Molecular Signature of Subtypes of Non-Small-Cell Lung Cancer by Large-Scale Transcriptional Profiling: Identification of Key Modules and Genes by Weighted Gene Co-Expression Network Analysis (WGCNA)

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Baseline Characteristic
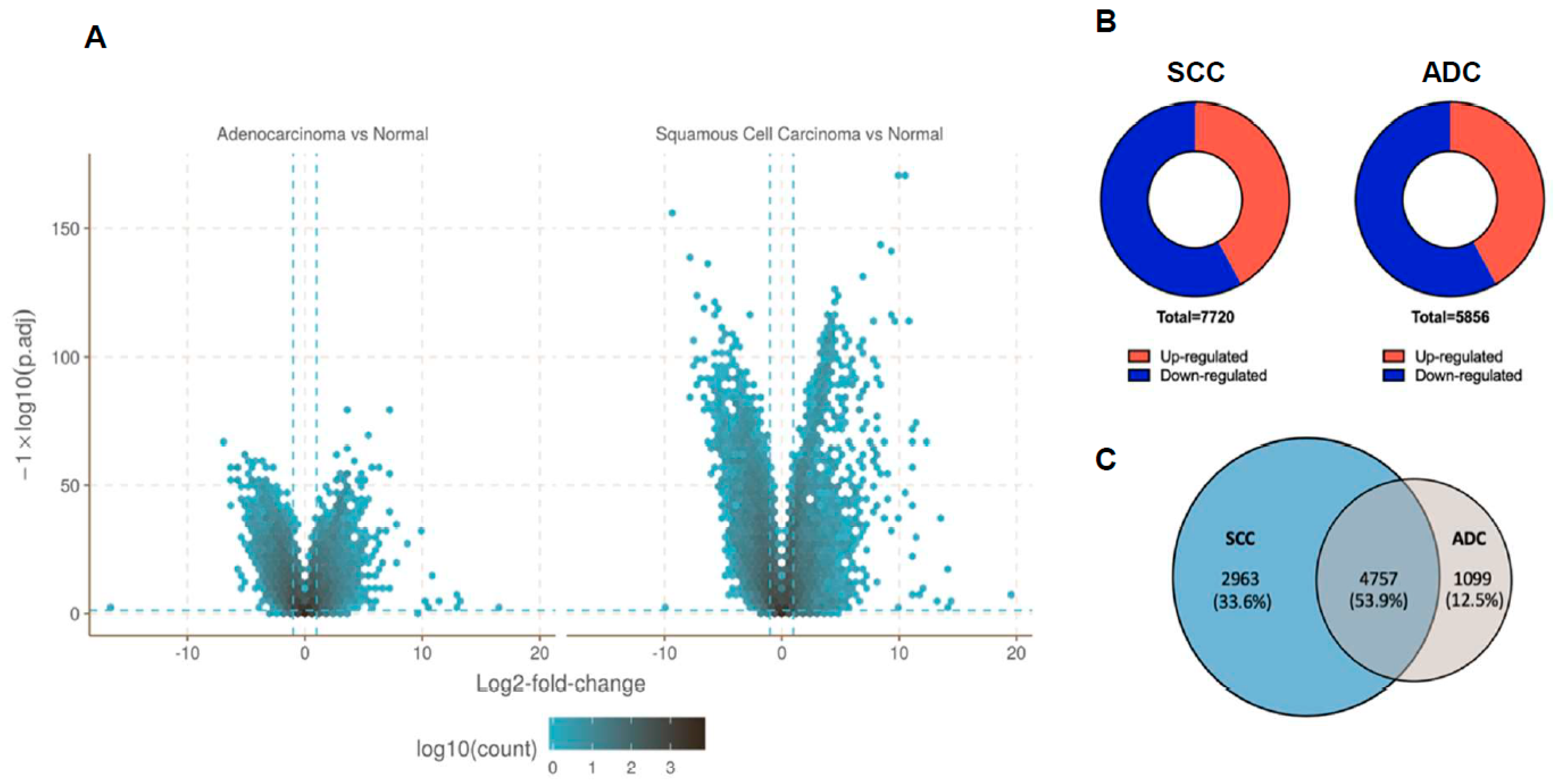
2.2. Gene Expression Profile Data in NSCLC Subtypes
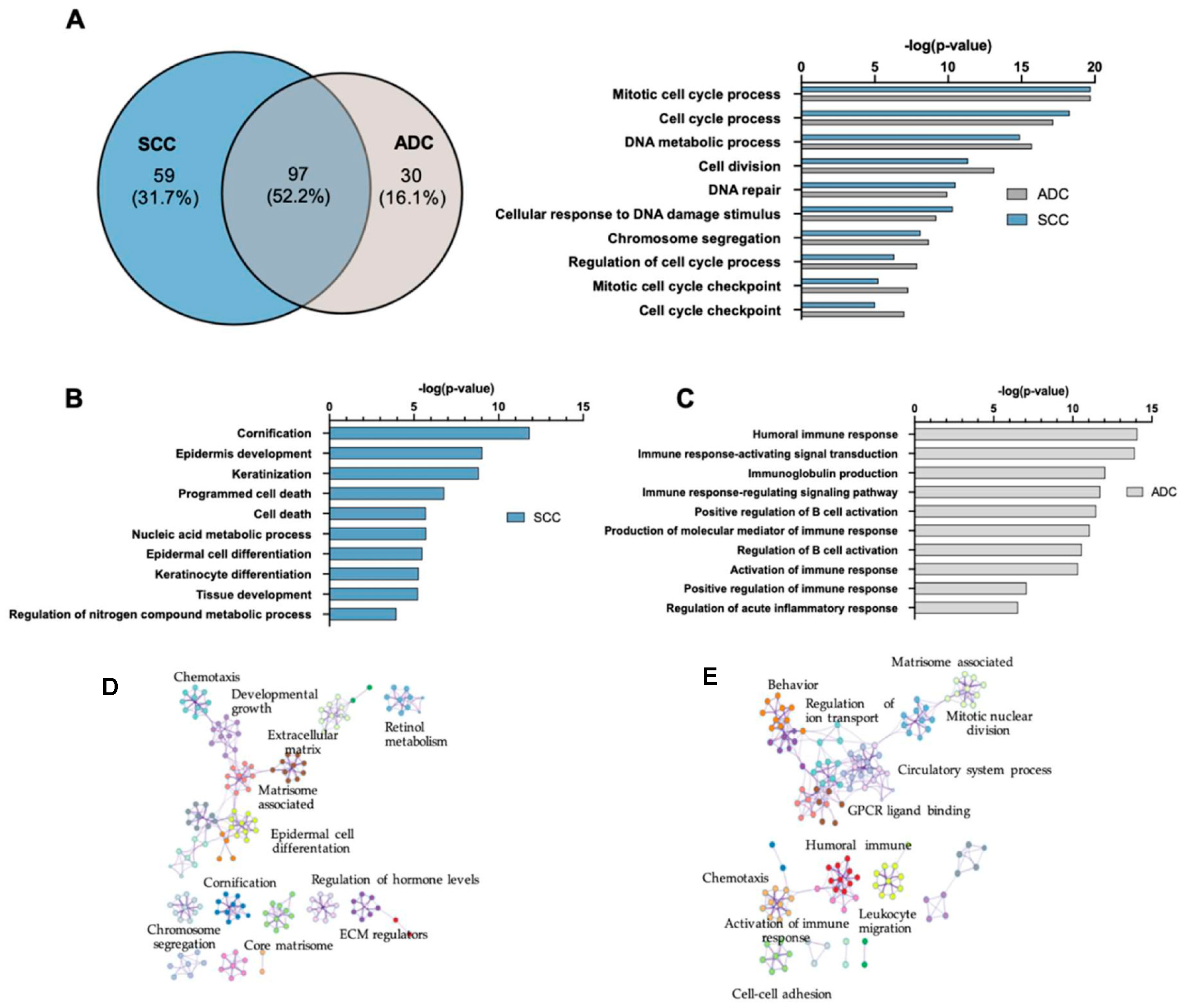
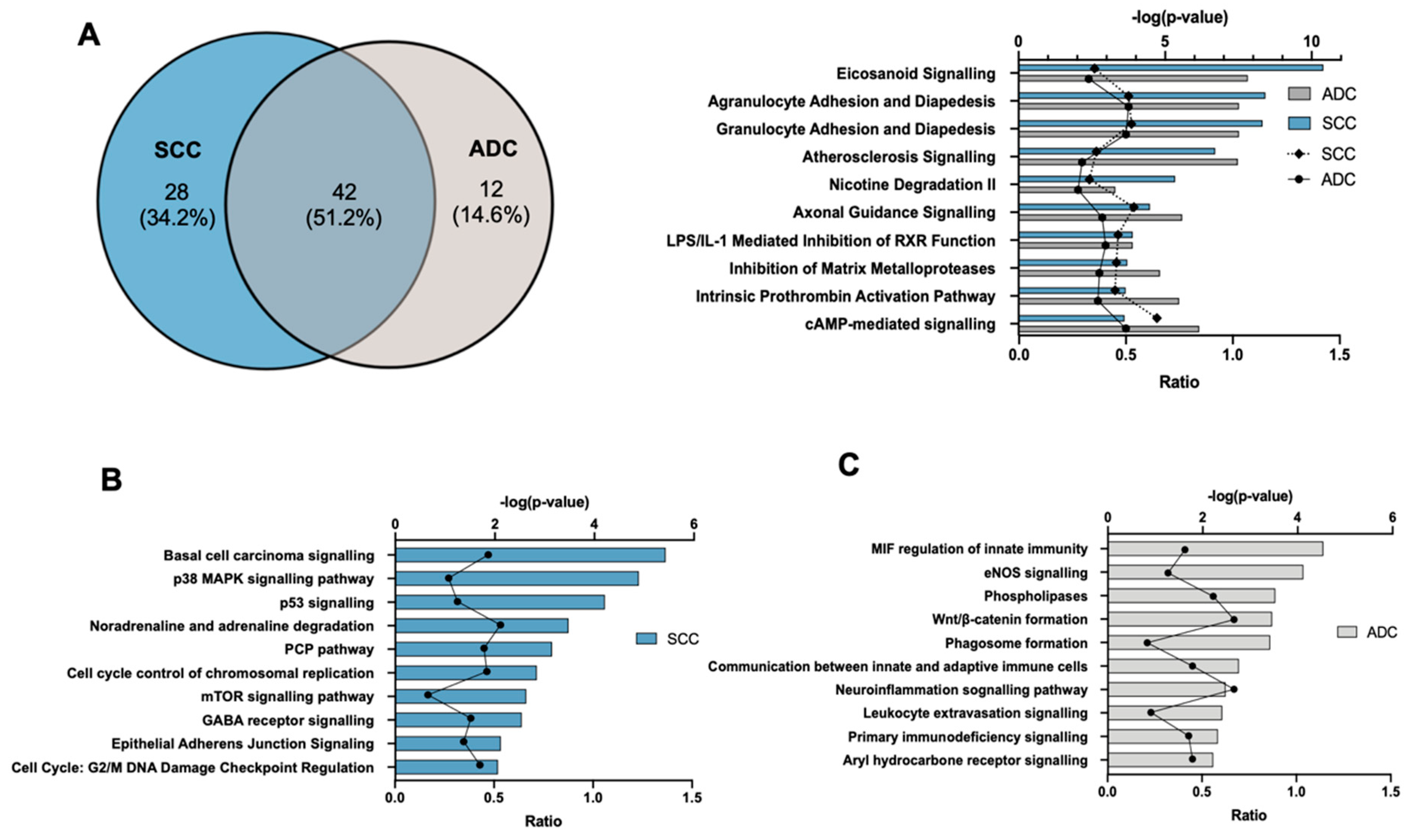
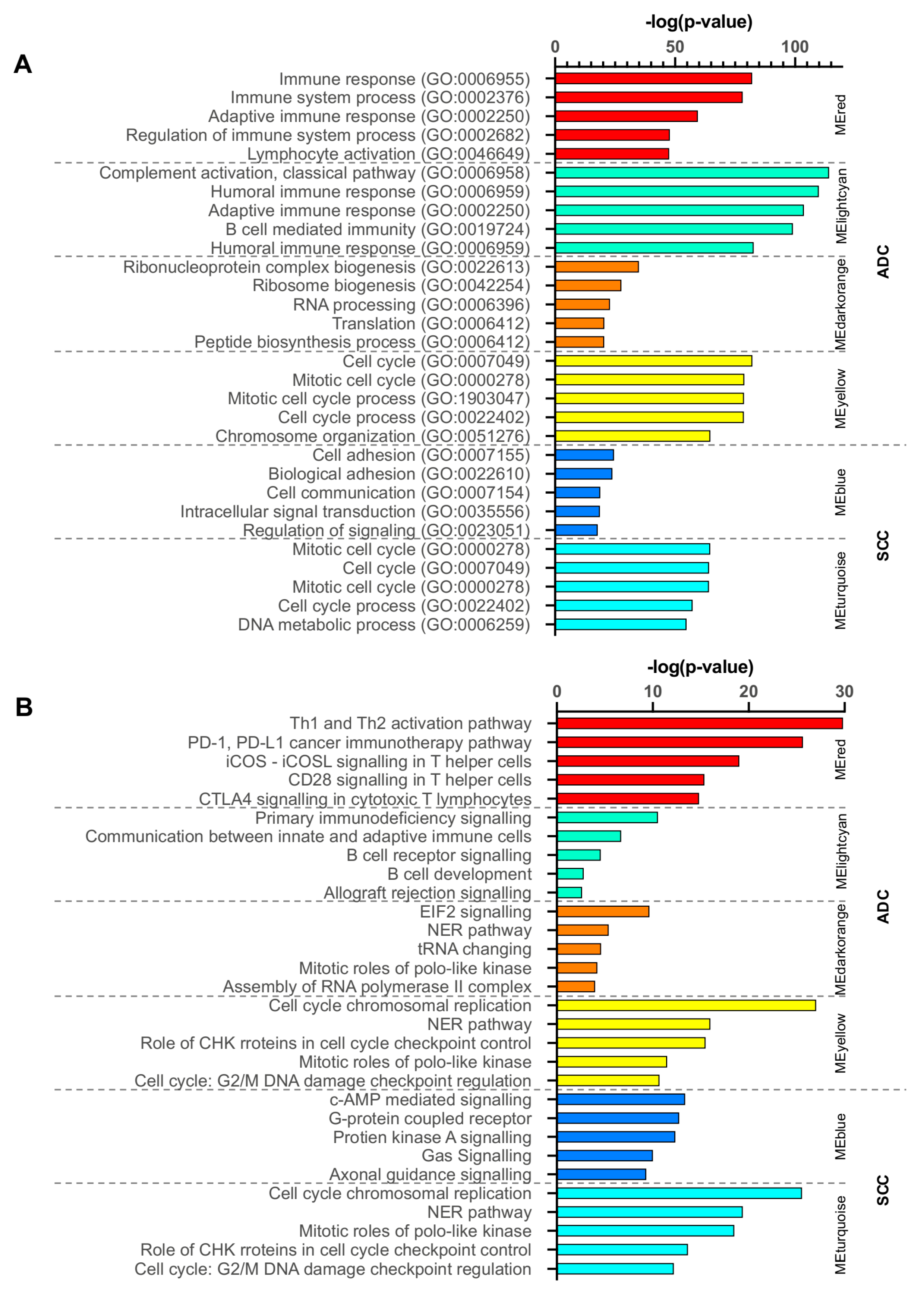
2.3. Gene Ontology and Canonical Pathway Analysis
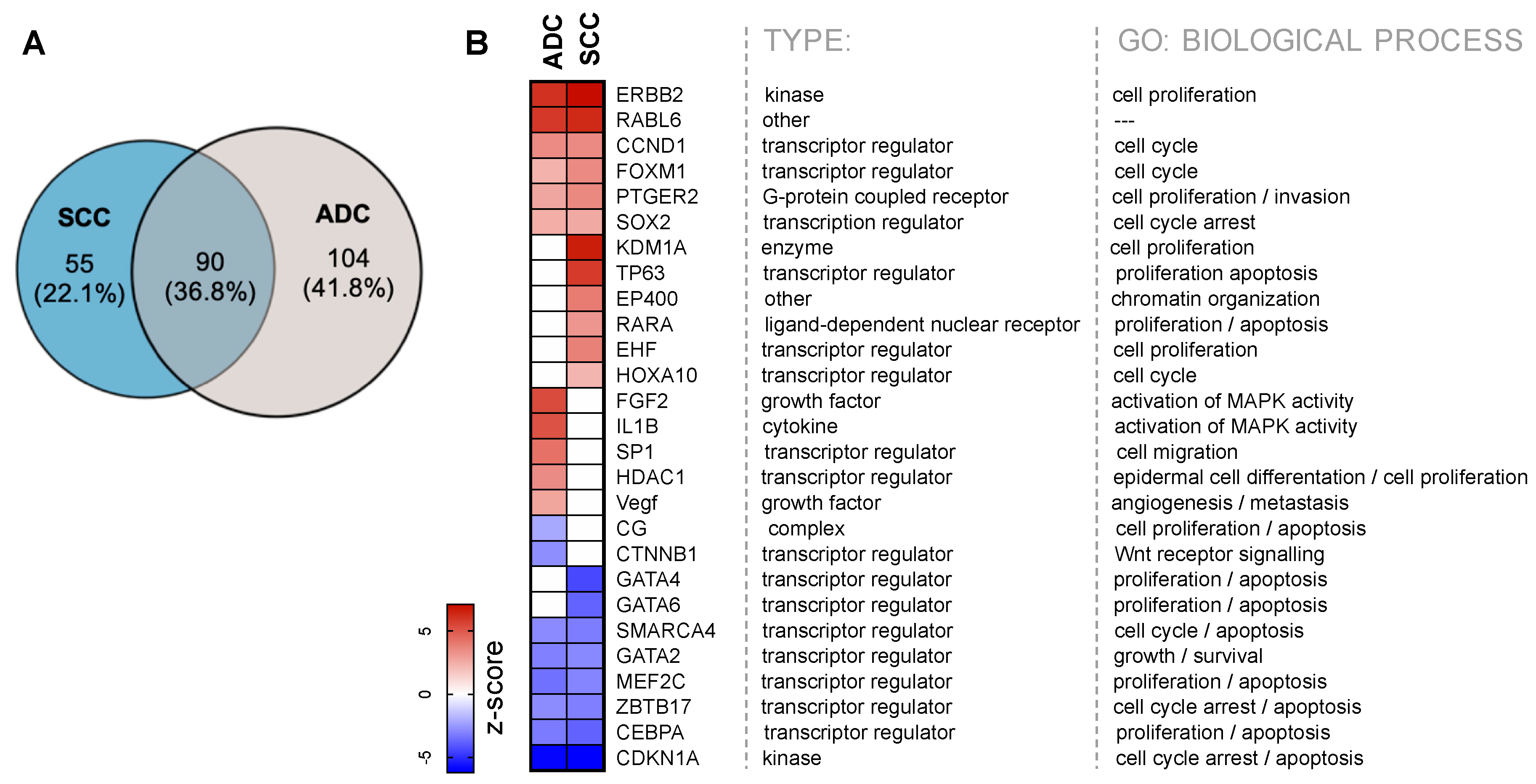
2.4. Upstream Transcription Regulators
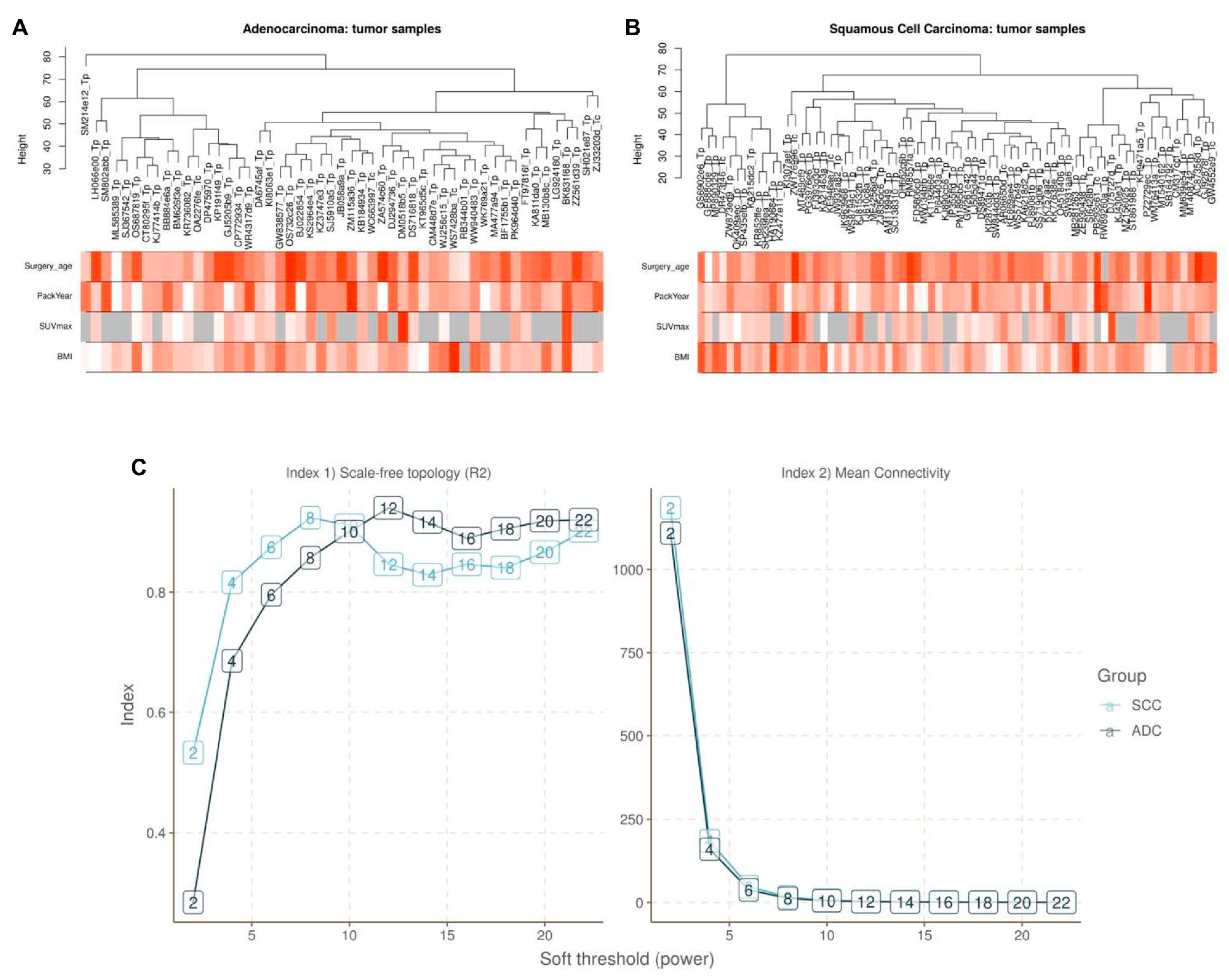
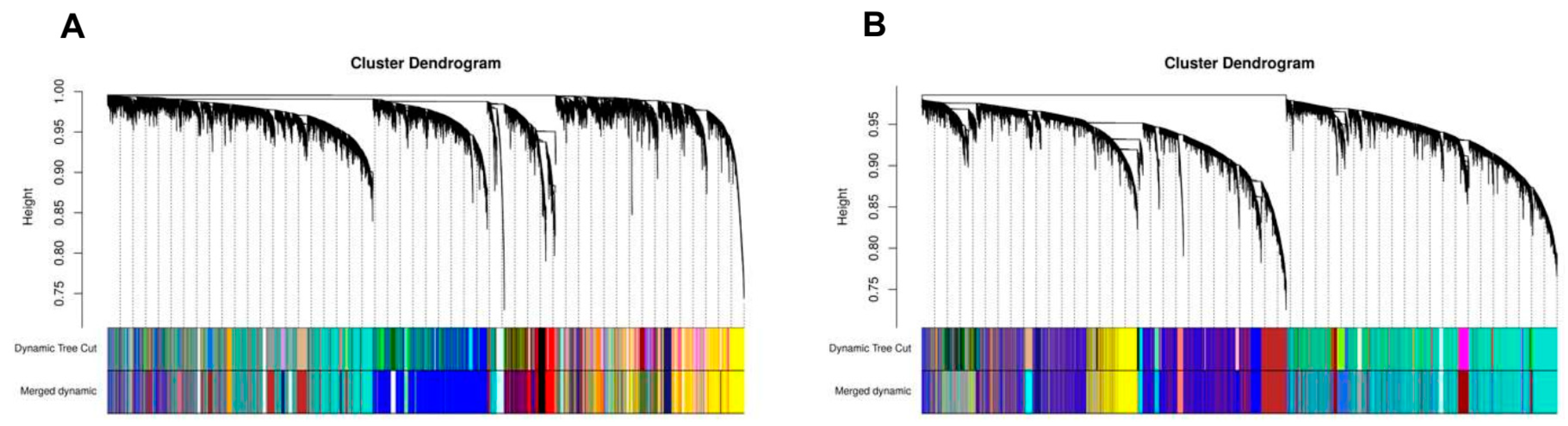
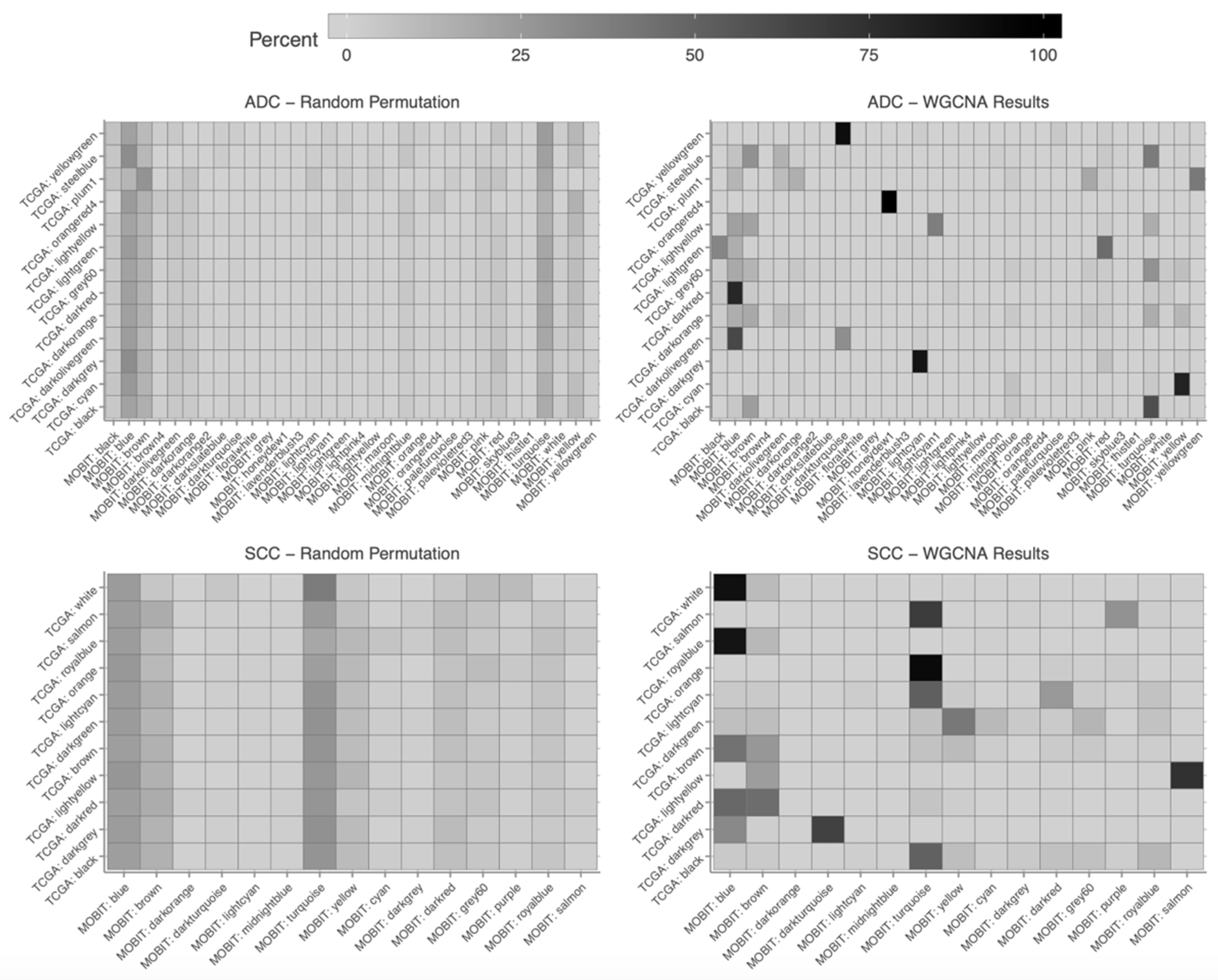
2.5. Construction of Weighted Gene Co-Expression Network
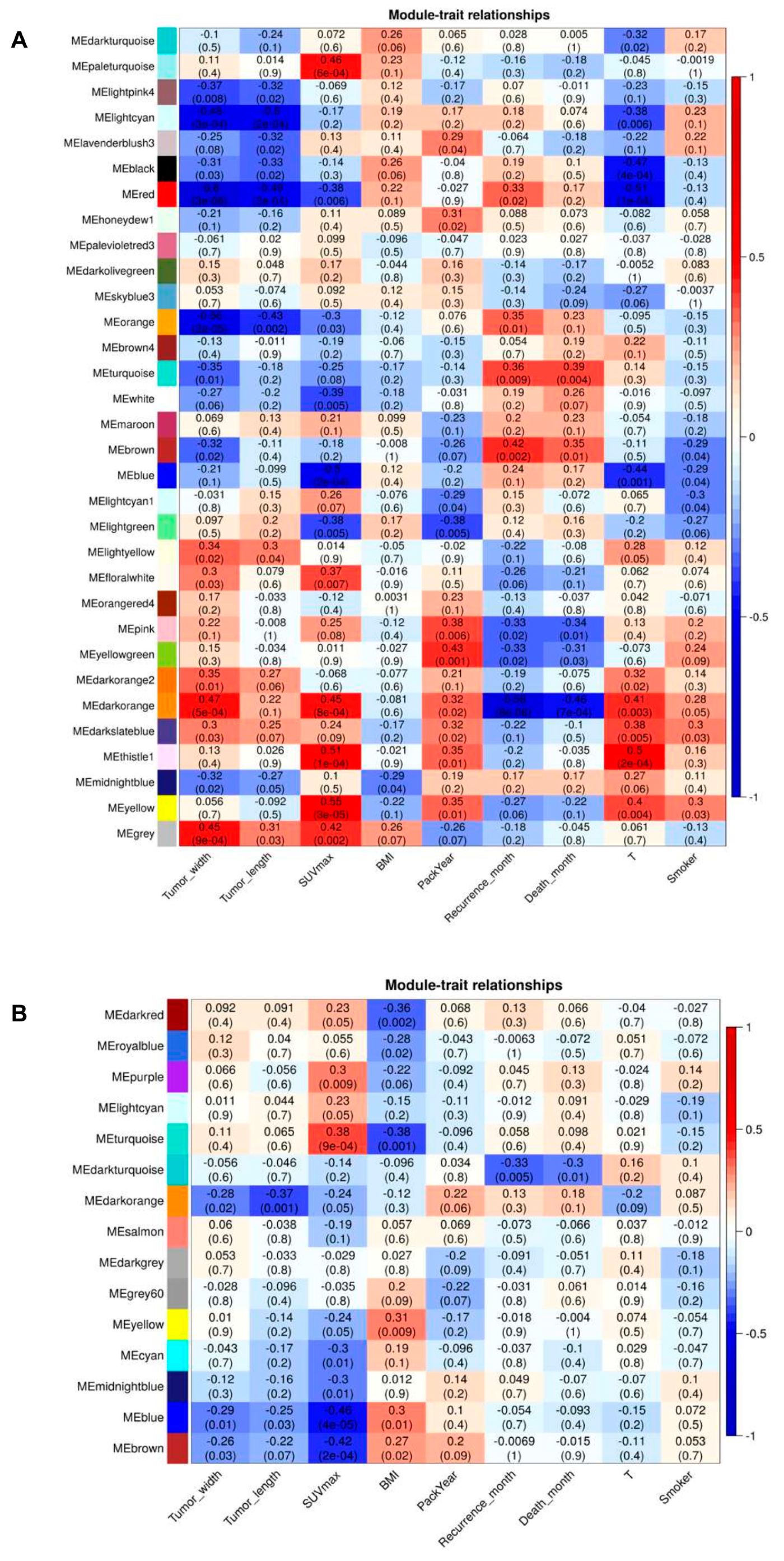
2.6. Association of Modules with Clinical Traits
2.7. Functional Enrichment Analysis of Critical Modules
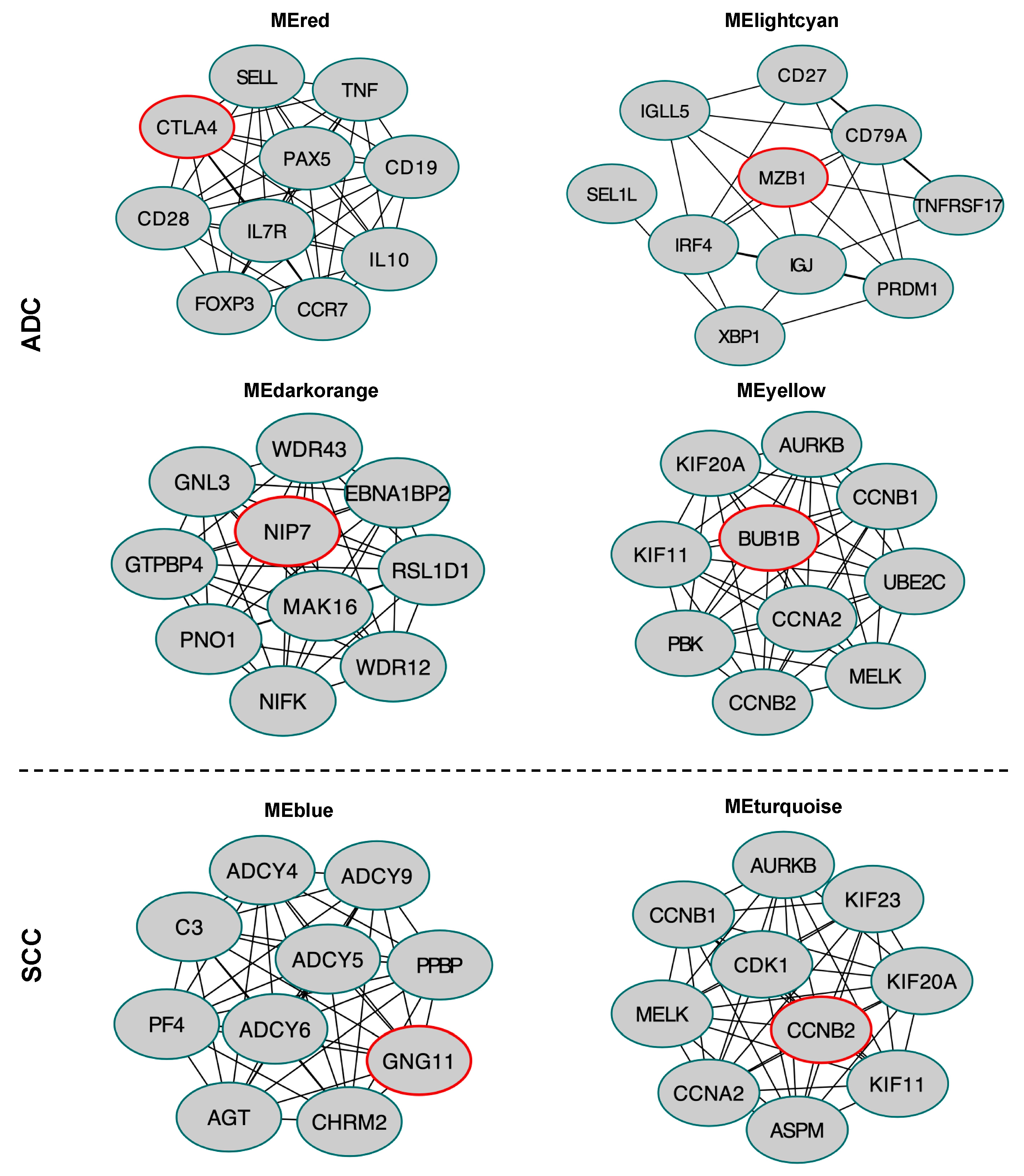
2.8. Hub Gene Identification in the Selected Module
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. RNA Sample Preparation and Sequencing
4.3. Functional Enrichment Analysis and Identification of Upstream Regulators
4.4. Construction of Weighted Gene Co-Expression Networks and Identification of Modules Associated with Clinical Traits
4.5. Protein-Protein Interaction Network Construction for Selected Modules and Hub Genes Identification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019: {Cancer} {Statistics}, 2019. CA. Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faruki, H.; Mayhew, G.M.; Serody, J.S.; Hayes, D.N.; Perou, C.M.; Lai-Goldman, M. Lung Adenocarcinoma and Squamous Cell Carcinoma Gene Expression Subtypes Demonstrate Significant Differences in Tumor Immune Landscape. J. Thorac. Oncol. 2017, 12, 943–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentles, A.J.; Bratman, S.V.; Lee, L.J.; Harris, J.P.; Feng, W.; Nair, R.V.; Shultz, D.B.; Nair, V.S.; Hoang, C.D.; West, R.B.; et al. Integrating Tumor and Stromal Gene Expression Signatures With Clinical Indices for Survival Stratification of Early-Stage Non-Small Cell Lung Cancer. J. Natl. Cancer Inst. 2015, 107, djv211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charkiewicz, R.; Niklinski, J.; Claesen, J.; Sulewska, A.; Kozlowski, M.; Michalska-Falkowska, A.; Reszec, J.; Moniuszko, M.; Naumnik, W.; Niklinska, W. Gene Expression Signature Differentiates Histology But Not Progression Status of Early-Stage NSCLC. Transl. Oncol. 2017, 10, 450–458. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Yin, X.; Walter, V.; Zhao, N.; Cabanski, C.R.; Hayward, M.C.; Miller, C.R.; Socinski, M.A.; Parsons, A.M.; Thorne, L.B.; et al. Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PLoS ONE 2012, 7, e36530. [Google Scholar] [CrossRef] [Green Version]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef]
