HERC Ubiquitin Ligases in Cancer

 , , , , and
, , , , and
Abstract
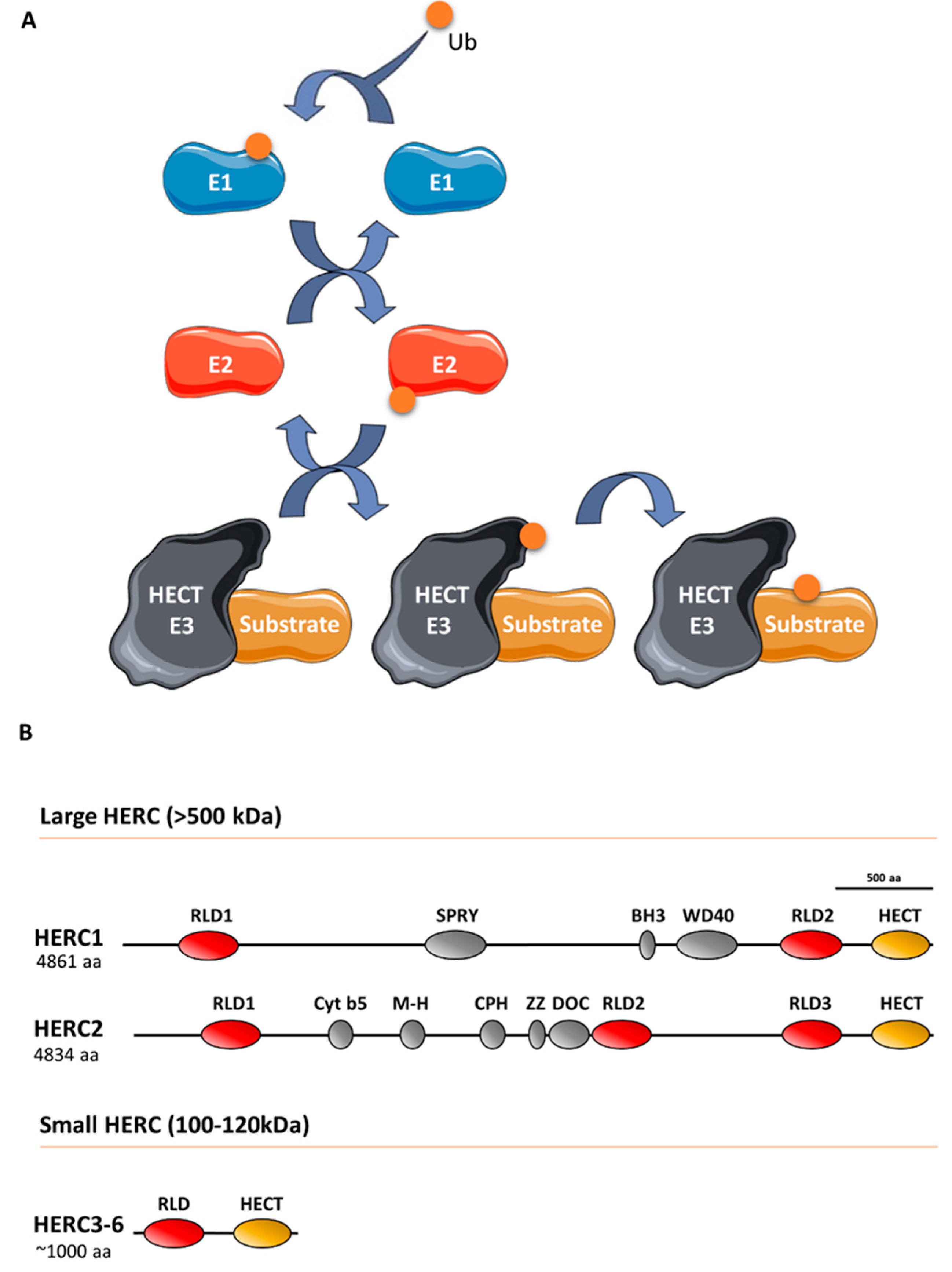
:1. Introduction
2. The Role of HERCs in Cancer
3. Signaling Pathways Regulated by HERCs
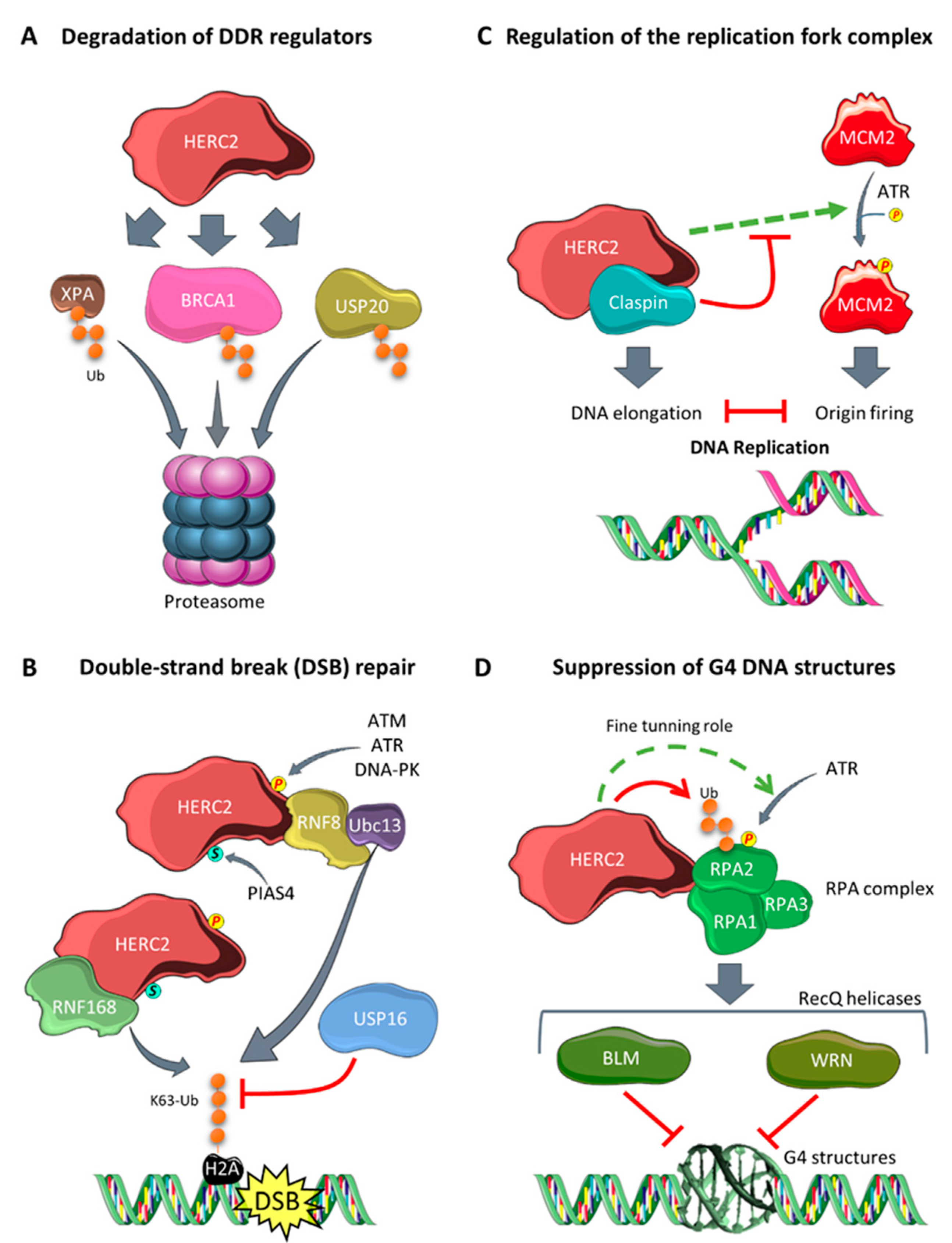
3.1. Regulation of Genomic Stability
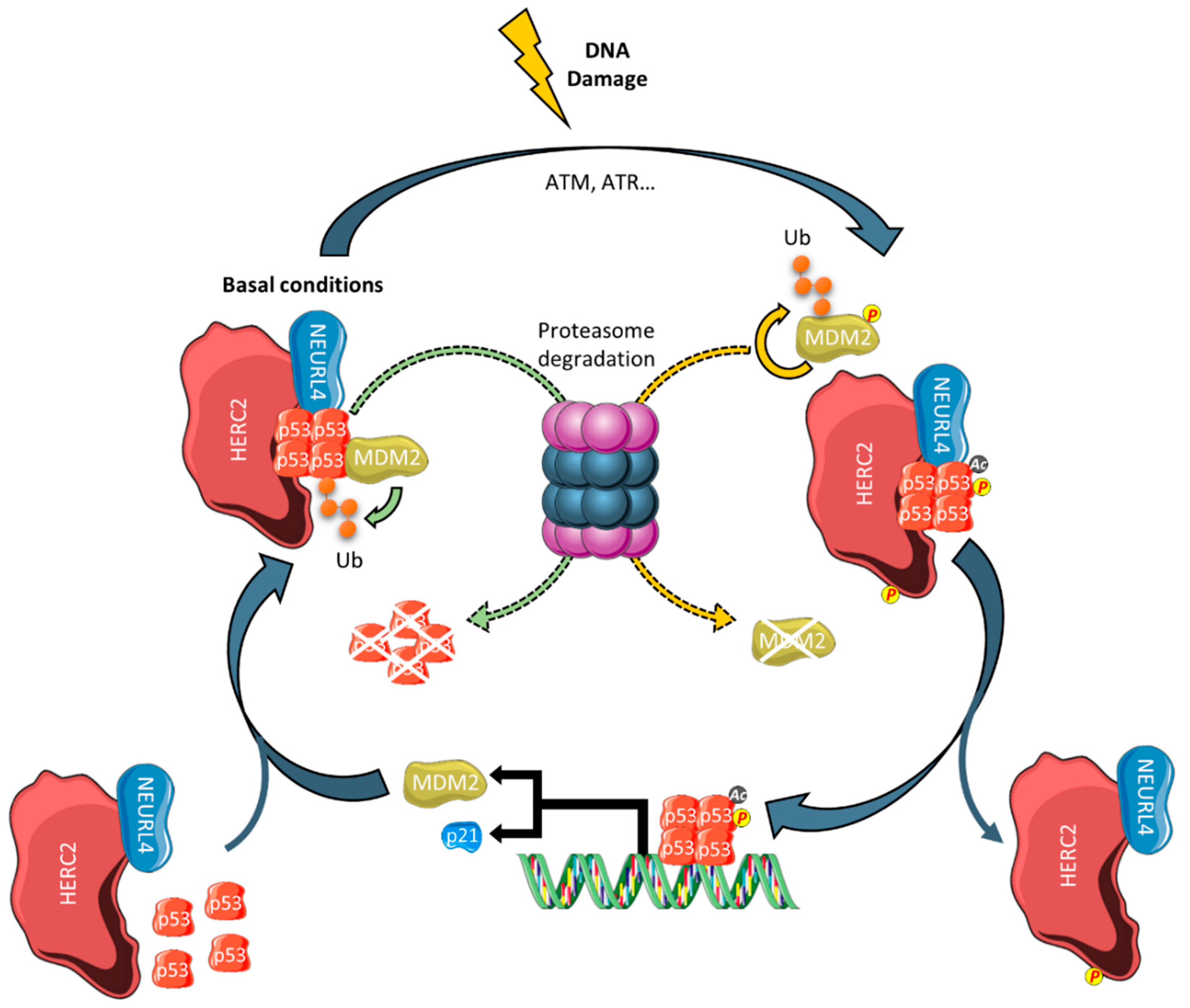
3.2. Regulation of p53 Transcriptional Programming
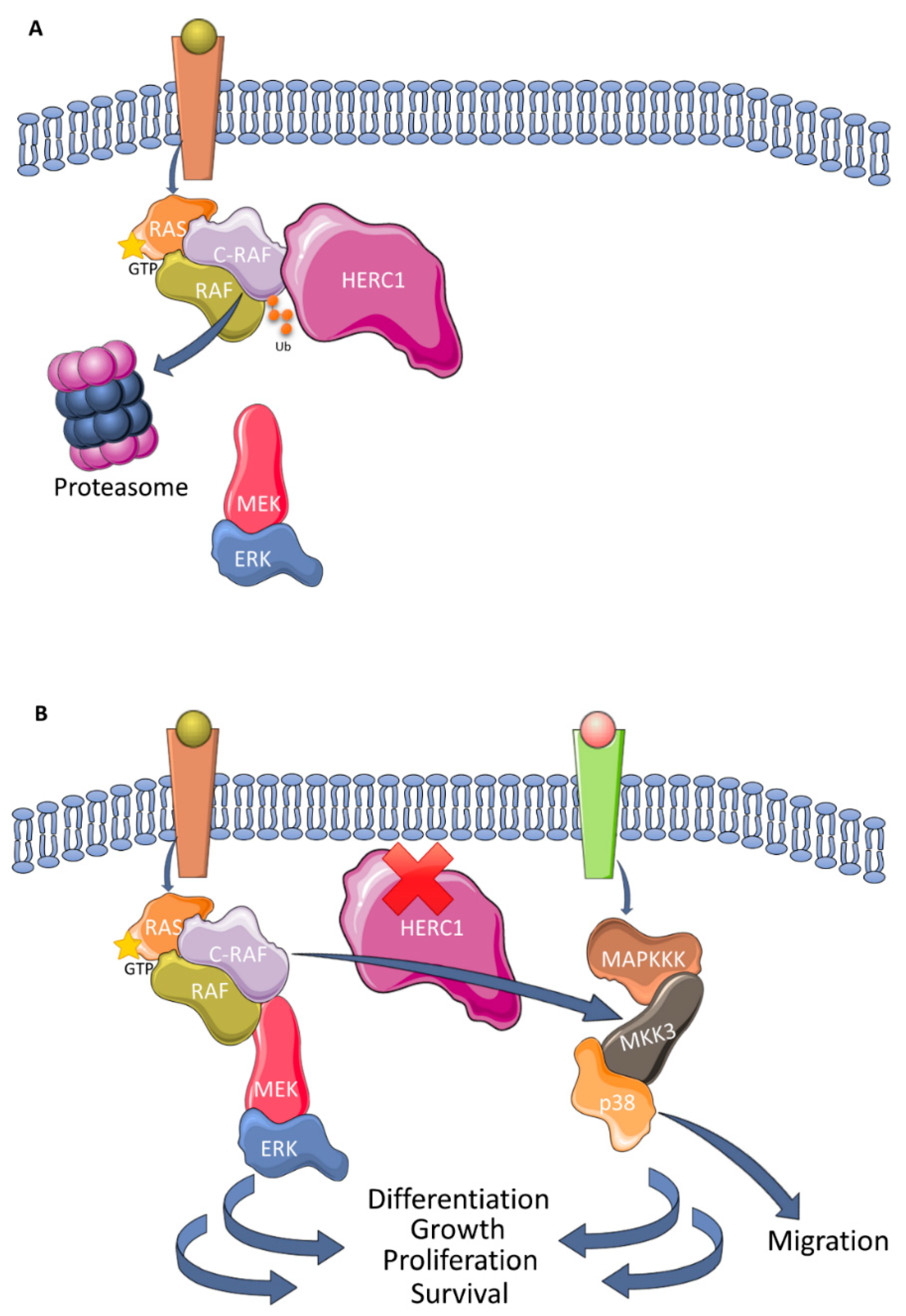
3.3. Regulation of MAPK Signaling
4. Perspective and Therapeutic Implications
5. Conclusions
Funding
Conflicts of Interest
References
- Ciechanover, A. The unravelling of the ubiquitin system. Nat. Rev. Mol. Cell Biol. 2015, 16, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalo, F.R.; Rosa, J.L. The HERC proteins: Functional and evolutionary insights. Cell. Mol. Life Sci. 2005, 62, 1826–1838. [Google Scholar] [CrossRef] [PubMed]
- García-Cano, J.; Martinez-Martinez, A.; Sala-Gaston, J.; Pedrazza, L.; Rosa, J.L. HERCing: Structural and Functional Relevance of the Large HERC Ubiquitin Ligases. Front. Physiol. 2019, 10, 1014. [Google Scholar] [CrossRef] [PubMed]
- Hadjebi, O.; Casas-Terradellas, E.; Garcia-Gonzalo, F.R.; Rosa, J.L. The RCC1 superfamily: From genes, to function, to disease. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 1467–1479. [Google Scholar] [CrossRef] [Green Version]
- Marin, I. Animal HECT ubiquitin ligases: Evolution and functional implications. BMC Evol. Biol. 2010, 10, 56. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Tena, S.; Cubillos-Rojas, M.; Schneider, T.; Rosa, J.L. Functional and pathological relevance of HERC family proteins: A decade later. Cell. Mol. Life Sci. 2016, 73, 1955–1968. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Guerra, S.; Sánchez-Madrid, F. ISGylation—A key to lock the cell gates for preventing the spread of threats. J. Cell Sci. 2017, 130, 2961–2969. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Sethi, G.; Zhang, Z.; Wang, Q. The Emerging Roles of the HERC Ubiquitin Ligases in Cancer. Curr. Pharm. Des. 2018, 24, 1676–1681. [Google Scholar] [CrossRef]
- Walz, C.; Grimwade, D.; Saussele, S.; Lengfelder, E.; Haferlach, C.; Schnittger, S.; Lafage-Pochitaloff, M.; Hochhaus, A.; Cross, N.C.P.; Reiter, A. Atypical mRNA fusions in PML-RARA positive, RARA-PML negative acute promyelocytic leukemia. Genes Chromosom. Cancer 2010, 49, 471–479. [Google Scholar]
- Opatz, S.; Bamopoulos, S.A.; Metzeler, K.H.; Herold, T.; Ksienzyk, B.; Bräundl, K.; Tschuri, S.; Vosberg, S.; Konstandin, N.P.; Wang, C.; et al. The clinical mutatome of core binding factor leukemia. Leukemia 2020, 34, 1553–1562. [Google Scholar] [CrossRef] [Green Version]
