Inhibition of Methyltransferase DOT1L Sensitizes to Sorafenib Treatment AML Cells Irrespective of MLL-Rearrangements: A Novel Therapeutic Strategy for Pediatric AML

 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Results
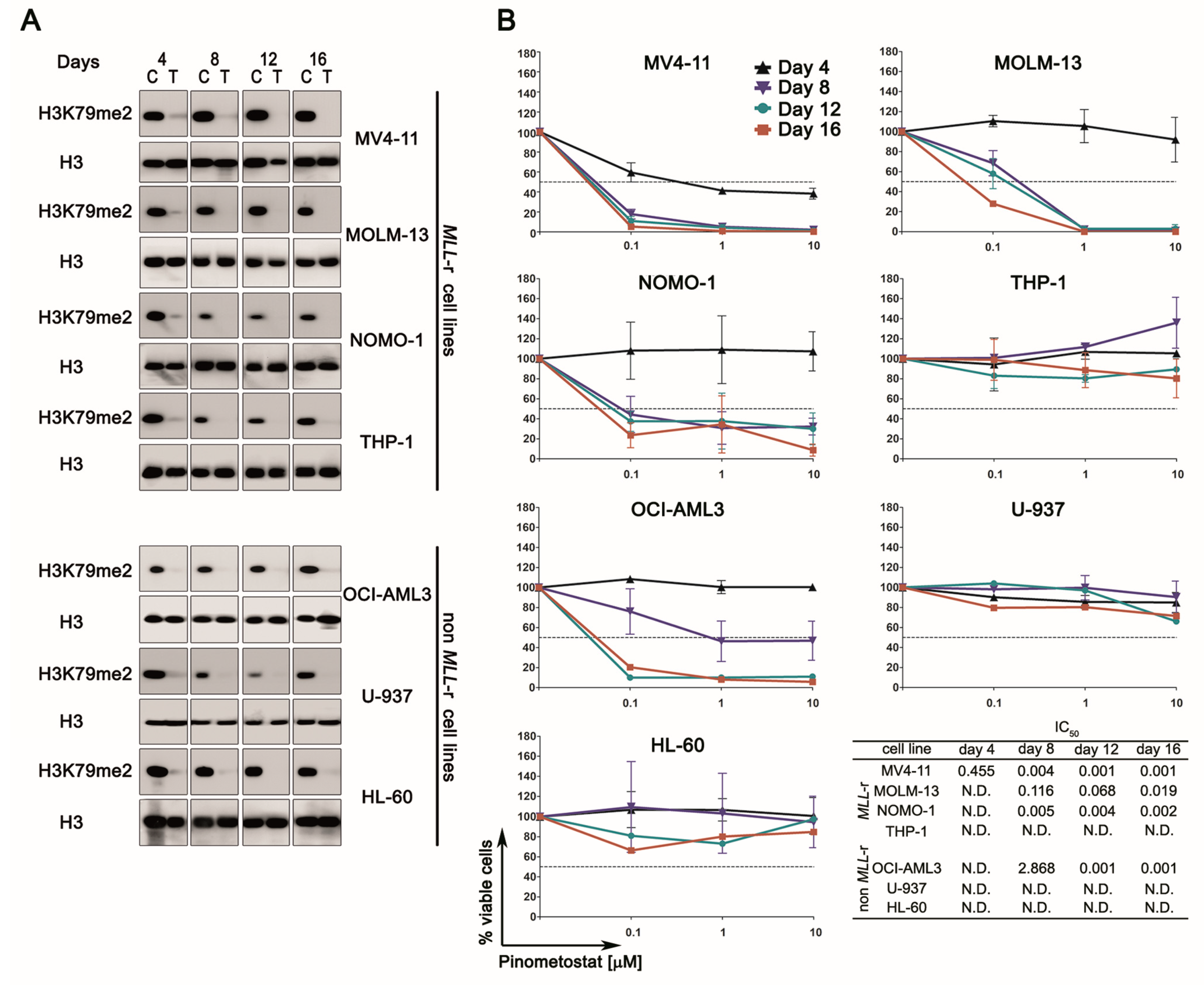
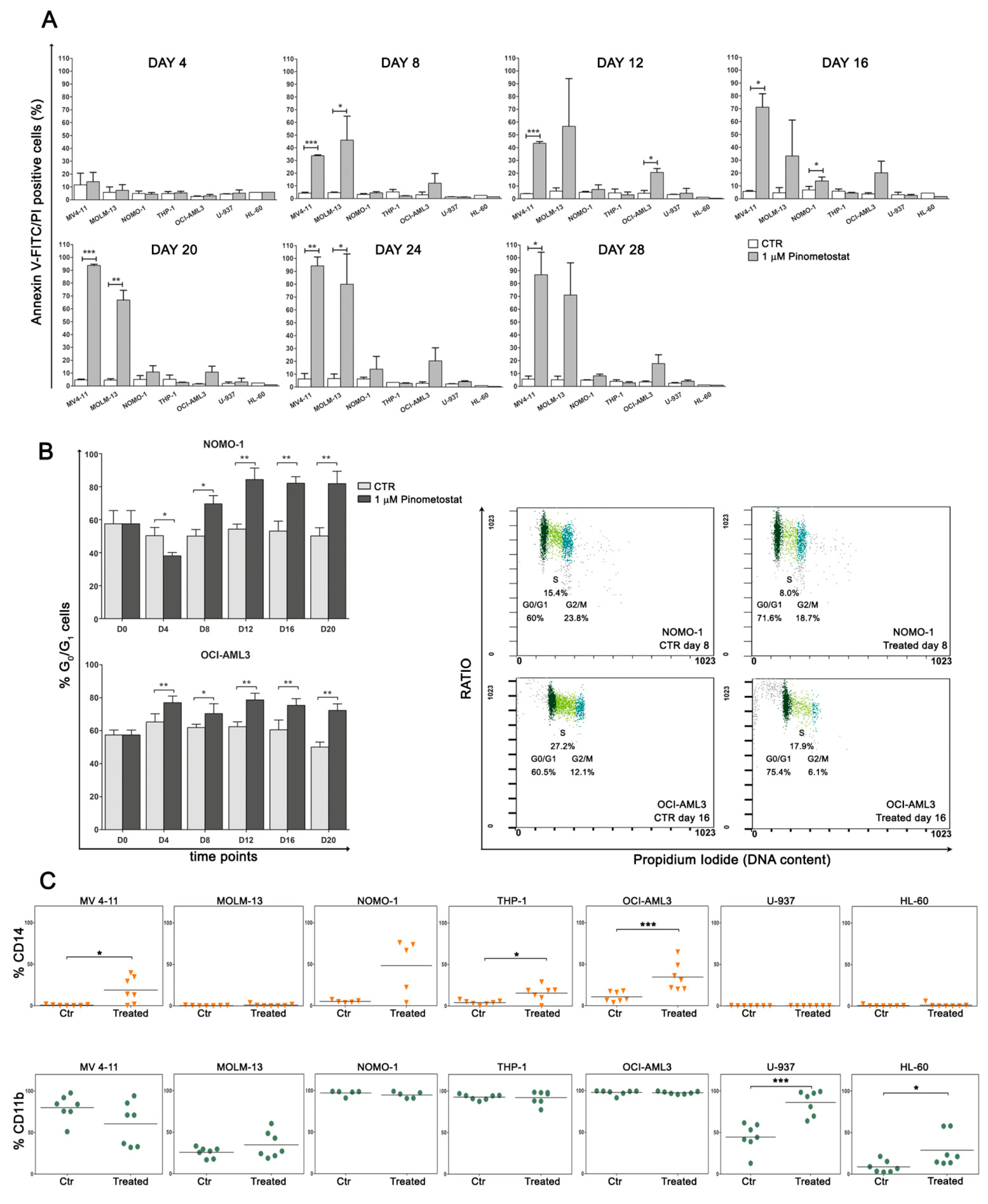
2.1. DOT1L Inhibition Impairs Proliferation and Induces Myeloid Differentiation Irrespective of MLL-r
2.2. DOT1L Inhibition Impacts Proliferation Pathways Which Are Essential in Sustaining AML Cells
2.3. Primary MLL-r AML Cells Are Barely Affected by DOT1L Inhibition
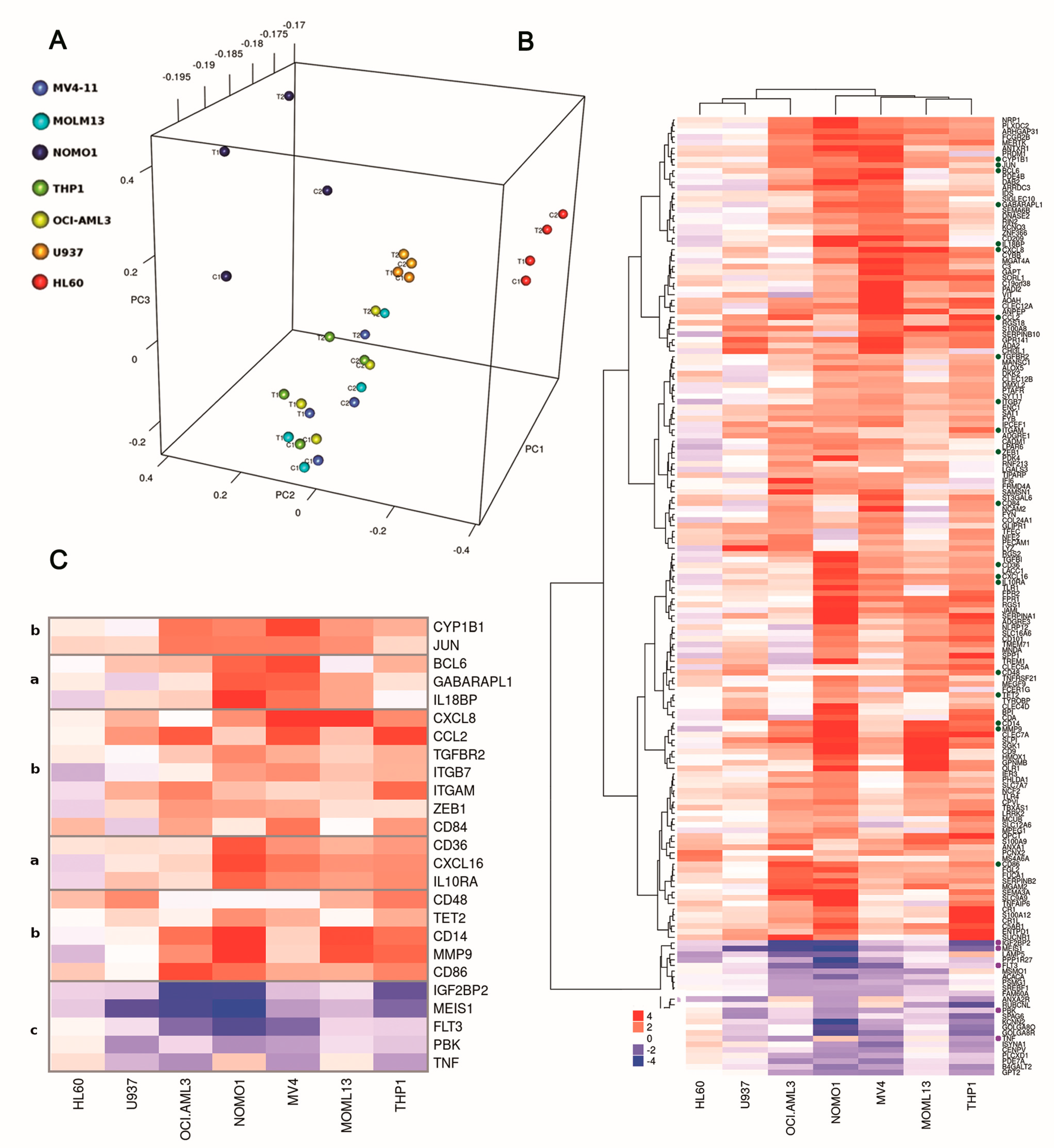
2.4. DOT1L Inhibition Induces Gene Disregulation Not Limited to MLL-r Cells
2.5. Pharmacological Inhibition of DOT1L Sensitizes Both MLL-r and Non-MLL-r AML to Sorafenib Treatment
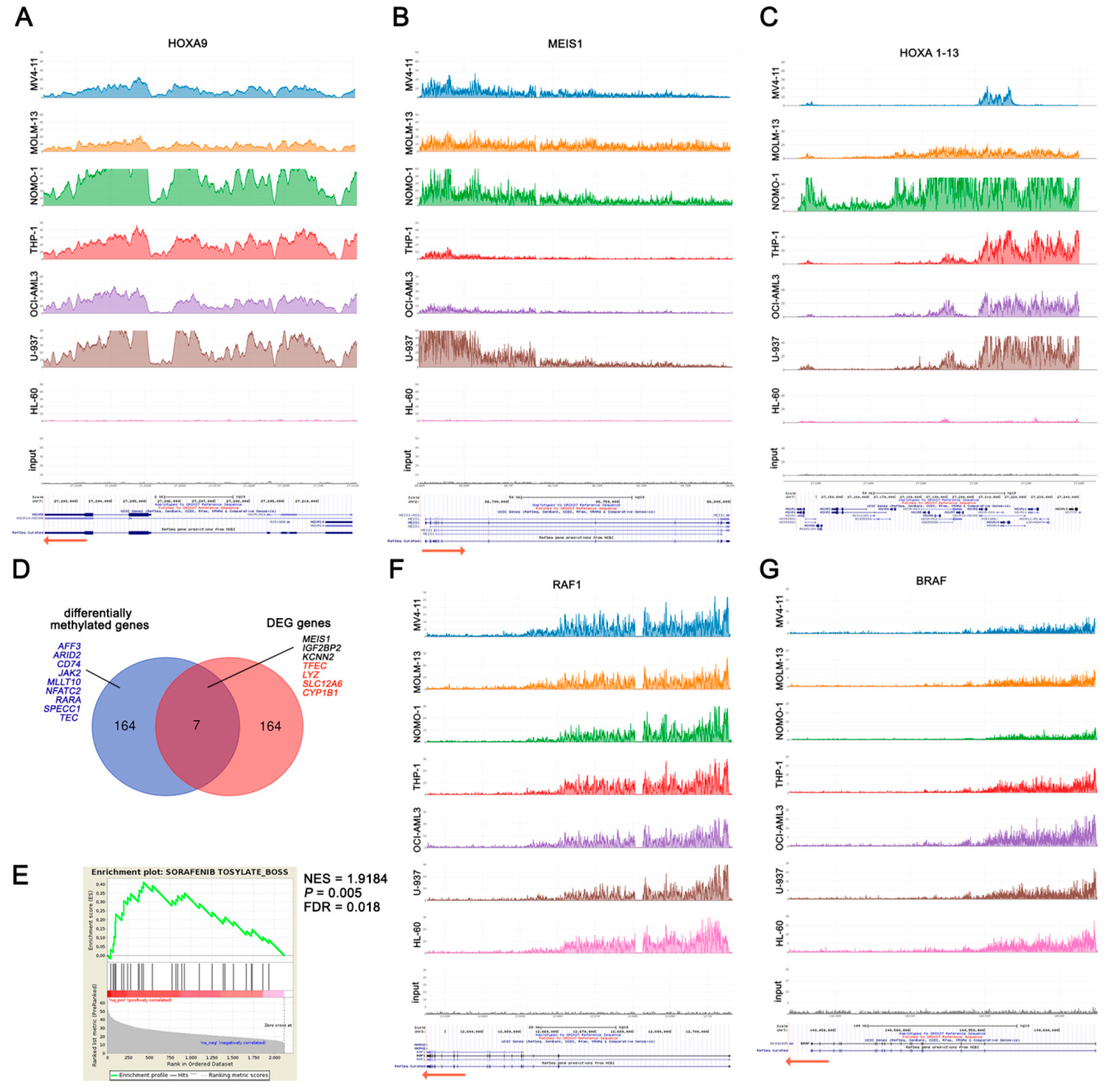
2.6. DOT1L Target Genes Are Mainly Involved in AML Maintenance Irrespective to MLL Fusions
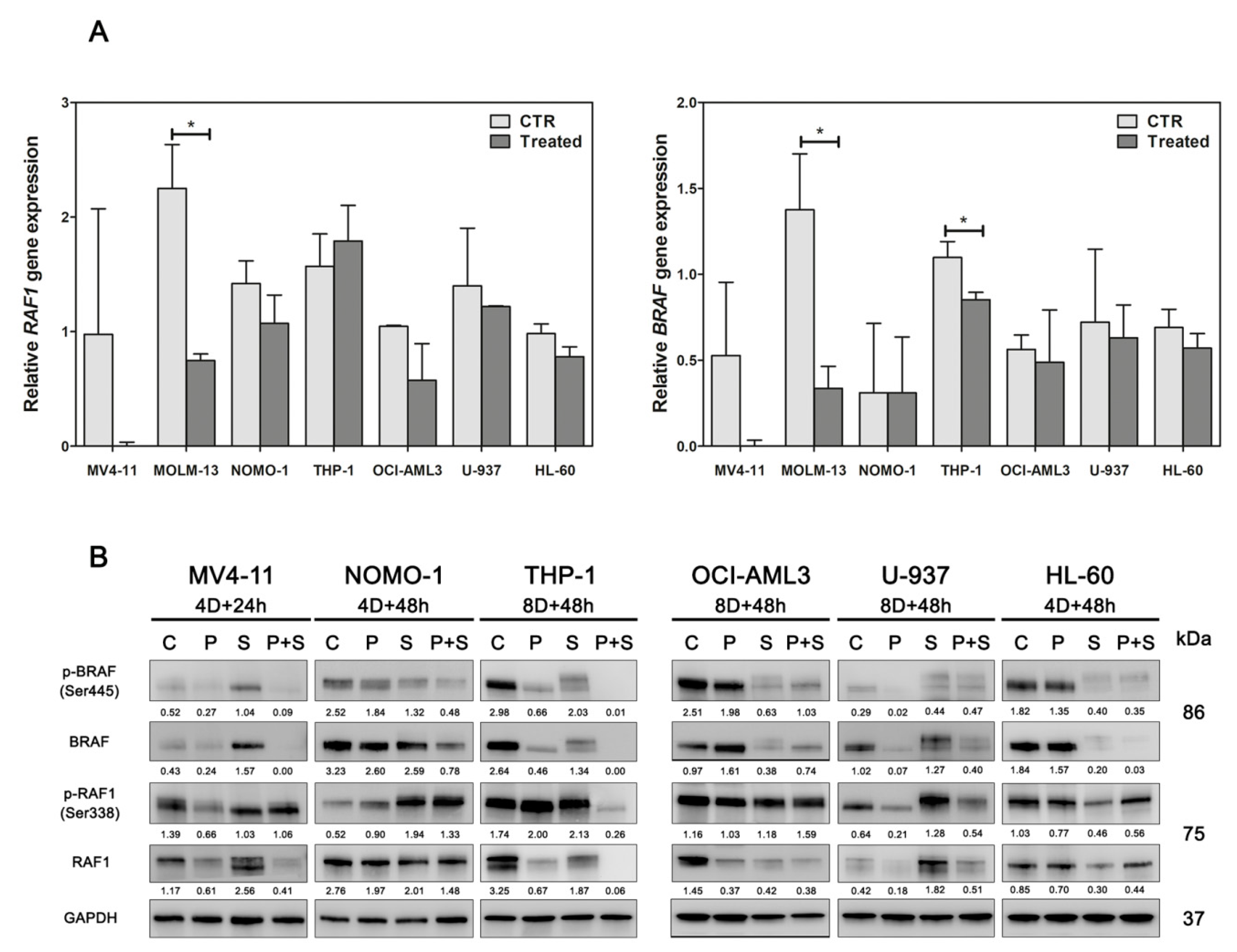
2.7. DOT1L Inhibition Enhances Sorafenib Efficacy through Down-Modulation of BRAF
3. Discussion
4. Material and Methods
4.1. Cell Lines and Primary Cultures of AML
4.2. Inhibitors and Treatments
4.3. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Availability of Data and Materials
References
- Pui, C.H.; Carroll, W.L.; Meshinchi, S.; Arceci, R.J. Biology, risk stratification, and therapy of pediatric acute leukemias: An update. J. Clin. Oncol. 