PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies


1
Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02215-5450, USA
2
Division of Gynecologic Oncology, Dana-Farber Cancer Institute, Boston, MA 02215-5450, USA
*
Author to whom correspondence should be addressed.
Cancers 2020, 12(8), 2054; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082054
Submission received: 1 July 2020
/
Revised: 21 July 2020
/
Accepted: 22 July 2020
/
Published: 25 July 2020
(This article belongs to the Special Issue Preclinical and Clinical Advances in Ovarian Cancer)
{kind=link}
{kind=link}
Abstract
:The use of PARP inhibitors (PARPi) is growing widely as FDA approvals have shifted its use from the recurrence setting to the frontline setting. In parallel, the population developing PARPi resistance is increasing. Here we review the role of PARP, DNA damage repair, and synthetic lethality. We discuss mechanisms of resistance to PARP inhibition and how this informs on novel combinations to re-sensitize cancer cells to PARPi.
1. Introduction
Poly(ADP-ribose)polymerase (PARP) inhibitors (PARPi) have changed the treatment landscape of epithelial ovarian cancers (EOC). PARPi are FDA approved across all lines of treatment of EOC. While this expands the treatment armamentarium, this also raises the question of how best to treat patients who progress on a PARPi. Effectively, these patients are considered PARPi resistant, and there arises an urgent need to understand and clinically validate the mechanisms of PARPi resistance, allowing us to determine appropriately matched post-PARPi-progression combination therapies. Additionally, these strategies will also likely be applicable to homologous recombination proficient (HRP) and BRCA wild-type (BRCAwt) cancers, where only minimal benefit is seen with single agent PARPi. Here, we review the role of PARP in DNA repair pathways, the effects of PARPi, mechanisms of resistance, and strategies for subsequent combination therapies.
2. DNA Damage Repair and the Role of PARP
High fidelity DNA repair is integral to cell survival. Therefore, there are several pathways by which DNA damage can be repaired. PARP plays multiple roles in several DNA repair pathways, highlighting where PARPi exerts its cytotoxic function, but also demonstrates the points at which PARPi resistance may arise.
Double strand breaks (DSBs) in DNA are highly toxic lesions for which there are several DNA repair processes, the choice of which is influenced by the cell cycle phase during which the DSB occurs, the availability of a DNA template, and competition between functional effector proteins [1].
Homologous recombination (HR) is one means of repairing DSBs and is restricted to the S and G2 phases when a sister chromatid is present as a repair template. DSBs are recognized by PARP1, whereupon poly(ADP-ribos)ylation (PARylation) initiates a cascade of protein recruitment, including that of BRCA1 and the MRN complex, composed of the endonucleases MRE11, RAD50, and NBS1. The 3′ strand end-resection is initiated by the MRN complex and is continued by other nucleases, such as CTIP, DNA2, and EXO-1. The resultant overhanging single stranded DNA (ssDNA) is coated by replication protein A (RPA). BRCA1, PALB2, and BRCA2 facilitate the replacement of RPA with RAD51, generating a RAD51-loaded nucleoprotein filament; this subsequently facilitates strand invasion of the sister chromatid and error-free, fidelitous DNA synthesis [2].
End-resection is a crucial step in HR and is balanced by antagonism between BRCA1 and 53BP1 [2]. The 53BP1 interacts with the shieldin complex (which includes a REV7 subunit [3,4]) to bind the initial 3′ ssDNA overhangs. The shieldin complex protects against further end-resection, preventing the completion of HR and diverting DNA repair toward the classical non-homologous end-joining (cNHEJ) pathway [3,4,5,6]. Furthermore, DYNL11 (dynein light chain 1) promotes the formation of 53BP1 complexes at DSB sites and represses the activity of the MRN complex [1]. Another negative regulator of HR is DNA helicase B (HELB), which is recruited by RPA to coated ssDNA and represses further end resection, serving as another means to divert DNA repair from HR toward NHEJ.
cNHEJ, the other primary means of DSB repair, is a mechanism that is preferentially employed during the G1 phase [1,7], occurs without a repair template, and is inherently error-prone. Here, the free ends of DNA are bound by the Ku70/Ku80 complex, leading to the recruitment of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and the formation of a DNA-PK complex that recruits downstream ligation proteins. Competition between PARP1 and the Ku70/80 binding of the free DNA ends underlies the anti-cNHEJ role of PARP [8]. The limited amount of end-processing required for end to end ligation leads to deletions and inaccurate gap-filling and explains the mutagenic nature of NHEJ.
