Non-Small-Cell Lung Cancer Signaling Pathways, Metabolism, and PD-1/PD-L1 Antibodies

 , ,
, , 
Abstract
:1. Introduction
2. Anti-PD-1 and Anti-PD-L1 Antibodies and Driver Alterations in NSCLC
3. Anti-PD-1 and Anti-PD-L1 Antibodies and Endocytosis
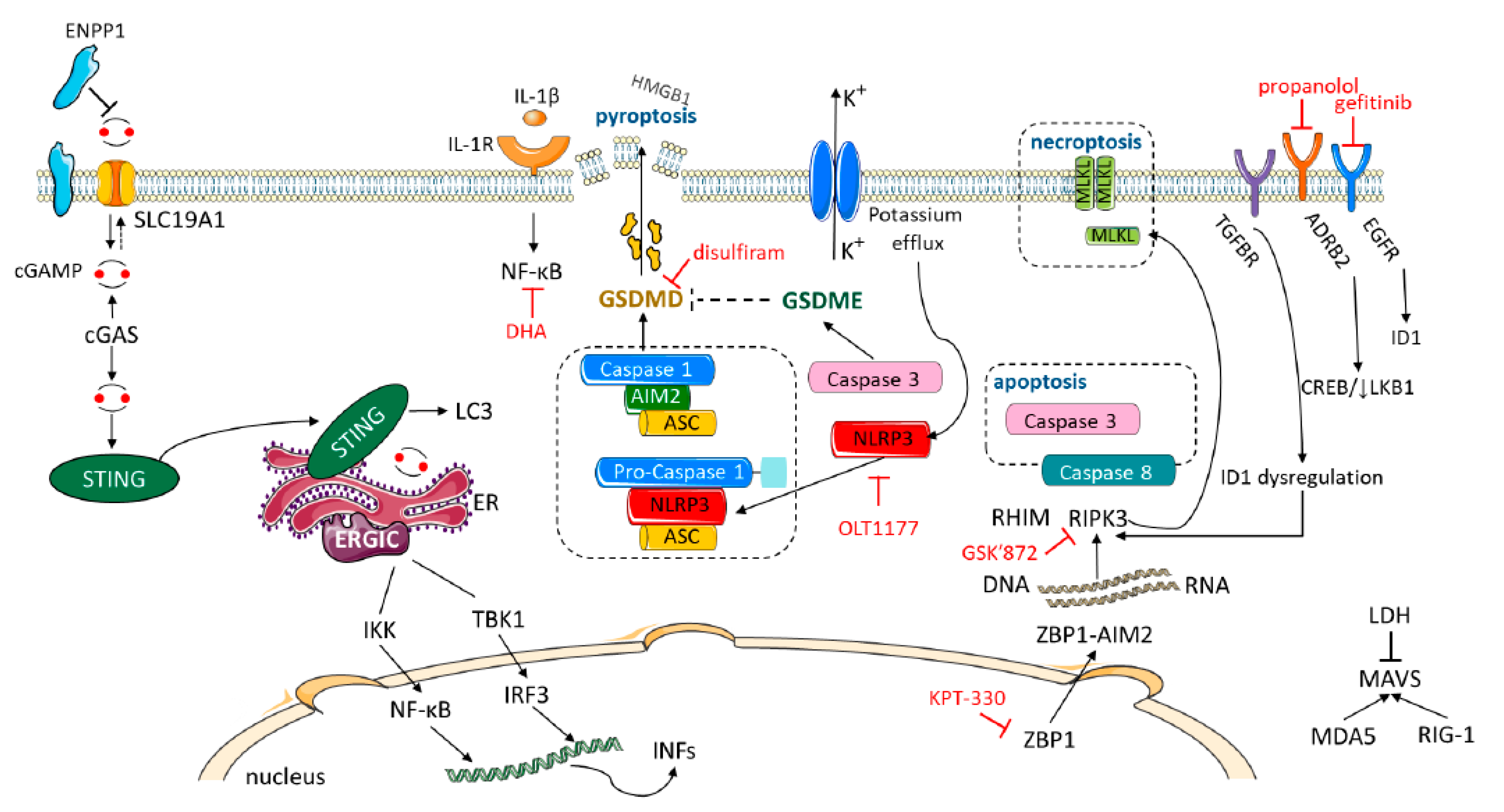
4. Inhibitor of DNA Binding I (ID1), a Read-Out of Loss of LKB1 and a Predictor of Sensitivity to Gefitinib
5. NLRP3 and IL-1β
6. CFTR and Mucins
7. Neoadjuvant Anti-PD-1 or Anti-PD-L1 Monoclonal Antibodies
8. Post-Translational Modifications of PD-L1: N-Linked Glycosylation of PD-L1 as Predictive Biomarker and Therapeutic Target
9. Glutathione Synthesis and Ferroptosis: Cystine-Glutamate Antiporter (xCT)-GSH/GPX Pathway and p53
10. Metabolic Rewiring
11. Immunotransmitters: the cGAS-STING Pathway
12. Neurotransmitters and Associated Receptors: β2-Adrenergic Receptor, NTRKs, Neurokin-1 Receptor, 5-HT Receptors and Dopamine Receptors
13. Gasdermin D and Gasdermin E
14. K-Ras Mutations
15. Transforming Growth Factor-β Signaling in K-Ras Mutations
16. Metabolism-Based Therapy for LUAD with K-Ras and LKB1 Mutations
17. BACH1 and NRF2-KEAP Alterations in K-Ras Driven LUAD
18. Perspectives: Potential Role for PDCD1 and PDCD1LG1 Genes, MICA and MICB, Exosomes and Other Checkpoints
19. Conclusions
Funding
Conflicts of Interest
References
- Topalian, S.L.; Taube, J.M.; Pardoll, D.M. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 2020, 367, 6477. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, K.P.; Johnson, M.L.; Lockhart, A.C.; Moore, K.; Falchook, G.S.; Formenti, S.C.; Naing, A.; Carvajal, R.D.; Rosen, L.S.; Weiss, G.J.; et al. First-in-human study of cemiplimab alone or in combination with radiotherapy and/or low-dose cyclophosphamide in patients with advanced malignancies. Clin. Cancer Res. 2020, 26, 1025–1033. [Google Scholar] [CrossRef] [Green Version]
- Hanna, N.H.; Schneider, B.J.; Temin, S.; Baker, S., Jr.; Brahmer, J.; Ellis, P.M.; Gaspar, L.E.; Haddad, R.Y.; Hesketh, P.J.; Jain, D.; et al. Therapy for stage IV non-small-cell lung cancer without driver alterations: ASCO and OH (CCO) joint guideline update. J. Clin. Oncol. 2020, 38, 1608–1632. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Sanmamed, M.F.; Hastings, K.; Politi, K.; Rimm, D.L.; Chen, L.; Melero, I.; Schalper, K.A.; Herbst, R.S. Immunotherapy in non-small cell lung cancer: Facts and hopes. Clin. Cancer Res. 2019, 25, 4592–4602. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Niknafs, N.; Marrone, K.; Bruhm, D.C.; White, J.R.; Naidoo, J.; Hummelink, K.; Monkhorst, K.; Lalezari, F.; Lanis, M.; et al. Multimodal genomic features predict outcome of immune checkpoint blockade in non-small-cell lung cancer. Nat. Cancer 2020, 1, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Park, S.; Park, H.; Kim, S.; Lee, J.; Lee, J.; Youk, J.; Yi, K.; An, Y.; Park, I.K.; et al. Tracing oncogene rearrangements in the mutational history of lung adenocarcinoma. Cell 2019, 177, 1842–1857.e1821. [Google Scholar] [CrossRef]
- Chen, K.; Liu, J.; Liu, S.; Xia, M.; Zhang, X.; Han, D.; Jiang, Y.; Wang, C.; Cao, X. Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell 2017, 170, 492–506.e414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.-C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of anifrolumab in active systemic lupus erythematosus. N. Engl. J. Med. 2019, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.X.; Li, X.; Nan, F.; Jiang, S.; Gao, X.; Guo, S.K.; Xue, W.; Cui, Y.; Dong, K.; Ding, H.; et al. Structure and degradation of circular RNAs regulate PKR activation in innate immunity. Cell 2019, 177, 865–880.e821. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E. Circle the wagons: Circular RNAs control innate immunity. Cell 2019, 177, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Golji, J.; Brodeur, L.K.; Chung, F.S.; Chen, J.T.; de Beaumont, R.S.; Bullock, C.P.; Jones, M.D.; Kerr, G.; Li, L.; et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat. Med. 2019, 25, 95–102. [Google Scholar] [CrossRef]
- Liu, Z.; Han, C.; Dong, C.; Shen, A.; Hsu, E.; Ren, Z.; Lu, C.; Liu, L.; Zhang, A.; Timmerman, C.; et al. Hypofractionated EGFR tyrosine kinase inhibitor limits tumor relapse through triggering innate and adaptive immunity. Sci. Immunol. 2019, 4, eaav6473. [Google Scholar] [CrossRef]
- Dutta, D.; Donaldson, J.G. Search for inhibitors of endocytosis: Intended specificity and unintended consequences. Cell. Logist. 2012, 2, 203–208. [Google Scholar] [CrossRef] [Green Version]
- Grassart, A.; Cheng, A.T.; Hong, S.H.; Zhang, F.; Zenzer, N.; Feng, Y.; Briner, D.M.; Davis, G.D.; Malkov, D.; Drubin, D.G. Actin and dynamin2 dynamics and interplay during clathrin-mediated endocytosis. J. Cell Biol. 2014, 205, 721–735. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [Green Version]
- Zahavi, D.J.; Aldeghaither, D.S.; O’Connell, A.; Weiner, L.M. Enhancing antibody-dependent cell-mediated cytotoxicity: A strategy for improving antibody-based immunotherapy. Antib. Ther. 2018, 1, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.R.; Gaffney, D.; Barry, R.; Hu, L.; Banushi, B.; Wells, J.W.; Lambie, D.; Strutton, G.; Porceddu, S.V.; Burmeister, B.; et al. An ex vivo human tumor assay shows distinct patterns of EGFR trafficking in squamous cell carcinoma correlating to therapeutic outcomes. J. Investig. Dermatol. 2019, 139, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellman, I.; Yarden, Y. Endocytosis and cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a016949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, A.I. Exocytosis and endocytosis. Preface. Methods Mol. Biol. 2008, 440, v–vi. [Google Scholar] [CrossRef] [PubMed]
