Establishment and Characterization of Humanized Mouse NPC-PDX Model for Testing Immunotherapy

 , , , and
, , , and 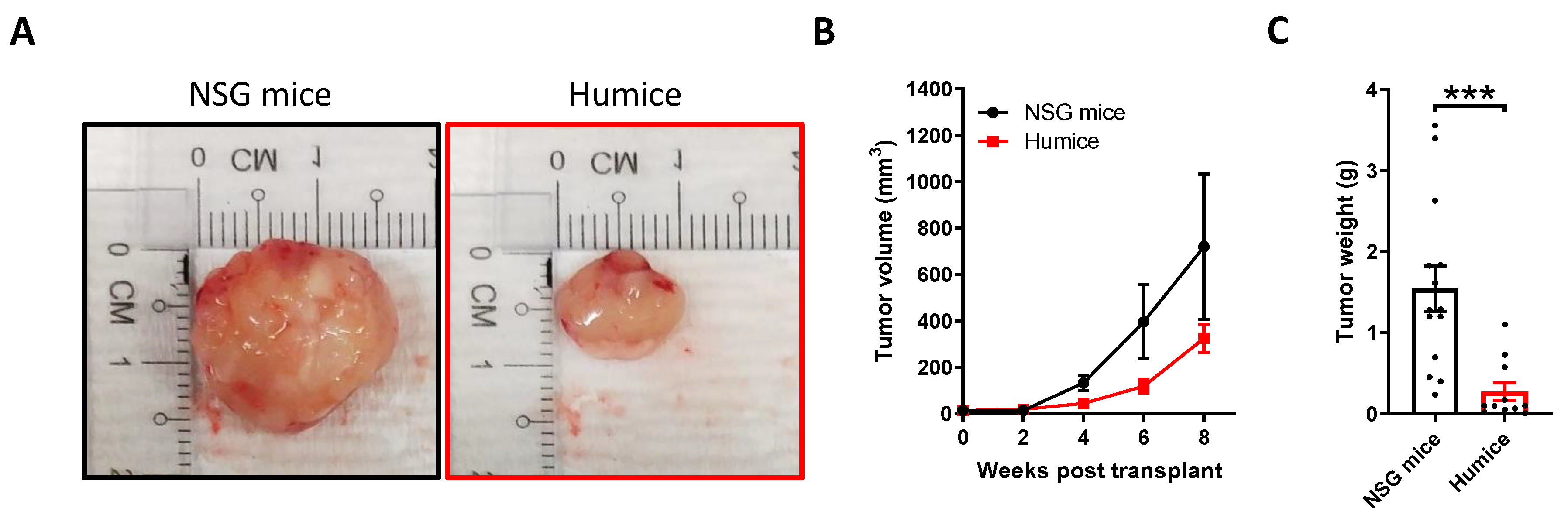{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
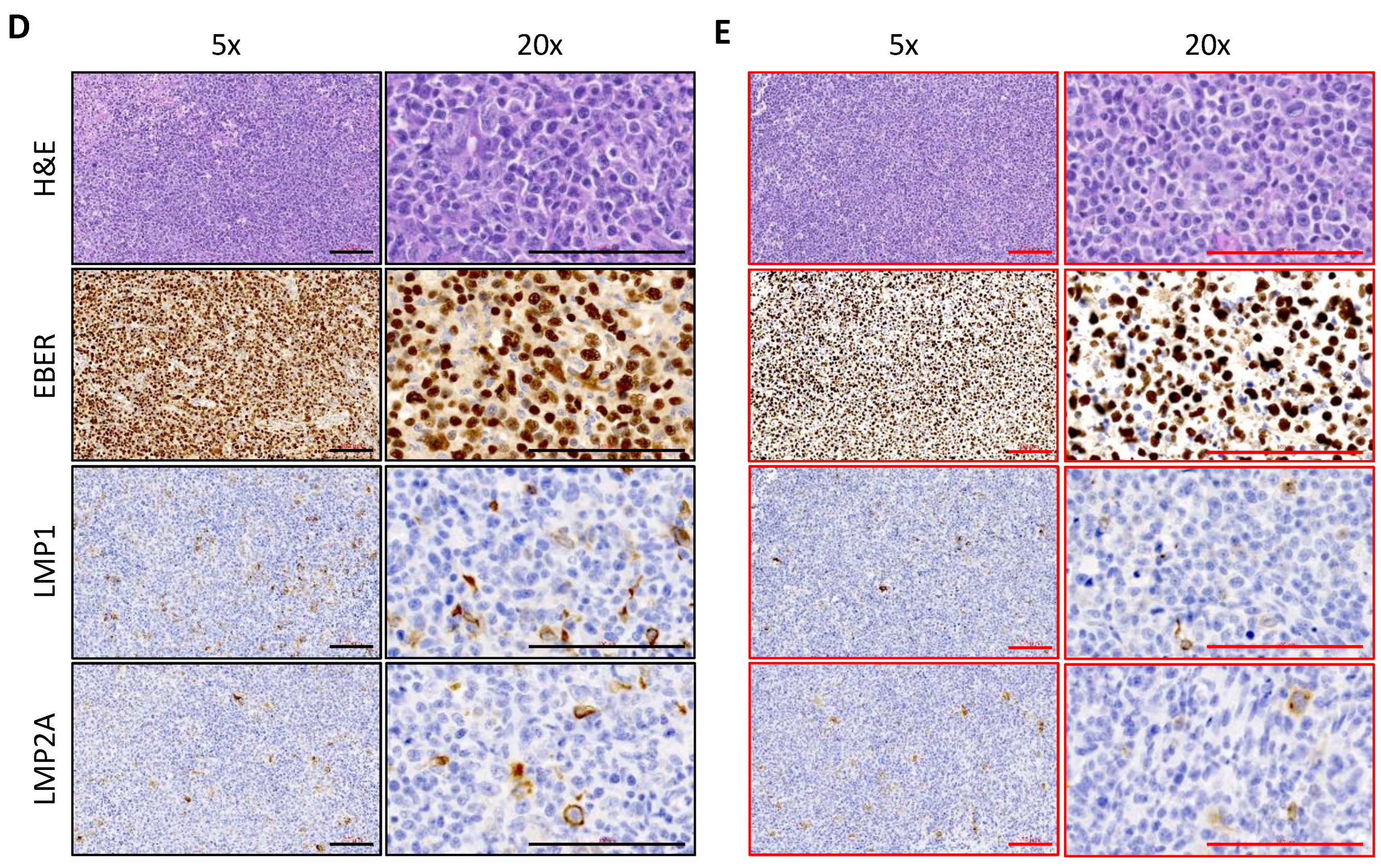
2.1. Establishment and Characterization of NPC-PDX in Humanized Mice
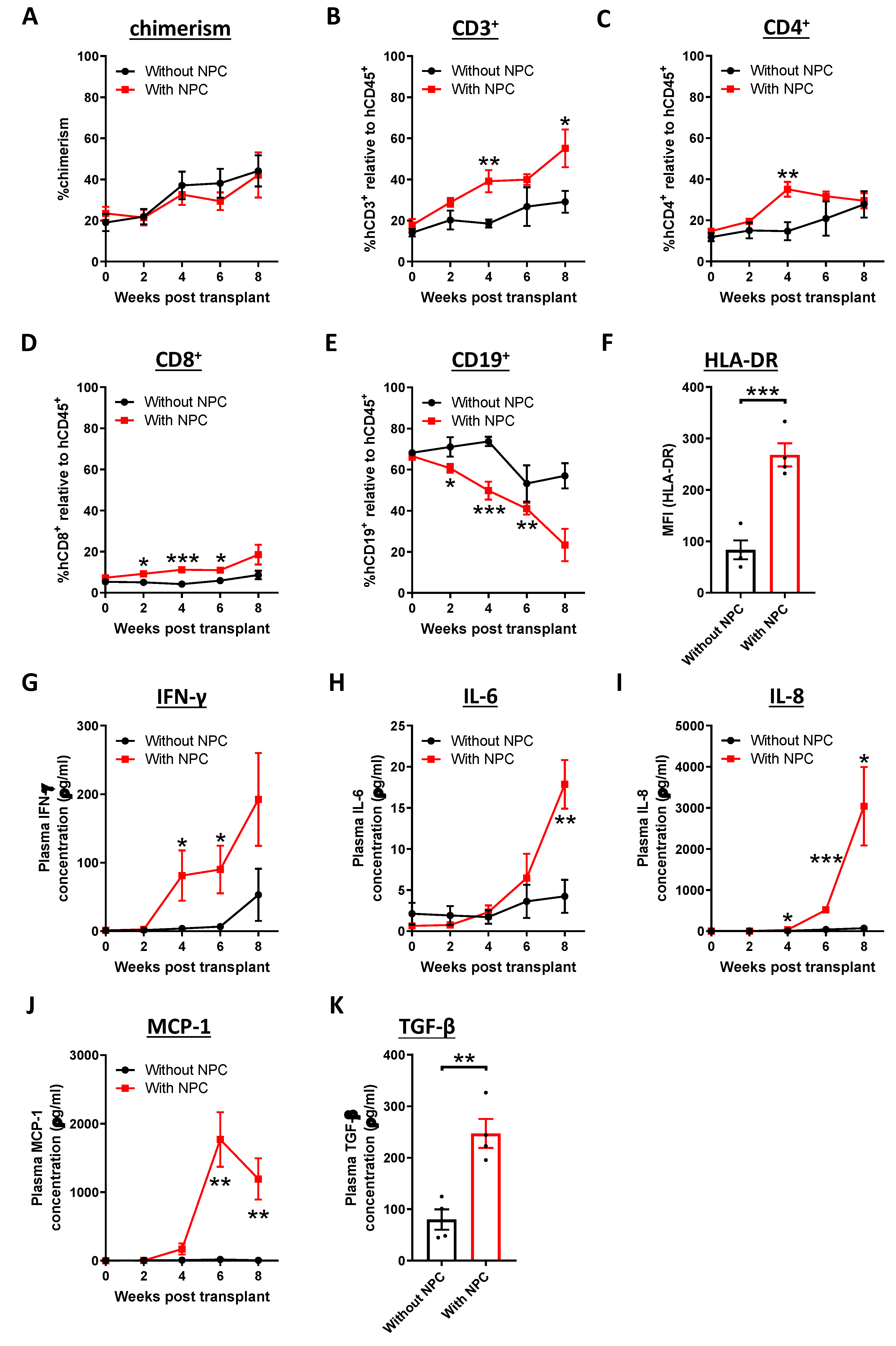
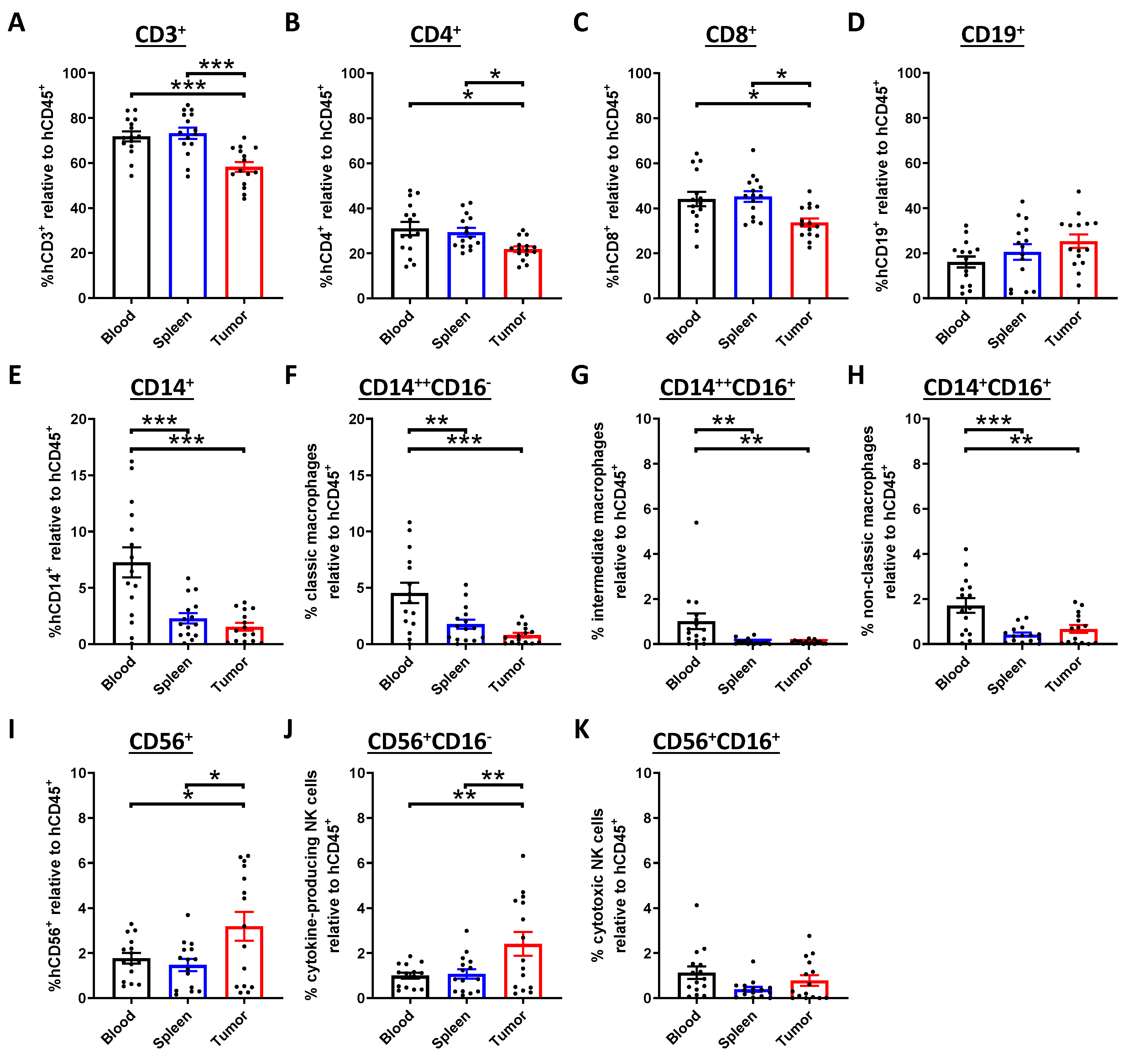
2.2. Activation of the Immune Response in NPC-transplanted Humanized Mice
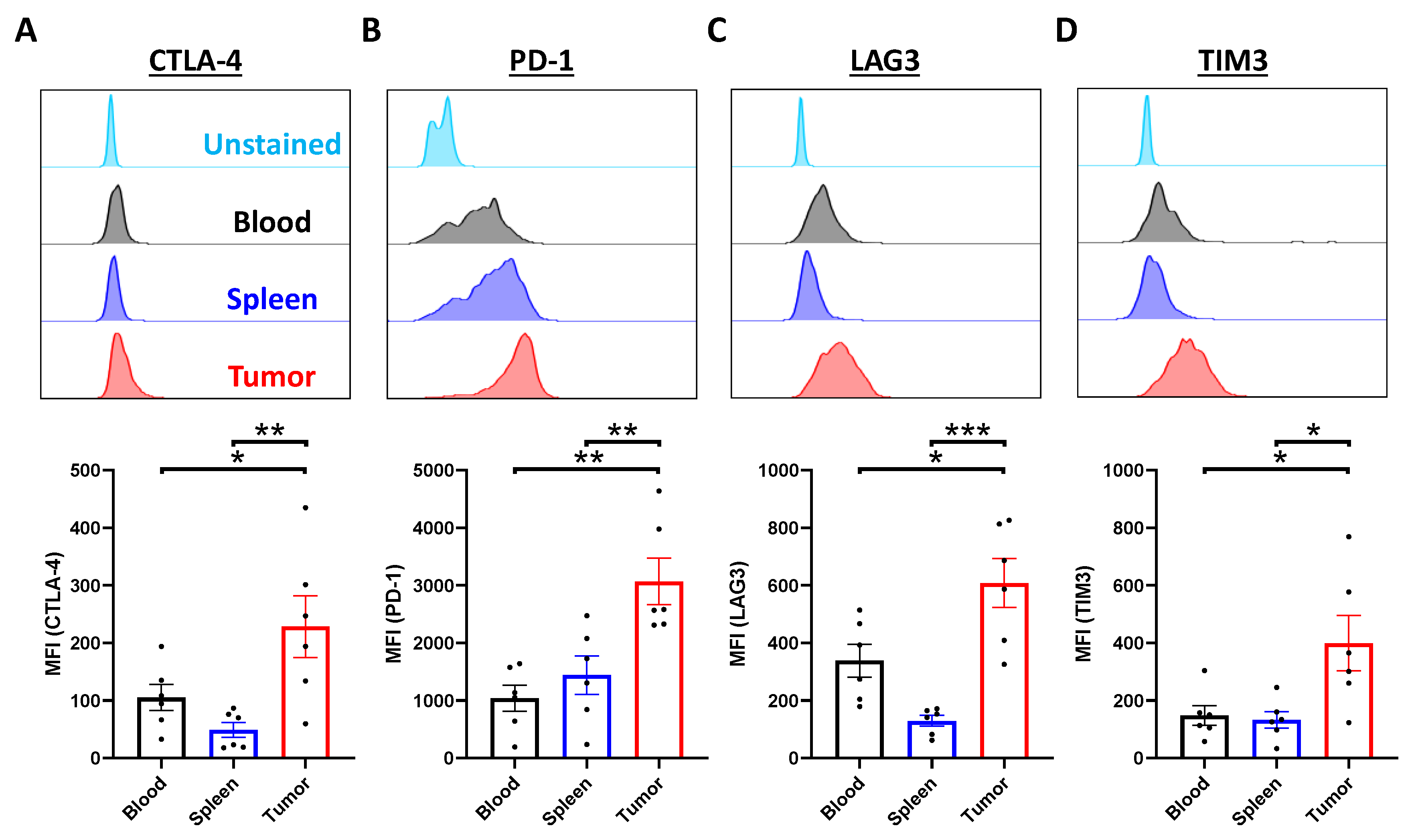
2.3. Tumor-Infiltrating T Cells Display Exhausted Phenotypes
2.4. Examination of Anti-Tumor Efficacy Using ICB in Humanized Mouse NPC-PDX Model
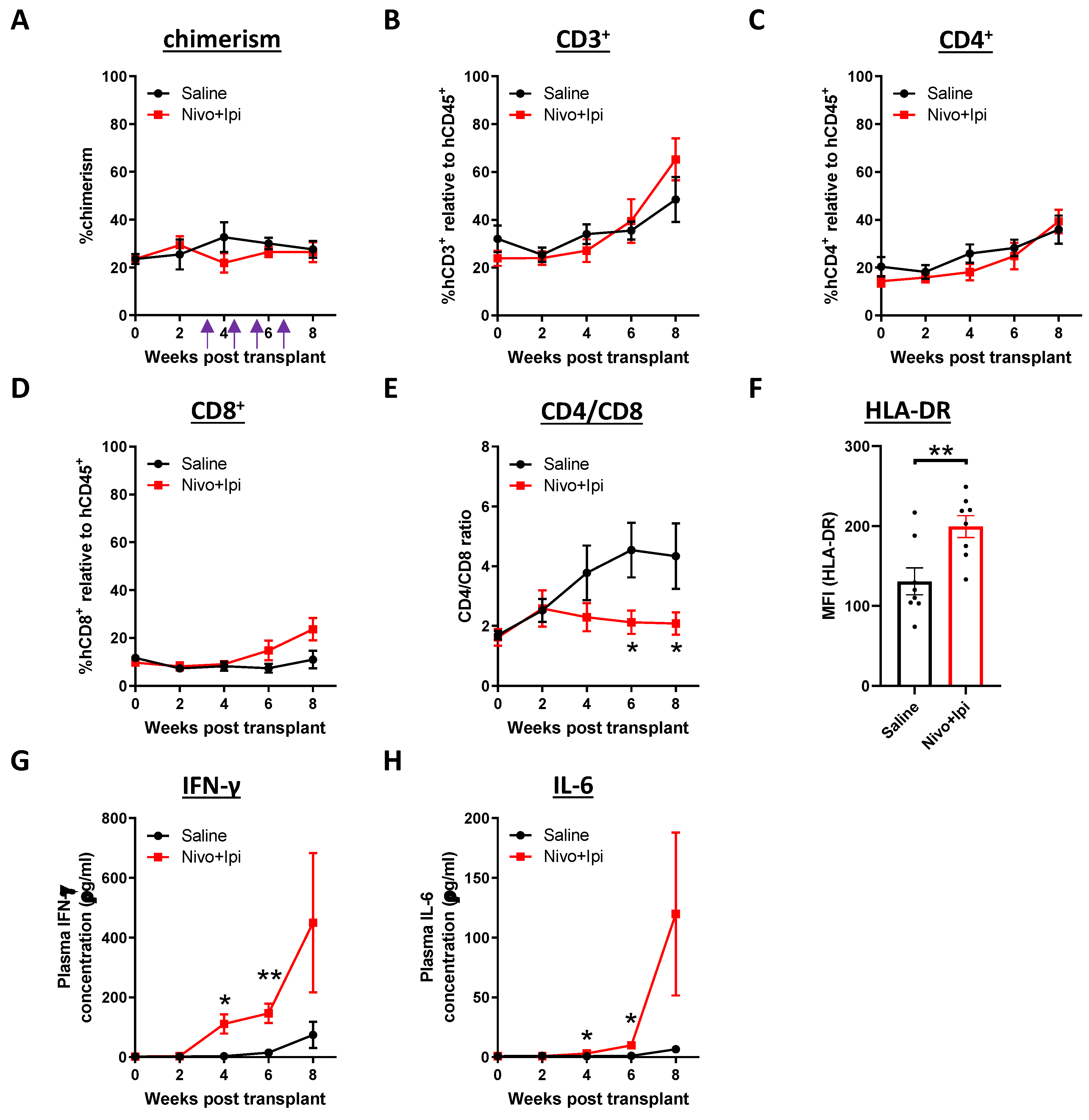
2.5. Modulation of Immune Cell and Cytokine Profile after Treatment
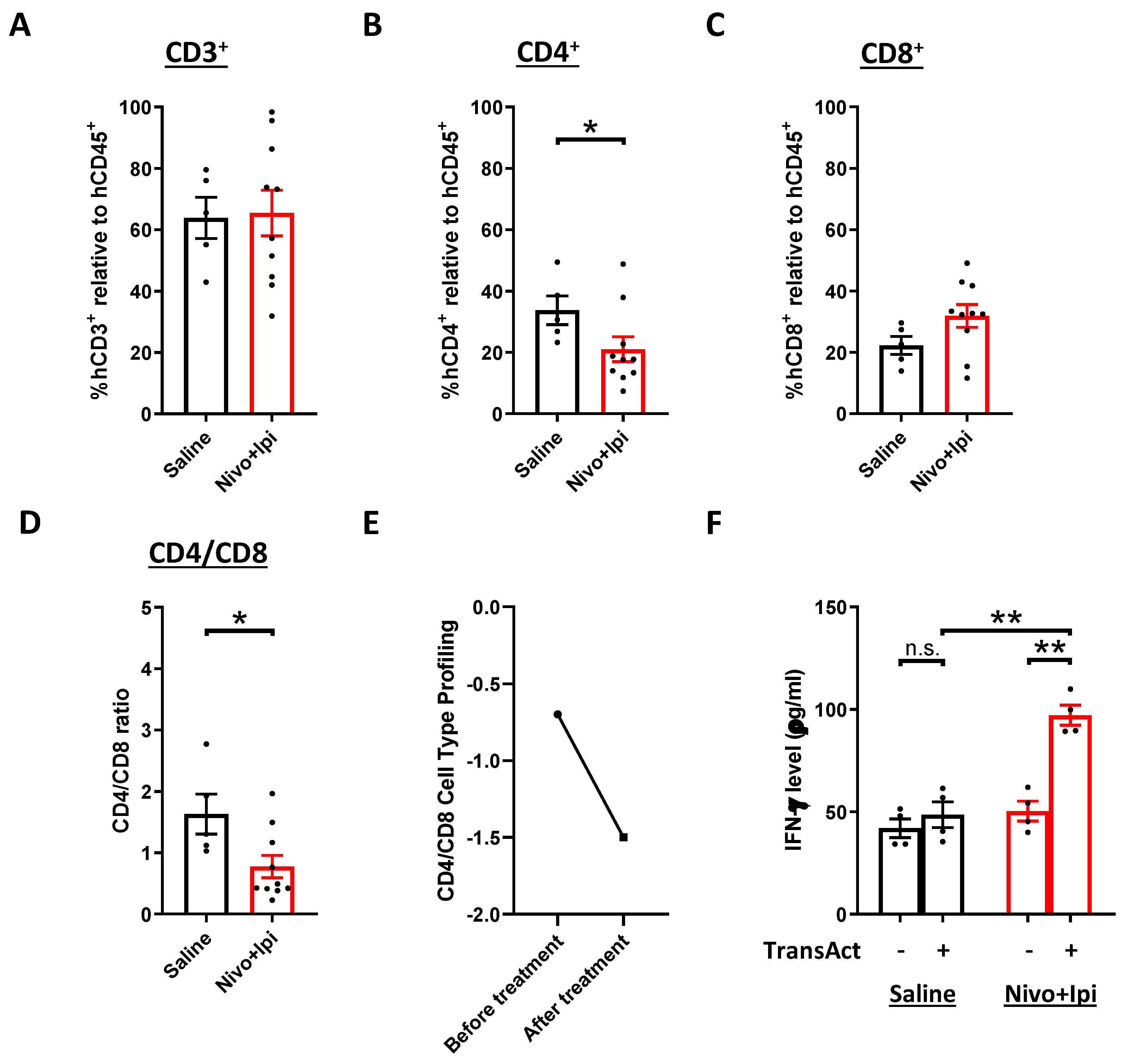
2.6. Presence of Re-Activated Tumor-Infiltrating T Cells after Treatment
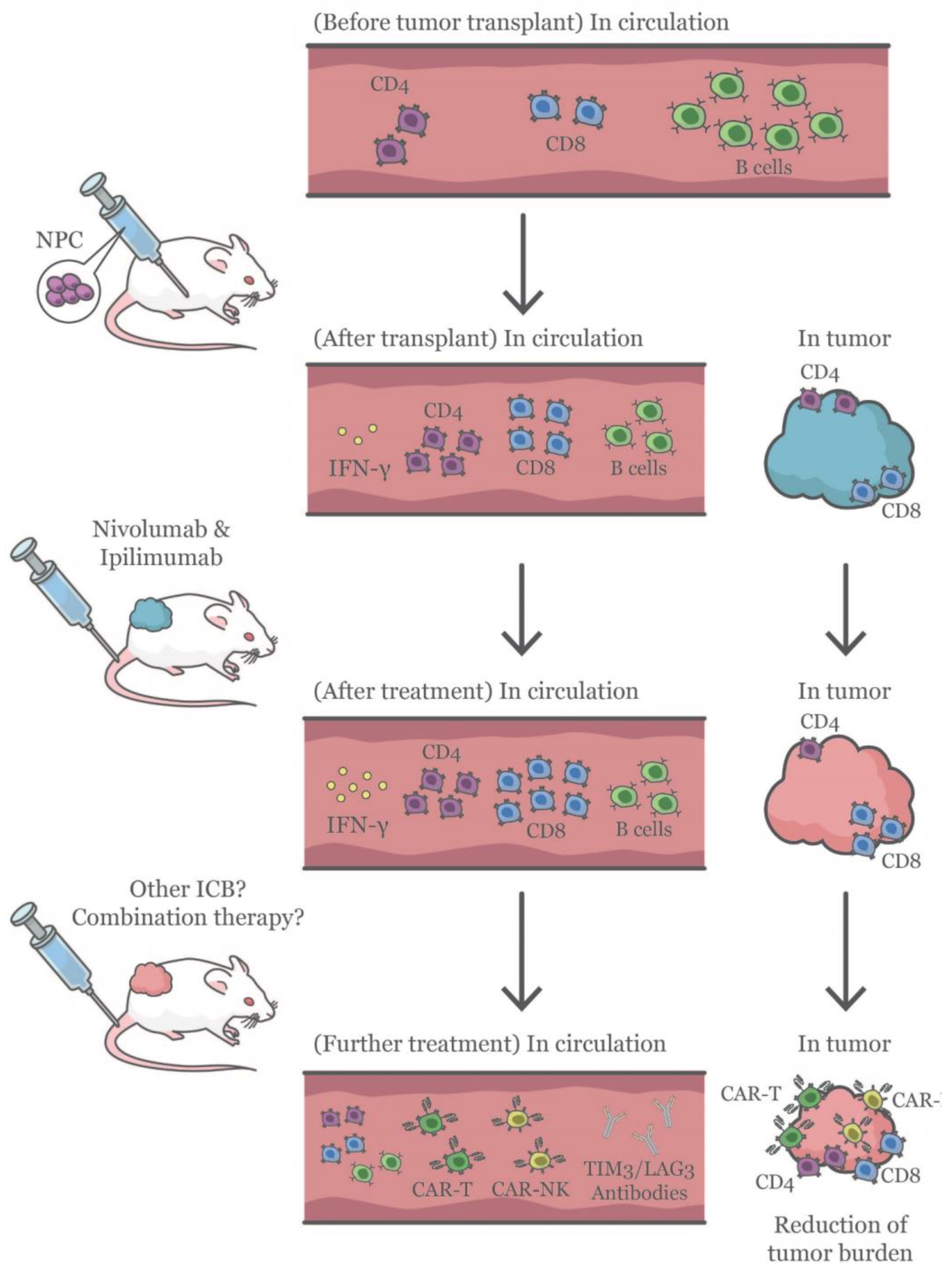
3. Discussion
4. Materials and Methods
4.1. Cord Blood Processing and Isolation of Human CD34+ HSC
4.2. Mice and Transplantation of CD34+ Cells
4.3. Transplantation of NPC-PDX and Drug Treatment
4.4. H&E, EBER ISH and IHC Staining
4.5. Isolation of Mononuclear Cells from Blood, Spleen and Tumor
4.6. Flow Cytometry
4.7. Cytokines and Chemokines Quantification
4.8. Nanostring Analysis
4.9. ELISA for Human TGF-β1 and IFN-γ
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein–Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.-M.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.P.; Chan, A.T.C.; Le, Q.T.; Blanchard, P.; Sun, Y.; Ma, J. Nasopharyngeal Carcinoma. Lancet 2019, 394, 64–80. [Google Scholar] [CrossRef]
- Ia, W.-H.; Qin, H.-D. Non-viral environmental risk factors for nasopharyngeal carcinoma: A systematic review. Semin. Cancer Boil. 2012, 22, 117–126. [Google Scholar]
- Jain, A.; Chia, W.K.; Toh, H.C. Immunotherapy for nasopharyngeal cancer—A review. Chin. Clin. Oncol. 2016, 5, 22. [Google Scholar] [CrossRef]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein–Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. B Boil. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Chin, Y.-M.; Mushiroda, T.; Takahashi, A.; Kubo, M.; Krishnan, G.; Yap, L.F.; Teo, S.-H.; Lim, P.V.-H.; Yap, Y.-Y.; Pua, K.-C.; et al. HLA-ASNPs and amino acid variants are associated with nasopharyngeal carcinoma in Malaysian Chinese. Int. J. Cancer 2014, 136, 678–687. [Google Scholar] [CrossRef]
