Nanosecond Pulsed Electric Fields Induce Endoplasmic Reticulum Stress Accompanied by Immunogenic Cell Death in Murine Models of Lymphoma and Colorectal Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
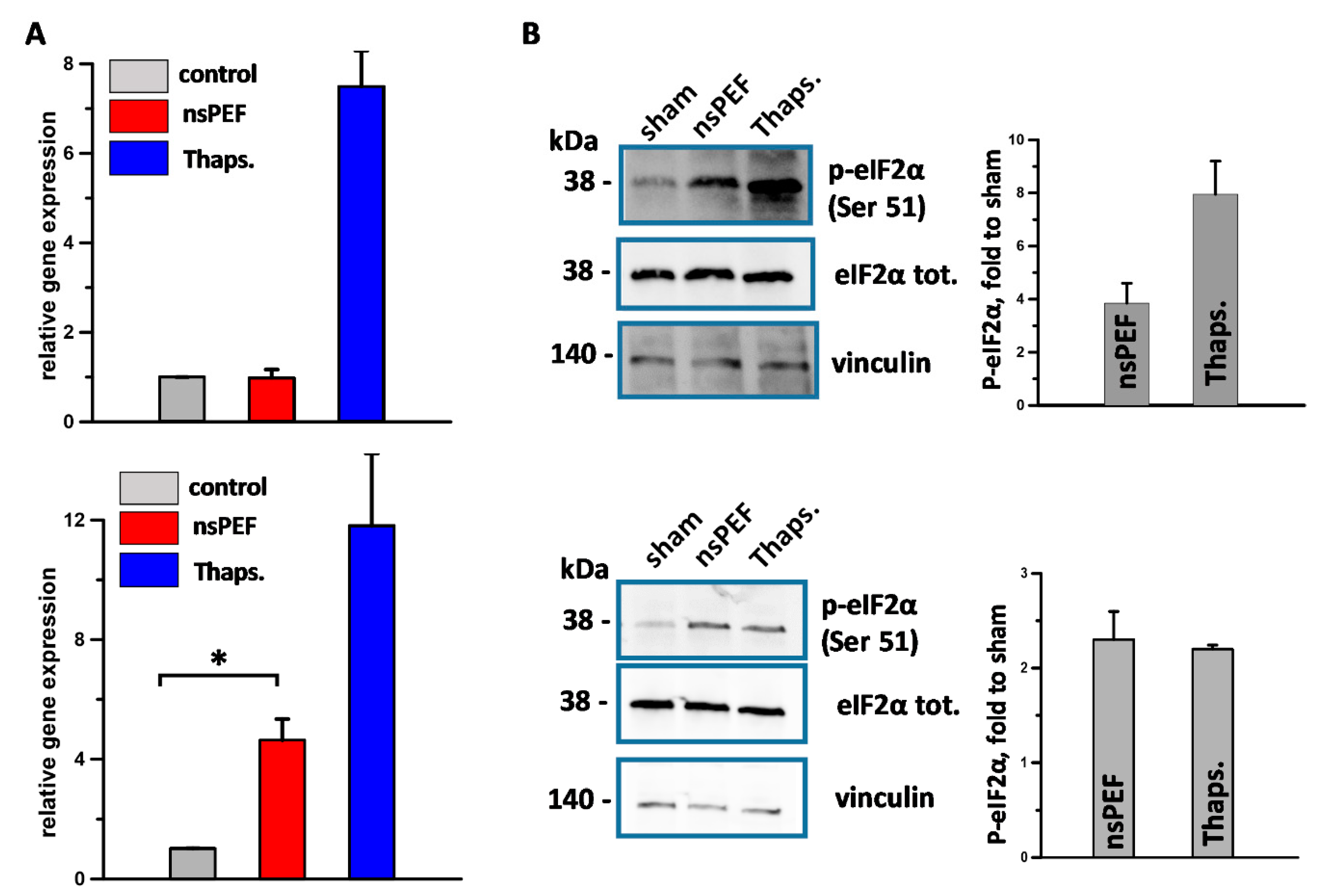
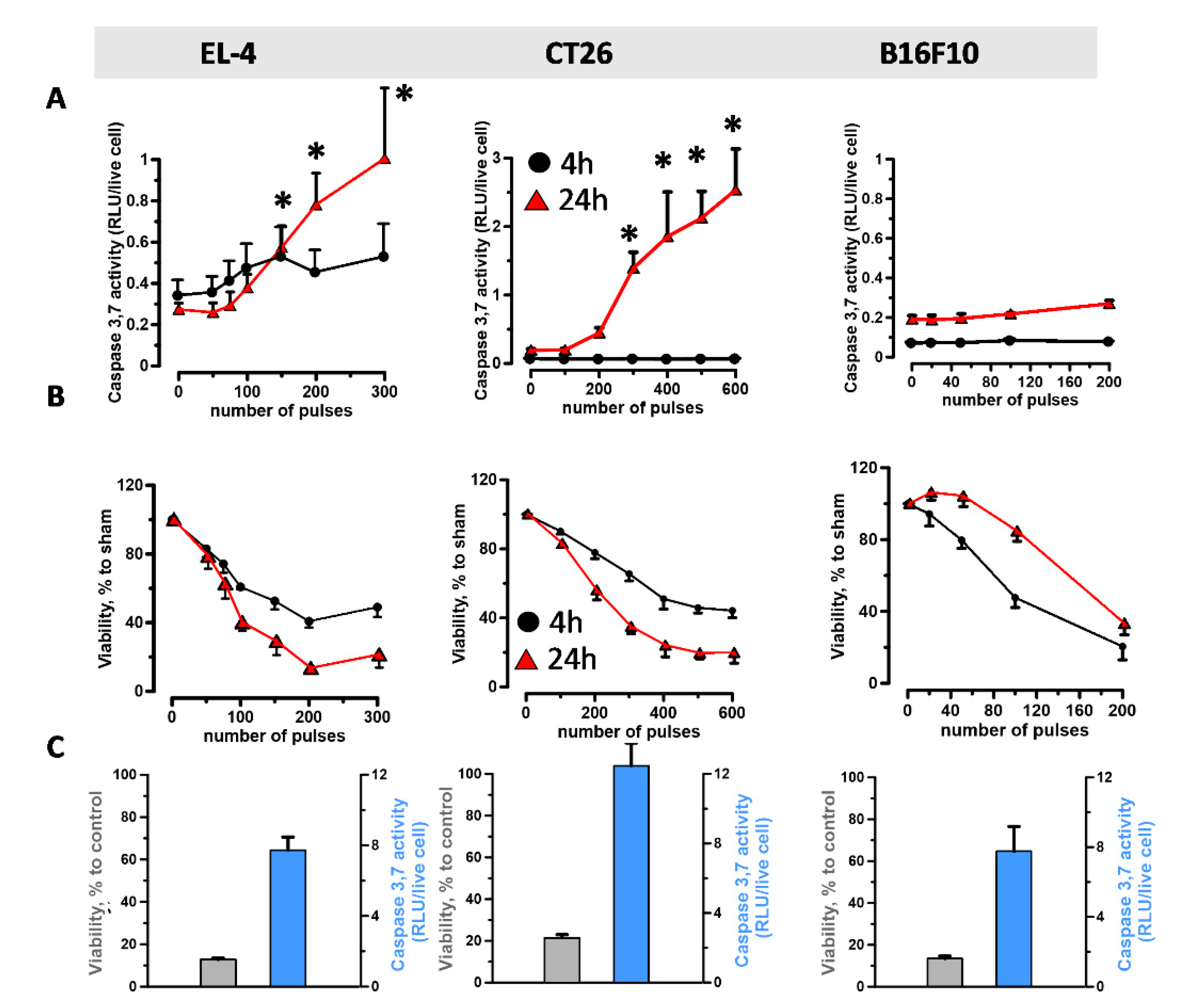
2.1. nsPEF Induce ER-Stress in Cancer Cells
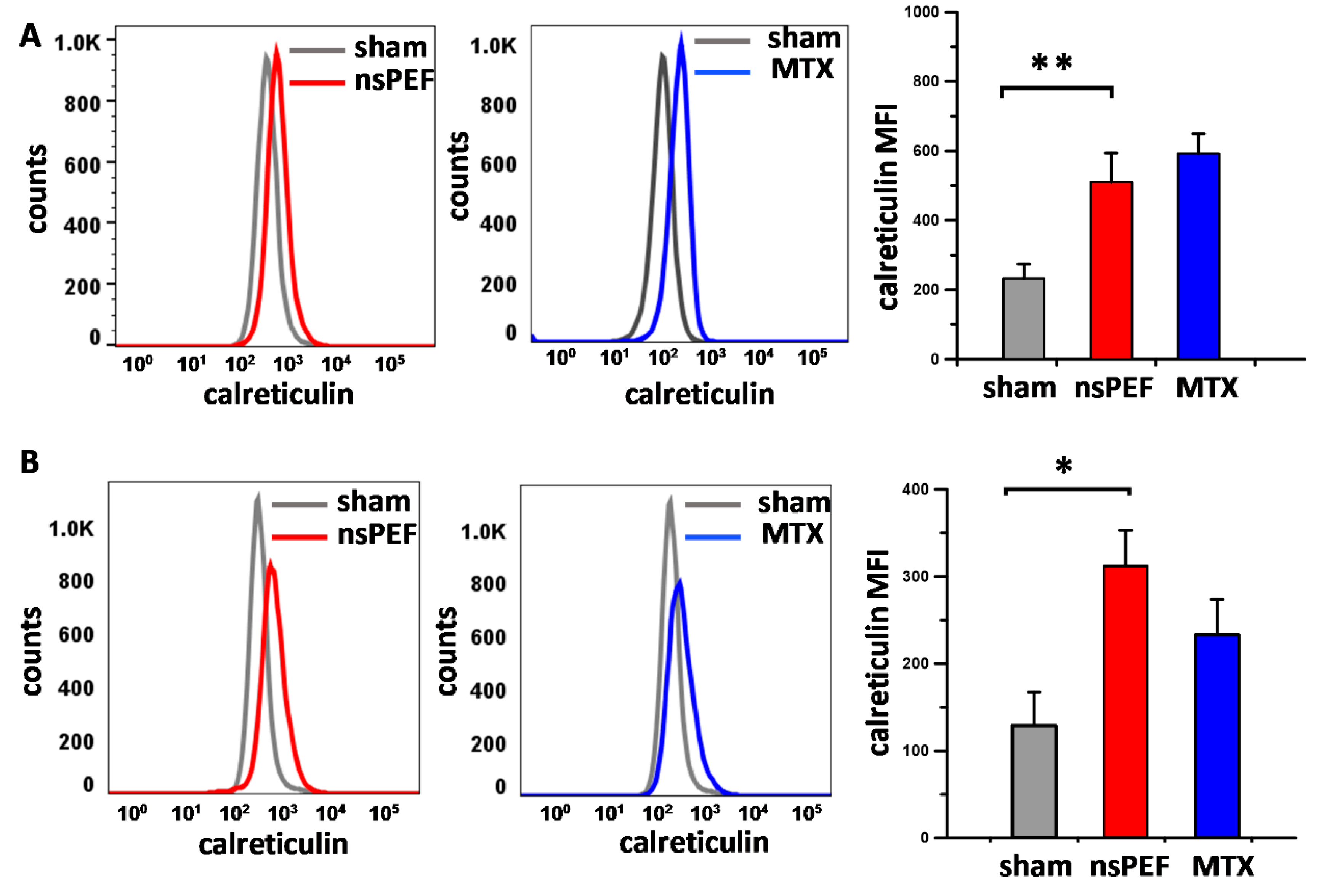
2.2. PERK Activation Correlates with Apoptosis Induction and CRT Translocation onto the Cell Plasma Membrane of Nanoporated Tumor Cells
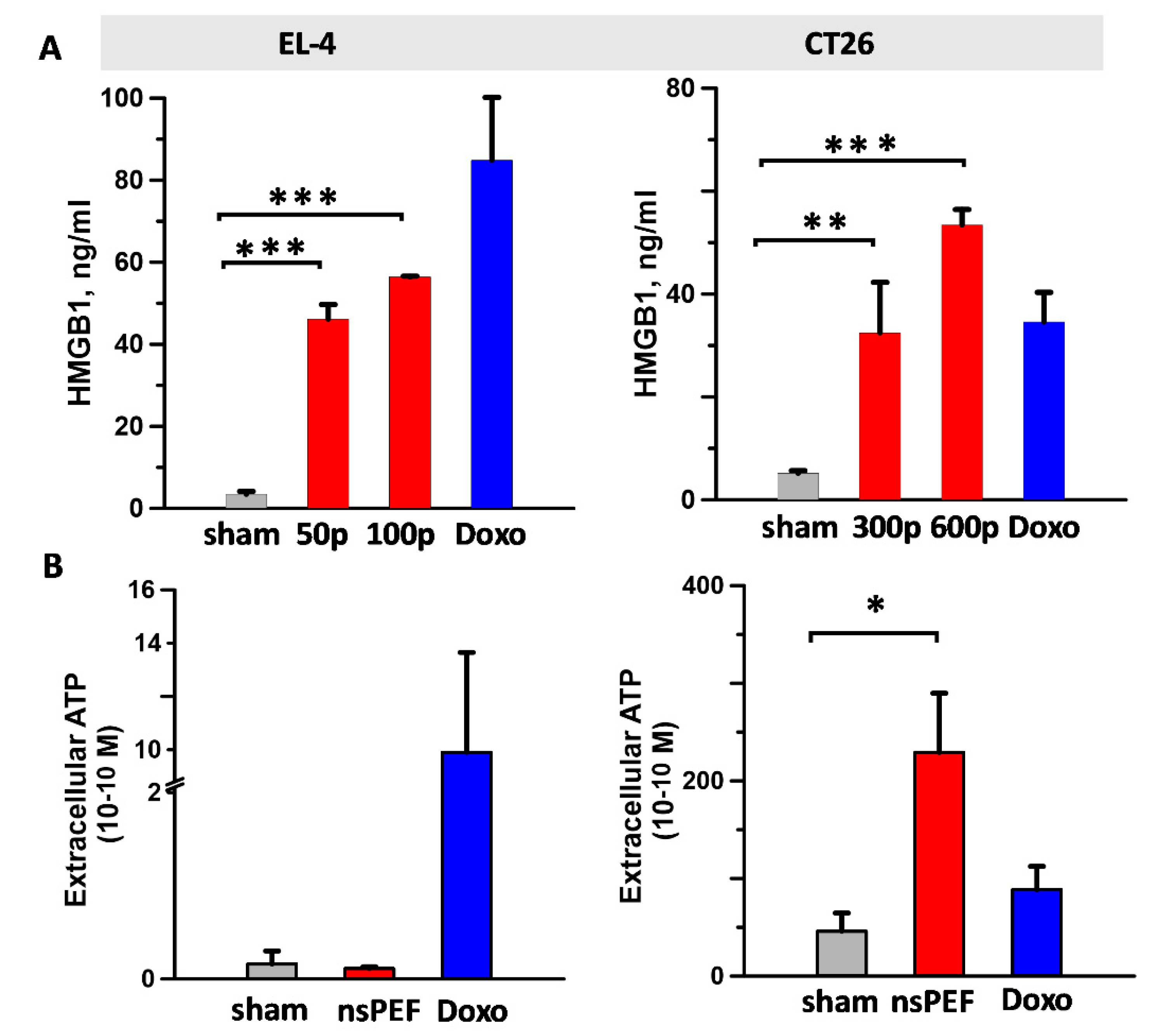
2.3. Effect of nsPEF on Secreted Immunogenic DAMPs
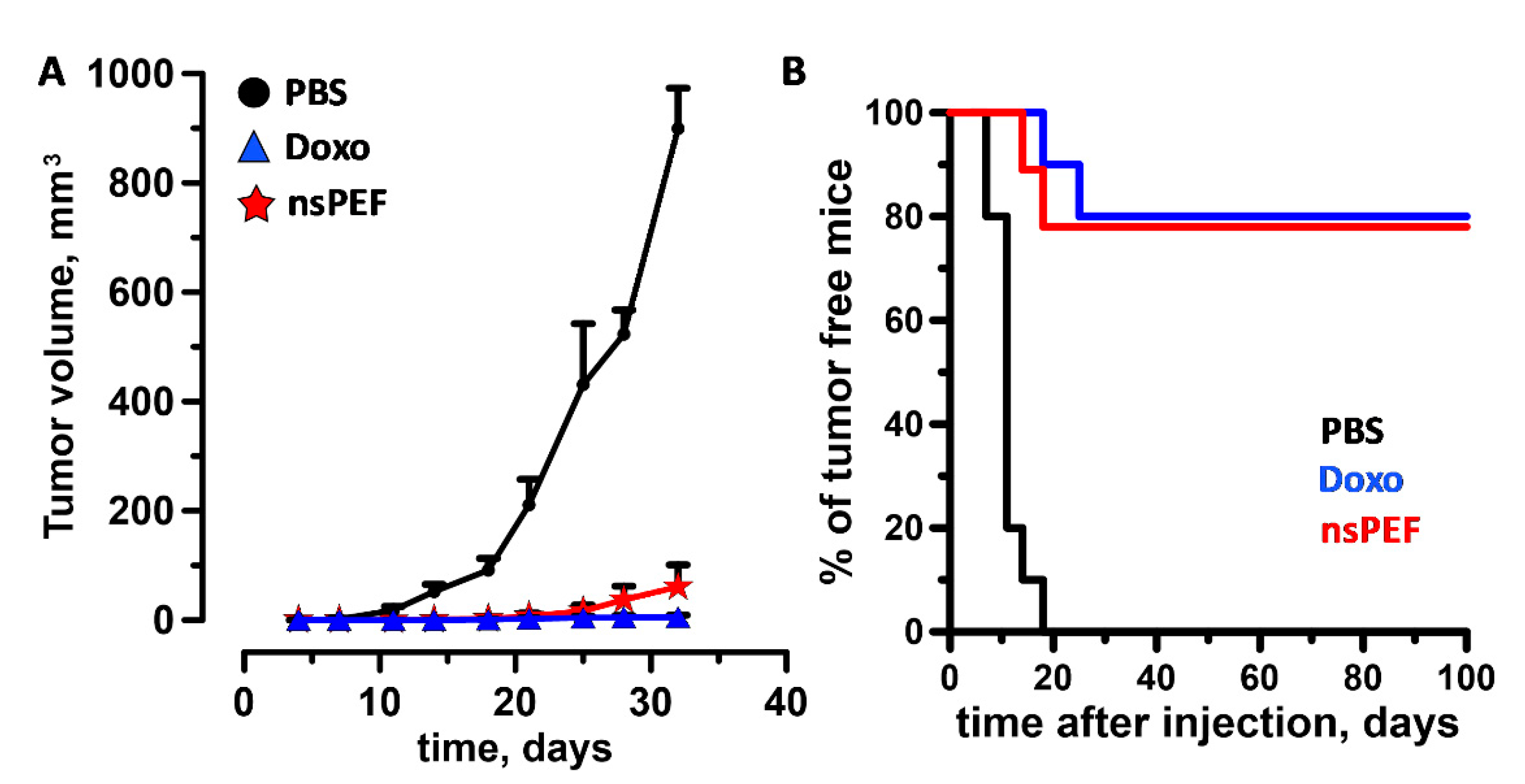
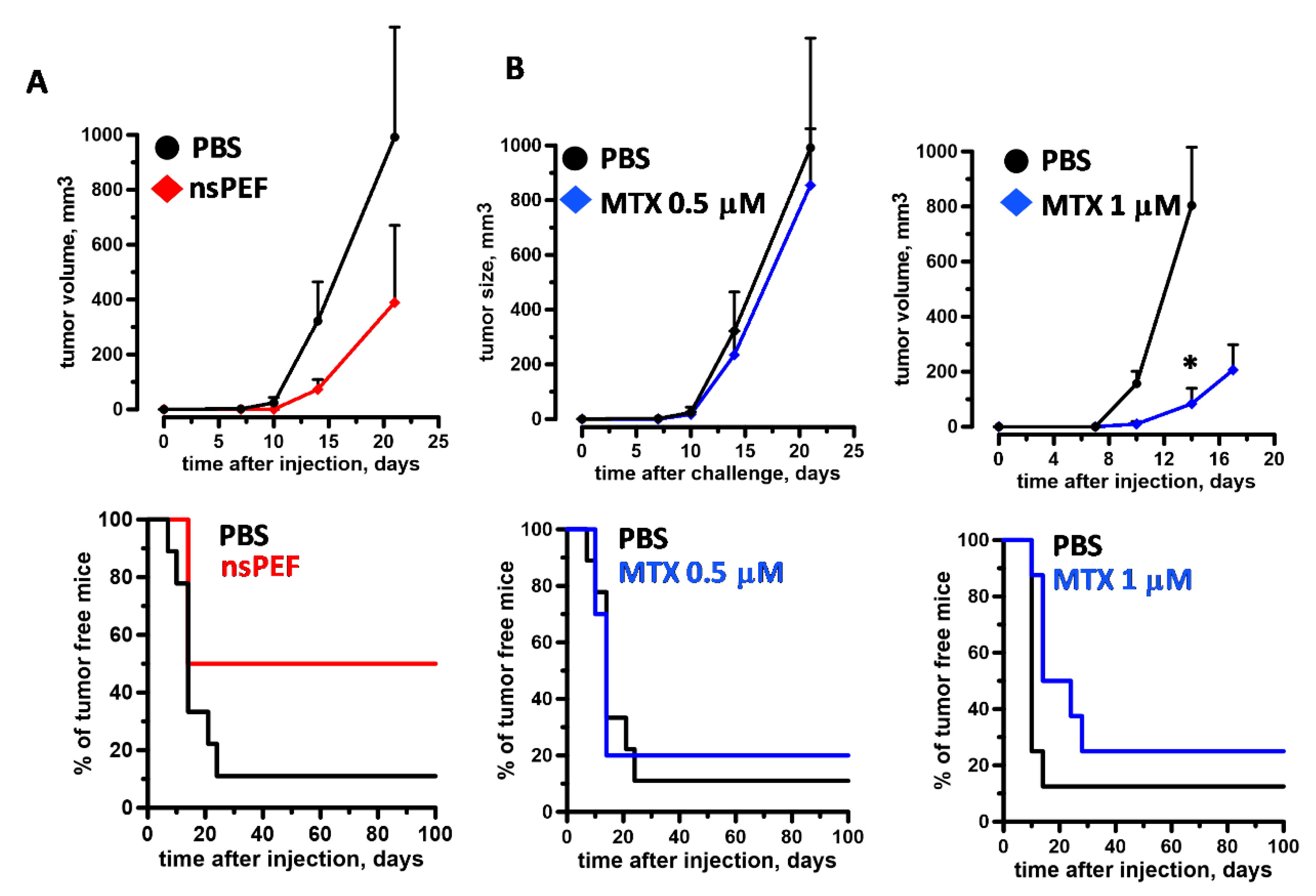
2.4. Immunogenicity of nsPEF-Induced Cell Death
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Pulsed Electric Field Exposure Methods
4.3. Viability and Caspase-3/7 Activity Assays
4.4. RT-PCR and Western Blot
4.5. DAMPs Detection
4.6. Vaccination Experiments
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kepp, O.; Marabelle, A.; Zitvogel, L.; Kroemer, G. Oncolysis without viruses—Inducing systemic anticancer immune responses with local therapies. Nat. Rev. Clin. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The immune response to secondary necrotic cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamrekelashvili, J.; Ormandy, L.A.; Heimesaat, M.M.; Kirschning, C.J.; Manns, M.P.; Korangy, F.; Greten, T.F. Primary sterile necrotic cells fail to cross-prime CD8 (+) T cells. Oncoimmunology 2012, 1, 1017–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffer, S.R.; Nave, H.; Korangy, F.; Schlote, K.; Pabst, R.; Jaffee, E.M.; Manns, M.P.; Greten, T.F. Apoptotic, but not necrotic, tumor cell vaccines induce a potent immune response in vivo. Int. J. Cancer 2003, 103, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Adjemian, S.; Yang, H.; Catani, J.P.; Hannani, D.; Martins, I.; Michaud, M.; Kepp, O.; Sukkurwala, A.Q.; Vacchelli, E.; et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology 2013, 2, e24568. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Agostinis, P. ER stress, autophagy and immunogenic cell death in photodynamic therapy-induced anti-cancer immune responses. Photochem. Photobiol. Sci. 2014, 13, 474–487. [Google Scholar] [CrossRef]
- Rufo, N.; Garg, A.D.; Agostinis, P. The Unfolded Protein Response in Immunogenic Cell Death and Cancer Immunotherapy. Trends Cancer 2017, 3, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; McGrath, E.P.; Mnich, K.; Healy, S.; Chevet, E.; Samali, A. Controlling the unfolded protein response-mediated life and death decisions in cancer. Semin. Cancer Biol. 2015, 33, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Garg, A.D.; Dudek-Peric, A.M.; Romano, E.; Agostinis, P. Immunogenic cell death. Int. J. Dev. Biol. 2015, 59, 131–140. [Google Scholar] [CrossRef]