- Chew, H.Y.; De Lima, P.O.; Gonzalez Cruz, J.L.; Banushi, B.; Echejoh, G.; Hu, L.; Joseph, S.R.; Lum, B.; Rae, J.; O’Donnell, J.S.; et al. Endocytosis inhibition in humans to improve responses to ADCC-mediating antibodies. Cell 2020, 180, 895–914.e827. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, A.; Daniel, J.A.; Hadzic, G.; Chau, N.; Clayton, E.L.; Mariana, A.; Whiting, A.; Gorgani, N.N.; Lloyd, J.; Quan, A.; et al. Building a better dynasore: The dyngo compounds potently inhibit dynamin and endocytosis. Traffic 2013, 14, 1272–1289. [Google Scholar] [CrossRef]
- Daniel, J.A.; Chau, N.; Abdel-Hamid, M.K.; Hu, L.; von Kleist, L.; Whiting, A.; Krishnan, S.; Maamary, P.; Joseph, S.R.; Simpson, F.; et al. Phenothiazine-derived antipsychotic drugs inhibit dynamin and clathrin-mediated endocytosis. Traffic 2015, 16, 635–654. [Google Scholar] [CrossRef]
- Boucrot, E.; Ferreira, A.P.; Almeida-Souza, L.; Debard, S.; Vallis, Y.; Howard, G.; Bertot, L.; Sauvonnet, N.; McMahon, H.T. Endophilin marks and controls a clathrin-independent endocytic pathway. Nature 2015, 517, 460–465. [Google Scholar] [CrossRef]
- Chaib, I.; Cai, X.; Llige, D.; Santarpia, M.; Jantus-Lewintre, E.; Filipska, M.; Pedraz, C.; Cui, J.; Yang, J.; Miao, J.; et al. Osimertinib and dihydroartemisinin: A novel drug combination targeting head and neck squamous cell carcinoma. Ann. Transl. Med. 2019, 7, 651. [Google Scholar] [CrossRef]
- Tan, H.Y.; Wang, N.; Chan, Y.T.; Zhang, C.; Guo, W.; Chen, F.; Zhong, Z.; Li, S.; Feng, Y. ID1 overexpression increases gefitinib sensitivity in non-small cell lung cancer by activating RIP3/MLKL-dependent necroptosis. Cancer Lett. 2020, 475, 109–118. [Google Scholar] [CrossRef]
- Rodon, L.; Svensson, R.U.; Wiater, E.; Chun, M.G.H.; Tsai, W.W.; Eichner, L.J.; Shaw, R.J.; Montminy, M. The CREB coactivator CRTC2 promotes oncogenesis in LKB1-mutant non-small cell lung cancer. Sci. Adv. 2019, 5, eaaw6455. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, M.B.; Sun, H.; Diao, L.; Tong, P.; Liu, D.; Li, L.; Fan, Y.; Poteete, A.; Lim, S.O.; Howells, K.; et al. Stress hormones promote EGFR inhibitor resistance in NSCLC: Implications for combinations with beta-blockers. Sci. Transl. Med. 2017, 9, eaao4307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, F.; Luo, M.; Rong, Q.X.; Zhang, H.; Chen, Z.; Wang, F.; Zhao, H.Y.; Fu, L.W. Niclosamide, an antihelmintic drug, enhances efficacy of PD-1/PD-L1 immune checkpoint blockade in non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 245. [Google Scholar] [CrossRef] [PubMed]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.L.; Hoss, F.; Buhmann, R.; et al. The DNA inflammasome in human myeloid cells is initiated by a STING-Cell death program upstream of NLRP3. Cell 2017, 171, 1110–1124.e1118. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Liang, X.; Ma, L.; Shen, L.; Li, T.; Zheng, L.; Sun, A.; Shang, W.; Chen, C.; Zhao, W.; et al. MiR-22 sustains NLRP3 expression and attenuates H. pylori-induced gastric carcinogenesis. Oncogene 2018, 37, 884–896. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Group, C.T. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Qu, J.; Zhao, M.; Xu, Q.; Sun, Y. Targeting SHP2 as a promising strategy for cancer immunotherapy. Pharmacol. Res. 2020, 152, 104595. [Google Scholar] [CrossRef]
- Ji, X.; Qian, J.; Rahman, S.M.J.; Siska, P.J.; Zou, Y.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Heidi, C.; Eisenberg, R.; et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018, 37, 5007–5019. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padro, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; van’t Veer, L.J.; et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 2013, 24, 450–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miess, H.; Dankworth, B.; Gouw, A.M.; Rosenfeldt, M.; Schmitz, W.; Jiang, M.; Saunders, B.; Howell, M.; Downward, J.; Felsher, D.W.; et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene 2018, 37, 5435–5450. [Google Scholar] [CrossRef] [PubMed]
- Arensman, M.D.; Yang, X.S.; Leahy, D.M.; Toral-Barza, L.; Mileski, M.; Rosfjord, E.C.; Wang, F.; Deng, S.; Myers, J.S.; Abraham, R.T.; et al. Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 9533–9542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Lee, D.H.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-induced endoplasmic reticulum stress: Cross-talk between ferroptosis and apoptosis. Mol. Cancer Res. 2018, 16, 1073–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Zhang, Z.; Chen, L.; Zhou, Y.; Zou, P.; Feng, C.; Wang, L.; Liang, G. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 2016, 381, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Yi, M.; Qin, S.; Song, Y.; Chu, Q.; Wu, K. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 35. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Ding, C.; Song, Z.; Shen, A.; Chen, T.; Zhang, A. Small molecules targeting the innate immune cGAS‒STING‒TBK1 signaling pathway. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef]
- White, S.M.; Murakami, S.; Yi, C. The complex entanglement of Hippo-Yap/Taz signaling in tumor immunity. Oncogene 2019, 38, 2899–2909. [Google Scholar] [CrossRef]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Van Tassell, B.W.; Mezzaroma, E.; Del Buono, M.G.; Prestamburgo, A.; Potere, N.; Abbate, A. The NLRP3 inflammasome inhibitor, OLT1177 (dapansutrile), reduces infarct size and preserves contractile function after ischemia reperfusion injury in the mouse. J. Cardiovasc. Pharmacol. 2019, 73, 215–222. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datar, I.; Sanmamed, M.F.; Wang, J.; Henick, B.S.; Choi, J.; Badri, T.; Dong, W.; Mani, N.; Toki, M.; Mejias, L.D.; et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin. Cancer Res. 2019, 25, 4663–4673. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Bono, P.; Bhatia, S.; Melero, I.; Nyakas, M.S.; Svane, I.-M.; Larkin, J.; Gomez-Roca, C.; Schadendorf, D.; Dummer, D.; et al. Efficacy of BMS-986016, a monoclonal antibody that targets Lymphocyte Activation Gene-3 (LAG-3), in combination with nivolumab in pts with melanoma who progressed during prior anti–PD-1/PD-L1 therapy (mel prior IO) in all-comer and biomarker-enriched populations. Ann. Oncol. 2017, 28 (Suppl. S5), v605–v649. [Google Scholar]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell 2020, 180, 862–877.e822. [Google Scholar] [CrossRef]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Nunez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Lan, X.; Wang, T.; Lu, H.; Cao, M.; Yan, S.; Cui, Y.; Jia, D.; Cai, L.; Xing, Y. Targeting the IL-1beta/EHD1/TUBB3 axis overcomes resistance to EGFR-TKI in NSCLC. Oncogene 2020, 39, 1739–1755. [Google Scholar] [CrossRef]