- Jin, Y.; Shi, Y.X.; Cai, X.Y.; Xia, X.Y.; Cai, Y.C.; Cao, Y.; Zhang, W.D.; Hu, W.H.; Jiang, W.Q. Erratum To: Comparison of Five Cisplatin-Based Regimens Frequently Used as the First-Line Protocols in Metastatic Nasopharyngeal Carcinoma. J. Cancer Res. Clin. Oncol. 2015, 141, 767. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, L.; Hu, G.Q.; Zhang, N.; Zhu, X.D.; Yang, K.Y.; Jin, F.; Shi, M.; Chen, Y.P.; Hu, W.H.; et al. Gemcitabine and Cisplatin Induction Chemotherapy in Nasopharyngeal Carcinoma. N. Engl. J. Med. 2019, 381, 1124–1135. [Google Scholar] [CrossRef]
- Le, Q.-T.; Colevas, A.D.; O’Sullivan, B.; Lee, A.W.M.; Lee, N.; Ma, B.; Siu, L.L.; Waldron, J.; Lim, C.-M.; Riaz, N.; et al. Current Treatment Landscape of Nasopharyngeal Carcinoma and Potential Trials Evaluating the Value of Immunotherapy. J. Natl. Cancer Inst. 2019, 111, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, W.K.J.; Chan, J.Y.K. Recent advances in the management of nasopharyngeal carcinoma. F1000Research 2018, 7, 1829. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-S.; Lin, S.-Y.; Lee, P.-F.; Chung, H.-C.; Tsai, M.-S. Establishment and Characterization of a Tumor Cell Line from Human Nasopharyngeal Carcinoma Tissue. Cancer Res. 1989, 49, 6752–6757. [Google Scholar] [PubMed]
- Huang, D.P.; Ho, J.H.C.; Poon, Y.F.; Chew, E.C.; Saw, D.; Lui, M.; Li, C.L.; Mak, L.S.; Lai, S.H.; Lau, W.H. Establishment of a cell line (NPC/HK1) from a differentiated squamous carcinoma of the nasopharynx. Int. J. Cancer 1980, 26, 127–132. [Google Scholar] [CrossRef]
- Cheung, S.T.; Huang, D.P.; Hui, A.B.; Lo, K.W.; Ko, C.W.; Tsang, Y.S.; Wong, N.; Whitney, B.M.; Lee, J.C. Nasopharyngeal carcinoma cell line (C666-1) consistently harbouring Epstein-Barr virus. Int. J. Cancer 1999, 83, 121–126. [Google Scholar] [CrossRef]
- Sacks, P.G.; Parnes, S.M.; Gallick, G.E.; Mansouri, Z.; Lichtner, R.; Satya-Prakash, K.L.; Pathak, S.; Parsons, D.F. Establishment and characterization of two new squamous cell carcinoma cell lines derived from tumors of the head and neck. Cancer Res. 1988, 48, 2858–2866. [Google Scholar]
