A Germline Mutation in the POT1 Gene Is a Candidate for Familial Non-Medullary Thyroid Cancer

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
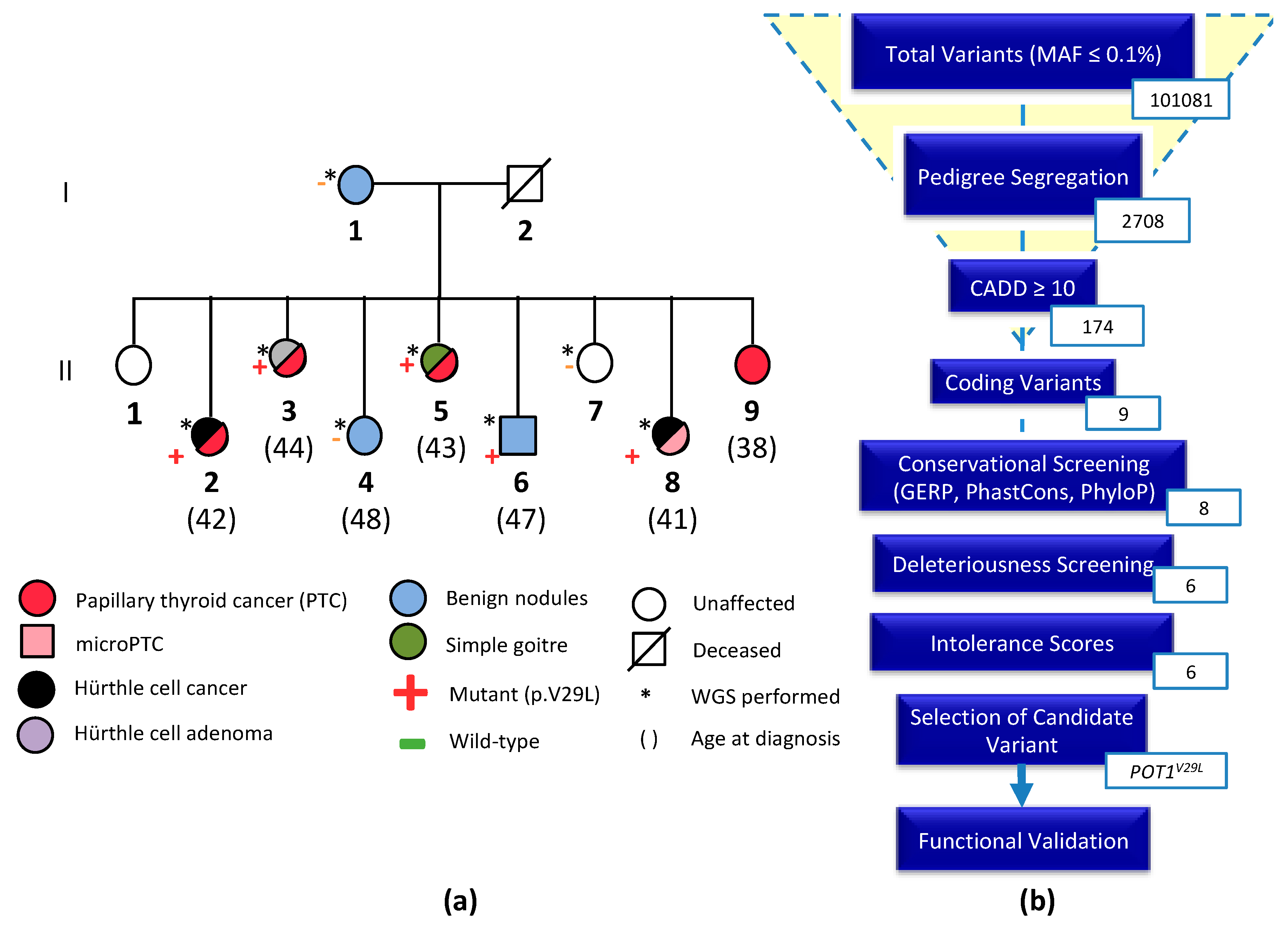
2.1. Clinical Characteristics of the NMTC Family
2.2. Whole-Genome Sequencing and Variant Prioritization
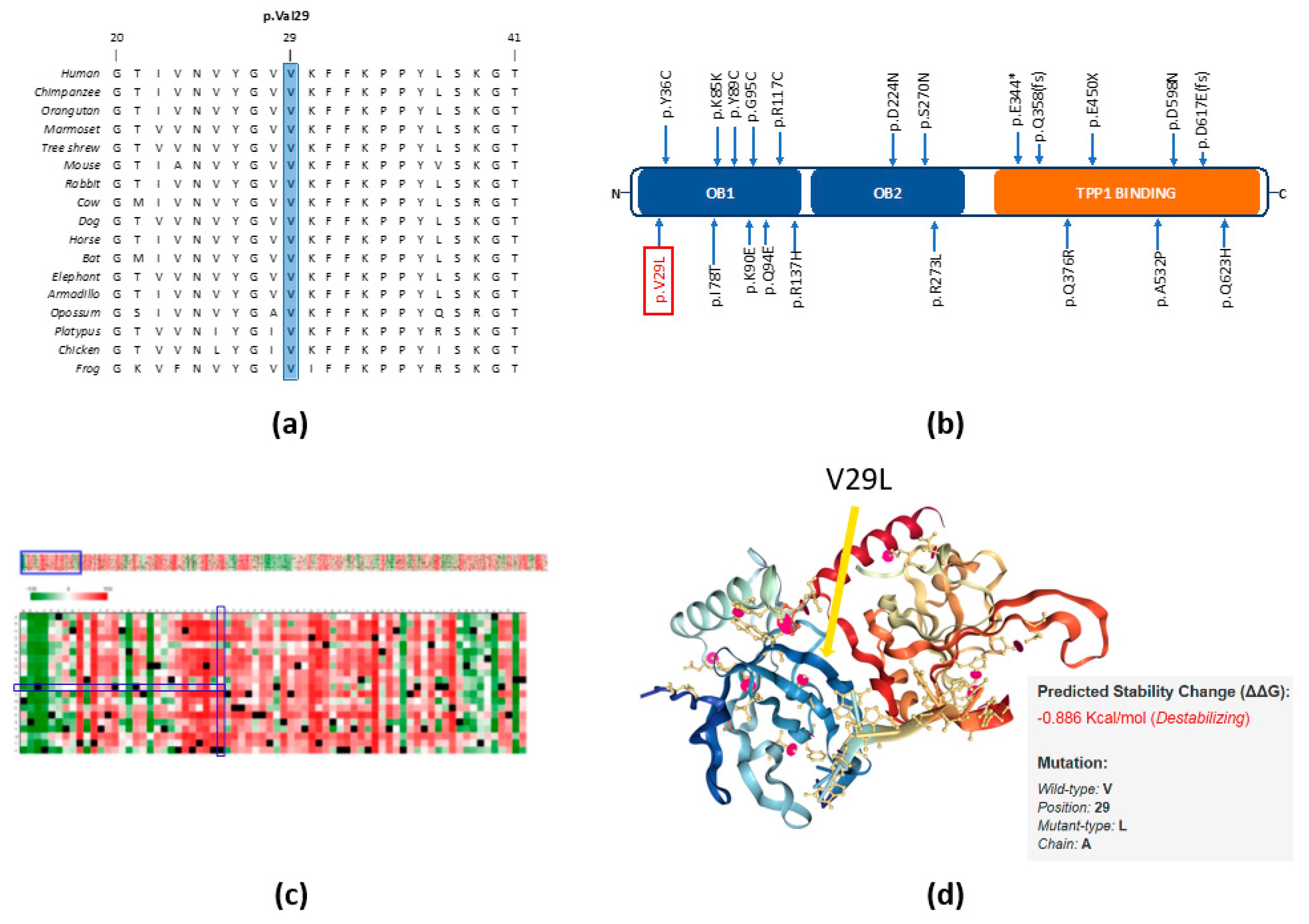
2.3. In Silico Studies Predict the Importance of the p.V29L Mutation to POT1 Protein Function
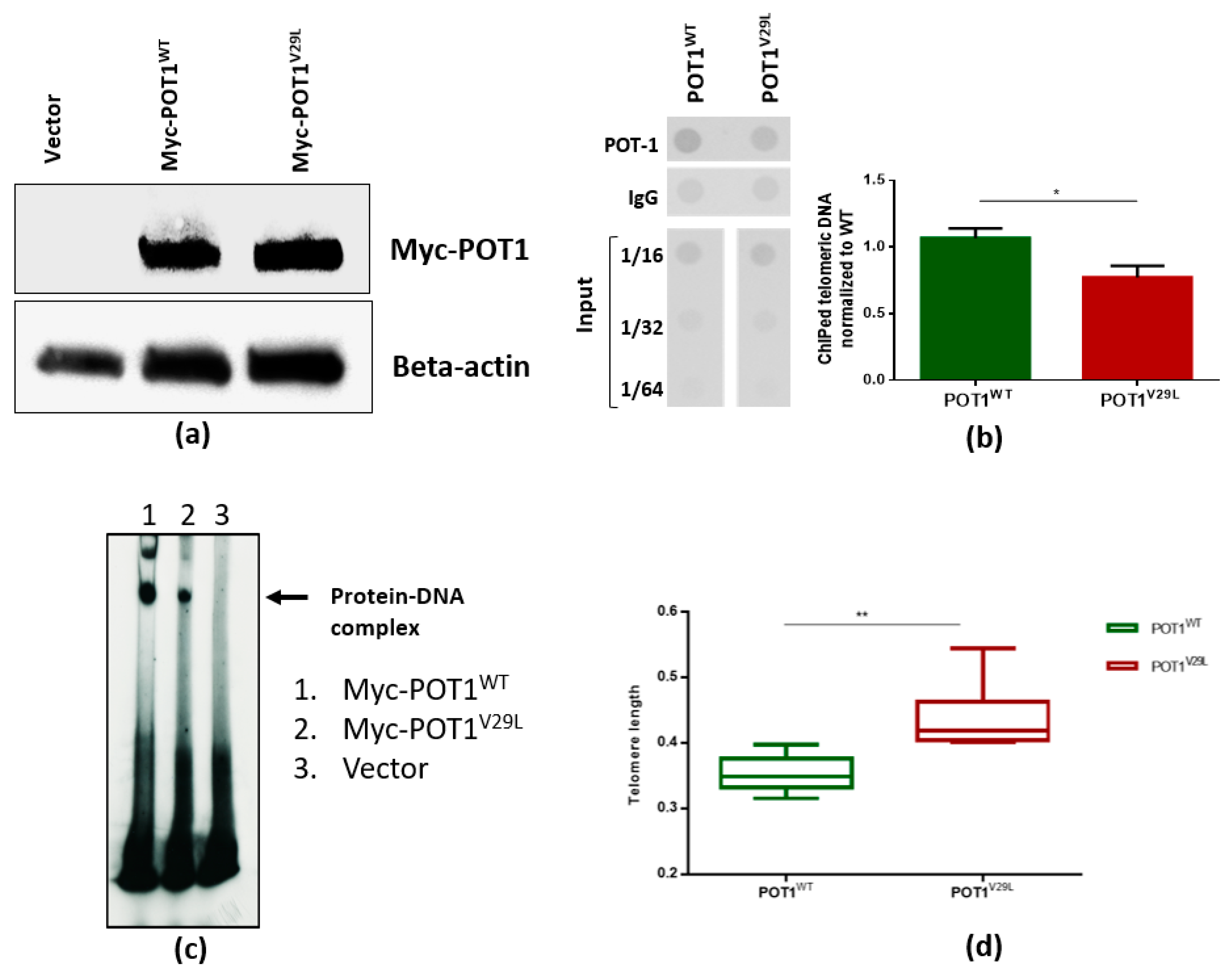
2.4. The V29L Mutation Aggravates DNA-Dependent Functions of POT1
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Whole-Genome Sequencing
4.3. Data Analysis and Variant Prioritization
4.4. Candidate Variant Selection and Validation
4.5. Further In Silico Studies
4.6. Protein Alignment and Structural Modeling
4.7. Cell Culture
4.8. Construction of Expression Plasmids, Transfection, and Selection of Stable POT1 Clones
4.9. Measurement of Relative Telomere Length (TL)
4.10. Western Blot
4.11. Chromatin Immunoprecipitation (ChIP) Assay and Telomere Dot Blots
4.12. Electrophoretic Mobility Shift Assay (EMSA)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Preprint
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hincza, K.; Kowalik, A.; Kowalska, A. Current Knowledge of Germline Genetic Risk Factors for the Development of Non-Medullary Thyroid Cancer. Genes 2019, 10, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiling Yang, S.; Ngeow, J. Familial non-medullary thyroid cancer: Unraveling the genetic maze. Endocr. Relat. Cancer 2016, 23, R577–R595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallah, M.; Pukkala, E.; Tryggvadottir, L.; Olsen, J.H.; Tretli, S.; Sundquist, K.; Hemminki, K. Risk of thyroid cancer in first-degree relatives of patients with non-medullary thyroid cancer by histology type and age at diagnosis: A joint study from five Nordic countries. J. Med. Genet. 2013, 50, 373–382. [Google Scholar] [CrossRef]