2011, 29, 551–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwaan, C.M.; Kolb, E.A.; Reinhardt, D.; Abrahamsson, J.; Adachi, S.; Aplenc, R.; de Bont, E.S.J.M.; de Moerloose, B.; Dworzak, M.; Gibson, B.; et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 2949–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steger, D.J.; Lefterova, M.I.; Ying, L.; Stonestrom, A.J.; Schupp, M.; Zhuo, D.; Vakoc, A.L.; Kim, J.E.; Chen, J.; Lazar, M.A.; et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol. 2008, 28, 2825–2839. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.T.; Taranova, O.; He, J.; Zhang, Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood 2011, 117, 6912–6922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Deshpande, A.; Banka, D.; Bernt, K.M.; Dias, S.; Buske, C.; Olhava, E.J.; Daigle, S.R.; Richon, V.M.; Pollock, R.M.; et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia 2013, 27, 813–822. [Google Scholar] [CrossRef]
- Mueller, D.; Bach, C.; Zeisig, D.; Garciacuellar, M.P.; Monroe, S.; Sreekumar, A.; Zhou, R.; Nesvizhskii, A.I.; Chinnaiyan, A.M.; Hess, J.L.; et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110, 4445–4454. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Feng, Z.; Lemieux, M.E.; Faber, J.; Vempati, S.; Sinha, A.U.; Xia, X.; Jesneck, J.L.; Bracken, A.P.; Silverman, L.B.; et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 2008, 14, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.; Wu, H.; Achille, N.J.; Reisenauer, M.R.; Chou, C.; Zeleznikle, N.J.; Hemenway, C.S.; Zhang, W. Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res. 2010, 70, 10234–10242. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, A.; Chen, L.; Fazio, M.; Sinha, A.U.; Bernt, K.M.; Banka, D.; Dias, S.; Chang, J.; Olhava, E.J.; Daigle, S.R.; et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 2013, 121, 2533–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Jiang, Q.; Lemieux, M.; Jeannotte, L.; Su, L.; Zhang, Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L. Nat. Cell Biol. 2006, 8, 1017–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, A.E.; Scotland, P.B.; Lavau, C.P.; Wechsler, D.S. A CALM-derived nuclear export signal is essential for CALM-AF10-mediated leukemogenesis. Blood 2013, 121, 4758–4768. [Google Scholar] [CrossRef] [Green Version]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriacksjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.M.; Garciamanero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongenlavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef]
- Kuhn, M.W.M.; Hadler, M.; Daigle, S.R.; Koche, R.; Krivtsov, A.V.; Olhava, E.J.; Caligiuri, M.A.; Huang, G.; Bradner, J.E.; Pollock, R.M.; et al. MLL partial tandem duplication leukemia cells are sensitive to small molecule DOT1L inhibition. Haematologica 2015, 100, e190–e193. [Google Scholar] [CrossRef] [Green Version]
- Sarkaria, S.M.; Christopher, M.J.; Klco, J.M.; Ley, T.J. Primary acute myeloid leukemia cells with IDH1 or IDH2 mutations respond to a DOT1L inhibitor in vitro. Leukemia 2014, 28, 2403–2406. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M.W.M.; Song, E.; Feng, Z.; Sinha, A.U.; Chen, C.; Deshpande, A.; Cusan, M.; Farnoud, N.; Mupo, A.; Grove, C.S.; et al. Targeting Chromatin Regulators Inhibits Leukemogenic Gene Expression in NPM1 Mutant Leukemia. Cancer Discov. 