- Dittmer, D.P.; Hilscher, C.J.; Gulley, M.L.; Yang, E.V.; Chen, M.; Glaser, R. Multiple pathways for Epstein-Barr virus episome loss from nasopharyngeal carcinoma. Int. J. Cancer 2008, 123, 2105–2112. [Google Scholar] [CrossRef] [Green Version]
- Busson, P.; Ganem, G.; Flores, P.; Mugneret, F.; Clausse, B.; Caillou, B.; Braham, K.; Wakasugi, H.; Lipiński, M.; Tursz, T. Establishment and characterization of three transplantable EBV-containing nasopharyngeal carcinomas. Int. J. Cancer 1988, 42, 599–606. [Google Scholar] [CrossRef]
- Huang, D.P.; Ho, J.H.C.; Chan, W.K.; Lau, W.H.; Lui, M. Cytogenetics of undifferentiated nasopharyngeal carcinoma xenografts from southern chinese. Int. J. Cancer 1989, 43, 936–939. [Google Scholar] [CrossRef]
- Lin, W.; Yip, Y.L.; Jia, L.; Deng, W.; Zheng, H.; Dai, W.; Ko, J.M.Y.; Lo, K.W.; Chung, G.T.Y.; Yip, K.Y.; et al. Establishment and Characterization of New Tumor Xenografts and Cancer Cell Lines from Ebv-Positive Nasopharyngeal Carcinoma. Nat. Commun. 2018, 9, 4663. [Google Scholar] [CrossRef]
- Decker, W.K.; da Silva, R.F.; Sanabria, M.H.; Angelo, L.S.; Guimaraes, F.; Burt, B.M.; Kheradmand, F.; Paust, S. Cancer Immunotherapy: Historical Perspective of a Clinical Revolution and Emerging Preclinical Animal Models. Front Immunol. 2017, 8, 829. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Pitt, J.M.; Daillère, R.; Smyth, M.J.; Kroemer, G. Mouse models in oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Rodriguez, I.; Schalper, K.A.; Onate, C.; Azpilikueta, A.; Rodriguez-Ruiz, M.E.; Morales-Kastresana, A.; Labiano, S.; Perez-Gracia, J.L.; Martin-Algarra, S.; et al. Nivolumab and Urelumab Enhance Antitumor Activity of Human T Lymphocytes Engrafted in Rag2-/-Il2rgammanull Immunodeficient Mice. Cancer Res. 2015, 75, 3466–3478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Shuen, T.W.H.; Toh, T.B.; Chan, X.Y.; Liu, M.; Tan, S.Y.; Fan, Y.; Yang, H.; Lyer, S.G.; Bonney, G.K.; et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut 2018, 67, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Tramcourt, L.; Leloup, C.; Molens, J.