- Dudek, A.M.; Garg, A.D.; Krysko, D.V.; De Ruysscher, D.; Agostinis, P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013, 24, 319–333. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef]
- Kepp, O.; Semeraro, M.; Bravo-San Pedro, J.M.; Bloy, N.; Buque, A.; Huang, X.; Zhou, H.; Senovilla, L.; Kroemer, G.; Galluzzi, L. eIF2alpha phosphorylation as a biomarker of immunogenic cell death. Semin. Cancer Biol. 2015, 33, 86–92. [Google Scholar] [CrossRef]
- Bezu, L.; Sauvat, A.; Humeau, J.; Gomes-da-Silva, L.C.; Iribarren, K.; Forveille, S.; Garcia, P.; Zhao, L.; Liu, P.; Zitvogel, L.; et al. eIF2alpha phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ. 2018, 25, 1375–1393. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teissie, J.; Eynard, N.; Gabriel, B.; Rols, M.P. Electropermeabilization of cell membranes. Adv. Drug Deliv. Rev. 1999, 35, 3–19. [Google Scholar] [CrossRef]
- Neumann, E.; Rosenheck, K. Permeability changes induced by electric impulses in vesicular membranes. J. Membr. Biol. 1972, 10, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Kinosita, K., Jr.; Tsong, T.Y. Voltage-induced pore formation and hemolysis of human erythrocytes. Biochim. Biophys. Acta 1977, 471, 227–242. [Google Scholar] [CrossRef]
- Neumann, E.; Schaefer-Ridder, M.; Wang, Y.; Hofschneider, P.H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1982, 1, 841–845. [Google Scholar] [CrossRef]
- Heller, L.C.; Heller, R. In vivo electroporation for gene therapy. Hum. Gene 2006, 17, 890–897. [Google Scholar] [CrossRef]
- Daud, A.I.; DeConti, R.C.; Andrews, S.; Urbas, P.; Riker, A.I.; Sondak, V.K.; Munster, P.N.; Sullivan, D.M.; Ugen, K.E.; Messina, J.L.; et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J. Clin. Oncol. 2008, 26, 5896–5903. [Google Scholar] [CrossRef] [Green Version]
- Belehradek, J., Jr.; Orlowski, S.; Poddevin, B.; Paoletti, C.; Mir, L.M. Electrochemotherapy of spontaneous mammary tumours in mice. Eur. J. Cancer 1991, 27, 73–76. [Google Scholar] [CrossRef]
- Mir, L.M.; Orlowski, S.; Belehradek, J., Jr.; Paoletti, C. Electrochemotherapy potentiation of antitumour effect of bleomycin by local electric pulses. Eur J. Cancer 1991, 27, 68–72. [Google Scholar] [CrossRef]
- Miklavcic, D.; Mali, B.; Kos, B.; Heller, R.; Sersa, G. Electrochemotherapy: From the drawing board into medical practice. Biomed. Eng. Online 2014, 13, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campana, L.G.; Kis, E.; Bottyan, K.; Orlando, A.; de Terlizzi, F.; Mitsala, G.; Careri, R.; Curatolo, P.; Snoj, M.; Sersa, G.; et al. Electrochemotherapy for advanced cutaneous angiosarcoma: A European register-based cohort study from the International Network for Sharing Practices of Electrochemotherapy (InspECT). Int. J. Surg. 2019. [Google Scholar] [CrossRef] [PubMed]