- Wang, X.; Yin, H.; Zhang, H.; Hu, J.; Lu, H.; Li, C.; Cao, M.; Yan, S.; Cai, L. NF-kappaB-driven improvement of EHD1 contributes to erlotinib resistance in EGFR-mutant lung cancers. Cell Death Dis. 2018, 9, 418. [Google Scholar] [CrossRef]
- Meng, Q.; Xing, Y.; Ren, T.; Lu, H.; Xi, Y.; Jiang, Z.; Hu, J.; Li, C.; Sun, L.; Sun, D.; et al. Mammalian Eps15 homology domain 1 promotes metastasis in non-small cell lung cancer by inducing epithelial-mesenchymal transition. Oncotarget 2017, 8, 22433–22442. [Google Scholar] [CrossRef] [Green Version]
- Rosell, R.; Bivona, T.G.; Karachaliou, N. Genetics and biomarkers in personalisation of lung cancer treatment. Lancet 2013, 382, 720–731. [Google Scholar] [CrossRef]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Reja, R.; Zhang, Y.; Roose-Girma, M.; Modrusan, Z.; Sagolla, M.S.; Webster, J.D.; et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 2019, 575, 679–682. [Google Scholar] [CrossRef]
- Fritsch, M.; Gunther, S.D.; Schwarzer, R.; Albert, M.C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 beta-a friend or foe in malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 inflammasome in neurological diseases, from functions to therapies. Front. Cell. Neurosci. 2017, 11, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Song, Q.; Zhou, C.; Li, X.; Pi, L.; Ma, X.; Li, H.; Lu, X.; Shen, Y. Dihydroartemisinin as a putative STAT3 inhibitor, suppresses the growth of head and neck squamous cell carcinoma by targeting Jak2/STAT3 signaling. PLoS ONE 2016, 11, e0147157. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.C.; Peters, H.L.; Taguchi, A.; Katayama, H.; Wang, H.; Momin, A.; Jolly, M.K.; Celiktas, M.; Rodriguez-Canales, J.; Liu, H.; et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc. Natl. Acad. Sci. USA 2016, 113, E1555–E1564. [Google Scholar] [CrossRef] [Green Version]
- Karachaliou, N.; Gonzalez-Cao, M.; Crespo, G.; Drozdowskyj, A.; Aldeguer, E.; Gimenez-Capitan, A.; Teixido, C.; Molina-Vila, M.A.; Viteri, S.; De Los Llanos Gil, M.; et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther. Adv. Med. Oncol. 2018, 10, 1–23. [Google Scholar] [CrossRef]
- Dai, J.; Huang, Y.J.; He, X.; Zhao, M.; Wang, X.; Liu, Z.S.; Xue, W.; Cai, H.; Zhan, X.Y.; Huang, S.Y.; et al. Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell 2019, 176, 1447–1460.e1414. [Google Scholar] [CrossRef] [Green Version]
- Ochi, A.; Nguyen, A.H.; Bedrosian, A.S.; Mushlin, H.M.; Zarbakhsh, S.; Barilla, R.; Zambirinis, C.P.; Fallon, N.C.; Rehman, A.; Pylayeva-Gupta, Y.; et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J. Exp. Med. 2012, 209, 1671–1687. [Google Scholar] [CrossRef] [PubMed]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Boucher, R.C. Muco-obstructive lung diseases. N. Engl. J. Med. 2019, 380, 1941–1953. [Google Scholar] [CrossRef]
- Clunes, L.A.; Davies, C.M.; Coakley, R.D.; Aleksandrov, A.A.; Henderson, A.G.; Zeman, K.L.; Worthington, E.N.; Gentzsch, M.; Kreda, S.M.; Cholon, D.; et al. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J. 2012, 26, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Maeda, T.; Hiraki, M.; Jin, C.; Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Hu, X.; Suzuki, Y.; Miyo, M.; et al. MUC1-C induces PD-L1 and immune evasion in triple-negative breast cancer. Cancer Res. 2018, 78, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouillez, A.; Rajabi, H.; Jin, C.; Samur, M.; Tagde, A.; Alam, M.; Hiraki, M.; Maeda, T.; Hu, X.; Adeegbe, D.; et al. MUC1-C integrates PD-L1 induction with repression of immune effectors in non-small-cell lung cancer. Oncogene 2017, 36, 4037–4046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Blake, S.J.; Yong, M.C.; Harjunpaa, H.; Ngiow, S.F.; Takeda, K.; Young, A.; O’Donnell, J.S.; Allen, S.; Smyth, M.J.; et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 2016, 6, 1382–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; O’Donnell, J.S.; Yan, J.; Madore, J.; Allen, S.; Smyth, M.J.; Teng, M.W.L. Timing of neoadjuvant immunotherapy in relation to surgery is crucial for outcome. Oncoimmunology 2019, 8, e1581530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.A.; Gainor, J.F.; Awad, M.M. Neoadjuvant atezolizumab and chemotherapy in patients with resectable non-small cell lung cancer: A single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 786–795. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for early triple-negative breast cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Ruiz-Patino, A.; Arrieta, O.; Cardona, A.F.; Martin, C.; Raez, L.E.; Zatarain-Barron, Z.L.; Barron, F.; Ricaurte, L.; Bravo-Garzon, M.A.; Mas, L.; et al. Immunotherapy at any line of treatment improves survival in patients with advanced metastatic non-small cell lung cancer (NSCLC) compared with chemotherapy (Quijote-CLICaP). Thorac. Cancer 2020, 11, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Zou, J.; Li, S.; Topper, M.J.; Tao, Y.; Zhang, H.; Jiao, X.; Xie, W.; Kong, X.; Vaz, M.; et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020, 579, 284–290. [Google Scholar] [CrossRef]
- Marasco, M.; Berteotti, A.; Weyershaeuser, J.; Thorausch, N.; Sikorska, J.; Krausze, J.; Brandt, H.J.; Kirkpatrick, J.; Rios, P.; Schamel, W.W.; et al. Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci. Adv. 2020, 6, eaay4458. [Google Scholar] [CrossRef] [Green Version]