-P.; Leccia, M.; Charles, J.; Plumas, J. Imiquimod Inhibits Melanoma Development by Promoting pDC Cytotoxic Functions and Impeding Tumor Vascularization. J. Investig. Dermatol. 2014, 134, 2551–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, M.D.; Harui, A. Human tumor infiltrating lymphocytes cooperatively regulate prostate tumor growth in a humanized mouse model. J. Immunother. Cancer 2015, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.-K.; Moniz, R.J.; Xu, Z.; Sun, J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Human anti-CAIX antibodies mediate immune cell inhibition of renal cell carcinoma in vitro and in a humanized mouse model in vivo. Mol. Cancer 2015, 14, 119. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-L.; Li, J.; Mo, H.-Y.; Qiu, F.; Zheng, L.; Qian, C.-N.; Zeng, Y.-X. Different subsets of tumor infiltrating lymphocytes correlate with NPC progression in different ways. Mol. Cancer 2010, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Lakhdar, M.; Ben Aribia, M.H.; Maalej, M.; Ladgham, A. Selective homing of phenotypically lytic cells within nasopharyngeal carcinoma biopsies: Numerous CD8- and CD16-positive cells in the tumor. Int. J. Cancer 1991, 48, 57–61. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.; Lee, S.-H.; Ejadi, S.; Even, C.; Cohen, R.B.; Le Tourneau, C.; Mehnert, J.M.; Algazi, A.; Van Brummelen, E.M.; Saraf, S.; et al. Safety and Antitumor Activity of Pembrolizumab in Patients With Programmed Death-Ligand 1–Positive Nasopharyngeal Carcinoma: Results of the KEYNOTE-028 Study. J. Clin. Oncol. 2017, 35, 4050–4056. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.B.Y.; Lim, W.T.; Goh, B.C.; Hui, E.P.; Lo, K.W.; Pettinger, A.; Foster, N.R.; Riess, J.W.; Agulnik, M.; Chang, A.Y.C.; et al. Antitumor Activity of Nivolumab in Recurrent and Metastatic Nasopharyngeal Carcinoma: An International, Multicenter Study of the Mayo Clinic Phase 2 Consortium (Nci-9742). J. Clin. Oncol. 2018, 36, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Yang, Y.; Ma, Y.; Hong, S.; Lin, L.; He, X.; Xiong, J.; Li, P.; Zhao, H.; Huang, Y.; et al. Camrelizumab (SHR-1210) alone or in combination with gemcitabine plus cisplatin for nasopharyngeal carcinoma: Results from two single-arm, phase 1 trials. Lancet Oncol. 2018, 19, 1338–1350. [Google Scholar] [CrossRef]