- Davalos, R.V.; Mir, I.L.M.; Rubinsky, B. Tissue ablation with irreversible electroporation. Ann. Biomed. Eng. 2005, 33, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.; Leor, J.; Rubinsky, B. Cancer cells ablation with irreversible electroporation. Technol Cancer Res. Treat. 2005, 4, 699–705. [Google Scholar] [CrossRef]
- Scheffer, H.J.; Nielsen, K.; de Jong, M.C.; van Tilborg, A.A.; Vieveen, J.M.; Bouwman, A.R.; Meijer, S.; van Kuijk, C.; van den Tol, P.M.; Meijerink, M.R. Irreversible electroporation for nonthermal tumor ablation in the clinical setting: A systematic review of safety and efficacy. J. Vasc. Interv. Radiol. 2014, 25, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Batista Napotnik, T.; Wu, Y.H.; Gundersen, M.A.; Miklavcic, D.; Vernier, P.T. Nanosecond electric pulses cause mitochondrial membrane permeabilization in Jurkat cells. Bioelectromagnetics 2012, 33, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Beebe, S.J.; Fox, P.M.; Rec, L.J.; Willis, E.L.; Schoenbach, K.H. Nanosecond, high-intensity pulsed electric fields induce apoptosis in human cells. FASEB J. 2003, 17, 1493–1495. [Google Scholar] [CrossRef] [Green Version]
- Nesin, O.M.; Pakhomova, O.N.; Xiao, S.; Pakhomov, A.G. Manipulation of cell volume and membrane pore comparison following single cell permeabilization with 60- and 600-ns electric pulses. Biochim. Biophys. Acta 2011, 1808, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Semenov, I.; Xiao, S.; Pakhomov, A.G. Primary pathways of intracellular Ca(2+) mobilization by nanosecond pulsed electric field. Biochim. Biophys. Acta 2013, 1828, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Semenov, I.; Xiao, S.; Pakhomova, O.N.; Pakhomov, A.G. Recruitment of the intracellular Ca2+ by ultrashort electric stimuli: The impact of pulse duration. Cell Calcium 2013, 54, 145–150. [Google Scholar] [CrossRef] [Green Version]
- White, J.A.; Blackmore, P.F.; Schoenbach, K.H.; Beebe, S.J. Stimulation of capacitative calcium entry in HL-60 cells by nanosecond pulsed electric fields. J. Biol. Chem. 2004, 279, 22964–22972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernier, P.; Li, A.; Marcu, L.; Craft, C.; Gundersen, M. Ultrashort pulsed electric fields induce membrane phospholipid translocation and caspase activation: Differential sensitivities of Jurkat T lymphoblasts and rat glioma C6 cells. IEEE Trans. Dielectr. Electr. Insul. 2003, 10, 795–809. [Google Scholar] [CrossRef]
- Schoenbach, K.H.; Beebe, S.J.; Buescher, E.S. Intracellular effect of ultrashort electrical pulses. Bioelectromagnetics 2001, 22, 440–448. [Google Scholar] [CrossRef]