- Ramani, V.; Qiu, R.; Shendure, J. High-throughput determination of RNA structure by proximity ligation. Nat. Biotechnol. 2015, 33, 980–984. [Google Scholar] [CrossRef] [Green Version]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, e559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Zhao, J.; Fan, X.; Kang, Z. Weighted gene co-expression network analysis in identification of metastasis-related genes of lung squamous cell carcinoma based on the Cancer Genome Atlas database. J. Thorac. Dis. 2017, 9, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.C.N.; Li, C.Y.; Cai, J.-H.; Sheu, P.C.-Y.; Tsai, J.J.P.; Wu, M.-Y.; Li, C.-J.; Hou, M.-F. Identification of Prognostic Candidate Genes in Breast Cancer by Integrated Bioinformatic Analysis. J. Clin. Med. 2019, 8, e1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Cai, Z.; Zhu, B.; Xu, C. Identification of Key Pathways and Genes in the Dynamic Progression of HCC Based on WGCNA. Genes (Basel) 2018, 9, e92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Wang, J.; Zhang, J. Identification of biomarkers of chromophobe renal cell carcinoma by weighted gene co-expression network analysis. Cancer Cell Int. 2018, 18, e206. [Google Scholar] [CrossRef]
- Zhou, X.; Huang, X.; Liang, S.; Tang, S.; Wu, S.; Huang, T.; Mo, Z.; Wang, Q. Identifying miRNA and gene modules of colon cancer associated with pathological stage by weighted gene co-expression network analysis. Onco Targets. Ther. 2018, 11, 2815–2830. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Fujii, K.; Gupta, V.; Hata, H.; Koizumu, H.; Hoshikawa, M.; Naruki, S.; Miyata, Y.; Takahashi, I.; Miyazawa, T.; et al. Identification of key modules and hub genes for small-cell lung carcinoma and large-cell neuroendocrine lung carcinoma by weighted gene co-expression network analysis of clinical tissue-proteomes. PLoS ONE 2019, 14, e0217105. [Google Scholar] [CrossRef]
- Niklinski, J.; Kretowski, A.; Moniuszko, M.; Reszec, J.; Michalska-Falkowska, A.; Niemira, M.; Ciborowski, M.; Charkiewicz, R.; Jurgilewicz, D.; Kozlowski, M.; et al. Systematic biobanking, novel imaging techniques, and advanced molecular analysis for precise tumor diagnosis and therapy: The Polish MOBIT project. Adv. Med. Sci. 2017, 62, 405–413. [Google Scholar] [CrossRef]
- Marko-Varga, G. BioBanking as the central tool for translational medicine CTM issue 2013. Clin. Transl. Med. 2013, 2, e4. [Google Scholar] [CrossRef] [Green Version]
