Insights into Genetic Susceptibility to Melanoma by Gene Panel Testing: Potential Pathogenic Variants in ACD, ATM, BAP1, and POT1

 ,
,  ,
,  , , , , , and add
Show full author list
, , , , , and add
Show full author list
Abstract
:1. Introduction
2. Results
2.1. Panel Validation
2.2. Detection Yield and Variant Interpretation in Established and Candidate Melanoma Predisposition Genes
2.2.1. BAP1 (2.2–2.5%)
2.2.2. POT1 (0.7–2.9%)
2.2.3. ACD (0.36%), TERF2IP (0.36%), and TERT (0%)
2.2.4. MITF (2.6%)
2.2.5. PC-Associated Candidate Genes: ATM (3.3–5.5%) and PALB2
2.2.6. CDKN2A/ARF and CDK4
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. Panel Design
4.3. Targeted Sequencing and Variant Selection and Interpretation
4.4. Next-Generation Sequencing (NGS) Analysis
4.5. BAP1 LOH/Protein Expression by Immunohistochemistry (IHC)
4.6. Splicing Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aoude, L.; Wadt, K.; Pritchard, A.; Hayward, N.K. Genetics of familial melanoma: 20 years afterCDKN2A. Pigment. Cell Melanoma Res. 2015, 28, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Bruno, W.; Pastorino, L.; Ghiorzo, P.; Andreotti, V.; Martinuzzi, C.; Menin, C.; Elefanti, L.; Stagni, C.; Vecchiato, A.; Rodolfo, M.; et al. Multiple primary melanomas (MPMs) and criteria for genetic assessment: MultiMEL, a multicenter study of the Italian Melanoma Intergroup. J. Am. Acad. Dermatol. 2016, 74, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Bruno, W.; Ghiorzo, P.; Battistuzzi, L.; Ascierto, P.A.; Barile, M.; Gargiulo, S.; Gensini, F.; Gliori, S.; Guida, M.; Lombardo, M.; et al. Clinical genetic testing for familial melanoma in Italy: A cooperative study. J. Am. Acad. Dermatol. 2009, 61, 775–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiorzo, P.; Pastorino, L.; Bonelli, L.; Cusano, R.; Nicora, A.; Zupo, S.; Queirolo, P.; Sertoli, M.; Pugliese, V.; Bianchi Scarrà, G. INK4/ARF germline alterations in pancreatic cancer patients. Ann. Oncol. 2004, 15, 70–78. [Google Scholar] [CrossRef]
- Ghiorzo, P.; Fornarini, G.; Sciallero, S.; Battistuzzi, L.; Belli, F.; Bernard, L.; Bonelli, L.; Borgonovo, G.; Bruno, W.; De Cian, F.; et al. CDKN2A is the main susceptibility gene in Italian pancreatic cancer families. J. Med. Genet. 2012, 49, 164–170. [Google Scholar] [CrossRef]
- Puntervoll, H.E.; Yang, X.R.; Vetti, H.H.; Bachmann, I.M.; Avril, M.-F.; Benfodda, M.; Catricala, C.; Dalle, S.; Duval-Modeste, A.B.; Ghiorzo, P.; et al. Melanoma prone families with CDK4germline mutation: Phenotypic profile and associations with MC1Rvariants. J. Med. Genet. 2013, 50, 264–270. [Google Scholar] [CrossRef] [Green Version]
- Bottillo, I.; La Starza, R.; Radio, F.C.; Pedace, L.; Pierini, T.; De Bernardo, C.; Stingeni, L.; Bargiacchi, S.; Paiardini, A.; Janson, G.; et al. A novel germline mutation in CDK4 codon 24 associated to familial melanoma. Clin. Genet. 2017, 93, 934–935. [Google Scholar] [CrossRef]
- Leachman, S.A.; Lucero, O.M.; Sampson, J.E.; Cassidy, P.; Bruno, W.; Queirolo, P.; Ghiorzo, P. Identification, genetic testing, and management of hereditary melanoma. Cancer Metastasis Rev. 2017, 36, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Yang, X.R.; Ballew, B.; Rotunno, M.; Calista, D.; Fargnoli, M.C.; Ghiorzo, P.; Paillerets, B.B.-D.; Nagore, E.; Avril, M.-F.; et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat. Genet. 2014, 46, 482–486. [Google Scholar] [CrossRef]