- Bracht, J.W.P.; Karachaliou, N.; Bivona, T.; Lanman, R.B.; Faull, I.; Nagy, R.J.; Drozdowskyj, A.; Berenguer, J.; Fernandez-Bruno, M.; Molina-Vila, M.A.; et al. BRAF mutations classes I, II, and III in NSCLC patients included in the SLLIP trial: The need for a new pre-clinical treatment rationale. Cancers 2019, 11, 1381. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.L.; Hsu, W.M.; Huang, M.C.; Kadomatsu, K.; Nakagawara, A. Protein glycosylation in cancers and its potential therapeutic applications in neuroblastoma. J. Hematol. Oncol. 2016, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Feng, D.; Yao, Y.; Li, P.; Sun, H.; Yang, H.; Li, C.; Jiang, R.; Sun, B.; Chen, Y. Listeria-based hepatocellular carcinoma vaccine facilitates anti-PD-1 therapy by regulating macrophage polarization. Oncogene 2020, 39, 1429–1444. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Kang, Y.; Song, X.; Xu, Y.; Chen, H.; Gong, X.; Zhang, W.; Xu, Y.; Xia, X.; Gao, X.; et al. PDL1-targeted vaccine exhibits potent antitumor activity by simultaneously blocking PD1/PDL1 pathway and activating PDL1-specific immune responses. Cancer Lett. 2020, 476, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Codony-Servat, J.; Garcia-Roman, S.; Molina-Vila, M.A.; Bertran-Alamillo, J.; Gimenez-Capitan, A.; Viteri, S.; Cardona, A.F.; D’Hondt, E.; Karachaliou, N.; Rosell, R. Anti-epidermal growth factor vaccine antibodies enhance the efficacy of tyrosine kinase inhibitors and delay the emergence of resistance in EGFR mutant lung cancer cells. J. Thorac. Oncol. 2018, 13, 1324–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.M.; Li, C.W.; Lai, Y.J.; Hung, M.C. Posttranslational modifications of PD-L1 and their applications in cancer therapy. Cancer Res. 2018, 78, 6349–6353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gooijer, M.C.; Guillen Navarro, M.; Bernards, R.; Wurdinger, T.; van Tellingen, O. An experimenter’s guide to glioblastoma invasion pathways. Trends Mol. Med. 2018, 24, 763–780. [Google Scholar] [CrossRef]
- Hsu, J.M.; Xia, W.; Hsu, Y.H.; Chan, L.C.; Yu, W.H.; Cha, J.H.; Chen, C.T.; Liao, H.W.; Kuo, C.W.; Khoo, K.H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef]
- Lim, S.O.; Li, C.W.; Xia, W.; Cha, J.H.; Chan, L.C.; Wu, Y.; Chang, S.S.; Lin, W.C.; Hsu, J.M.; Hsu, Y.H.; et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [Green Version]
- Burr, M.L.; Sparbier, C.E.; Chan, Y.C.; Williamson, J.C.; Woods, K.; Beavis, P.A.; Lam, E.Y.N.; Henderson, M.A.; Bell, C.C.; Stolzenburg, S.; et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 2017, 549, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Mezzadra, R.; Sun, C.; Jae, L.T.; Gomez-Eerland, R.; de Vries, E.; Wu, W.; Logtenberg, M.E.W.; Slagter, M.; Rozeman, E.A.; Hofland, I.; et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 2017, 549, 106–110. [Google Scholar] [CrossRef]
- Lee, H.H.; Wang, Y.N.; Xia, W.; Chen, C.H.; Rau, K.M.; Ye, L.; Wei, Y.; Chou, C.K.; Wang, S.C.; Yan, M.; et al. Removal of N-linked glycosylation enhances PD-L1 detection and predicts anti-PD-1/PD-L1 therapeutic efficacy. Cancer Cell 2019, 36, 168–178.e164. [Google Scholar] [CrossRef]
- Saito, Y.; Takahashi, T.; Obata, Y.; Nishida, T.; Ohkubo, S.; Nakagawa, F.; Serada, S.; Fujimoto, M.; Ohkawara, T.; Nishigaki, T.; et al. TAS-116 inhibits oncogenic KIT signalling on the Golgi in both imatinib-naive and imatinib-resistant gastrointestinal stromal tumours. Br. J. Cancer 2020, 122, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Cui, J.; Xia, T.; Xie, D.; Gao, Y.; Jia, Z.; Wei, D.; Wang, L.; Huang, S.; Quan, M.; Xie, K. HGF/Met and FOXM1 form a positive feedback loop and render pancreatic cancer cells resistance to met inhibition and aggressive phenotypes. Oncogene 2016, 35, 4708–4718. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.; Wang, X.; Liu, Y.; Wang, M.; Yan, B.; Jiang, Y.; Shi, Y.; Shen, Y.; Liu, X.; Lai, W.; et al. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 2018, 78, 3484–3496. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Mao, C.; Wang, M.; Liu, N.; Ouyang, L.; Liu, S.; Tang, H.; Cao, Y.; Liu, S.; Wang, X.; et al. Cancer progression is mediated by proline catabolism in non-small cell lung cancer. Oncogene 2020, 39, 2358–2376. [Google Scholar] [CrossRef]
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell 2020, 37, 168–182.e164. [Google Scholar] [CrossRef] [Green Version]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef]
- Xiao, R.; Ding, C.; Zhu, H.; Liu, X.; Gao, J.; Liu, Q.; Lu, D.; Zhang, N.; Zhang, A.; Zhou, H. Suppression of asparagine synthetase enhances the antitumor potency of ART and artemalogue SOMCL-14-221 in non-small cell lung cancer. Cancer Lett. 2020, 475, 22–33. [Google Scholar] [CrossRef]
- Ojha, R.; Leli, N.M.; Onorati, A.; Piao, S.; Verginadis, I.I.; Tameire, F.; Rebecca, V.W.; Chude, C.I.; Murugan, S.; Fennelly, C.; et al. ER translocation of the MAPK pathway drives therapy resistance in BRAF-mutant melanoma. Cancer Discov. 2019, 9, 396–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-Del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell 2018, 33, 91–107.e106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.; Levantini, E.; Teo, J.T.; Goggi, J.; Clohessy, J.G.; Wu, C.S.; Chen, L.; Yang, H.; Krishnan, I.; Kocher, O.; et al. Fatty acid synthase mediates EGFR palmitoylation in EGFR mutated non-small cell lung cancer. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Sun, Y.; Chen, B.; Zheng, B.; Jarugumilli, G.K.; Walker, S.R.; Hata, A.N.; Mino-Kenudson, M.; Frank, D.A.; Wu, X. Fatty acids and cancer-amplified ZDHHC19 promote STAT3 activation through S-palmitoylation. Nature 2019, 573, 139–143. [Google Scholar] [CrossRef]
- Amendola, C.R.; Mahaffey, J.P.; Parker, S.J.; Ahearn, I.M.; Chen, W.C.; Zhou, M.; Court, H.; Shi, J.; Mendoza, S.L.; Morten, M.J.; et al. KRAS4A directly regulates hexokinase 1. Nature 2019, 576, 482–486. [Google Scholar] [CrossRef]