- Neumeister, V.M.; Juhl, H. Tumor Pre-Analytics in Molecular Pathology: Impact on Protein Expression and Analysis. Curr. Pathobiol. Rep. 2018, 6, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, K.A.; Unger, F.T.; Uhlig, P.; Juhl, H.; Moore, H.M.; Compton, C.; Nashan, B.; Dörner, A.; de Weerth, A.; Zornig, C. Surgical procedures and postsurgical tissue processing significantly affect expression of genes and EGFR-pathway proteins in colorectal cancer tissue. Oncotarget 2014, 5, 11017–11028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, e550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weon, J.L.; Potts, P.R. The MAGE protein family and cancer. Curr. Opin. Cell Biol. 2015, 37, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Kong, M.; Zhang, Z.; Xu, S.; Yan, F.; Wei, L.; Zhou, J. FGF19 genetic amplification as a potential therapeutic target in lung squamous cell carcinomas. Thorac. Cancer 2017, 8, 655–665. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Zhang, J.J.; Song, H.J.; Li, D.W. Overexpression of CST4 promotes gastric cancer aggressiveness by activating the ELFN2 signaling pathway. Am. J. Cancer Res. 2017, 7, 2290–2304. [Google Scholar]
- Yin, Y.; Betsuyaku, T.; Garbow, J.R.; Miao, J.; Govindan, R.; Ornitz, D.M. Rapid induction of lung adenocarcinoma by fibroblast growth factor 9 signaling through FGF receptor 3. Cancer Res. 2013, 73, 5730–5741. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Wang, Z.; Chen, W.; Yang, Z.; Zheng, L.; Wang, S.; Li, S. Pseudogene CHIAP2 inhibits proliferation and invasion of lung adenocarcinoma cells by means of the WNT pathway. J. Cell. Physiol. 2019, 234, 13735–13746. [Google Scholar] [CrossRef]
- Zhai, S.; Xu, H.; Jiang, X.; Sun, S.; Qin, L.; Wei, D.; Hu, L. Expression Depression of CD300LG-γ in Human Pulmonary Carcinoma. Monoclon. Antib. Immunodiagn. Immunother. 2016, 35, 94–99. [Google Scholar] [CrossRef]
- Schürch, D.; Ospina, O.L.; Cruz, A.; Pérez-Gil, J. Combined and Independent Action of Proteins SP-B and SP-C in the Surface Behavior and Mechanical Stability of Pulmonary Surfactant Films. Biophys. J. 2010, 99, 3290–3299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, e1523. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Chuang, J.C.; Stehr, H.; Liang, Y.; Das, M.; Huang, J.; Diehn, M.; Wakelee, H.A.; Neal, J.W. ERBB2-Mutated Metastatic Non-Small Cell Lung Cancer: Response and Resistance to Targeted Therapies. J. Thorac. Oncol. 2017, 12, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Li, S.; Hai, J.; Wang, X.; Chen, T.; Quinn, M.M.; Gao, P.; Zhang, Y.; Ji, H.; Cross, D.A.E.; et al. Targeting HER2 Aberrations in Non–Small Cell Lung Cancer with Osimertinib. Clin. Cancer Res. 2018, 24, 2594–2604. [Google Scholar] [CrossRef] [Green Version]
- LaBaer, J.; Garrett, M.D.; Stevenson, L.F.; Slingerland, J.M.; Sandhu, C.; Chou, H.S.; Fattaey, A.; Harlow, E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997, 11, 847–862. [Google Scholar] [CrossRef] [Green Version]
- Hayami, S.; Kelly, J.D.; Cho, H.-S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.J.; et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef]