- Tekle, E.; Oubrahim, H.; Dzekunov, S.M.; Kolb, J.F.; Schoenbach, K.H.; Chock, P.B. Selective field effects on intracellular vacuoles and vesicle membranes with nanosecond electric pulses. Biophys. J. 2005, 89, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craviso, G.L.; Choe, S.; Chatterjee, P.; Chatterjee, I.; Vernier, P.T. Nanosecond electric pulses: A novel stimulus for triggering Ca2+ influx into chromaffin cells via voltage-gated Ca2+ channels. Cell Mol. Neurobiol. 2010, 30, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Vernier, P.T.; Sun, Y.; Marcu, L.; Salemi, S.; Craft, C.M.; Gundersen, M.A. Calcium bursts induced by nanosecond electric pulses. Biochem. Biophys. Res. Commun. 2003, 310, 286–295. [Google Scholar] [CrossRef]
- Pakhomova, O.N.; Khorokhorina, V.A.; Bowman, A.M.; Rodaite-Riseviciene, R.; Saulis, G.; Xiao, S.; Pakhomov, A.G. Oxidative effects of nanosecond pulsed electric field exposure in cells and cell-free media. Arch. Biochem. Biophys. 2012, 527, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Nuccitelli, R.; Lui, K.; Kreis, M.; Athos, B.; Nuccitelli, P. Nanosecond pulsed electric field stimulation of reactive oxygen species in human pancreatic cancer cells is Ca(2+)-dependent. Biochem. Biophys. Res. Commun. 2013, 435, 580–585. [Google Scholar] [CrossRef] [Green Version]
- Vernier, P.T.; Sun, Y.; Marcu, L.; Craft, C.M.; Gundersen, M.A. Nanoelectropulse-induced phosphatidylserine translocation. Biophys. J. 2004, 86, 4040–4048. [Google Scholar] [CrossRef] [Green Version]
- Vincelette, R.L.; Roth, C.C.; McConnell, M.P.; Payne, J.A.; Beier, H.T.; Ibey, B.L. Thresholds for phosphatidylserine externalization in Chinese hamster ovarian cells following exposure to nanosecond pulsed electrical fields (nsPEF). PLoS ONE 2013, 8, e63122. [Google Scholar] [CrossRef]
- Muratori, C.; Pakhomov, A.G.; Gianulis, E.; Meads, J.; Casciola, M.; Mollica, P.A.; Pakhomova, O.N. Activation of the phospholipid scramblase TMEM16F by nanosecond pulsed electric field (nsPEF) facilitates its diverse cytophysiological effects. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morotomi-Yano, K.; Uemura, Y.; Katsuki, S.; Akiyama, H.; Yano, K. Activation of the JNK pathway by nanosecond pulsed electric fields. Biochem. Biophys. Res. Commun. 2011, 408, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Morotomi-Yano, K.; Akiyama, H.; Yano, K. Nanosecond pulsed electric fields activate MAPK pathways in human cells. Arch. Biochem. Biophys. 2011, 515, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Pakhomova, O.N.; Gregory, B.W.; Semenov, I.; Pakhomov, A.G. Two modes of cell death caused by exposure to nanosecond pulsed electric field. PLoS ONE 2013, 8, e70278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibey, B.L.; Pakhomov, A.G.; Gregory, B.W.; Khorokhorina, V.A.; Roth, C.C.; Rassokhin, M.A.; Bernhard, J.A.; Wilmink, G.J.; Pakhomova, O.N. Selective cytotoxicity of intense nanosecond-duration electric pulses in mammalian cells. Biochim. Biophys. Acta 2010, 1800, 