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Van Eck, M.; Auphan-Anezin, N.; et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab. 2019, 29, 1376–1389.e1374. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Zhu, Y.; Lin, X.; Tan, X.; Lu, B.; Li, Y. Stabilization of FASN by ACAT1-mediated GNPAT acetylation promotes lipid metabolism and hepatocarcinogenesis. Oncogene 2020, 39, 2437–2449. [Google Scholar] [CrossRef]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef]
- Dear, A.E. Epigenetic modulators and the new immunotherapies. N. Engl. J. Med. 2016, 374, 684–686. [Google Scholar] [CrossRef]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, G.; Xu, Z.G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell 2019, 178, 176–189.e115. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Carozza, J.A.; Böhnert, V.; Nguyen, K.C.; Skariah, G.; Shaw, K.E.; Brown, J.A.; Rafat, M.; von Eyben, R.; Graves, E.E.; Glenn, J.S.; et al. Extracellular cGAMP is a cancer-cell-produced immunotransmitter involved in radiation-induced anticancer immunity. Nat. Cancer 2020, 1, 184–196. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef]
- Ritchie, C.; Cordova, A.F.; Hess, G.T.; Bassik, M.C.; Li, L. SLC19A1 is an importer of the immunotransmitter cGAMP. Mol. Cell 2019, 75, 372–381.e375. [Google Scholar] [CrossRef]
- Luteijn, R.D.; Zaver, S.A.; Gowen, B.G.; Wyman, S.K.; Garelis, N.E.; Onia, L.; McWhirter, S.M.; Katibah, G.E.; Corn, J.E.; Woodward, J.J.; et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 2019, 573, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vanpouille-Box, C.; Bakhoum, S.F.; Demaria, S. SnapShot: CGAS-STING signaling. Cell 2018, 173, 276–276.e271. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor neurobiology and the war of nerves in cancer. Cancer Discov. 2019, 9, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Mohammadpour, H.; Bucsek, M.J.; Hylander, B.L.; Repasky, E.A. Depression stresses the immune response and promotes prostate cancer growth. Clin. Cancer Res. 2019, 25, 2363–2365. [Google Scholar] [CrossRef]
- Cheng, Y.; Tang, X.Y.; Li, Y.X.; Zhao, D.D.; Cao, Q.H.; Wu, H.X.; Yang, H.B.; Hao, K.; Yang, Y. Depression-induced neuropeptide Y secretion promotes prostate cancer growth by recruiting myeloid cells. Clin. Cancer Res. 2019, 25, 2621–2632. [Google Scholar] [CrossRef] [PubMed]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Urbanucci, A.; Nielsen, H.K.; Guldvik, I.J.; Engedal, A.; Ketola, K.; Wang, W.; Svindland, A.; et al. The beta2-adrenergic receptor is a molecular switch for neuroendocrine transdifferentiation of prostate cancer cells. Mol. Cancer Res. 2019, 17, 2154–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Gao, X.H.; Li, X.J.; Cao, Q.H.; Zhao, D.D.; Zhou, J.R.; Wu, H.X.; Wang, Y.; You, L.J.; Yang, H.B.; et al. Depression promotes prostate cancer invasion and metastasis via a sympathetic-cAMP-FAK signaling pathway. Oncogene 2018, 37, 2953–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell 2018, 33, 75–90.e77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Liang, Y.; He, Z.; An, Y.; Zhao, W.; Wu, J. Autocrine activity of BDNF induced by the STAT3 signaling pathway causes prolonged TrkB activation and promotes human non-small-cell lung cancer proliferation. Sci. Rep. 2016, 6, 30404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Shen, H.; Buckley, B.; Chen, Y.; Yang, N.; Mussell, A.L.; Chernov, M.; Kobzik, L.; Frangou, C.; Han, S.X.; et al. NTRK1 is a positive regulator of YAP oncogenic function. Oncogene 2019, 38, 2778–2787. [Google Scholar] [CrossRef]
- Ge, C.; Huang, H.; Huang, F.; Yang, T.; Zhang, T.; Wu, H.; Zhou, H.; Chen, Q.; Shi, Y.; Sun, Y.; et al. Neurokinin-1 receptor is an effective target for treating leukemia by inducing oxidative stress through mitochondrial calcium overload. Proc. Natl. Acad. Sci. USA 2019, 116, 19635–19645. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.H.; Hu, L.P.; Wang, X.; Li, J.; Zhang, Z.G. Neurotransmitters: Emerging targets in cancer. Oncogene 2020, 39, 503–515. [Google Scholar] [CrossRef]
- Sola-Penna, M.; Paixao, L.P.; Branco, J.R.; Ochioni, A.C.; Albanese, J.M.; Mundim, D.M.; Baptista-de-Souza, D.; Figueiredo, C.P.; Coelho, W.S.; Marcondes, M.C.; et al. Serotonin activates glycolysis and mitochondria biogenesis in human breast cancer cells through activation of the Jak1/STAT3/ERK1/2 and adenylate cyclase/PKA, respectively. Br. J. Cancer 2020, 122, 194–208. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.Y.; Li, C.G.; Shu, J.X.; Xu, L.H.; Ouyang, D.Y.; Mai, F.Y.; Zeng, Q.Z.; Zhang, C.C.; Li, R.M.; He, X.H. ATP induces caspase-3/gasdermin E-mediated pyroptosis in NLRP3 pathway-blocked murine macrophages. Apoptosis 2019, 24, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.; Wu, H.; Kagan, J.C. Gasdermin D activity in inflammation and host defense. Sci. Immunol. 2019, 4, eaav1447. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.J.; Liu, X.; Zhao, J.; Xia, S.; Ruan, J.; Luo, X.; Kim, J.; Lieberman, J.; Wu, H. Identification of pyroptosis inhibitors that target a reactive cysteine in gasdermin D. bioRxiv 2018, bioRxiv:365908. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Liu, T.; Jiang, L.; Tavana, O.; Gu, W. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res. 2019, 79, 1913–1924. [Google Scholar] [CrossRef]
- Nakada, S.; Tai, I.; Panier, S.; Al-Hakim, A.; Iemura, S.; Juang, Y.C.; O’Donnell, L.; Kumakubo, A.; Munro, M.; Sicheri, F.; et al. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature 2010, 466, 941–946. [Google Scholar] [CrossRef]
- Jang, J.H.; Kim, D.H.; Lim, J.M.; Lee, J.W.; Jeong, S.J.; Kim, K.P.; Surh, Y.J. Breast cancer cell-derived soluble CD44 promotes tumor progression by triggering macrophage IL1beta production. Cancer Res. 2020, 80, 1342–1356. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Liu, H.; Kuang, X.; Zhang, Y.; Ye, Y.; Li, J.; Liang, L.; Xie, Z.; Weng, L.; Guo, J.; Li, H.; et al. ADORA1 inhibition promotes tumor immune evasion by regulating the ATF3-PD-L1 Axis. Cancer Cell 2020, 37, 324–339.e328. [Google Scholar] [CrossRef]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47(+)/CD73(+)/PDL1(+) immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239–E1248. [Google Scholar] [CrossRef] [Green Version]