- El Lakis, M.; Giannakou, A.; Nockel, P.J.; Wiseman, D.; Gara, S.K.; Patel, D.; Sater, Z.A.; Kushchayeva, Y.Y.; Klubo-Gwiezdzinska, J.; Nilubol, N.; et al. Do patients with familial nonmedullary thyroid cancer present with more aggressive disease? Implications for initial surgical treatment. Surgery 2019, 165, 50–57. [Google Scholar] [CrossRef]
- Capezzone, M.; Cantara, S.; Marchisotta, S.; Filetti, S.; De Santi, M.M.; Rossi, B.; Ronga, G.; Durante, C.; Pacini, F. Short telomeres, telomerase reverse transcriptase gene amplification, and increased telomerase activity in the blood of familial papillary thyroid cancer patients. J. Clin. Endocrinol. Metab. 2008, 93, 3950–3957. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Kumar, A.; Giangiobbe, S.; Bonora, E.; Hemminki, K.; Forsti, A.; Bandapalli, O.R. Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways. Biomolecules 2019, 9, 605. [Google Scholar] [CrossRef] [Green Version]
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- Robles-Espinoza, C.D.; Harland, M.; Ramsay, A.J.; Aoude, L.G.; Quesada, V.; Ding, Z.; Pooley, K.A.; Pritchard, A.L.; Tiffen, J.C.; Petljak, M.; et al. POT1 loss-of-function variants predispose to familial melanoma. Nat. Genet. 2014, 46, 478–481. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Yang, X.R.; Ballew, B.; Rotunno, M.; Calista, D.; Fargnoli, M.C.; Ghiorzo, P.; Bressac-de Paillerets, B.; Nagore, E.; Avril, M.F.; et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat. Genet. 2014, 46, 482–486. [Google Scholar] [CrossRef]
- Potrony, M.; Puig-Butille, J.A.; Ribera-Sola, M.; Iyer, V.; Robles-Espinoza, C.D.; Aguilera, P.; Carrera, C.; Malvehy, J.; Badenas, C.; Landi, M.T.; et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br. J. Dermatol. 