- Diouf, B.; Pei, D.; Fan, Y.; Cheng, C.; Krynetskiy, E.Y.; Geng, H.; Chen, S.; Thierfelder, W.E.; Mullighan, C.G.; Downing, J.R.; et al. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat. Med. 2012, 17, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Vosberg, S.; Schlee, C.; Heesch, S.; Schwartz, S.; Gökbuget, N.; Hoelzer, D.; Graf, A.; Krebs, S.; Bartram, I.; et al. Mutational spectrum of adult T-ALL. Oncotarget 2015, 6, 2754–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, P.; Klein-Hitpass, L.; Choidas, A.; Habenberger, P.; Mahboubi, B.; Kim, B.; Bergmann, A.; Scholtysik, R.; Brauser, M.; Lollies, A.; et al. SAMHD1 is recurrently mutated in T-cell prolymphocytic leukemia. Blood Cancer J. 2018, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloway, A.; Simmonds, M.; Azad, A.; Fox, J.L.; Storey, A. Resistance to UV-induced apoptosis by β-HPV5 E6 involves targeting of activated BAK for proteolysis by recruitment of the HERC1 ubiquitin ligase. Int. J. Cancer 2015, 136, 2831–2843. [Google Scholar] [CrossRef]
- Fan, X.; Lin, L.; Wang, J.; Wang, Y.; Feng, A.; Nie, L.; Wu, H.; Meng, F.; Xu, H. Genome profile in a extremely rare case of pulmonary sclerosing pneumocytoma presenting with diffusely-scattered nodules in the right lung. Cancer Biol. Ther. 2018, 19, 13–19. [Google Scholar] [CrossRef]
- Ping, Z.; Siegal, G.P.; Harada, S.; Eltoum, I.E.; Youssef, M.; Shen, T.; He, J.; Huang, Y.; Chen, D.; Li, Y.; et al. ERBB2 mutation is associated with a worse prognosis in patients with CDH1 altered invasive lobular cancer of the breast. Oncotarget 2016, 7, 80655–80663. [Google Scholar] [CrossRef] [Green Version]
- Craig, D.W.; O’Shaughnessy, J.A.; Kiefer, J.A.; Aldrich, J.; Sinari, S.; Moses, T.M.; Wong, S.; Dinh, J.; Christoforides, A.; Blum, J.L.; et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol. Cancer Ther. 2013, 12, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Yang, D.; Li, X.; Feng, F. Heterogeneous expression and biological function of SOX18 in osteosaroma. J. Cell. Biochem. 2018, 119, 4184–4192. [Google Scholar] [CrossRef]
- Suh, Y.J.; Choe, J.Y.; Park, H.J. Malignancy in Pheochromocytoma or Paraganglioma: Integrative Analysis of 176 Cases in TCGA. Endocr. Pathol. 2017, 28, 159–164. [Google Scholar] [CrossRef]
- Ibarrola-Villava, M.; Fernandez, L.P.; Pita, G.; Bravo, J.; Floristan, U.; Sendagorta, E.; Feito, M.; Avilés, J.A.; Martin-Gonzalez, M.; Lázaro, P.; et al. Genetic analysis of three important genes in pigmentation and melanoma susceptibility: CDKN2A, MC1R and HERC2/OCA2. Exp. Dermatol. 2010, 19, 836–844. [Google Scholar] [CrossRef]
- Amos, C.I.; Wang, L.E.; Lee, J.E.; Gershenwald, J.E.; Chen, W.V.; Fang, S.; Kosoy, R.; Zhang, M.; Qureshi, A.A.; Vattathil, S.; et al. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Hum. Mol. Genet. 2011, 20, 5012–5023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fesenko, D.O.; Chudinov, A.V.; Surzhikov, S.A.; Zasedatelev, A.S. Biochip-Based Genotyping Assay for Detection of Polymorphisms in Pigmentation Genes Associated with Cutaneous Melanoma. Genet. Test. Mol. Biomarkers 2016, 20, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Laino, A.M.; Berry, E.G.; Jagirdar, K.; Lee, K.J.; Duffy, D.L.; Soyer, H.P.; Sturm, R.A. Iris pigmented lesions as a marker of cutaneous melanoma risk: An Australian case–control study. Br. J. Dermatol. 2018, 178, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Ma, J.; Cai, G.; Fang, S.; Lee, J.E.; Wei, Q.; Amos, C.I. Natural and orthogonal model for estimating gene-gene interactions applied to cutaneous melanoma. Hum. Genet. 2014, 133, 559–574. [Google Scholar] [CrossRef] [Green Version]