1210–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, W.; Beebe, S.J. An apoptosis targeted stimulus with nanosecond pulsed electric fields (nsPEFs) in E4 squamous cell carcinoma. Apoptosis 2011, 16, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Pakhomova, O.N.; Pakhomov, A.G.; Weygandt, S.; Bulysheva, A.A.; Murray, L.E.; Mollica, P.A.; Muratori, C. Mechanisms and immunogenicity of nsPEF-induced cell death in B16F10 melanoma tumors. Sci. Rep. 2019, 9, 431. [Google Scholar] [CrossRef]
- Ibey, B.L.; Roth, C.C.; Pakhomov, A.G.; Bernhard, J.A.; Wilmink, G.J.; Pakhomova, O.N. Dose-dependent thresholds of 10-ns electric pulse induced plasma membrane disruption and cytotoxicity in multiple cell lines. PLoS ONE 2011, 6, e15642. [Google Scholar] [CrossRef] [Green Version]
- Muratori, C.; Pakhomov, A.G.; Xiao, S.; Pakhomova, O.N. Electrosensitization assists cell ablation by nanosecond pulsed electric field in 3D cultures. Sci. Rep. 2016, 6, 23225. [Google Scholar] [CrossRef] [Green Version]
- Gianulis, E.C.; Labib, C.; Saulis, G.; Novickij, V.; Pakhomova, O.N.; Pakhomov, A.G. Selective susceptibility to nanosecond pulsed electric field (nsPEF) across different human cell types. Cell Mol. Life Sci. 2017, 74, 1741–1754. [Google Scholar] [CrossRef]
- Nuccitelli, R.; Huynh, J.; Lui, K.; Wood, R.; Kreis, M.; Athos, B.; Nuccitelli, P. Nanoelectroablation of human pancreatic carcinoma in a murine xenograft model without recurrence. Int. J. Cancer 2013, 132, 1933–1939. [Google Scholar] [CrossRef] [PubMed]
- Nuccitelli, R.; Tran, K.; Sheikh, S.; Athos, B.; Kreis, M.; Nuccitelli, P. Optimized nanosecond pulsed electric field therapy can cause murine malignant melanomas to self-destruct with a single treatment. Int. J. Cancer 2010, 127, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Nuccitelli, R.; Chen, X.; Pakhomov, A.G.; Baldwin, W.H.; Sheikh, S.; Pomicter, J.L.; Ren, W.; Osgood, C.; Swanson, R.J.; Kolb, J.F.; et al. A new pulsed electric field therapy for melanoma disrupts the tumor’s blood supply and causes complete remission without recurrence. Int. J. Cancer 2009, 125, 438–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuccitelli, R.; Pliquett, U.; Chen, X.; Ford, W.; James Swanson, R.; Beebe, S.J.; Kolb, J.F.; Schoenbach, K.H. Nanosecond pulsed electric fields cause melanomas to self-destruct. Biochem. Biophys. Res. Commun. 2006, 343, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Sain, N.M.; Harlow, K.T.; Chen, Y.J.; Shires, P.K.; Heller, R.; Beebe, S.J. A protective effect after clearance of orthotopic rat hepatocellular carcinoma by nanosecond pulsed electric fields. Eur. J. Cancer 2014, 50, 2705–2713. [Google Scholar] [CrossRef]