- Hsu, C.-L.; Lui, K.-W.; Chi, L.-M.; Kuo, Y.-C.; Chao, Y.-K.; Yeh, C.-N.; Lee, L.-Y.; Huang, Y.; Lin, T.-L.; Huang, M.-Y.; et al. Integrated genomic analyses in PDX model reveal a cyclin-dependent kinase inhibitor Palbociclib as a novel candidate drug for nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 233. [Google Scholar] [CrossRef]
- Cho, S.-Y.; Kang, W.; Han, J.Y.; Min, S.; Kang, J.; Lee, A.; Kwon, J.Y.; Lee, C.; Park, H. An Integrative Approach to Precision Cancer Medicine Using Patient-Derived Xenografts. Mol. Cells 2016, 39, 77–86. [Google Scholar]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interf. Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Dighe, A.S.; Richards, E.; Old, L.J.; Schreiber, R.D. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity 1994, 1, 447–456. [Google Scholar] [CrossRef]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Schreiber, R.D. Demonstration of an interferon γ-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The Il-8/Il-8r Axis: A Double Agent in Tumor Immune Resistance. Vaccines (Basel) 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massague, J. Tgfbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Menezes, J.; Prasad, U.; Ahmad, A. Elevated serum levels of transforming growth factor beta1 in Epstein-Barr virus-associated nasopharyngeal carcinoma patients. Int. J. Cancer 1999, 84, 396–399. [Google Scholar] [CrossRef]
- Larbcharoensub, N.; Mahaprom, K.; Jiarpinitnun, C.; Trachu, N.; Tubthong, N.; Pattaranutaporn, P.; Sirachainan, E.; Ngaimphaiboon, N. Characterization of PD-L1 and PD-1 Expression and CD8+ Tumor-infiltrating Lymphocyte in Epstein-Barr Virus-associated Nasopharyngeal Carcinoma. Am. J. Clin. Oncol. 2018, 41, 1204–1210. [Google Scholar] [CrossRef]
- Wang, Y.-Q.; Chen, Y.-P.; Zhang, Y.; Jiang, W.; Liu, N.; Yun, J.-P.; Sun, Y.; He, Q.-M.; Tang, X.-R.; Wen, X.; et al. Prognostic significance of tumor-infiltrating lymphocytes in nondisseminated nasopharyngeal carcinoma: A large-scale cohort study. Int. J. Cancer 2018, 142, 2558–2566. [Google Scholar] [CrossRef]