- Robles-Espinoza, C.D.; Harland, M.; Ramsay, A.J.; Aoude, L.; Quesada, V.; Ding, Z.; Pooley, K.A.; Pritchard, A.; Tiffen, J.C.; Petljak, M.; et al. POT1 loss-of-function variants predispose to familial melanoma. Nat. Genet. 2014, 46, 478–481. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, T.; Obenauf, A.C.; Murali, R.; Fried, I.; Griewank, K.G.; Ulz, P.; Windpassinger, C.; Wackernagel, W.; Loy, S.; Wolf, I.; et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat. Genet. 2011, 43, 1018–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoude, L.; Pritchard, A.; Robles-Espinoza, C.D.; Wadt, K.; Harland, M.; Choi, J.; Gartside, M.; Quesada, V.; Johansson, P.; Palmer, J.M.; et al. Nonsense Mutations in the Shelterin Complex Genes ACD and TERF2IP in Familial Melanoma. J. Natl. Cancer Inst. 2014, 107, 107. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A. The Importance of Multidisciplinary Approach in Early Detection of BAP1 Tumor Predisposition Syndrome: Clinical Management and Risk Assessment. Clin. Med. Insights Oncol. 2014, 8, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Harland, M.; Petljak, M.; Robles-Espinoza, C.D.; Ding, Z.; Gruis, N.A.; Van Doorn, R.; Pooley, K.A.; Dunning, A.M.; Aoude, L.; Wadt, K.A.W.; et al. Germline TERT promoter mutations are rare in familial melanoma. Fam. Cancer 2016, 15, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; de Lichy, M.; Bille, K.; Dessen, P.; dHayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef]
- Yokoyama, S.; Woods, S.; Boyle, G.M.; Aoude, L.; MacGregor, S.; Zismann, V.; Gartside, M.; Cust, A.E.; Haq, R.; Harland, M.; et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature 2011, 480, 99–103. [Google Scholar] [CrossRef]
- Ghiorzo, P.; Pastorino, L.; Queirolo, P.; Bruno, W.; Tibiletti, M.G.; Nasti, S.; Andreotti, V.; Paillerets, B.B.-D.; Scarrà, G.B. Prevalence of the E318K MITF germline mutation in Italian melanoma patients: Associations with histological subtypes and family cancer history. Pigment. Cell Melanoma Res. 2012, 26, 259–262. [Google Scholar] [CrossRef]
- Read, J.; Wadt, K.A.W.; Hayward, N.K. Melanoma genetics. J. Med. Genet. 2015, 53, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Potrony, M.; Puig-Butille, J.A.; Aguilera, P.; Badenas, C.; Tell-Martí, G.; Carrera, C.; Del Pozo, L.J.; Conejo-Mir, J.; Malvehy, J.; Puig, S. Prevalence ofMITFp.E318K in Patients with Melanoma Independent of the Presence ofCDKN2ACausative Mutations. JAMA Dermatol. 2016, 152, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Ciccarese, G.; Pastorino, L.; Andreotti, V.; Spagnolo, F.; Tanda, E.; Ponti, G.; Massone, C.; Drago, F.; Parodi, A.; Ghigliotti, G.; et al. Clinical, pathological and dermoscopic phenotype of MITF p.E318K carrier cutaneous melanoma patients. J. Transl. Med. 2020, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Potrony, M.; Badenas, C.; Aguilera, P.; Puig-Butille, J.A.; Carrera, C.; Malvehy, J.; Puig, S. Update in genetic susceptibility in melanoma. Ann. Transl. Med. 2015, 3, 210. [Google Scholar] [PubMed]