2019, 181, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Robles-Espinoza, C.D.; Rodriguez, D.; Rudat, S.S.; Puig, S.; Potrony, M.; Wong, C.C.; Hewinson, J.; Aguilera, P.; Puig-Butille, J.A.; et al. Association of the POT1 Germline Missense Variant p.I78T with Familial Melanoma. JAMA Dermatol. 2019, 155, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, M.N.; Armstrong, G.N.; Gramatges, M.M.; Bertuch, A.A.; Jhangiani, S.N.; Doddapaneni, H.; Lewis, L.; Tombrello, J.; Tsavachidis, S.; Liu, Y.; et al. Germline mutations in shelterin complex genes are associated with familial glioma. J. Natl.Cancer Inst. 2015, 107, 384. [Google Scholar] [CrossRef] [PubMed]
- Calvete, O.; Martinez, P.; Garcia-Pavia, P.; Benitez-Buelga, C.; Paumard-Hernández, B.; Fernandez, V.; Dominguez, F.; Salas, C.; Romero-Laorden, N.; Garcia-Donas, J.; et al. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li–Fraumeni-like families. Nat. Commun. 2015, 6, 8383. [Google Scholar] [CrossRef] [Green Version]
- Chubb, D.; Broderick, P.; Dobbins, S.E.; Frampton, M.; Kinnersley, B.; Penegar, S.; Price, A.; Ma, Y.P.; Sherborne, A.L.; Palles, C.; et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat. Commun. 2016, 7, 11883. [Google Scholar] [CrossRef]
- Speedy, H.E.; Kinnersley, B.; Chubb, D.; Broderick, P.; Law, P.J.; Litchfield, K.; Jayne, S.; Dyer, M.J.S.; Dearden, C.; Follows, G.A.; et al. Germ line mutations in shelterin complex genes are associated with familial chronic lymphocytic leukemia. Blood 2016, 128, 2319–2326. [Google Scholar] [CrossRef] [Green Version]
- McMaster, M.L.; Sun, C.; Landi, M.T.; Savage, S.A.; Rotunno, M.; Yang, X.R.; Jones, K.; Vogt, A.; Hutchinson, A.; Zhu, B.; et al. Germline mutations in Protection of Telomeres 1 in two families with Hodgkin lymphoma. Br. J. Haematol. 2018, 181, 372–377. [Google Scholar] [CrossRef] [Green Version]
- Lei, M.; Podell, E.R.; Baumann, P.; Cech, T.R. DNA self-recognition in the structure of Pot1 bound to telomeric single-stranded DNA. Nature 2003, 426, 198–203. [Google Scholar] [CrossRef]