- Zhu, Q.; Cai, M.-Y.; Chen, C.-L.; Hu, H.; Lin, H.-X.; Li, M.; Weng, D.-S.; Zhao, J.-J.; Guo, L.; Xia, J. Tumor cells PD-L1 expression as a favorable prognosis factor in nasopharyngeal carcinoma patients with pre-existing intratumor-infiltrating lymphocytes. OncoImmunology 2017, 6, e1312240. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- He, Q.-F.; Xu, Y.; Li, J.; Huang, Z.-M.; Li, X.-H.; Wang, X. CD8+ T-cell exhaustion in cancer: Mechanisms and new area for cancer immunotherapy. Briefings Funct. Genom. 2018, 18, 99–106. [Google Scholar] [CrossRef]
- Callahan, M.K.; Wolchok, J.D. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J. Leukoc. Boil. 2013, 94, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.-Y.; Guo, S.-S.; Zhang, Y.; Lu, J.-B.; Chen, Q.-Y.; Tang, L.-Q.; Zhang, L.; Liu, L.-T.; Zhang, L.; Mai, H.-Q. Tumor CTLA-4 overexpression predicts poor survival in patients with nasopharyngeal carcinoma. Oncotarget 2016, 7, 13060–13068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, D.T.W.; Wang, H.M.; Li, S.H.; Ngan, R.; Dechaphunkul, A.; Zhang, L.; Yen, C.J.; Chan, P.C.; Chakrabandhu, S.; Ma, B.; et al. Phase Ii Study of Spartalizumab (Pdr001) Vs Chemotherapy (Ct) in Patients with Recurrent/Metastatic Nasopharyngeal Cancer (Npc). Cancer Res. 2019, 79, 615. [Google Scholar]
- Andersson, P.; Ostheimer, C. Editorial: Combinatorial Approaches to Enhance Anti-tumor Immunity: Focus on Immune Checkpoint Blockade Therapy. Front. Immunol. 2019, 10, 2083. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.S.M.; Her, Z.; Chen, Q. Humanized Mice as Unique Tools for Human-Specific Studies. Arch. Immunol. Ther. Exp. 2018, 66, 245–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traggiai, E.; Chicha, L.; Mazzucchelli, L.; Bronz, L.; Piffaretti, J.-C.; Lanzavecchia, A.; Manz, M.G. Development of a Human Adaptive Immune System in Cord Blood Cell-Transplanted Mice. Science 2004, 304, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, F.; Yasukawa, M.; Lyons, B.; Yoshida, S.; Miyamoto, T.; Yoshimoto, G.; Watanabe, T.; Akashi, K.; Shultz, L.D.; Harada, M. Development of Functional Human Blood and Immune Systems in Nod/Scid/Il2 Receptor {Gamma} Chain(Null) Mice. Blood 2005, 106, 1565–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, E.; Xu, H.; Wang, L.; Kryczek, I.; Wu, K.; Hu, Y.; Wang, G.; Zou, W. Bone marrow and the control of immunity. Cell. Mol. Immunol. 2011, 9, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef]