- Ghiorzo, P.; Gargiulo, S.; Pastorino, L.; Nasti, S.; Cusano, R.; Bruno, W.; Gliori, S.; Sertoli, M.R.; Burroni, A.; Savarino, V.; et al. Impact of E27X, a novel CDKN2A germ line mutation, on p16 and p14ARF expression in Italian melanoma families displaying pancreatic cancer and neuroblastoma. Hum. Mol. Genet. 2006, 15, 2682–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, A.M.; Chan, M.; Harland, M.; Gillanders, E.M.; Hayward, N.K.; Avril, M.-F.; Azizi, E.; Scarrà, G.B.; Newton-Bishop, J.; Paillerets, B.B.-D.; et al. High-risk Melanoma Susceptibility Genes and Pancreatic Cancer, Neural System Tumors, and Uveal Melanoma across GenoMEL. Cancer Res. 2006, 66, 9818–9828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, M.A.; Elder, D.E.; Curry, M.; Fraser, M.C.; Pichler, V.; Zametkin, D.; Yang, X.; Goldstein, A.M. Risks of Melanoma and Other Cancers in Melanoma-Prone Families over 4 Decades. J. Investig. Dermatol. 2018, 138, 1620–1626. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, A.M.; Xiao, Y.; Sampson, J.; Rotunno, M.; Bennett, H.; Wen, Y.; Jones, K.; Vogt, A.; Burdette, L.; Luo, W.; et al. Rare germline variants in known melanoma susceptibility genes in familial melanoma. Hum. Mol. Genet. 2017, 26, 4886–4895. [Google Scholar] [CrossRef]
- Rai, K.; Pilarski, R.; Cebulla, C.; Abdel-Rahman, M. Comprehensive review of BAP1 tumor predisposition syndrome with report of two new cases. Clin. Genet. 2015, 89, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Bainbridge, M.N.; Armstrong, G.N.; Gramatges, M.M.; Bertuch, A.A.; Jhangiani, S.N.; Doddapaneni, H.; Lewis, L.; Tombrello, J.; Tsavachidis, S.; Liu, Y.; et al. Germline mutations in shelterin complex genes are associated with familial glioma. J. Natl. Cancer Inst. 2014, 107, 384. [Google Scholar] [CrossRef]
- Wilson, T.L.-S.; Hattangady, N.; Lerario, A.; Williams, C.; Koeppe, E.; Quinonez, S.; Osborne, J.; Else, T.; Cha, K.B. A new POT1 germline mutation—Expanding the spectrum of POT1-associated cancers. Fam. Cancer 2017, 53, 1–566. [Google Scholar] [CrossRef]
- Calvete, O.; Garcia, C.L.; Dominguez, F.; Bougeard, G.; Kunze, K.; Braeuninger, A.; Teule, A.; Lasa, A.; Cajal, T.R.Y.; Llort, G.; et al. The wide spectrum of POT1 gene variants correlates with multiple cancer types. Eur. J. Hum. Genet. 2017, 25, 1278–1281. [Google Scholar] [CrossRef] [Green Version]
- Chubb, D.; Broderick, P.; Dobbins, S.E.; Frampton, M.; Kinnersley, B.; Penegar, S.; Price, A.; Ma, Y.P.; Sherborne, A.L.; Palles, C.; et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat. Commun. 2016, 7, 11883. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.J.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.L.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L.; et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2011, 2, 41–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilad, S.; Chessa, L.; Khosravi, R.; Russell, P.; Galanty, Y.; Piane, M.; Gatti, R.A.; Jorgensen, T.J.; Shiloh, Y.; Bar-Shira, A. Genotype-phenotype relationships in ataxia-telangiectasia and variants. Am. J. Hum. Genet. 1998, 62, 551–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Jones, S.; Hruban, R.H.; Hruban, R.H.; Kamiyama, M.; Kamiyama, M.; Borges, M.; Borges, M.; Zhang, X.; Zhang, X.; et al. Exomic Sequencing Identifies PALB2 as a Pancreatic Cancer Susceptibility Gene. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, D.; Rabe, K.G.; Gallinger, S.; Syngal, S.; Schwartz, A.G.; Goggins, M.G.; Hruban, R.H.; Cote, M.L.; McWilliams, R.R.; Roberts, N.J.; et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: A PACGENE study. Genet. Med. 2014, 17, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombernowsky, S.L.; Weischer, M.; Allin, K.H.; Bojesen, S.E.; Tybjjrg-Hansen, A.; Nordestgaard, B.G. Risk of Cancer by ATMMissense Mutations in the General Population. J. Clin. Oncol. 2008, 26, 3057–3062. [Google Scholar] [CrossRef]