2016, 6, 1166–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Wang, P.; Parton, T.; Zhou, Y.; Chrysovergis, K.; Rockowitz, S.; Chen, W.; Abdelwahab, O.; Wade, P.A.; Zheng, D.; et al. Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer Cell 2016, 30, 92–107. [Google Scholar] [CrossRef]
- Rau, R.E.; Rodriguez, B.; Luo, M.; Jeong, M.; Rosen, A.; Rogers, J.H.; Campbell, C.T.; Daigle, S.R.; Deng, L.; Song, Y.; et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 2016, 128, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Deng, L.; Song, Y.; Redell, M. DOT1L inhibition sensitizes MLL-rearranged AML to chemotherapy. PLoS ONE 2014, 9, e98270. [Google Scholar] [CrossRef] [PubMed]
- Klaus, C.R.; Iwanowicz, D.; Johnston, L.D.; Campbell, C.A.; Smith, J.J.; Moyer, M.P.; Copeland, R.A.; Olhava, E.J.; Scott, M.P.; Pollock, R.M.; et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J. Pharmacol. Exp. Ther. 2014, 350, 646–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borthakur, G.; Kantarjian, H.M.; Ravandi, F.; Zhang, W.; Konopleva, M.; Wright, J.J.; Faderl, S.; Verstovsek, S.; Mathews, S.; Andreeff, M.; et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica 2011, 96, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, D.; Li, M.; Cao, C.; Wan, D.; Xi, B.; Li, W.; Tan, J.; Wang, J.; Wu, Z.; et al. Prognostic and therapeutic value of disruptor of telomeric silencing-1-like (DOT1L) expression in patients with ovarian cancer. J. Hematol. Oncol. 