- Bergholz, J.; Xiao, Z.-X. Role of p63 in Development, Tumorigenesis and Cancer Progression. Cancer Microenviron. 2012, 5, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Grose, R.; Dickson, C. Fibroblast growth factor signaling in tumorigenesis. Cytokine Growth Factor Rev. 2005, 16, 179–186. [Google Scholar] [CrossRef]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, e2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flach, H.; Rosenbaum, M.; Duchniewicz, M.; Kim, S.; Zhang, S.L.; Cahalan, M.D.; Mittler, G.; Grosschedl, R. Mzb1 Protein Regulates Calcium Homeostasis, Antibody Secretion, and Integrin Activation in Innate-like B Cells. Immunity 2010, 33, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Stålberg, P.; Grimfjärd, P.; Santesson, M.; Zhou, Y.; Lindberg, D.; Gobl, A.; Öberg, K.; Westin, G.; Rastad, J.; Wang, S.; et al. Transfection of the Multiple Endocrine Neoplasia Type 1 Gene to a Human Endocrine Pancreatic Tumor Cell Line Inhibits Cell Growth and Affects Expression of JunD, δ-Like Protein 1/Preadipocyte Factor-1, Proliferating Cell Nuclear Antigen, and QM/Jif-1. J. Clin. Endocrinol. Metab. 2004, 89, 2326–2337. [Google Scholar] [CrossRef] [Green Version]
- Bolanos-Garcia, V.M.; Blundell, T.L. BUB1 and BUBR1: Multifaceted kinases of the cell cycle. Trends Biochem. Sci. 2011, 36, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Giotti, B.; Chen, S.-H.; Barnett, M.W.; Regan, T.; Ly, T.; Wiemann, S.; Hume, D.A.; Freeman, T.C. Assembly of a parts list of the human mitotic cell cycle machinery. J. Mol. Cell Biol. 2019, 11, 703–718. [Google Scholar] [CrossRef]
- Ni, Z.; Wang, X.; Zhang, T.; Li, L.; Li, J. Comprehensive analysis of differential expression profiles reveals potential biomarkers associated with the cell cycle and regulated by p53 in human small cell lung cancer. Exp. Ther. Med. 2018, 15, 3273–3282. [Google Scholar] [CrossRef]
- Hossain, M.N.; Sakemura, R.; Fujii, M.; Ayusawa, D. G-protein gamma subunit GNG11 strongly regulates cellular senescence. Biochem. Biophys. Res. Commun. 2006, 351, 645–650. [Google Scholar] [CrossRef]
- Meek, D.W.; Marcar, L. MAGE-A antigens as targets in tumour therapy. Cancer Lett. 2012, 324, 126–132. [Google Scholar] [CrossRef]
- Bergeron, A.; Picard, V.; LaRue, H.; Harel, F.; Hovington, H.; Lacombe, L.; Fradet, Y. High frequency of MAGE-A4 and MAGE-A9 expression in high-risk bladder cancer. Int. J. Cancer 2009, 125, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Eichmüller, S.; Usener, D.; Thiel, D.; Schadendorf, D. Tumor-specific antigens in cutaneous T-cell lymphoma: Expression and sero-reactivity. Int. J. Cancer 2003, 104, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lin, L.; Thomas, D.G.; Greenson, J.K.; Giordano, T.J.; Robinson, G.S.; Barve, R.A.; Weishaar, F.A.; Taylor, J.M.G.; Orringer, M.B.; et al. Melanoma-Associated Antigens in Esophageal Adenocarcinoma: Identification of Novel MAGE-A10 Splice Variants. Clin. Cancer Res. 2004, 10, 5708–5716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatiboglu, G.; Pritsch, M.; Macher-Goeppinger, S.; Zöller, M.; Huber, J.; Haferkamp, A.; Pahernik, S.; Wagener, N.; Hohenfellner, M. Prognostic value of melanoma-associated antigen A9 in renal cell carcinoma. Scand. J. Urol. 2013, 47, 311–322. [Google Scholar] [CrossRef]