- Shultz, L.D.; Saito, Y.; Najima, Y.; Tanaka, S.; Ochi, T.; Tomizawa, M.; Doi, T.; Sone, A.; Suzuki, N.; Fujiwara, H.; et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc. Natl. Acad. Sci. USA 2010, 107, 13022–13027. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Khoury, M.; Chen, J. Expression of human cytokines dramatically improves reconstitution of specific human-blood lineage cells in humanized mice. Proc. Natl. Acad. Sci. USA 2009, 106, 21783–21788. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; He, F.; Kwang, J.; Chan, J.K.Y.; Chen, J. GM-CSF and IL-4 stimulate antibody responses in humanized mice by promoting T, B, and dendritic cell maturation. J. Immunol. 2012, 189, 5223–5229. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Keck, J.; Burzenski, L.; Jangalwe, S.; Vaidya, S.; Greiner, D.L.; Brehm, M.A. Humanized mouse models of immunological diseases and precision medicine. Mamm. Genome 2019, 30, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Malaney, P.; Nicosia, S.V.; Davé, V. One mouse, one patient paradigm: New avatars of personalized cancer therapy. Cancer Lett. 2013, 344, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Rochère, P.; Guil-Luna, S.; Decaudin, D.; Azar, G.; Sidhu, S.S.; Piaggio, E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol. 2018, 39, 748–763. [Google Scholar] [CrossRef]
- Taoka, K.; Arai, S.; Kataoka, K.; Hosoi, M.; Miyauchi, M.; Yamazaki, S.; Honda, A.; Aixinjueluo, W.; Kobayashi, T.; Kumano, K.; et al. Using patient-derived iPSCs to develop humanized mouse models for chronic myelomonocytic leukemia and therapeutic drug identification, including liposomal clodronate. Sci. Rep. 2018, 8, 15855. [Google Scholar] [CrossRef]
- Keng, C.T.; Sze, C.W.; Zheng, D.; Zheng, Z.; Yong, K.S.; Tan, S.Q.; Ong, J.J.; Tan, S.Y.; Loh, E.; Upadya, M.H.; et al. Characterisation of Liver Pathogenesis, Human Immune Responses and Drug Testing in a Humanised Mouse Model of Hcv Infection. Gut 2016, 65, 1744–1753. [Google Scholar] [CrossRef] [Green Version]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef]