- Zierhut, C.; Yamaguchi, N.; Paredes, M.; Luo, J.D.; Carroll, T.; Funabiki, H. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 2019, 178, 302–315.e323. [Google Scholar] [CrossRef] [PubMed]
- Gujral, T.S.; Kirschner, M.W. Hippo pathway mediates resistance to cytotoxic drugs. Proc. Natl. Acad. Sci. USA 2017, 114, E3729–E3738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ye, L.; Shi, X.; Chen, J.; Feng, F.; Chen, Y.; Xiao, Y.; Shen, J.; Li, P.; Jiang, W.G.; et al. Repulsive guidance molecule B inhibits metastasis and is associated with decreased mortality in non-small cell lung cancer. Oncotarget 2016, 7, 15678–15689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.H.; Hu, J.; Chen, F.; Lecomte, N.; Basnet, H.; David, C.J.; Witkin, M.D.; Allen, P.J.; Leach, S.D.; Hollmann, T.J.; et al. ID1 mediates escape from TGFbeta tumor suppression in pancreatic cancer. Cancer Discov. 2020, 10, 142–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.B.; Jang, H.R.; Xu, R.; Won, J.Y.; Yim, H. Active PLK1-driven metastasis is amplified by TGF-beta signaling that forms a positive feedback loop in non-small cell lung cancer. Oncogene 2020, 39, 767–785. [Google Scholar] [CrossRef] [PubMed]
- Guillen Diaz-Maroto, N.; Sanz-Pamplona, R.; Berdiel-Acer, M.; Cimas, F.J.; Garcia, E.; Goncalves-Ribeiro, S.; Albert, N.; Garcia-Vicien, G.; Capella, G.; Moreno, V.; et al. Noncanonical TGFbeta pathway relieves the blockade of IL1beta/TGFbeta-mediated crosstalk between tumor and stroma: TGFBR1 and TAK1 inhibition in colorectal cancer. Clin. Cancer Res. 2019, 25, 4466–4479. [Google Scholar] [CrossRef] [Green Version]
- Freudlsperger, C.; Bian, Y.; Contag Wise, S.; Burnett, J.; Coupar, J.; Yang, X.; Chen, Z.; Van Waes, C. TGF-beta and NF-kappaB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene 2013, 32, 1549–1559. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, H.; Lee, N.Y. Multifaceted roles of TAK1 signaling in cancer. Oncogene 2020, 39, 1402–1413. [Google Scholar] [CrossRef]
- Huang, S.; Holzel, M.; Knijnenburg, T.; Schlicker, A.; Roepman, P.; McDermott, U.; Garnett, M.; Grernrum, W.; Sun, C.; Prahallad, A.; et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell 2012, 151, 937–950. [Google Scholar] [CrossRef] [Green Version]
- Marwitz, S.; Depner, S.; Dvornikov, D.; Merkle, R.; Szczygiel, M.; Muller-Decker, K.; Lucarelli, P.; Wasch, M.; Mairbaurl, H.; Rabe, K.F.; et al. Downregulation of the TGFbeta pseudoreceptor BAMBI in non-small cell lung cancer enhances TGFbeta signaling and invasion. Cancer Res. 2016, 76, 3785–3801. [Google Scholar] [CrossRef] [Green Version]
- Rosell, R. Mediating resistance in oncogene-driven cancers. N. Engl. J. Med. 2013, 368, 1551–1552. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.A.; Wei, Y.; Montas, G.; Leong, D.; Golden, J.A.; Trinh, B.N.; Wolters, P.J.; Le Saux, C.J.; Jones, K.D.; Hills, N.K.; et al. Reversal of TGFbeta1-driven profibrotic state in patients with pulmonary fibrosis. N. Engl. J. Med. 2020, 382, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Kim, T.J.; Peng, D.H.; Duan, D.; Gibbons, D.L.; Yamauchi, M.; Jackson, J.R.; Le Saux, C.J.; Calhoun, C.; Peters, J.; et al. Fibroblast-specific inhibition of TGF-beta1 signaling attenuates lung and tumor fibrosis. J. Clin. Investig. 2017, 127, 3675–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.L.; Huang, M.S.; Hung, J.Y.; Chang, W.A.; Tsai, Y.M.; Pan, Y.C.; Lin, Y.S.; Tsai, H.P.; Kuo, P.L. Bone-marrow-derived cell-released extracellular vesicle miR-92a regulates hepatic pre-metastatic niche in lung cancer. Oncogene 2020, 39, 739–753. [Google Scholar] [CrossRef]
- Gonzalez-Cao, M.; Morán, T.; Dalmau, J.; Garcia-Corbacho, J.; Bracht, J.W.P.; Bernabe, R.; Juan, O.; de Castro, J.; Blanco, R.; Drozdowskyj, A.; et al. Assessment of the feasibility and safety of durvalumab for treatment of solid tumors in patients with HIV-1 infection: The phase 2 DURVAST study. JAMA Oncol. 2020. [Google Scholar] [CrossRef]
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Bragelmann, J.; Kron, A.; Abedpour, N.; Ueckeroth, F.; et al. K-ras mutation subtypes in NSCLC and associated co-occuring mutations in other oncogenic pathways. J. Thorac. Oncol. 2019, 14, 606–616. [Google Scholar] [CrossRef] [Green Version]
- Arrieta, O.; Barron, F.; Padilla, M.S.; Aviles-Salas, A.; Ramirez-Tirado, L.A.; Arguelles Jimenez, M.J.; Vergara, E.; Zatarain-Barron, Z.L.; Hernandez-Pedro, N.; Cardona, A.F.; et al. Effect of metformin plus tyrosine kinase inhibitors compared with tyrosine kinase inhibitors alone in patients with epidermal growth factor receptor-mutated lung adenocarcinoma: A phase 2 randomized clinical trial. JAMA Oncol. 2019, 5, e192553. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [Green Version]
- Groenendijk, F.H.; Mellema, W.W.; van der Burg, E.; Schut, E.; Hauptmann, M.; Horlings, H.M.; Willems, S.M.; van den Heuvel, M.M.; Jonkers, J.; Smit, E.F.; et al. Sorafenib synergizes with metformin in NSCLC through AMPK pathway activation. Int. J. Cancer 2015, 136, 1434–1444. [Google Scholar] [CrossRef]