- Zhang, S.; Zhai, X.; Wang, G.; Feng, J.; Zhu, H.; Xu, L.; Mao, G.; Huang, J. High expression of MAGE-A9 in tumor and stromal cells of non-small cell lung cancer was correlated with patient poor survival. Int. J. Clin. Exp. Pathol. 2015, 8, 541–550. [Google Scholar]
- Li, B.; Meng, Y.-Q.; Li, Z.; Yin, C.; Lin, J.-P.; Zhu, D.-J.; Zhang, S.-B. MiR-629-3p-induced downregulation of SFTPC promotes cell proliferation and predicts poor survival in lung adenocarcinoma. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3286–3296. [Google Scholar] [CrossRef] [Green Version]
- Djureinovic, D.; Hallström, B.M.; Horie, M.; Mattsson, J.S.M.; La Fleur, L.; Fagerberg, L.; Brunnström, H.; Lindskog, C.; Madjar, K.; Rahnenführer, J.; et al. Profiling cancer testis antigens in non-small-cell lung cancer. JCI Insight 2016, 1, e86837. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-C.; Su, L.-J.; Wang, H.-W.; Jeff Lin, C.-F.; Hsu, W.-H.; Chou, T.-Y.; Huang, C.-Y.F.; Lu, C.-L.; Hsueh, C.-T. Co-Overexpression of Cyclooxygenase-2 and Microsomal Prostaglandin E Synthase-1 Adversely Affects the Postoperative Survival in Non-small Cell Lung Cancer. J. Thorac. Oncol. 2010, 5, 1167–1174. [Google Scholar] [CrossRef] [Green Version]
- Strell, C.; Entschladen, F. Extravasation of leukocytes in comparison to tumor cells. Cell Commun. Signal. 2008, 6, e10. [Google Scholar] [CrossRef] [Green Version]
- Peeters, C.F.J.M.; Ruers, T.J.M.; Westphal, J.R.; de Waal, R.M.W. Progressive loss of endothelial P-selectin expression with increasing malignancy in colorectal cancer. Lab. Investig. 2005, 85, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Steege, J.C.A.B.-T.; Baeten, C.I.M.; Thijssen, V.L.J.L.; Satijn, S.A.; Verhoeven, I.C.L.; Hillen, H.F.P.; Wagstaff, J.; Griffioen, A.W. Angiogenic Profile of Breast Carcinoma Determines Leukocyte Infiltration. Clin. Cancer Res. 2004, 10, 7171–7178. [Google Scholar] [CrossRef] [Green Version]
- Hellwig, S.M.; Damen, C.A.; van Adrichem, N.P.; Blijham, G.H.; Groenewegen, G.; Griffioen, A.W. Endothelial CD34 is suppressed in human malignancies: Role of angiogenic factors. Cancer Lett. 1997, 120, 203–211. [Google Scholar] [CrossRef]
- Languino, L.R.; Singh, A.; Prisco, M.; Inman, G.J.; Luginbuhl, A.; Curry, J.M.; South, A.P. Exosome-mediated transfer from the tumor microenvironment increases TGFβ signaling in squamous cell carcinoma. Am. J. Transl. Res. 2016, 8, 2432–2437. [Google Scholar]
- Park, H.J.; Cha, Y.-J.; Kim, S.H.; Kim, A.; Kim, E.Y.; Chang, Y.S. Keratinization of Lung Squamous Cell Carcinoma Is Associated with Poor Clinical Outcome. Tuberc. Respir. Dis. (Seoul) 2017, 80, 179–186. [Google Scholar] [CrossRef]