- Gu, P.; Wang, Y.; Bisht, K.K.; Wu, L.; Kukova, L.; Smith, E.M.; Xiao, Y.; Bailey, S.M.; Lei, M.; Nandakumar, J.; et al. Pot1 OB-fold mutations unleash telomere instability to initiate tumorigenesis. Oncogene 2017, 36, 1939–1951. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, A.J.; Quesada, V.; Foronda, M.; Conde, L.; Martinez-Trillos, A.; Villamor, N.; Rodriguez, D.; Kwarciak, A.; Garabaya, C.; Gallardo, M.; et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat. Genet. 2013, 45, 526–530. [Google Scholar] [CrossRef]
- Cantara, S.; Pisu, M.; Frau, D.V.; Caria, P.; Dettori, T.; Capezzone, M.; Capuano, S.; Vanni, R.; Pacini, F. Telomere Abnormalities and Chromosome Fragility in Patients Affected by Familial Papillary Thyroid Cancer. J. Clin. Endocrinol. Metab. 2012, 97, E1327–E1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvete, O.; Garcia-Pavia, P.; Dominguez, F.; Bougeard, G.; Kunze, K.; Braeuninger, A.; Teule, A.; Lasa, A.; Ramon, Y.C.T.; Llort, G.; et al. The wide spectrum of POT1 gene variants correlates with multiple cancer types. Eur. J. Hum. Genet. 2017, 25, 1278–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Bian, B.; Gesuwan, K.; Gulati, N.; Zhang, L.; Nilubol, N.; Kebebew, E. Telomere length is shorter in affected members of families with familial nonmedullary thyroid cancer. Thyroid 2013, 23, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; An, C.; Zheng, H.; Lei, T.; Zhang, N.; Zheng, Y.; Yang, M. Leukocyte Telomere Length and Risk of Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2019, 104, 2712–2718. [Google Scholar] [CrossRef]
- McNally, E.J.; Luncsford, P.J.; Armanios, M. Long telomeres and cancer risk: The price of cellular immortality. J. Clin. Investig. 2019, 130, 3474–3481. [Google Scholar] [CrossRef] [Green Version]