- Hammerlindl, H.; Ravindran Menon, D.; Hammerlindl, S.; Emran, A.A.; Torrano, J.; Sproesser, K.; Thakkar, D.; Xiao, M.; Atkinson, V.G.; Gabrielli, B.; et al. Acetylsalicylic acid governs the effect of sorafenib in RAS-mutant cancers. Clin. Cancer Res. 2018, 24, 1090–1102. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hu, Z.; Cai, L.; Li, K.; Choi, E.; Faubert, B.; Bezwada, D.; Rodriguez-Canales, J.; Villalobos, P.; Lin, Y.F.; et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 2017, 546, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.W.; Hughey, C.C.; Lantier, L.; Sundelin, E.I.; Peggie, M.; Zeqiraj, E.; Sicheri, F.; Jessen, N.; Wasserman, D.H.; Sakamoto, K. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat. Med. 2018, 24, 1395–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, C.W.; Brady, J.J.; Tsai, M.K.; Li, C.; Winters, I.P.; Tang, R.; Andrejka, L.; Ma, R.K.; Kunder, C.A.; Chu, P.; et al. An LKB1-SIK axis suppresses lung tumor growth and controls differentiation. Cancer Discov. 2019, 9, 1590–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollstein, P.E.; Eichner, L.J.; Brun, S.N.; Kamireddy, A.; Svensson, R.U.; Vera, L.I.; Ross, D.S.; Rymoff, T.J.; Hutchins, A.; Galvez, H.M.; et al. The AMPK-related kinases SIK1 and SIK3 mediate key tumor-suppressive effects of LKB1 in NSCLC. Cancer Discov. 2019, 9, 1606–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Chun, J.; Pan, C.; Kumar, A.; Zhang, G.; Ha, Y.; Li, D.; Alesi, G.N.; Kang, Y.; Zhou, L.; et al. The PLAG1-GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2-AMPK signaling in LKB1-deficient lung cancer. Mol. Cell 2018, 69, 87–99.e87. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhou, B.; Oshiro-Rapley, N.; Li, M.; Paulo, J.A.; Webster, C.M.; Mou, F.; Kacergis, M.C.; Talkowski, M.E.; Carr, C.E.; et al. An ancient, unified mechanism for metformin growth inhibition in C. elegans and cancer. Cell 2016, 167, 1705–1718.e1713. [Google Scholar] [CrossRef] [Green Version]
- Mazur, P.K.; Reynoird, N.; Khatri, P.; Jansen, P.W.; Wilkinson, A.W.; Liu, S.; Barbash, O.; Van Aller, G.S.; Huddleston, M.; Dhanak, D.; et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 2014, 510, 283–287. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, B.-H.; Lin, W.-H.; Huang, Y.-H.; Ni, J.-Y.; Hu, J.; Cui, W.; Zhou, J.; Shen, L.; Xu, L.-F.; et al. Amplification of SMYD3 promotes tumorigenicity and intrahepatic metastasis of hepatocellular carcinoma via upregulation of CDK2 and MMP2. Oncogene 2019, 38, 4948–4961. [Google Scholar] [CrossRef]
- Lee, J.; Yesilkanal, A.E.; Wynne, J.P.; Frankenberger, C.; Liu, J.; Yan, J.; Elbaz, M.; Rabe, D.C.; Rustandy, F.D.; Tiwari, P.; et al. Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature 2019, 568, 254–258. [Google Scholar] [CrossRef]
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell 2019, 178, 316–329.e318. [Google Scholar] [CrossRef]
- Sanghvi, V.R.; Leibold, J.; Mina, M.; Mohan, P.; Berishaj, M.; Li, Z.; Miele, M.M.; Lailler, N.; Zhao, C.; de Stanchina, E.; et al. The oncogenic action of NRF2 depends on de-glycation by fructosamine-3-kinase. Cell 2019, 178, 807–819.e821. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Braas, D.; Go, J.; Graeber, T.G.; Parlati, F.; Demo, S.; Li, R.; et al. The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell 2018, 33, 905–921.e905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; van den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crinò, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: The SELECT-1 randomized clinical trial. JAMA 2017, 317, 1844–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Cai, H.; Wu, T.; Sobhian, B.; Huo, Y.; Alcivar, A.; Mehta, M.; Cheung, K.L.; Ganesan, S.; Kong, A.N.; et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol. Cell. Biol. 2012, 32, 1506–1517. [Google Scholar] [CrossRef] [Green Version]
- Karachaliou, N.; Bracht, J.W.P.; Fernandez Bruno, M.; Drozdowskyj, A.; Gimenez Capitan, A.; Moran, T.; Carcereny, E.; Cobo, M.; Domine, M.; Chaib, I.; et al. Association of PALB2 messenger RNA expression with platinum-docetaxel efficacy in advanced non-small cell lung cancer. J. Thorac. Oncol. 2019, 14, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Kemper, E.K.; Suciu, R.M.; Vinogradova, E.V.; Backus, K.M.; Horning, B.D.; Paul, T.A.; Ichu, T.A.; Svensson, R.U.; Olucha, J.; et al. Chemical proteomics identifies druggable vulnerabilities in a genetically defined cancer. Cell 2017, 171, 696–709.e623. [Google Scholar] [CrossRef] [Green Version]
- Visconti, R.; Morra, F.; Guggino, G.; Celetti, A. The between Now and Then of Lung Cancer Chemotherapy and Immunotherapy. Int. J. Mol. Sci. 2017, 18, 1374. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Huang, Q.; Luster, T.A.; Hu, H.; Zhang, H.; Ng, W.L.; Khodadadi-Jamayran, A.; Wang, W.; Chen, T.; Deng, J.; et al. In vivo epigenetic CRISPR screen identifies Asf1a as an immunotherapeutic target in kras-mutant lung adenocarcinoma. Cancer Discov. 2020, 10, 270–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lantuejoul, S.; Sound-Tsao, M.; Cooper, W.A.; Girard, N.; Hirsch, F.R.; Roden, A.C.; Lopez-Rios, F.; Jain, D.; Chou, T.-Y.; Motoi, N.; et al. PD-L1 testing for lung cancer in 2019: Perspective from the IASLC pathology committee. J. Thorac. Oncol. 2020, 15, 499–519. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yang, X.; Zhang, C.; Wang, Y.; Cheng, T.; Duan, L.; Tong, Z.; Tan, S.; Zhang, H.; Saw, P.E.; et al. Tumor cell-intrinsic PD-1 receptor is a tumor suppressor and mediates resistance to PD-1 blockade therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 6640–6650. [Google Scholar] [CrossRef]
- Du, S.; McCall, N.; Park, K.; Guan, Q.; Fontina, P.; Ertel, A.; Zhan, T.; Dicker, A.P.; Lu, B. Blockade of tumor-expressed PD-1 promotes lung cancer growth. Oncoimmunology 2018, 7, e1408747. [Google Scholar] [CrossRef]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef]