- Egolf, S.; Aubert, Y.; Doepner, M.; Anderson, A.; Maldonado-Lopez, A.; Pacella, G.; Lee, J.; Ko, E.K.; Zou, J.; Lan, Y.; et al. LSD1 Inhibition Promotes Epithelial Differentiation through Derepression of Fate-Determining Transcription Factors. Cell Rep. 2019, 28, 1981–1992.e7. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Zhang, P.; Li, W.; Yang, Y.; Tian, Y.; Wang, X.; Chen, S.; Yang, Y.; Huang, T.; Zhao, T.; et al. KDM1A promotes tumor cell invasion by silencing TIMP3 in non-small cell lung cancer cells. Oncotarget 2016, 7, 27959–27974. [Google Scholar] [CrossRef] [Green Version]
- Grieb, G.; Merk, M.; Bernhagen, J.; Bucala, R. Macrophage migration inhibitory factor (MIF): A promising biomarker. Drug News Perspect. 2010, 23, e257. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.-T.; Guo, L.-L.; Feng, N.; Zhang, L.; Zhou, N.; Ma, L.-L.; Shen, L.; Tong, G.; Yan, Q.-W.; Zhu, S.; et al. MIF, secreted by human hepatic sinusoidal endothelial cells, promotes chemotaxis and outgrowth of colorectal cancer in liver prometastasis. Oncotarget 2015, 6, 22410–22423. [Google Scholar] [CrossRef] [Green Version]
- Dave, M.; Islam, A.B.M.M.K.; Jensen, R.V.; Rostagno, A.; Ghiso, J.; Amin, A.R. Proteomic Analysis Shows Constitutive Secretion of MIF and p53-associated Activity of COX-2−/− Lung Fibroblasts. Genomics. Proteom. Bioinform. 2017, 15, 339–351. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Chanvorachote, P. Nitric Oxide and Aggressive Behavior of Lung Cancer Cells. Anticancer Res. 2015, 35, 4585–4592. [Google Scholar]
- Pardo, O.E.; Wellbrock, C.; Khanzada, U.K.; Aubert, M.; Arozarena, I.; Davidson, S.; Bowen, F.; Parker, P.J.; Filonenko, V.V.; Gout, I.T.; et al. FGF-2 protects small cell lung cancer cells from apoptosis through a complex involving PKCɛ, B-Raf and S6K2. EMBO J. 2006, 25, 3078–3088. [Google Scholar] [CrossRef]
- Kwabi-Addo, B.; Ozen, M.; Ittmann, M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr. Relat. Cancer 2004, 11, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Barajon, I.; Garlanda, C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol. Rev. 2018, 281, 57–61. [Google Scholar] [CrossRef]
- Zheng, X.; Hu, Y.; Yao, C. The paradoxical role of tumor-infiltrating immune cells in lung cancer. Intractable Rare Dis. Res. 2017, 6, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.; Na, K.J. Integrative analysis of imaging and transcriptomic data of the immune landscape associated with tumor metabolism in lung adenocarcinoma: Clinical and prognostic implications. Theranostics 2018, 8, 1956–1965. [Google Scholar] [CrossRef] [Green Version]
- Ohue, Y.; Kurose, K.; Nozawa, R.; Isobe, M.; Nishio, Y.; Tanaka, T.; Doki, Y.; Hori, T.; Fukuoka, J.; Oka, M.; et al. Survival of Lung Adenocarcinoma Patients Predicted from Expression of PD-L1, Galectin-9, and XAGE1 (GAGED2a) on Tumor Cells and Tumor-Infiltrating T Cells. Cancer Immunol. Res. 2016, 4, 1049–1060. [Google Scholar] [CrossRef] [Green Version]