- Guo, S.; Jing, Y.; Burcus, N.I.; Lassiter, B.P.; Tanaz, R.; Heller, R.; Beebe, S.J. Nano-pulse stimulation induces potent immune responses, eradicating local breast cancer while reducing distant metastases. Int. J. Cancer 2018, 142, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuccitelli, R.; Wood, R.; Kreis, M.; Athos, B.; Huynh, J.; Lui, K.; Nuccitelli, P.; Epstein, E.H., Jr. First-in-human trial of nanoelectroablation therapy for basal cell carcinoma: Proof of method. Exp. Derm. 2014, 23, 135–137. [Google Scholar] [CrossRef]
- Skeate, J.G.; Da Silva, D.M.; Chavez-Juan, E.; Anand, S.; Nuccitelli, R.; Kast, W.M. Nano-Pulse Stimulation induces immunogenic cell death in human papillomavirus-transformed tumors and initiates an adaptive immune response. PLoS ONE 2018, 13, e0191311. [Google Scholar] [CrossRef] [Green Version]
- Nuccitelli, R.; Berridge, J.C.; Mallon, Z.; Kreis, M.; Athos, B.; Nuccitelli, P. Nanoelectroablation of Murine Tumors Triggers a CD8-Dependent Inhibition of Secondary Tumor Growth. PLoS ONE 2015, 10, e0134364. [Google Scholar] [CrossRef] [Green Version]
- Nuccitelli, R.; Tran, K.; Lui, K.; Huynh, J.; Athos, B.; Kreis, M.; Nuccitelli, P.; De Fabo, E.C. Non-thermal nanoelectroablation of UV-induced murine melanomas stimulates an immune response. Pigment. Cell Melanoma Res. 2012, 25, 618–629. [Google Scholar] [CrossRef] [Green Version]
- Nuccitelli, R.; McDaniel, A.; Anand, S.; Cha, J.; Mallon, Z.; Berridge, J.C.; Uecker, D. Nano-Pulse Stimulation is a physical modality that can trigger immunogenic tumor cell death. J. Immunother. Cancer 2017, 5, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.G.; Karimi, S.S.; Barry-Holson, K.; Angell, T.E.; Murphy, K.A.; Church, C.H.; Ohlfest, J.R.; Hu, P.; Epstein, A.L. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J. Immunother. 2013, 36, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, R.A.; Flies, D.B.; Zhu, G.; Johnson, A.J.; Tamada, K.; Chapoval, A.I.; Strome, S.E.; Pease, L.R.; Chen, L. Provision of antigen and CD137 signaling breaks immunological ignorance, promoting regression of poorly immunogenic tumors. J. Clin. Investig. 2002, 109, 651–659. [Google Scholar] [CrossRef]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Morotomi-Yano, K.; Oyadomari, S.; Akiyama, H.; Yano, K. Nanosecond pulsed electric fields act as a novel cellular stress that induces translational suppression accompanied by eIF2alpha phosphorylation and 4E-BP1 dephosphorylation. Exp. Cell Res. 2012, 318, 1733–1744. [Google Scholar] [CrossRef]
- Agostinis, P.; Vantieghem, A.; Merlevede, W.; de Witte, P.A. Hypericin in cancer treatment: More light on the way. Int. J. Biochem. Cell Biol. 2002, 34, 221–241. [Google Scholar] [CrossRef]
- Diwu, Z.; Lown, J.W. Photosensitization with anticancer agents. 17. EPR studies of photodynamic action of hypericin: Formation of semiquinone radical and activated oxygen species on illumination. Free Radic Biol. Med. 1993, 14, 209–215. [Google Scholar] [CrossRef]