- Barrett, J.; Iles, M.M.; Harland, M.; Taylor, J.C.; Aitken, J.F.; Andresen, P.A.; Akslen, L.A.; Armstrong, B.K.; Avril, M.-F.; Azizi, E.; et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat. Genet. 2011, 43, 1108–1113. [Google Scholar] [CrossRef]
- Fang, S.; Lu, J.; Zhou, X.; Wang, Y.; Ross, M.I.; Gershenwald, J.E.; Cormier, J.N.; Wargo, J.; Sui, D.; Amos, C.I.; et al. Functional annotation of melanoma risk loci identifies novel susceptibility genes. Carcinogenesis 2019. [Google Scholar] [CrossRef]
- Yang, X.R.; Rotunno, M.; Xiao, Y.; Ingvar, C.; Helgadottir, H.; Pastorino, L.; Van Doorn, R.; Bennett, H.; Graham, C.; Sampson, J.N.; et al. Multiple rare variants in high-risk pancreatic cancer-related genes may increase risk for pancreatic cancer in a subset of patients with and without germline CDKN2A mutations. Qual. Life Res. 2016, 135, 1241–1249. [Google Scholar] [CrossRef] [Green Version]
- Landi, M.T.; Bishop, T.D.; MacGregor, S.; Machiela, M.J.; Stratigos, A.J.; Ghiorzo, P.; Brossard, M.; Calista, D.; Choi, J.; Fargnoli, M.C.; et al. Genome-wide association meta-analyses combining multiple risk phenotypes provides insights into the genetic architecture of cutaneous melanoma susceptibility. Nat. Genet. 2020. [Google Scholar] [CrossRef]
- Fang, S.; Han, J.; Zhang, M.; Wang, L.-E.; Wei, Q.; Amos, C.I.; Lee, J.E. Joint Effect of Multiple Common SNPs Predicts Melanoma Susceptibility. PLoS ONE 2013, 8, e85642. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Chen, T.-H.; Pfeiffer, R.M.; Fargnoli, M.C.; Calista, N.; Ghiorzo, P.; Peris, K.; Puig, S.; Menin, C.; De Nicolo, A.; et al. Combining common genetic variants and non-genetic risk factors to predict risk of cutaneous melanoma. Hum. Mol. Genet. 2018, 27, 4145–4156. [Google Scholar] [CrossRef] [PubMed]
- Schlafly, A.; Pfeiffer, R.M.; Nagore, E.; Puig, S.; Calista, D.; Ghiorzo, P.; Menin, C.; Fargnoli, M.C.; Peris, K.; Song, L.; et al. Contribution of Common Genetic Variants to Familial Aggregation of Disease and Implications for Sequencing Studies. PLoS Genet. 2019, 15, e1008490. [Google Scholar] [CrossRef] [PubMed]
- Walpole, S.; Pritchard, A.; Cebulla, C.; Pilarski, R.; Stautberg, M.; Davidorf, F.H.; De La Fouchardiere, A.; Cabaret, O.; Golmard, L.; Stoppa-Lyonnet, D.; et al. Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide. J. Natl. Cancer Inst. 2018, 110, 1328–1341. [Google Scholar] [CrossRef]
- Potrony, M.; Puig-Butille, J.A.; Ribera-Sola, M.; Iyer, V.; Robles-Espinoza, C.D.; Aguilera, P.; Carrera, C.; Malvehy, J.; Badenas, C.; Landi, M.T.; et al. POT1germline mutations but not TERTpromoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br. J. Dermatol. 2019, 3, 210–219. [Google Scholar]
- Ramsay, A.J.; Quesada, V.; Foronda, M.; Conde, L.; Martínez-Trillos, A.; Villamor, N.; Rodríguez, D.; Kwarciak, A.; Garabaya, C.; Gallardo, M.; et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat. Genet. 2013, 45, 526–530. [Google Scholar] [CrossRef]