If the DSB occurs in a region of DNA with areas of microhomology, and if the Ku complex is absent or out-competed for binding, alternative NHEJ (altNHEJ) can be employed for repair. This process involves PARP1-mediated recruitment of the MRN complex [9], alignment of ssDNA at areas of microhomology, and POLθ- and LIG3-mediated gap-filling and ligation. POLθ is an error-prone polymerase that antagonizes RAD51-mediated recombination, thereby suppressing HR [10]. Similarly, DSBs occurring in areas with longer stretches of homology may rely on the single strand annealing (SSA) pathway of repair [11], which is also inherently mutagenic.
3. Replication Fork Dynamics and the Role of PARP
PARP1 has been shown to modulate replication fork dynamics in situations of replication stress. PARP1 binds the sites of DNA damage, forming a PARP–DNA complex. Replication forks that encounter these complexes become stalled and undergo fork reversal. In this process, nascent ssDNA overhangs are protected by RAD51 filaments, thought to be facilitated by BRCA1 and BRCA2 [2,17]. PARylated PARP, bound to damaged DNA, concomitantly inhibits the DNA helicase Q1 (RECQ1) to prevent premature fork restart [9]. Once the DNA lesion is repaired and PARP1 has dissociated, RECQ1 is no longer inhibited and promotes replication fork restart. There is conflicting evidence that limited end-resection may be required for fork restart, partly facilitated by PARP1-mediated recruitment of MRE11 [9,18]. If the coordination of these steps does not occur, progression of the replication fork into DNA lesions causes fork collapse and the generation of toxic DSBs.
4. Effects of PARP Inhibitors
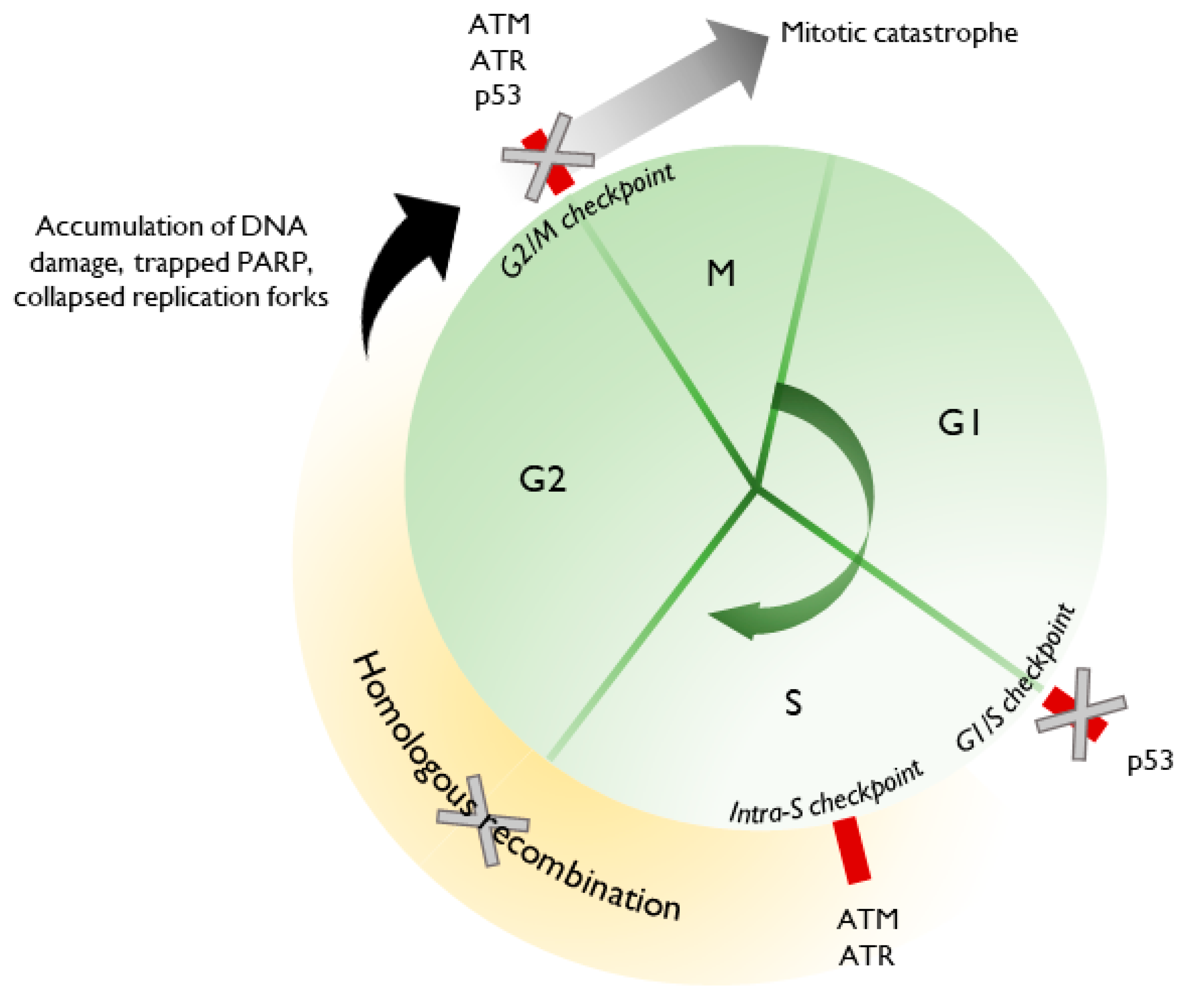
PARPi can be highly injurious to a cancer cell due to its multiple roles in DNA repair and synthesis, which is tightly coordinated with the cell cycle. The deleterious effects of PARPi are further amplified in the context of underlying alterations affecting DNA repair or cell cycle regulation, such as in high grade serous ovarian cancer (HGSOC), the most