2017, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Widemann, B.C.; Kim, A.R.; Fox, E.; Baruchel, S.; Adamson, P.C.; Ingle, A.M.; Bender, J.L.G.; Burke, M.J.; Weigel, B.J.; Stempak, D.; et al. A phase I trial and pharmacokinetic study of sorafenib in children with refractory solid tumors or leukemias: A Children’s Oncology Group Phase I Consortium report. Clin. Cancer Res. 2012, 18, 6011–6022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skucha, A.; Ebner, J.; Grebien, F. Roles of SETD2 in Leukemia-Transcription, DNA-Damage, and Beyond. Int. J. Mol. Sci. 2019, 20, 1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masetti, R.; Pigazzi, M.; Togni, M.; Astolfi, A.; Indio, V.; Manara, E.; Casadio, R.; Pession, A.; Basso, G.; Locatelli, F. CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 2013, 121, 3469–3472. [Google Scholar] [CrossRef] [Green Version]
- Castelli, G.; Pelosi, E.; Testa, U. Targeting histone methyltransferase and demethylase in acute myeloid leukemia therapy. Onco Targets Ther. 2018, 11, 131–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobucci, I.; Iraci, N.; Messina, M.; Lonetti, A.; Chiaretti, S.; Valli, E.; Ferrari, A.; Papayannidis, C.; Paoloni, F.; Vitale, A.; et al. IKAROS deletions dictate a unique gene expression signature in patients with adult B-cell acute lymphoblastic leukemia. PLoS ONE 2012, 7, e40934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilan, O.; Lam, E.Y.N.; Becher, I.; Lugo, D.; Cannizzaro, E.; Joberty, G.; Ward, A.; Wiese, M.; Fong, C.Y.; Ftouni, S.; et al. Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat. Struct. Mol. Biol. 2016, 23, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Nakatsugawa, M.; Saso, K.; Guo, T.; Anczurowski, M.; Wang, C.; Butler, M.O.; Arrowsmith, C.H.; Hirano, N. DOT1L inhibition attenuates graft-versus-host disease by allogeneic T cells in adoptive immunotherapy models. Nat. Commun. 2018, 9, 1915. [Google Scholar] [CrossRef] [PubMed]
- Staffas, A.; Arabanian, L.; Wei, S.Y.; Jansson, A.; Stahlman, S.; Johansson, P.; Fogelstrand, L.; Cammenga, J.; Kuchenbauer, F.; Palmqvist, L. Upregulation of Flt3 is a passive event in Hoxa9/Meis1-induced acute myeloid leukemia in mice. Oncogene 2017, 36, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Pasillas, M.P.; Kamps, M.P. Meis1 programs transcription of FLT3 and cancer stem cell character, using a mechanism that requires interaction with Pbx and a novel function of the Meis1 C-terminus. Blood 2005, 106, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Stirewalt, D.L.; Radich, J.P. The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer 2003, 3, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qin, Y.; Yang, S.; Wang, Y.; Chang, Y.; Zhao, T.; Jiang, Q.; Huang, X. Meis1 is critical to the maintenance of human acute myeloid leukemia cells independent of MLL rearrangements. Ann. Hematol. 2017, 96, 567–574. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Dinardo, C.D.; Gharibyan, V.; Yang, H.; Wei, Y.; Pierce, S.; Kantarjian, H.M.; Garciamanero, G.; Rytting, M.E. Impact of aberrant DNA methylation patterns including CYP1B1 methylation in adolescents and young adults with acute lymphocytic leukemia. Am. J. Hematol. 2013, 88, 784–789. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Li, W.; Liang, X.; Zhu, X.; Zhang, L.; Huang, Y.; Yu, T.; Li, S.; Chen, Z. IGF2BP2 Overexpression Indicates Poor Survival in Patients with Acute Myelocytic Leukemia. Cell. Physiol. Biochem. 2018, 51, 1945–1956. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell division cycle (CDC) genes |
| CDC123 CDC25A CDC27 CDC34 CDC37 CDC40 CDC42 CDC45 CDC6 CDC73 CDCA4 CDCA5 CDCA8 |
| Cyclin-dependent kinases |
| CDK1 CDK12 CDK13 CDK4 CDK6 CDK9 CDKN1B CDKN2C |
| Gene involved in transcription regulation |
| E2F1 E2F3 E2F4 ELOF1 ELL EFTUD2 ELP2 EPC1 EPC2 ERH EZH2 EEF2K EEF1B2 EEF1D EEF1E1 EEF1G EEF2 EIF2S2 EIF2A EIF2B4 EIF2B5 EIF2C2 EIF3B EIF3D EIF3E EIF3H EIF3J EIF3L EIF3M EIF4G1 EIF4G3 EIF4A3 EIF4B EIF4E EIF4EBP1 EIF4EBP2 EIF4H EIF5 EIF5A EIF5B EIF6 ETF1 |
| Bcl-2 family members |
| BCL2 BNIP2 BAK1 BAG1 BAG5 BAG6 BCLAF1 BCL2L11 BID |
| p53 signaling components |
| TP53RK TP53BP2 MDM2 MDM4 |
| Methyltransferases |
| EHMT1 METTL10 METTL11A METTL14 METTL16 METTL19 MLL MLLT1 MLL2 MLL3 MLL5 |
| Members of MAP kinases |
| MAPK1 MAPK1IP1L MAPK14 MAPK6 MAPKAP1 MAP2K2 MAP2K3 MAP3K1 MAP3K4 MAP3K7 MAP4K5 MAPKAPK2 MAPKAPK3 MAPKAPK5 |
| Cancer related genes |
| PDGFRA MYC PIM3 PARP1 POU2F1 CXCR4 CDKN1B IGF2R |
| Genes encoding for proteins with a reported role in leukemia [27,28,29,30] |