- Sandoval, N. Characterization of ATM gene mutations in 66 ataxia telangiectasia families. Hum. Mol. Genet. 1999, 8, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Andreotti, V.; Bisio, A.; Paillerets, B.B.-D.; Harland, M.; Cabaret, O.; Newton-Bishop, J.; Pastorino, L.; Bruno, W.; Bertorelli, R.; De Sanctis, V.; et al. TheCDKN2A/p16INK4a5′UTR sequence and translational regulation: Impact of novel variants predisposing to melanoma. Pigment. Cell Melanoma Res. 2015, 29, 210–221. [Google Scholar] [CrossRef]
- Bisio, A.; Nasti, S.; Jordan, J.J.; Gargiulo, S.; Pastorino, L.; Provenzani, A.; Quattrone, A.; Queirolo, P.; Bianchi-Scarra, G.; Ghiorzo, P.; et al. Functional analysis of CDKN2A/p16INK4a 5′-UTR variants predisposing to melanoma. Hum. Mol. Genet. 2010, 19, 1479–1491. [Google Scholar] [CrossRef] [Green Version]
- Chau, C.; Van Doorn, R.; Van Poppelen, N.M.; Van Der Stoep, N.; Mensenkamp, A.R.; Sijmons, R.; Van Paassen, B.W.; Ouweland, A.M.W.V.D.; Naus, N.C.; Van Der Hout, A.H.; et al. Families with BAP1-Tumor Predisposition Syndrome in The Netherlands: Path to Identification and a Proposal for Genetic Screening Guidelines. Cancers 2019, 11, 1114. [Google Scholar] [CrossRef] [Green Version]
- Avril, M.-F.; Bahadoran, P.; Cabaret, O.; Caron, O.; de la Fouchardière, A.; Demenais, F.; Desjardins, L.; Frébourg, T.; Hammel, P.; Leccia, M.-T.; et al. Recommendations for genetic testing and management of individuals genetically at-risk of cutaneous melanoma. Ann. Dermatol. Venereol. 2015, 142, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Ghiorzo, P.; Pensotti, V.; Fornarini, G.; Sciallero, S.; Battistuzzi, L.; Belli, F.; Bonelli, L.; Borgonovo, G.; Bruno, W.; Gozza, A.; et al. Contribution of germline mutations in the BRCA and PALB2 genes to pancreatic cancer in Italy. Fam. Cancer 2011, 11, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Aoude, L.; Xu, M.; Zhao, Z.Z.; Kovacs, M.; Palmer, J.M.; Johansson, P.; Symmons, J.; Trent, J.M.; Martin, N.G.; Montgomery, G.W.; et al. Assessment of PALB2 as a Candidate Melanoma Susceptibility Gene. PLoS ONE 2014, 9, e100683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.-L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370. [Google Scholar] [CrossRef] [Green Version]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [CrossRef] [Green Version]
- Daly, M.B.; Pilarski, R.; Berry, M.; Buys, S.S.; Farmer, M.; Friedman, S.; Garber, J.E.; Kauff, N.D.; Khan, S.; Klein, C.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J. Natl. Compr. Cancer Netw. 2017, 15, 9–20. [Google Scholar] [CrossRef]
- Jerzak, K.J.; Mancuso, T.; Eisen, A. Ataxia–telangiectasia gene (ATM) mutation heterozygosity in breast cancer: A narrative review. Curr. Oncol. 2018, 25, e176–e180. [Google Scholar] [CrossRef] [Green Version]
- Moretta, J.; Berthet, P.; Bonadona, V.; Caron, O.; Cohen-Haguenauer, O.; Colas, C.; Corsini, C.; Cusin, V.; De Pauw, A.; Delnatte, C.; et al. The French Genetic and Cancer Consortium guidelines for multigene panel analysis in hereditary breast and ovarian cancer predisposition. Bull. Cancer (Paris) 2018, 105, 907–917. [Google Scholar] [CrossRef]
- Potjer, T.P.; Bollen, S.; Grimbergen, A.J.E.M.; Doorn, R.; Gruis, N.A.; Asperen, C.J.; Hes, F.J.; Stoep, N.; on behalf of the Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4melanoma families. Int. J. Cancer J. Int. Cancer 2019, 144, 2453–2464. [Google Scholar] [CrossRef] [Green Version]