- Kosiniak-Kamysz, A.; Marczakiewicz-Lustig, A.; Marcińska, M.; Skowron, M.; Wojas-Pelc, A.; Pośpiech, E.; Branicki, W. Increased risk of developing cutaneous malignant melanoma is associated with variation in pigmentation genes and VDR, and may involve epistatic effects. Melanoma Res. 2014, 24, 388–396. [Google Scholar] [CrossRef]
- Wei, L.; Allain, D.C.; Bernhardt, M.N.; Gillespie, J.L.; Peters, S.B.; Iwenofu, O.H.; Nelson, H.H.; Arron, S.T.; Toland, A.E. Variants at the OCA2/HERC2 locus affect time to first cutaneous squamous cell carcinoma in solid organ transplant recipients collected using two different study designs. Br. J. Dermatol. 2017, 177, 1066–1073. [Google Scholar] [CrossRef]
- Ferguson, R.; Vogelsang, M.; Ucisik-Akkaya, E.; Rai, K.; Pilarski, R.; Martinez, C.N.; Rendleman, J.; Kazlow, E.; Nagdimov, K.; Osman, I.; et al. Genetic markers of pigmentation are novel risk loci for uveal melanoma. Sci. Rep. 2016, 6, 31191. [Google Scholar] [CrossRef]
- Bonanno, L.; Costa, C.; Majem, M.; Sanchez, J.J.; Rodriguez, I.; Gimenez-Capitan, A.; Molina-Vila, M.A.; Vergnenegre, A.; Massuti, B.; Favaretto, A.; et al. Combinatory effect of BRCA1 and HERC2 expression on outcome in advanced non-small-cell lung cancer. BMC Cancer 2016, 16, 312. [Google Scholar] [CrossRef]
- Wu, W.; Sato, K.; Koike, A.; Nishikawa, H.; Koizumi, H.; Venkitaraman, A.R.; Ohta, T. HERC2 is an E3 ligase that targets BRCA1 for degradation. Cancer Res. 2010, 70, 6384–6392. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Dai, H.; Wang, E.; Lin, C.C.J.; Mo, W.; Peng, G.; Lin, S.Y. TUSC4 functions as a tumor suppressor by regulating BRCA1 stability. Cancer Res. 2015, 75, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Yoo, N.J.; Park, S.W.; Lee, S.H. Frameshift mutations of ubiquitinationrelated genes HERC2, HERC3, TRIP12, UBE2Q1 and UBE4B in gastric and colorectal carcinomas with microsatellite instability. Pathology 2011, 43, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, J.; Chen, L.; Qi, S.; Yu, S.; Weng, Z.; Hu, Z.; Zhou, Q.; Xin, Z.; Shi, L.; et al. HERC3-Mediated SMAD7 ubiquitination degradation promotes autophagy-induced EMT and chemoresistance in Glioblastoma. Clin. Cancer Res. 2019, 25, 3602–3616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Tong, J.; Tang, X.; Juan, J.; Cao, B.; Hurren, R.; Chen, G.; Taylor, P.; Xu, X.; Shi, C.X.; et al. The ubiquitin ligase HERC4 mediates c-Maf ubiquitination and delays the growth of multiple myeloma xenografts in nude mice. Blood 2016, 127, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.L.; Chen, Y.W.; Zhou, H.; Zhou, J.Y.; Wei, M.; Shi, R. Expression of HERC4 in lung cancer and its correlation with clinicopathological parameters. Asian Pac. J. Cancer Prev. 2015, 16, 513–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Yao, X.; Shan, Z.; Li, W.; Gao, Y.; Zhang, Q. E3 ligase Herc4 regulates Hedgehog signalling through promoting Smoothened degradation. J. Mol. Cell Biol. 2019, 11, 791–803. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Sun, B.; Cui, M.; Zhou, Z. HERC4 exerts an anti-tumor role through destabilizing the oncoprotein Smo. Biochem. Biophys. Res. Commun. 2019, 513, 1013–1018. [Google Scholar] [CrossRef]
- Zhou, H.; Shi, R.; Wei, M.; Zheng, W.L.; Zhou, J.Y.; Ma, W.L. The expression and clinical significance of HERC4 in breast cancer. Cancer Cell Int. 2013, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Ji, K.; Wu, M.; Hao, B.; Yao, K.T.; Xu, Y. A miRNA-HERC4 pathway promotes breast tumorigenesis by inactivating tumor suppressor LATS1. Protein Cell 2019, 10, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Li, J.; Pan, C.; Zhou, G.; Zhuge, L.; Jin, L.; Fang, P. HERC4 Is Overexpressed in Hepatocellular Carcinoma and Contributes to the Proliferation and Migration of Hepatocellular Carcinoma Cells. DNA Cell Biol. 2017, 36, 490–500. [Google Scholar] [CrossRef]
- Wang, H.W.; Wu, Y.H.; Hsieh, J.Y.; Liang, M.L.; Chao, M.E.; Liu, D.J.; Hsu, M.T.; Wong, T.T. Pediatric primary central nervous system germ cell tumors of different prognosis groups show characteristic miRNome traits and chromosome copy number variations. BMC Genom. 2010, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, S.; Patil, V.; Mahalingam, K.; Somasundaram, K. Elucidation of the genetic and epigenetic landscape alterations in RNA binding proteins in glioblastoma. Oncotarget 2017, 8, 16650–16668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, P.; Yao, B.; Wei, L.; Zhu, H.; Fang, C.; Zhao, Y. Construction of prognostic risk prediction model based on high-throughput sequencing expression profile data in childhood acute myeloid leukemia. Blood Cells Mol. Dis. 2019, 77, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.B.; Khora, S.S.; Suresh, A. Molecular prognosticators in clinically and pathologically distinct cohorts of head and neck squamous cell carcinoma—A meta-analysis approach. PLoS ONE 2019, 14, e0218989. [Google Scholar] [CrossRef] [PubMed]
- Wrage, M.; Hagmann, W.; Kemming, D.; Uzunoglu, F.G.; Riethdorf, S.; Effenberger, K.; Westphal, M.; Lamszus, K.; Kim, S.Z.; Becker, N.; et al. Identification of HERC5 and its potential role in NSCLC progression. Int. J. Cancer 2015, 136, 2264–2272. [Google Scholar] [CrossRef]
- Tang, J.; Yang, Q.; Cui, Q.; Zhang, D.; Kong, D.; Liao, X.; Ren, J.; Gong, Y.; Wu, G. Weighted gene correlation network analysis identifies RSAD2, HERC5, and CCL8 as prognostic candidates for breast cancer. J. Cell. Physiol. 2020, 235, 394–407. [Google Scholar] [CrossRef]