- Hadjur, C.; Richard, M.J.; Parat, M.O.; Jardon, P.; Favier, A. Photodynamic effects of hypericin on lipid peroxidation and antioxidant status in melanoma cells. Photochem. Photobiol. 1996, 64, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, P.; Babusikova, E.; Lehotsky, J.; Dobrota, D. Free radical-induced protein modification and inhibition of Ca2+-ATPase of cardiac sarcoplasmic reticulum. Mol. Cell Biochem. 2003, 248, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Viner, R.I.; Williams, T.D.; Schoneich, C. Peroxynitrite modification of protein thiols: Oxidation, nitrosylation, and S-glutathiolation of functionally important cysteine residue(s) in the sarcoplasmic reticulum Ca-ATPase. Biochemistry 1999, 38, 12408–12415. [Google Scholar] [CrossRef] [PubMed]
- Buytaert, E.; Callewaert, G.; Hendrickx, N.; Scorrano, L.; Hartmann, D.; Missiaen, L.; Vandenheede, J.R.; Heirman, I.; Grooten, J.; Agostinis, P. Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. FASEB J. 2006, 20, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, B.; Teissie, J. Generation of reactive-oxygen species induced by electropermeabilization of Chinese hamster ovary cells and their consequence on cell viability. Eur. J. Biochem. 1994, 223, 25–33. [Google Scholar] [CrossRef]
- Bonnafous, P.; Vernhes, M.; Teissie, J.; Gabriel, B. The generation of reactive-oxygen species associated with long-lasting pulse-induced electropermeabilisation of mammalian cells is based on a non-destructive alteration of the plasma membrane. Biochim. Biophys. Acta 1999, 1461, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Maccarrone, M.; Rosato, N.; Agro, A.F. Electroporation enhances cell membrane peroxidation and luminescence. Biochem. Biophys. Res. Commun. 1995, 206, 238–245. [Google Scholar] [CrossRef]
- Benov, L.C.; Antonov, P.A.; Ribarov, S.R. Oxidative damage of the membrane lipids after electroporation. Gen. Physiol. Biophys. 1994, 13, 85–97. [Google Scholar]
- Breton, M.; Mir, L.M. Investigation of the chemical mechanisms involved in the electropulsation of membranes at the molecular level. Bioelectrochemistry 2018, 119, 76–83. [Google Scholar] [CrossRef]
- Maccarrone, M.; Bladergroen, M.R.; Rosato, N.; Finazzi Agro, A.F. Role of lipid peroxidation in electroporation-induced cell permeability. Biochem. Biophys. Res. Commun. 1995, 209, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Yang, R.; Liang, Q.; Zhang, W.; Hua, X.; Tang, Y. Electrochemical reaction and oxidation of lecithin under pulsed electric fields (PEF) processing. J. Agric. Food Chem. 2012, 60, 12204–12209. [Google Scholar] [CrossRef] [PubMed]