- Qian, M.; Geng, J.; Luo, K.; Huang, Z.; Zhang, Q.; Zhang, J.A.; Ji, L.; Wu, J. BCL11B regulates MICA/B-mediated immune response by acting as a competitive endogenous RNA. Oncogene 2020, 39, 1514–1526. [Google Scholar] [CrossRef]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal microbiota promote lung cancer development via gammadelta T cells. Cell 2019, 176, 998–1013.e1016. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Sha, H.; Qin, X.; Chen, Y.; Li, X.; Shi, M.; Feng, J. EGFR E746-A750 deletion in lung cancer represses antitumor immunity through the exosome-mediated inhibition of dendritic cells. Oncogene 2020, 39, 2643–2657. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Gui, J.; Zahedi, F.; Yu, P.; Cho, C.; Bhattacharya, S.; Carbone, C.J.; Yu, Q.; Katlinski, K.V.; Katlinskaya, Y.V.; et al. An interferon-driven oxysterol-based defense against tumor-derived extracellular vesicles. Cancer Cell 2019, 35, 33–45.e36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, T.V.; Hussmann, D.; Nielsen, A.L. PD-L1 and PD-L2 expression correlated genes in non-small-cell lung cancer. Cancer Commun. 2019, 39, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudnik, E.; Peled, N.; Nechushtan, H.; Wollner, M.; Onn, A.; Agbarya, A.; Moskovitz, M.; Kere, S.; Popovits-Hadari, N.; Urban, D.; et al. BRAF Mutant Lung Cancer: Programmed Death Ligand 1 Expression, Tumor Mutational Burden, Microsatellite Instability Status, and Response to Immune Check-Point Inhibitors. J. Thorac. Oncol. 2018, 13, 1128–1137. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Okamura, R.; Kato, S.; Lee, S.; Jiménez, R.E.; Sicklick, J.K.; Kurzrock, R. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J. Immunother. Cancer 2020, 8, e000438. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anti-PD1/PDL1 Antibody | FDA-Approved Indications |
|---|---|
| Pembrolizumab | Melanoma, NSCLC, SCLC, HNSCC, cHL, PMBCL, urothelial carcinoma, MSI-H or dMMR cancer, gastric cancer, esophageal cancer, cervical cancer, endometrial carcinoma, RCC, hepatocellular carcinoma and Merkel cell carcinoma |
| Nivolumab | Melanoma, NSCLC, SCLC, RCC, cHL, HNSCC, urothelial carcinoma, MSI-H or dMMR colorectal cancer and hepatocellular carcinoma. |
| Atezolizumab | Urothelial carcinoma, NSCLC, TNBC, SCLC |
| Durvalumab | Urothelial Carcinoma, NSCLC, SCLC |
| Avelumab | Merkel cell carcinoma, urothelial carcinoma, RCC |
| Cemiplimab | Cutaneous squamous cell carcinoma |
| Biomarker | Proposed Function | Targeted Agent | Status in Solid Tumors | Reference |
|---|---|---|---|---|
| Dynamin | Regulation of endocytosis (e.g., EGFR, PD-L1) through Src-FAK signaling | -Dyngo compounds | -Preclinical | [24,25,26,27,28] |
| -Prochlorperazine | ||||
| -FAK inhibitors (e.g., defactinib) | -Phase 1-2 studies | |||
| ID1 (Inhibitor of DNA binding I) | -Regulation of cancer stem cells and tumour aggressiveness; | -Gefitinib | -Preclinical | [29,30] |
| -Regulation of pathways related to inflammation-associated cell death: activation of necroptosis by triggering activation of RIP1/RIP3/MLKL pathway and minimal effect in inducing pyroptosis; | -ID1 Inhibitors (e.g., AGX-51, pimozide) | |||
| -ID1 overexpression can be correlated with KRAS and LKB1 mutations. | ||||
| β2-adrenergic receptor (β2-AR) | Activation of β2-AR by neurotransmitters, such as norepinephrine, inactivates LKB1, with upregulation of cAMP response element-binding protein (CREB) and interleukin-6 (IL6) | Propranolol | -Phase 1-2 studies | [31] |
| STAT3 | Mediator of tumor-induced immunosuppression | STAT inhibitors (e.g., niclosamide, dihydroartemisinin) | -Phase 1–2 studies | [28,32] |
| IL-1β | Regulation of tumorigenesis and mediator of immunosuppression through myeloid-derived suppressor cells (MDSCs) | IL-1β inhibitors (e.g., canakinumab, rilonacept, anakinra) | -Phase 1–3 studies | [33,34,35,36,37] |
| SHP2 (Src homology 2 domain containing phosphatase 2) | Regulation of signaling pathways, in cancer and immune cells, involved in inflammation and tumorigenesis (e.g., RTK, RAS and PD1) | SHP2 inhibitors (e.g., TNO155, RMC-4630, JAB-3068) | -Phase 1–2 studies | [38] |
| xCT (glutamate-cystine antiporter system) | Glutathione (GSH) synthesis, antioxidant response and ferroptosis | Inhibitors of xCT-GSH pathway (e.g., erastin, iImidazole ketone erastin [IKE], sulfasalazine, dihydroartemisinin [DHA], sorafenib, buthionine sulfoximine [BSO]) | -Preclinical (Erastin, IKE); | [28,39,40,41,42,43,44] |
| -Phase 1–3 (phase 3 for Sorafenib) | ||||
| Acetyl-CoA acetyltransferase (ACAT1) | Cholesterol esterification in T cells | ACAT1 inhibitor (e.g., avasimibe) | -Preclinical | [45] |
| STING | cGAS-STING pathway: key role in bridging cGAS-STcGAS-STING pathway: key role in cGAS-STING pathway: key role in bridging innate and adaptive anticancer immunity | STING agonists (e.g., ADU-S100, MK-1454, STING agonists (e.g. ADU-S100, MK-1454, GSK3745417) | -Phase 1–2 studies | [46,47,48] |
| YAP | Master transcriptional regulator involved in multiple cellular functions (activated by NTRK1/NTRK2) and immunosuppression | NTRK or YAP inhibitors (e.g., entrectinib, larotrectinib, repotrectinib, or dihydroartemisinin) | -Phase 1–3 (phase 3 for Entrectinib) | [28,49] |
| NLRP3 | NLRP3 inflammasome: key role in immune response | NLPR3 inhibitors (e.g., OLT1177-dapansutrile, CY-09, tranilast) | -Preclinical | [50,51] |
| LAG-3 | Immune checkpoint receptor modulating T-cell proliferation and activation | LAG-3 inhibitors (e.g., relatlimab) | Phase 1–2 studies | [52,53] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santarpia, M.; Aguilar, A.; Chaib, I.; Cardona, A.F.; Fancelli, S.; Laguia, F.; Bracht, J.W.P.; Cao, P.; Molina-Vila, M.A.; Karachaliou, N.; et al. Non-Small-Cell Lung Cancer Signaling Pathways, Metabolism, and PD-1/PD-L1 Antibodies. Cancers 2020, 12, 1475. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061475
Santarpia M, Aguilar A, Chaib I, Cardona AF, Fancelli S, Laguia F, Bracht JWP, Cao P, Molina-Vila MA, Karachaliou N, et al. Non-Small-Cell Lung Cancer Signaling Pathways, Metabolism, and PD-1/PD-L1 Antibodies. Cancers. 2020; 12(6):1475. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061475
Chicago/Turabian StyleSantarpia, Mariacarmela, Andrés Aguilar, Imane Chaib, Andrés Felipe Cardona, Sara Fancelli, Fernando Laguia, Jillian Wilhelmina Paulina Bracht, Peng Cao, Miguel Angel Molina-Vila, Niki Karachaliou, and et al. 2020. "Non-Small-Cell Lung Cancer Signaling Pathways, Metabolism, and PD-1/PD-L1 Antibodies" Cancers 12, no. 6: 1475. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061475