- Xue, F.; Higgs, B.W.; Huang, J.; Morehouse, C.; Zhu, W.; Yao, X.; Brohawn, P.; Xiao, Z.; Sebastian, Y.; Liu, Z.; et al. HERC5 is a prognostic biomarker for post-liver transplant recurrent human hepatocellular carcinoma. J. Transl. Med. 2015, 13, 379. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, Q.; Xu, T.; Li, C.Y.; Zhou, D.D.; Zhang, L. HZ-6d targeted HERC5 to regulate p53 ISGylation in human hepatocellular carcinoma. Toxicol. Appl. Pharmacol. 2017, 334, 180–191. [Google Scholar] [CrossRef]
- Januchowski, R.; Sterzyńska, K.; Zawierucha, P.; Ruciński, M.; Świerczewska, M.; Partyka, M.; Bednarek-Rajewska, K.; Brazert, M.; Nowicki, M.; Zabel, M.; et al. Microarray-based detection and expression analysis of new genes associated with drug resistance in ovarian cancer cell lines. Oncotarget 2017, 8, 49944–49958. [Google Scholar] [CrossRef] [Green Version]
- Swierczewska, M.; Klejewski, A.; Wojtowicz, K.; Brazert, M.; Izycki, D.; Nowicki, M.; Zabel, M.; Januchowski, R. New and old genes associated with primary and established responses to cisplatin and topotecan treatment in ovarian cancer cell lines. Molecules 2017, 22, 1717. [Google Scholar] [CrossRef] [Green Version]
- Schneider, T.; Martinez-Martinez, A.; Cubillos-Rojas, M.; Bartrons, R.; Ventura, F.; Rosa, J.L. Large HERCS function as tumor suppressors. Front. Oncol. 2019, 9, 524. [Google Scholar] [CrossRef]
- Tong, H.V.; Hoan, N.X.; Binh, M.T.; Quyen, D.T.; Meyer, C.G.; Hang, D.T.T.; Hang, D.T.D.; Son, H.A.; Luong, H.V.; Thuan, N.D.; et al. Upregulation of enzymes involved in isgylation and ubiquitination in patients with hepatocellular carcinoma. Int. J. Med. Sci. 2020, 17, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.H.; Lindsey-Boltz, L.A.; Reardon, J.T.; Sancar, A. Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2010, 107, 4890–4895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.H.; Park, J.M.; Leem, S.H.; Kang, T.H. Coordinated regulation of XPA stability by ATR and HERC2 during nucleotide excision repair. Oncogene 2014, 33, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Greenberg, R.A. Deciphering the BRCA1 tumor suppressor network. J. Biol. Chem. 2015, 290, 17724–17732. [Google Scholar] [CrossRef] [Green Version]
- Bekker-Jensen, S.; Danielsen, J.R.; Fugger, K.; Gromova, I.; Nerstedt, A.; Bartek, J.; Lukas, J.; Mailand, N. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell Biol. 2010, 12, 80–86. [Google Scholar] [CrossRef]
- Danielsen, J.R.; Povlsen, L.K.; Villumsen, B.H.; Streicher, W.; Nilsson, J.; Wikström, M.; Bekker-Jensen, S.; Mailand, N. DNA damage-inducible SUMOylation of HERC2 promotes RNF8 binding via a novel SUMO-binding Zinc finger. J. Cell Biol. 2012, 197, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yang, H.; Wang, H. The histone H2A deubiquitinase USP16 interacts with HERC2 and fine-tunes cellular response to DNA damage. J. Biol. Chem. 2014, 289, 32883–32894. [Google Scholar] [CrossRef] [Green Version]