- Rols, M.P.; Teissie, J. Electropermeabilization of mammalian cells. Quantitative analysis of the phenomenon. Biophys. J. 1990, 58, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margariti, A.; Li, H.; Chen, T.; Martin, D.; Vizcay-Barrena, G.; Alam, S.; Karamariti, E.; Xiao, Q.; Zampetaki, A.; Zhang, Z.; et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J. Biol. Chem. 2013, 288, 859–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullery, J.C.; Tarango, M.; Roth, C.C.; Ibey, B.L. Activation of autophagy in response to nanosecond pulsed electric field exposure. Biochem. Biophys. Res. Commun. 2015, 458, 411–417. [Google Scholar] [CrossRef]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [Green Version]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Garon, E.B.; Sawcer, D.; Vernier, P.T.; Tang, T.; Sun, Y.; Marcu, L.; Gundersen, M.A.; Koeffler, H.P. In vitro and in vivo evaluation and a case report of intense nanosecond pulsed electric field as a local therapy for human malignancies. Int. J. Cancer 2007, 121, 675–682. [Google Scholar] [CrossRef]
- Ren, W.; Sain, N.M.; Beebe, S.J. Nanosecond pulsed electric fields (nsPEFs) activate intrinsic caspase-dependent and caspase-independent cell death in Jurkat cells. Biochem. Biophys. Res. Commun. 2012, 421, 808–812. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Weng, D.; Li, F.; Zou, X.; Young, D.O.; Ji, J.; Shen, P. Involvement of JNK regulation in oxidative stress-mediated murine liver injury by microcystin-LR. Apoptosis 2008, 13, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med. 2018, 215, 115–140. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, M.; Musner, N.; Scapin, C.; Ungaro, D.; Del Carro, U.; Ron, D.; Feltri, M.L.; Wrabetz, L. Resetting translational homeostasis restores myelination in Charcot-Marie-Tooth disease type 1B mice. J. Exp. Med. 2013, 210, 821–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, A.; N. Pakhomova, O.; Mollica, P.A.; Casciola, M.; Mangalanathan, U.; G. Pakhomov, A.; Muratori, C. Nanosecond Pulsed Electric Fields Induce Endoplasmic Reticulum Stress Accompanied by Immunogenic Cell Death in Murine Models of Lymphoma and Colorectal Cancer. Cancers 2019, 11, 2034. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11122034
Rossi A, N. Pakhomova O, Mollica PA, Casciola M, Mangalanathan U, G. Pakhomov A, Muratori C. Nanosecond Pulsed Electric Fields Induce Endoplasmic Reticulum Stress Accompanied by Immunogenic Cell Death in Murine Models of Lymphoma and Colorectal Cancer. Cancers. 2019; 11(12):2034. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11122034
Chicago/Turabian StyleRossi, Alessandra, Olga N. Pakhomova, Peter A. Mollica, Maura Casciola, Uma Mangalanathan, Andrei G. Pakhomov, and Claudia Muratori. 2019. "Nanosecond Pulsed Electric Fields Induce Endoplasmic Reticulum Stress Accompanied by Immunogenic Cell Death in Murine Models of Lymphoma and Colorectal Cancer" Cancers 11, no. 12: 2034. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11122034