- Wee, Y.T.F.; Alkaff, S.M.F.; Lim, J.C.T.; Loh, J.J.H.; Hilmy, M.H.; Ong, C.; Nei, W.L.; Jain, A.; Lim, A.; Takano, A.; et al. An integrated automated multispectral imaging technique that simultaneously detects and quantitates viral RNA and immune cell protein markers in fixed sections from Epstein-Barr virus-related tumours. Ann. Diagn. Pathol. 2018, 37, 12–19. [Google Scholar] [CrossRef]
- Ke, Y.; Liu, W.N.; Her, Z.; Liu, M.; Tan, S.Y.; Tan, Y.W.; Chan, X.Y.; Fan, Y.; Huang, E.K.; Chen, H.; et al. Enterovirus A71 Infection Activates Human Immune Responses and Induces Pathological Changes in Humanized Mice. J. Virol. 2019, 93, e01066-18. [Google Scholar] [CrossRef] [Green Version]
- Cesano, A. Ncounter((R)) Pancancer Immune Profiling Panel (Nanostring Technologies, Inc., Seattle, Wa). J. Immunother Cancer 2015, 3, 42. [Google Scholar] [CrossRef] [Green Version]
- Danaher, P.; Warren, S.; Dennis, L.; D’Amico, L.; White, A.; Disis, M.L.; Geller, M.A.; Odunsi, K.; Beechem, J.; Fling, S.P. Gene expression markers of Tumor Infiltrating Leukocytes. J. Immunother. Cancer 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.N.; Fong, S.Y.; Tan, W.W.S.; Tan, S.Y.; Liu, M.; Cheng, J.Y.; Lim, S.; Suteja, L.; Huang, E.K.; Chan, J.K.Y.; et al. Establishment and Characterization of Humanized Mouse NPC-PDX Model for Testing Immunotherapy. Cancers 2020, 12, 1025. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12041025
Liu WN, Fong SY, Tan WWS, Tan SY, Liu M, Cheng JY, Lim S, Suteja L, Huang EK, Chan JKY, et al. Establishment and Characterization of Humanized Mouse NPC-PDX Model for Testing Immunotherapy. Cancers. 2020; 12(4):1025. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12041025
Chicago/Turabian StyleLiu, Wai Nam, Shin Yie Fong, Wilson Wei Sheng Tan, Sue Yee Tan, Min Liu, Jia Ying Cheng, Sherlly Lim, Lisda Suteja, Edwin Kunxiang Huang, Jerry Kok Yen Chan, and et al. 2020. "Establishment and Characterization of Humanized Mouse NPC-PDX Model for Testing Immunotherapy" Cancers 12, no. 4: 1025. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12041025