- Izawa, N.; Wu, W.; Sato, K.; Nishikawa, H.; Kato, A.; Boku, N.; Itoh, F.; Ohta, T. HERC2 interacts with claspin and regulates DNA origin firing and replication fork progression. Cancer Res. 2011, 71, 5621–5625. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Zhao, H.; Liao, J.; Xu, X. HERC2/USP20 coordinates CHK1 activation by modulating CLASPIN stability. Nucleic Acids Res. 2014, 42, 13074–13081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Luo, K.; Deng, M.; Li, Y.; Yin, P.; Gao, B.; Fang, Y.; Wu, P.; Liu, T.; Lou, Z. HERC2-USP20 axis regulates DNA damage checkpoint through Claspin. Nucleic Acids Res. 2014, 42, 13110–13121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Rokutanda, N.; Takeuchi, J.; Lai, Y.; Maruyama, R.; Togashi, Y.; Nishikawa, H.; Arai, N.; Miyoshi, Y.; Suzuki, N.; et al. HERC2 facilitates BLM and WRN helicase complex interaction with RPA to suppress G-quadruplex DNA. Cancer Res. 2018, 78, 6371–6385. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Zhu, M.; Wu, W.; Rokutanda, N.; Togashi, Y.; Liang, W.; Ohta, T. HERC2 regulates RPA2 by mediating ATR-induced Ser33 phosphorylation and ubiquitin-dependent degradation. Sci. Rep. 2019, 9, 14257. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Rojas, M.; Amair-Pinedo, F.; Peiró-Jordán, R.; Bartrons, R.; Ventura, F.; Rosa, J.L. The E3 ubiquitin protein ligase HERC2 modulates the activity of tumor protein p53 by regulating its oligomerization. J. Biol. Chem. 2014, 289, 14782–14795. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Rojas, M.; Schneider, T.; Bartrons, R.; Ventura, F.; Rosa, J.L. NEURL4 regulates the transcriptional activity of tumor suppressor protein p53 by modulating its oligomerization. Oncotarget 2017, 8, 61824–61836. [Google Scholar] [CrossRef] [Green Version]
- García-Cano, J.; Sánchez-Tena, S.; Sala-Gaston, J.; Figueras, A.; Viñals, F.; Bartrons, R.; Ventura, F.; Rosa, J.L. Regulation of the MDM2-p53 pathway by the ubiquitin ligase HERC2. Mol. Oncol. 2020, 14, 69–86. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, Á.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Montagut, C.; Settleman, J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009, 283, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Noel, G.; Galligan, J.T.; Sowa, M.E.; Arndt, V.; Overton, T.M.; Harper, J.W.; Howley, P.M. Identification and Proteomic Analysis of Distinct UBE3A/E6AP Protein Complexes. Mol. Cell. Biol. 2012, 32, 3095–3106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Noël, G.; Luck, K.; Kühnle, S.; Desbuleux, A.; Szajner, P.; Galligan, J.T.; Rodriguez, D.; Zheng, L.; Boyland, K.; Leclere, F.; et al. Network Analysis of UBE3A/E6AP-Associated Proteins Provides Connections to Several Distinct Cellular Processes. J. Mol. Biol. 2018, 430, 1024–1050. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Martinez-Martinez, A.; Cubillos-Rojas, M.; Bartrons, R.; Ventura, F.; Rosa, J.L. The E3 ubiquitin ligase HERC1 controls the ERK signaling pathway targeting C-RAF for degradation. Oncotarget 2018, 9, 31531–31548. [Google Scholar] [CrossRef]