- Richard, M.A.; Lupo, P.J.; Morton, L.M.; Yasui, Y.A.; Sapkota, Y.A.; Arnold, M.A.; Aubert, G.; Neglia, J.P.; Turcotte, L.M.; Leisenring, W.M.; et al. Genetic variation in POT1 and risk of thyroid subsequent malignant neoplasm: A report from the Childhood Cancer Survivor Study. PLoS ONE 2020, 15, e0228887. [Google Scholar] [CrossRef] [Green Version]
- Orois, A.; Badenas, C.; Reverter, J.L.; Lopez, V.; Potrony, M.; Mora, M.; Halperin, I.; Oriola, J. Lack of Mutations in POT1 Gene in Selected Families with Familial Non-Medullary Thyroid Cancer. Horm. Cancer 2020. [Google Scholar] [CrossRef]
- Jegerlehner, S.; Bulliard, J.L.; Aujesky, D.; Rodondi, N.; Germann, S.; Konzelmann, I.; Chiolero, A.; Group, N.W. Overdiagnosis and overtreatment of thyroid cancer: A population-based temporal trend study. PLoS ONE 2017, 12, e0179387. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.F.; Consortium, W.G.S.; Wilkie, A.O.M.; McVean, G.; Lunter, G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Exome Aggregation Consortium; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smigielski, E.M. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 2000, 28, 352–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bandapalli, O.R.; Paramasivam, N.; Giangiobbe, S.; Diquigiovanni, C.; Bonora, E.; Eils, R.; Schlesner, M.; Hemminki, K.; Forsti, A. Familial Cancer Variant Prioritization Pipeline version 2 (FCVPPv2) applied to a papillary thyroid cancer family. Sci. Rep. 2018, 8, 11635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Program, N.C.S.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, M.; Bromberg, Y.; Rost, B. Better prediction of functional effects for sequence variants. BMC Genom. 2015, 16, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yachdav, G.; Kloppmann, E.; Kajan, L.; Hecht, M.; Goldberg, T.; Hamp, T.; Honigschmid, P.; Schafferhans, A.; Roos, M.; Bernhofer, M.; et al. PredictProtein--an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014, 42, W337–W343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics (Oxford, England) 2014, 30, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Loayza, D.; De Lange, T. POT1 as a terminal transducer of TRF1 telomere length control. Nature 2003, 423, 1013–1018. [Google Scholar] [CrossRef]