| SETD2 CBFA2T3 CEBPA RUNX1 RUNX2 EZH2 IKZF1 CREB1 FBXW7 NPM1 LMO2 LYL1 |
| Genes encoding for proteins with a reported functional interdependence with DOT1L [31,32] |
| BRD4 DUSP6 DOT1L |
| PI3K signaling members |
| AKT2 MCL-1 RICTOR GSK3B PIK3C3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lonetti, A.; Indio, V.; Laginestra, M.A.; Tarantino, G.; Chiarini, F.; Astolfi, A.; Bertuccio, S.N.; Martelli, A.M.; Locatelli, F.; Pession, A.; et al. Inhibition of Methyltransferase DOT1L Sensitizes to Sorafenib Treatment AML Cells Irrespective of MLL-Rearrangements: A Novel Therapeutic Strategy for Pediatric AML. Cancers 2020, 12, 1972. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12071972
Lonetti A, Indio V, Laginestra MA, Tarantino G, Chiarini F, Astolfi A, Bertuccio SN, Martelli AM, Locatelli F, Pession A, et al. Inhibition of Methyltransferase DOT1L Sensitizes to Sorafenib Treatment AML Cells Irrespective of MLL-Rearrangements: A Novel Therapeutic Strategy for Pediatric AML. Cancers. 2020; 12(7):1972. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12071972
Chicago/Turabian StyleLonetti, Annalisa, Valentina Indio, Maria Antonella Laginestra, Giuseppe Tarantino, Francesca Chiarini, Annalisa Astolfi, Salvatore N. Bertuccio, Alberto M. Martelli, Franco Locatelli, Andrea Pession, and et al. 2020. "Inhibition of Methyltransferase DOT1L Sensitizes to Sorafenib Treatment AML Cells Irrespective of MLL-Rearrangements: A Novel Therapeutic Strategy for Pediatric AML" Cancers 12, no. 7: 1972. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12071972