- Casula, M.; Paliogiannis, P.; Ayala, F.; De Giorgi, V.; Stanganelli, I.; Mandalà, M.; Colombino, M.; Manca, A.; Sini, M.C.; Caracò, C.; et al. Germline and somatic mutations in patients with multiple primary melanomas: A next generation sequencing study. BMC Cancer 2019, 19, 772. [Google Scholar] [CrossRef]
- Teerlink, C.C.; Huff, C.; Stevens, J.; Yu, Y.; Holmen, S.L.; Silvis, M.R.; Trombetti, K.; Zhao, H.; Grossman, U.; Farnham, J.M.; et al. A Nonsynonymous Variant in the GOLM1 Gene in Cutaneous Malignant Melanoma. J. Natl. Cancer Inst. 2018, 48, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Aoude, L.; Heitzer, E.; Johansson, P.; Gartside, M.; Wadt, K.; Pritchard, A.; Palmer, J.M.; Symmons, J.; Gerdes, A.-M.; Montgomery, G.W.; et al. POLE mutations in families predisposed to cutaneous melanoma. Fam. Cancer 2015, 14, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, E.; Van Doorn, R.; Visser, M.; Teunisse, A.; Versluis, M.; Van Der Velden, P.; Hayward, N.K.; Jochemsen, A.; Gruis, N. NEK11 as a candidate high-penetrance melanoma susceptibility gene. J. Med. Genet. 2019, 57, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Artomov, M.; Stratigos, A.J.; Kim, I.; Kumar, R.; Lauss, M.; Reddy, B.Y.; Miao, B.; Robles-Espinoza, C.D.; Sankar, A.; Njauw, C.-N.; et al. Rare Variant, Gene-Based Association Study of Hereditary Melanoma Using Whole-Exome Sequencing. J. Natl. Cancer Inst. 2017, 109, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, J.E.; Cassidy, P.B.; Manga, P.; Boissy, R.E.; Goldgar, D.; Cannon-Albright, L.A.; Florell, S.R.; Leachman, S.A. Report of a novel OCA2 gene mutation and an investigation of OCA2 variants on melanoma risk in a familial melanoma pedigree. J. Dermatol. Sci. 2012, 69, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Nathan, V.; Johansson, P.; Palmer, J.M.; Howlie, M.; Hamilton, H.R.; Wadt, K.; Jönsson, G.; Brooks, K.M.; Pritchard, A.; Hayward, N.K. Germline variants in oculocutaneous albinism genes and predisposition to familial cutaneous melanoma. Pigment. Cell Melanoma Res. 2019, 32, 854–863. [Google Scholar] [CrossRef]
- Dalmasso, B.; Ghiorzo, P. Evolution of approaches to identify melanoma missing heritability. Expert Rev. Mol. Diagn. 2020, 1–9. [Google Scholar] [CrossRef]
- Bruno, W.; Andreotti, V.; Bisio, A.; Pastorino, L.; Fornarini, G.; Sciallero, S.; Bianchi-Scarrà, G.; Inga, A.; Ghiorzo, P. Functional analysis of a CDKN2A 5′UTR germline variant associated with pancreatic cancer development. PLoS ONE 2017, 12, e0189123. [Google Scholar] [CrossRef]
- Chang, X.; Wang, K. wANNOVAR: Annotating genetic variants for personal genomes via the web. J. Med. Genet. 2012, 49, 433–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]


{kind=link}
| Case Index (Age of Diagnosis) | Gene 1 | Description and Consequence | Type | ACMG Classification 2 | DbSNP 3 | gnomAD 4 | CADD 5 | SIFT 6 | Polyphen 7 | LRT 8 | Mutation Taster | Other Cancer in Family | Clin Var 9 | Co-Segregation 10 | Total No. of cm/um Cases in Family |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MPM (58) | ACD | c.866_867delCT; p.(Pro289ArgfsTer28) | frameshift | LP | rs777476880 | 2.44e-05 | - | - | - | - | - | BC, LC, PC | - | ND | 3 CM |
