NAD+ metabolism. Biochem. J. 2011, 439, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Khan, J.A.; Tao, X.; Tong, L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat. Struct. Mol. Biol. 2006, 13, 582–588. [Google Scholar] [CrossRef]
- Garten, A.; Petzold, S.; Korner, A.; Imai, S.; Kiess, W. Nampt: Linking NAD biology, metabolism and cancer. Trends Endocrinol. Metab. 2009, 20, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Shames, D.S.; Elkins, K.; Walter, K.; Holcomb, T.; Du, P.; Mohl, D.; Xiao, Y.; Pham, T.; Haverty, P.M.; Liederer, B. Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin. Cancer Res. 2013, 19, 6912–6923. [Google Scholar] [CrossRef] [Green Version]
- Neggers, J.E.; Kwanten, B.; Dierckx, T.; Noguchi, H.; Voet, A.; Bral, L.; Minner, K.; Massant, B.; Kint, N.; Delforge, M. Target identification of small molecules using large-scale CRISPR-Cas mutagenesis scanning of essential genes. Nat. Commun. 2018, 9, 502. [Google Scholar] [CrossRef]
- Aboukameel, A.; Muqbil, I.; Baloglu, E.; Senapedis, W.; Landesman, Y.; Argueta, C.; Kauffman, M.; Chang, H.; Kashyap, T.; Shacham, S.; et al. Down-regulation of AR splice variants through XPO1 suppression contributes to the inhibition of prostate cancer progression. Oncotarget 2018, 9, 35327–35342. [Google Scholar] [CrossRef] [PubMed]
- Wall, N.R.; Mohammad, R.M.; Nabha, S.M.; Pettit, G.R.; Al-Katib, A.M. Modulation of cIAP-1 by novel antitubulin agents when combined with bryostatin 1 results in increased apoptosis in the human early pre-B acute lymphoblastic leukemia cell line Reh. Biochem. Biophys. Res. Commun. 1999, 266, 76–80. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | Treatment Effect | p-Value |
|---|---|---|
| Kynurenic acid | −2.173 | 0.026 |
| Fructose 1,6-bisphosphate | −1.903 | 0.016 |
| Histidine | −2.356 | 0.023 |
| Phenylalanine | −1.076 | 0.044 |
| Arginine | −1.693 | 0.025 |
| Lysine (s) | −2.729 | 0.021 |
| dUMP | −1.043 | 0.025 |
| 1-Methylhistidine | −1.381 | 0.045 |
| 2-Ketohexanoic acid | 1.938 | 0.001 |
| N-Alpha-acetyllysine | −1.866 | 0.047 |
| ADP_new | 1.410 | 0.001 |
| CMP | 1.610 | 0.023 |
| DL-2-Aminooctanoic acid | −2.158 | 0.008 |
| Folic acid | −2.659 | 0.007 |
| Guanine | −1.380 | 0.010 |
| N-Acetylornithine | −2.706 | 0.046 |
| Phenyllactic acid | −1.514 | 0.047 |
| Pyridoxal 5-phosphate | −2.006 | 6.05 × 10−7 |
| pyridoxine | −1.682 | 0.046 |
| Thymine | −1.959 | 0.027 |
| Sarcosine (s) | −1.121 | 0.018 |
| CDP | 2.040 | 0.003 |
| GDP | 1.496 | 0.002 |
| UDP | 1.875 | 0.001 |
| dGMP_New 1 | 1.465 | 0.015 |
| dGDP_new | 1.452 | 0.001 |
| Cholesterol sulfate | −1.412 | 0.010 |
| Hippurate | −1.911 | 0.027 |
| p-cresyl sulfate | −1.536 | 0.049 |
| Linoleyl Carnitine | 1.224 | 0.033 |
| C14 Carnitine (Myristoyl-L-carnitine) | 1.307 | 0.040 |
| Metabolites | Treatment Effect | p-Value |
|---|---|---|
| Succinyl-CoA | −1.299 | 8.87 × 10−4 |
| Fructose 6-phosphate | 2.483 | 0.008 |
| Arginine | −1.179 | 0.008 |
| Picolinic acid | 1.633 | 0.034 |
| Nicotinamide | 1.695 | 0.033 |
| D-Ribose 5-phosphate | 2.741 | 6.27 × 10−6 |
| Adenosine | 3.164 | 2.51 × 10−6 |
| ADP_new | 1.805 | 5.26 × 10−8 |
| Aminoadipic acid | 1.352 | 0.026 |
| Cytosine | 1.806 | 0.007 |
| Glucose 6-phosphate | 2.506 | 0.008 |
| GMP_New | 2.775 | 2.08 × 10−4 |
| Ornithine | 1.342 | 0.025 |
| Uracil | 3.249 | 0.001 |
| Uridine | 3.731 | 0.001 |
| Adenosine triphosphate | −1.486 | 0.004 |
| Cytidine triphosphate | −1.884 | 0.003 |
| Guanosine triphosphate | −1.819 | 0.006 |
| Uridine triphosphate | −1.394 | 0.008 |
| dATP | −1.520 | 0.001 |
| dCTP | −1.011 | 0.008 |
| dGTP | −1.389 | 0.008 |
| dTTP | −1.052 | 0.002 |
| Argininosuccinic acid | −1.162 | 0.001 |
| Sarcosine (s) | −1.266 | 0.010 |
| Dimethylglycine+M191 | 1.571 | 0.001 |
| Choline | 1.509 | 0.001 |
| Cytidine | 2.923 | 0.001 |
| CDP | 2.720 | 2.06 × 10−6 |
| GDP | 1.622 | 0.004 |
| UDP | 2.185 | 0.001 |
| dGMP_New 1 | 3.419 | 3.40 × 10−8 |
| dGDP_new | 1.811 | 2.91 × 10−6 |
| D-Sedoheptulose 7-phosphate | 1.285 | 0.008 |
| N-Acetyl-glucosamine 1-phosphate | 1.210 | 0.008 |
| Citicoline | 3.320 | 0.005 |
| Adenine | 1.726 | 0.001 |
| Spermidine | 2.009 | 0.013 |
| C16:0 Carnitine (Palmitoyl-L carnitine) | 3.023 | 0.013 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mpilla, G.; Aboukameel, A.; Muqbil, I.; Kim, S.; Beydoun, R.; Philip, P.A.; Mohammad, R.M.; Kamgar, M.; Shidham, V.; Senapedis, W.; et al. PAK4-NAMPT Dual Inhibition as a Novel Strategy for Therapy Resistant Pancreatic Neuroendocrine Tumors. Cancers 2019, 11, 1902. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11121902
Mpilla G, Aboukameel A, Muqbil I, Kim S, Beydoun R, Philip PA, Mohammad RM, Kamgar M, Shidham V, Senapedis W, et al. PAK4-NAMPT Dual Inhibition as a Novel Strategy for Therapy Resistant Pancreatic Neuroendocrine Tumors. Cancers. 2019; 11(12):1902. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11121902
Chicago/Turabian StyleMpilla, Gabriel, Amro Aboukameel, Irfana Muqbil, Steve Kim, Rafic Beydoun, Philip A. Philip, Ramzi M. Mohammad, Mandana Kamgar, Vinod Shidham, William Senapedis, and et al. 2019. "PAK4-NAMPT Dual Inhibition as a Novel Strategy for Therapy Resistant Pancreatic Neuroendocrine Tumors" Cancers 11, no. 12: 1902. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11121902