- Kanda, M.; Tanaka, C.; Kobayashi, D.; Tanaka, H.; Shimizu, D.; Shibata, M.; Takami, H.; Hayashi, M.; Iwata, N.; Niwa, Y.; et al. Epigenetic suppression of the immunoregulator MZB1 is associated with the malignant phenotype of gastric cancer. Int. J. Cancer 2016, 139, 2290–2298. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Chen, Y.; Li, Q.; Zhang, L. Exploration of the hub genes and miRNAs in lung adenocarcinoma. Oncol. Lett. 2019, 18, 1713–1722. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Xiong, D.; Wang, Y.; You, M. Tumor intrinsic immunity related proteins may be novel tumor suppressors in some types of cancer. Sci. Rep. 2019, 9, e10918. [Google Scholar] [CrossRef]
- Kanteti, R.; Nallasura, V.; Loganathan, S.; Tretiakova, M.; Kroll, T.; Krishnaswamy, S.; Faoro, L.; Cagle, P.; Husain, A.N.; Vokes, E.E.; et al. PAX5 is expressed in small-cell lung cancer and positively regulates c-Met transcription. Lab. Investig. 2009, 89, 301–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, P.; Morin, P.; Ouellette, R.J.; Robichaud, G.A. The Pax-5 Gene: A Pluripotent Regulator of B-cell Differentiation and Cancer Disease. Cancer Res. 2011, 71, 7345–7350. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Li, S.; Gan, L.; Li, C.; Qiu, Z.; Feng, Y.; Li, J.; Li, L.; Li, C.; Peng, W.; et al. Paired box 5 is a frequently methylated lung cancer tumour suppressor gene interfering β-catenin signalling and GADD45G expression. J. Cell. Mol. Med. 2016, 20, 842–854. [Google Scholar] [CrossRef]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, R.; Liao, X.; Jiang, W. Comprehensive Bioinformatic Analysis Genes Associated to the Prognosis of Liposarcoma. Med. Sci. Monit. 2018, 24, 7329–7339. [Google Scholar] [CrossRef]
- Koksal, D.; Demirag, F.; Bayiz, H.; Ozmen, O.; Tatci, E.; Berktas, B.; Aydoğdu, K.; Yekeler, E. The correlation of SUVmax with pathological characteristics of primary tumor and the value of Tumor/ Lymph node SUVmax ratio for predicting metastasis to lymph nodes in resected NSCLC patients. J. Cardiothorac. Surg. 2013, 8, e63. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Song, X.; He, Y.; Yang, Z.; Sun, T.; Wang, J.; Zhu, G.; Xing, W.; You, C. CCNB2 overexpression is a poor prognostic biomarker in Chinese NSCLC patients. Biomed. Pharmacother. 2015, 74, 222–227. [Google Scholar] [CrossRef]
- Wang, T.; Chen, B.; Yang, Y.; Chen, H.; Wang, Y.; Cviko, A.; Quade, B.J.; Sun, D.; Yang, A.; McKeon, F.D.; et al. Histologic and immunophenotypic classification of cervical carcinomas by expression of the p53 homologue p63: A study of 250 cases. Hum. Pathol. 2001, 32, 479–486. [Google Scholar] [CrossRef]