| CM (31) | BAP1 | c.327_328insGA; p.(Pro110AspfsTer4) | frameshift | P | - | - | - | - | - | - | - | 2 MES | - | ND | 1 CM |
| UM (45), RCC (40) | BAP1 | c.639dupT; p.(lIe214TyrfsTer29) | frameshift | P | - | - | - | - | - | - | - | 2 KC, 2 UM | Y | 3 UM | |
| CM (53), MES (55) | BAP1 | c.799_800delCA; p.(Gln267AlafsTer16) | frameshift | P | - | - | - | - | - | - | - | 3 CM, LC, 2 MES | Y | 4 cCM | |
| MPM (41, 43) | BAP1 | c.1337delA; p.(Asn446ThrfsTer125) | frameshift | P | - | - | - | - | - | - | - | UM, BC | ND | 1 UM, 3 CM | |
| CM (52) | BAP1 | c.1507T>C; p.(Phe503Leu) | missense | VUS | rs745959970 | 3.99e-06 | 23.6 | D | D | D | D | CM | ND | 2 CM | |
| AST (23) | BAP1 | c.1777dupC; p.(Gln593ProfsTer50) | frameshift | P | - | - | - | - | - | - | RCC | - | ND | 1 AST | |
| AST (35), AGM | BAP1 | c.1939G>T; p.(Glu647Ter) | non-sense | P | - | - | - | - | - | - | BC, BCC, UM | Y | 1 AST, 1 UM | ||
| CM (26) | POT1 | c.158C>T;p.(Thr53Ile) | missense | VUS | - | - | 21.3 | D | D | D | D | 3 CM, GLB, PC, HL | - | Y | 4 CM, 1 UM |
| MPM (66, 66) | POT1 | c.158C>T; p.(Thr53Ile) | missense | VUS | - | - | 21.3 | D | D | D | D | CM | - | Y | 3 CM |
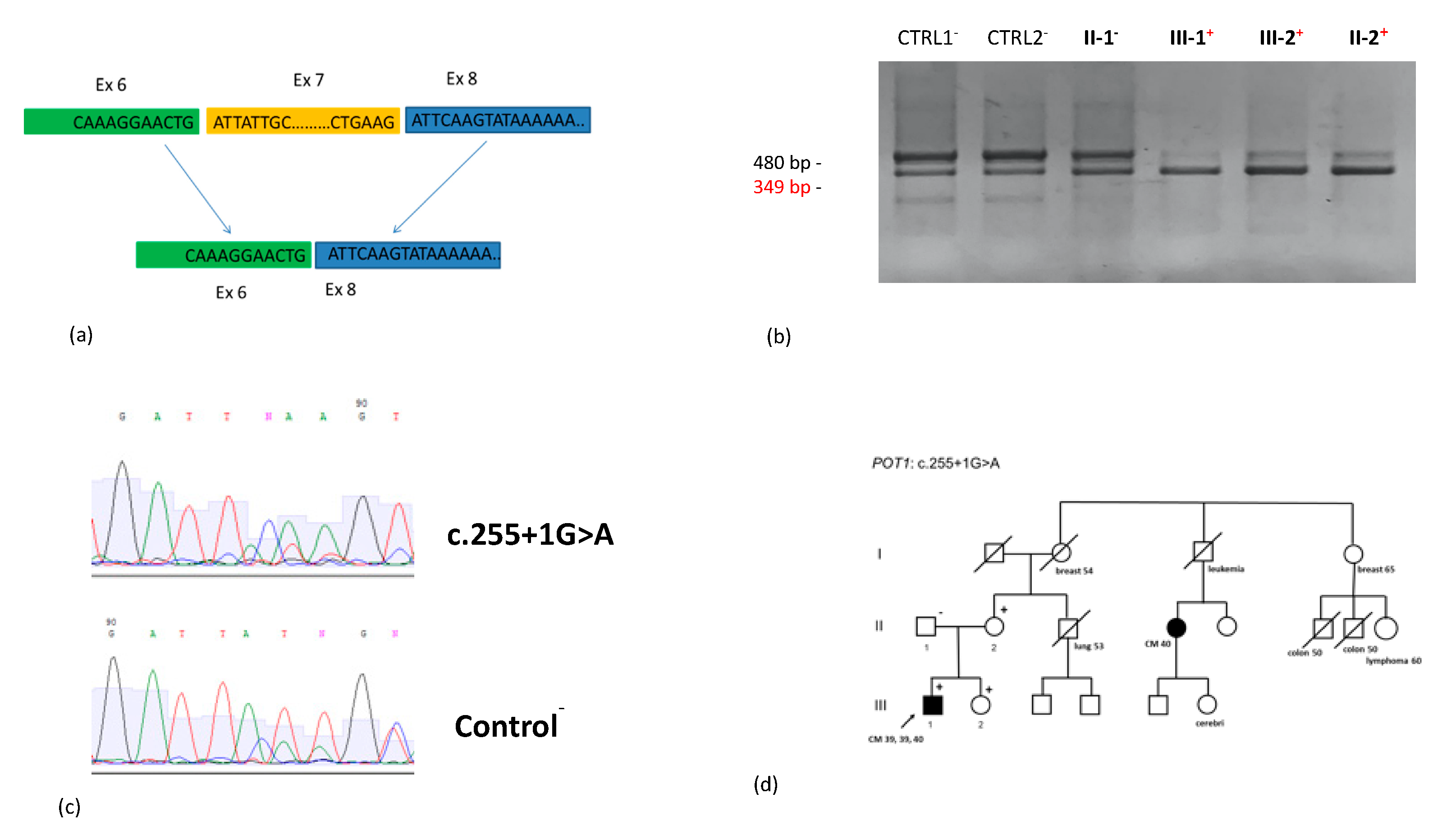
| MPM (39, 39, 40) | POT1 | c.255+1G>A | splicing | P | - | - | - | - | - | - | - | CM, BC, CC, LC, L | - | ND | 4 CM |
| CM (45), OC (23), MES (50) | POT1 | c.280C>A; p.(Gln94Lys) | missense | VUS | - | - | 22.8 | D | D | D | D | LC | - | ND | 1 CM |
| MPM (46, 52) | POT1 | c.314C>T; p.(Thr105Met) | missense | VUS | - | - | 28.8 | D | D | D | D | AST, BC, LC | - | ND | 2 CM |
| MPM (40), GL, MY | POT1 | c.809G>A; p.(Ser270Asn) | missense | VUS | rs587777477 | - | 23.2 | D | B | D | D | LC, S | RF | ND | 9 CM |
| MPM (40, 52) | POT1 | c.1400C>T;p.(Ser467Leu) | missense | VUS | rs1410842025 | - | 17.52 | D | PD | N | D | CM, GLB | - | ND | 3 CM |
| CM (17) | POT1 | c.1687-1G>A | splicing | P | rs587777473 | 4.28e-06 | - | - | - | - | - | CM, RCC | RF | Y | 2 CM |
| MPM (40, 51) | TERF2IP | c.258C>G; (p.Asp86Glu) | missense | VUS | rs752446617 | 6.14e-06 | 23.9 | T | B | D | D | BLC, BCC | - | ND | 2 CM |
| MPM (32) | MITF | c.952G>A; p.Glu318Lys | missense | LP | rs149617956 | 1.36e-03 | 27.9 | D | P | D | D | - | UNC | ND | 10 CM |
| CM (23) | MITF | c.952G>A; p.Glu318Lys | missense | LP | rs149617956 | 1.36e-03 | 27.9 | D | P | D | D | CM | UNC | Y | 2 CM |
| MPM (59) | MITF | c.952G>A; p.Glu318Lys | missense | LP | rs149617956 | 1.36e-03 | 27.9 | D | P | D | D | KD | UNC | ND | 3 CM |
| CM (39), TC (38) | MITF | c.952G>A; p.Glu318Lys | missense | LP | rs149617956 | 1.36e-03 | 27.9 | D | P | D | D | UNC | ND | 1 CM | |
| MPM (62, 62, 72) | MITF | c.952G>A; p.Glu318Lys | missense | LP | rs149617956 | 1.36e-03 | 27.9 | D | P | D | D | UNC | ND | 3 CM | |
| MPM (45, 46) | MITF | c.952G>A; p.Glu318Lys | missense | LP | rs149617956 | 1.36e-03 | 27.9 | D | P | D | D | BC | UNC | ND | 2 CM |
| MPM (52, 62, 65) | MITF | c.952G>A; p.Glu318Lys | missense | LP | rs149617956 | 1.36e-03 | 27.9 | D | P | D | D | CM | UNC | ND | 4 CM |
| Case Index (Age of Diagnosis) | Gene 1 | Description and Consequence | Type | ACMG Classification 2 | DbSNP 3 | gnomAD 4 | CADD 5 | SIFT 6 | Polyphen 7 | LRT 8 | Mutation Taster | Other Cancer in Family | Clin Var 9 | Co-Segregation 10 | Total No. of cm/um Cases in Family |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CM (24) | ATM | c.1516G>T:p.(Gly506Cys) | missense | VUS | rs587779816 | 29.0 | D | P | D | D | ON, PC | UNC | ND | 1 CM | |