- Hosen, I.; Rachakonda, P.S.; Heidenreich, B.; Sitaram, R.T.; Ljungberg, B.; Roos, G.; Hemminki, K.; Kumar, R. TERT promoter mutations in clear cell renal cell carcinoma. Int. J. Cancer 2015, 136, 2448–2452. [Google Scholar] [CrossRef]
- Liu, F.; Feng, X.; Ma, W. Analysis of Telomere Proteins by Chromatin Immunoprecipitation (ChIP). Methods Mol. Biol. 2017, 1587, 205–214. [Google Scholar] [CrossRef]
- Wilson, T.L.; Hattangady, N.; Lerario, A.M.; Williams, C.; Koeppe, E.; Quinonez, S.; Osborne, J.; Cha, K.B.; Else, T. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam. Cancer 2017, 16, 561–566. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, A.; Miao, B.; Skopelitou, D.; Kumar, V.; Kumar, A.; Paramasivam, N.; Bonora, E.; Hemminki, K.; Försti, A.; Bandapalli, O.R. A Germline Mutation in the POT1 Gene Is a Candidate for Familial Non-Medullary Thyroid Cancer. Cancers 2020, 12, 1441. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061441
Srivastava A, Miao B, Skopelitou D, Kumar V, Kumar A, Paramasivam N, Bonora E, Hemminki K, Försti A, Bandapalli OR. A Germline Mutation in the POT1 Gene Is a Candidate for Familial Non-Medullary Thyroid Cancer. Cancers. 2020; 12(6):1441. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061441
Chicago/Turabian StyleSrivastava, Aayushi, Beiping Miao, Diamanto Skopelitou, Varun Kumar, Abhishek Kumar, Nagarajan Paramasivam, Elena Bonora, Kari Hemminki, Asta Försti, and Obul Reddy Bandapalli. 2020. "A Germline Mutation in the POT1 Gene Is a Candidate for Familial Non-Medullary Thyroid Cancer" Cancers 12, no. 6: 1441. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061441