- Pedrazza, L.; Schneider, T.; Bartrons, R.; Ventura, F.; Rosa, J.L. The ubiquitin ligase HERC1 regulates cell migration via RAF-dependent regulation of MKK3/p38 signaling. Sci. Rep. 2020, 10, 824. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Sanclemente, M.; Francoz, S.; Esteban-Burgos, L.; Bousquet-Mur, E.; Djurec, M.; Lopez-Casas, P.P.; Hidalgo, M.; Guerra, C.; Drosten, M.; Musteanu, M.; et al. c-RAF Ablation Induces Regression of Advanced Kras/Trp53 Mutant Lung Adenocarcinomas by a Mechanism Independent of MAPK Signaling. Cancer Cell 2018, 33, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Blasco, M.T.; Navas, C.; Martín-Serrano, G.; Graña-Castro, O.; Lechuga, C.G.; Martín-Díaz, L.; Djurec, M.; Li, J.; Morales-Cacho, L.; Esteban-Burgos, L.; et al. Complete Regression of Advanced Pancreatic Ductal Adenocarcinomas upon Combined Inhibition of EGFR and C-RAF. Cancer Cell 2019, 35, 573–587. [Google Scholar] [CrossRef] [Green Version]
- Ashworth, A.; Lord, C.J.; Reis-Filho, J.S. Genetic interactions in cancer progression and treatment. Cell 2011, 145, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Ubhi, T.; Brown, G.W. Exploiting DNA replication stress for cancer treatment. Cancer Res. 2019, 79, 1730–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, H.; McCabe, H.; Lord, C.J.; Tutt, A.H.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Associated Cancers | Related Molecular Mechanisms | Reference |
|---|---|---|---|
| HERC1 | Acute promyelocytic leukemia | HERC1-PML genomic fusion | [9] |
| Acute Myeloid Leukemia | HERC1 mutations | [10] | |
| Acute lymphoblastic leukemia | Decreased MSH2 protein levels and HERC1 deletions | [11] | |
| Adult T-cell acute lymphoblastic leukemia | HERC1 mutations | [12] | |
| T-cell prolymphocytic leukemia | HERC1 mutations | [13] | |
| Non-melanoma skin cancer | Enhanced BAK protein degradation | [14] | |
| Pulmonary sclerosing pneumocytoma | HERC1 mutations | [15] | |
| Invasive lobular breast cancer | HERC1 mutations | [16] | |
| Metastatic triple-negative breast cancer | HERC1 mutations | [17] | |
| Sporadic colorectal cancer | Decreased MSH2 protein levels and HERC1 deletions | [11] | |
| Osteosarcoma | Negative correlation of SOX18 overexpression and HERC1 mRNA levels | [18] | |
| HERC2 | Pheochromocytoma and paraganglioma | HERC2 mutations | [19] |
| T-cell prolymphocytic leukemia | HERC2 mutations | [13] | |
| Cutaneous melanoma | SNPs in HERC2 gene increase susceptibility | [20,21,22,23] | |
| Gene-gene interactions between HERC2 gene and IL31RA and DDX4 genes | [24] | ||
| Epistatic effects between HERC2 and VDR genes | [25] | ||
| Cutaneous squamous cell carcinoma | SNPs in HERC2 gene impact on time to develop the tumor in organ transplant recipients | [26] | |