| CM (48) | ATM | c.1595G>A:p.(Cys532Tyr) | missense | VUS | rs35963548 | 21.0 | D | D | D | D | 2 CM, BC | UNC | ND | 3 CM | |
| CM (47) | ATM | c.3275C>A;p.(Ser1092Ter) | non sense | P | - | - | - | - | - | D | D | PC, LC | - | ND | 1 CM |
| MPM (42, 42) | ATM | c.3576G>A;p.(Ser1135_Lys1195del58) | splicing | LP | rs587776551 | 1.63e-05 | - | - | - | - | - | CM | P | ND | 3 CM |
| CM (49) | ATM | c.3576G>A;p.(Ser1135_Lys1195del58) | splicing | LP | rs587776551 | 1.63e-05 | - | - | - | - | - | CM, UC | P | ND | 2 CM |
| MPM (40, 40) | ATM | c.3576G>A;p.(Ser1135_Lys1195del58) | splicing | LP | rs587776551 | 1.63e-05 | - | - | - | - | - | MPM, BCC, PC, PR | P | Y | 4 CM |
| MPM (35, 63) | ATM | c.3934A>G:p.(Arg1312Gly) | missense | VUS | rs864622137 | 23.3 | D | D | D | D | LC, BR, GC | UNC | ND | 2 CM | |
| CM (45) | ATM | c.4049C>T:p.(Thr1350Met) | missense | VUS | rs587781785 | 27.0 | T | D | D | D | CM | UNC | ND | 3 CM | |
| CM (50) | ATM | c.4306C>T:p.(His1436Tyr) | missense | VUS | rs544891616 | 17.09 | T | D | D | D | 2 KD | UNC | ND | 1 CM | |
| HL (35), MPM (45, 45, 46), BCC (49), PC (50) | ATM | c.4451delT:p.(Met1484ArgfsTer15) | frameshift | P | - | - | - | - | - | - | CM, CC, LC, PC | - | ND | 4 CM | |
| CM (47) | ATM | c.5750G>C:p.(Arg1917Thr) | missense | LP | rs377289524 | 1.22e-05 | 25.6 | T | D | D | D | CM, UM, BC, BLC, PR, CC, GC | UNC | Y | 2 CM, 1 UM |
| MPM (45, 46) | ATM | c.5750G>C:p.(Arg1917Thr) | missense | LP | rs377289524 | 1.22e-05 | 25.6 | T | D | D | D | PR | UNC | ND | 2 CM |
| 2 UM (41), CM (51) | ATM | c.5979_5983delTAAAG; p.(Ser1993ArgfsTer23) | frameshift | P | rs876660134 | 8.13e-06 | - | - | - | - | - | BC, GC, LX | P | ND | 1 CM, 2 UM |
| MPM (48, 48, 53), BCC (49) | ATM | c.8319_8323dupTGTCC; p.(Pro2775LeufsTer33) | frameshift | P | rs1060501552 | - | - | - | - | - | MPM, BCC, PC, PR | P | ND | 5 CM | |
| CM (43) | ATM | c.8557A>G:p.(Thr2853Ala) | missense | VUS | - | 27.7 | D | D | D | D | MES | UNC | ND | 1 CM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastorino, L.; Andreotti, V.; Dalmasso, B.; Vanni, I.; Ciccarese, G.; Mandalà, M.; Spadola, G.; Pizzichetta, M.A.; Ponti, G.; Tibiletti, M.G.; et al. Insights into Genetic Susceptibility to Melanoma by Gene Panel Testing: Potential Pathogenic Variants in ACD, ATM, BAP1, and POT1. Cancers 2020, 12, 1007. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12041007
Pastorino L, Andreotti V, Dalmasso B, Vanni I, Ciccarese G, Mandalà M, Spadola G, Pizzichetta MA, Ponti G, Tibiletti MG, et al. Insights into Genetic Susceptibility to Melanoma by Gene Panel Testing: Potential Pathogenic Variants in ACD, ATM, BAP1, and POT1. Cancers. 2020; 12(4):1007. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12041007
Chicago/Turabian StylePastorino, Lorenza, Virginia Andreotti, Bruna Dalmasso, Irene Vanni, Giulia Ciccarese, Mario Mandalà, Giuseppe Spadola, Maria Antonietta Pizzichetta, Giovanni Ponti, Maria Grazia Tibiletti, and et al. 2020. "Insights into Genetic Susceptibility to Melanoma by Gene Panel Testing: Potential Pathogenic Variants in ACD, ATM, BAP1, and POT1" Cancers 12, no. 4: 1007. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12041007