- Shi, Y.; Li, Y.; Yan, C.; Su, H.; Ying, K. Identification of key genes and evaluation of clinical outcomes in lung squamous cell carcinoma using integrated bioinformatics analysis. Oncol. Lett. 2019, 18, 5859–5870. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Kong, W.; Huang, Z.; Xie, Z. Identification of key genes related to the mechanism and prognosis of lung squamous cell carcinoma using bioinformatics analysis. arXiv 2019, 1911, 05621. [Google Scholar]
- Hsu, Y.-L.; Hung, J.-Y.; Lee, Y.-L.; Chen, F.-W.; Chang, K.-F.; Chang, W.-A.; Tsai, Y.-M.; Chong, I.-W.; Kuo, P.-L. Identification of novel gene expression signature in lung adenocarcinoma by using next-generation sequencing data and bioinformatics analysis. Oncotarget 2017, 8, 104831–104854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, K.; Li, N.; Yang, M.; Li, W. Identification of Key Genes and Pathways in Female Lung Cancer Patients Who Never Smoked by a Bioinformatics Analysis. J. Cancer 2019, 10, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, e323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2015, 32, btv566. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | NSCLC patients (n = 114) |
|---|---|
| Age, year (mean ± SD) | 65.4 ± 7.9 |
| Male, n (%) | 80 (70.2) |
| Smoking, PY (mean ± SD) | 39.6 ± 21.0 |
| BMI (mean ± SD) | 26.9 ± 4.7 |
| SUV (max) (mean ± SD) | 8.8 ± 4.6 |
| Histology | |
| Adenocarcinoma (AC), n (%) | 45 (39.5) |
| Squamous Cell Carcinoma (SCC), n (%) | 69 (60.5) |
| TNM stage, n (%) | |
| I | 39 (34.2) |
| II | 52 (45.6) |
| III | 17 (14.9) |
| IV | 6 (5.3) |
| Recurrence (%) | 15.2 |
| Deaths (%) | 13.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niemira, M.; Collin, F.; Szalkowska, A.; Bielska, A.; Chwialkowska, K.; Reszec, J.; Niklinski, J.; Kwasniewski, M.; Kretowski, A. Molecular Signature of Subtypes of Non-Small-Cell Lung Cancer by Large-Scale Transcriptional Profiling: Identification of Key Modules and Genes by Weighted Gene Co-Expression Network Analysis (WGCNA). Cancers 2020, 12, 37. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12010037
Niemira M, Collin F, Szalkowska A, Bielska A, Chwialkowska K, Reszec J, Niklinski J, Kwasniewski M, Kretowski A. Molecular Signature of Subtypes of Non-Small-Cell Lung Cancer by Large-Scale Transcriptional Profiling: Identification of Key Modules and Genes by Weighted Gene Co-Expression Network Analysis (WGCNA). Cancers. 2020; 12(1):37. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12010037
Chicago/Turabian StyleNiemira, Magdalena, Francois Collin, Anna Szalkowska, Agnieszka Bielska, Karolina Chwialkowska, Joanna Reszec, Jacek Niklinski, Miroslaw Kwasniewski, and Adam Kretowski. 2020. "Molecular Signature of Subtypes of Non-Small-Cell Lung Cancer by Large-Scale Transcriptional Profiling: Identification of Key Modules and Genes by Weighted Gene Co-Expression Network Analysis (WGCNA)" Cancers 12, no. 1: 37. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12010037