| Uveal melanoma | SNPs in HERC2 gene increase susceptibility | [27] | |
| Non-small-cell lung cancer | Worse prognosis in patients expressing high HERC2 mRNA levels | [28] | |
| Breast cancer | Enhanced BRCA1 degradation | [29,30] | |
| Gastric and colorectal carcinomas | HERC2 mutations | [31] | |
| Osteosarcoma | Negative correlation of SOX18 overexpression and HERC2 mRNA levels | [18] | |
| HERC3 | Glioblastoma | Degradation of SMAD7 and activation of the TGFβ signaling | [32] |
| Gastric and colorectal carcinomas | HERC3 mutations | [31] | |
| Osteosarcoma | Negative correlation of SOX18 overexpression and HERC3 mRNA levels | [18] | |
| HERC4 | Multiple myeloma | Decreased c-Maf degradation | [33] |
| Lung cancer | HERC4 overexpression | [34] | |
| Non-small cell lung cancer | Increased Smo protein stability and Hh pathway activation | [35,36] | |
| Breast cancer | HERC4 upregulation | [37] | |
| Decreased expression of miRNAs targeting HERC4 expression and enhanced LATS1 degradation | [38] | ||
| Hepatocellular carcinoma | HERC4 overexpression | [39] | |
| Osteosarcoma | Negative correlation of SOX18 overexpression and HERC4 mRNA levels | [18] | |
| HERC5 | Pediatric germ cell tumors | Chromosome copy number variations (CNVs) at a region encompassing HERC5 gene | [40] |
| Glioblastoma | HERC5 upregulation | [41] | |
| Acute myeloid leukemia | HERC5 downregulation | [42] | |
| Oropharyngeal cancer | HERC5 gene expression is associated with overall survival | [43] | |
| Non-small cell lung cancer | HERC5 promoter hypermethylation | [44] | |
| Breast cancer | HERC5 upregulation | [45] | |
| Hepatocellular carcinoma | Negative correlation of CCL20 overexpression and HERC5 mRNA levels | [46] | |
| Reduced p53, p21 and Bax/Bcl-2 pathway activation | [47] | ||
| Ovarian cancer | HERC5 upregulation is associated with drug resistance | [48,49] | |
| Osteosarcoma | Negative correlation of SOX18 overexpression and HERC5 mRNA levels | [18] | |
| HERC6 | Osteosarcoma | Negative correlation of SOX18 overexpression and HERC6 mRNA levels | [18] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sala-Gaston, J.; Martinez-Martinez, A.; Pedrazza, L.; Lorenzo-Martín, L.F.; Caloto, R.; Bustelo, X.R.; Ventura, F.; Rosa, J.L. HERC Ubiquitin Ligases in Cancer. Cancers 2020, 12, 1653. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061653
Sala-Gaston J, Martinez-Martinez A, Pedrazza L, Lorenzo-Martín LF, Caloto R, Bustelo XR, Ventura F, Rosa JL. HERC Ubiquitin Ligases in Cancer. Cancers. 2020; 12(6):1653. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061653
Chicago/Turabian StyleSala-Gaston, Joan, Arturo Martinez-Martinez, Leonardo Pedrazza, L. Francisco Lorenzo-Martín, Rubén Caloto, Xosé R. Bustelo, Francesc Ventura, and Jose Luis Rosa. 2020. "HERC Ubiquitin Ligases in Cancer" Cancers 12, no. 6: 1653. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061653