common EOC histologic subtype. Approximately 50% of HGSOC are HR deficient [19,20], comprising germline (14%) and somatic (6%) BRCA1/2 mutations, BRCA1 promoter methylation (10%) and RAD51C promoter methylation and other RAD mutations (3–4%). Additionally, upwards of 90% of HGSOC patients harbor mutant TP53, which is critical for the appropriate regulation of the cell cycle, acting primarily at the G1/S checkpoint but also the G2/M checkpoint [21,22,23,24,25]. HGSOC is, therefore, particularly susceptible to additional perturbations in the DNA repair process, such as by PARPi.
This forms the basis for synthetic lethality with PARPi [19,26,27]. For example, PARP inhibition allows the accumulation of unrepaired SSBs, which are processed into DSBs during replication. In the absence of a functional HR pathway, and in the setting of unopposed Ku70/80 binding, DSBs are shunted toward error-prone cNHEJ. The accumulation of DNA errors leads to progressive genomic instability and cell death. Replication fork dynamics are also altered. PARPi cause the trapping of PARP1 at sites of DNA lesions, wherein the dissociation of PARP1 is impaired [28]. Without the resolution of these lesions, replication forks remain stalled. PARPi also disrupt the careful coordination of MRE11 and RECQ1, allowing strand degradation and inappropriate fork restart, leading to fork collapse. Furthermore, unresolved DNA lesions carried through a dysregulated cell cycle can precipitate mitotic catastrophe and cell death.
5. Biomarkers of PARP Inhibition
Determining predictive biomarkers of response to PARP inhibition is an area of significant interest and continued investigation [29,30]. The presence of a BRCA mutation [31] or a “BRCA-like” gene expression profile [32] both correlate with PARPi response. Among patients with a germline BRCA mutation, platinum sensitivity and fewer prior lines of therapy were associated with higher response rates and longer durations of response to PARPi [33]. The measurement of HR deficiency, based on genomic characteristics, such as loss of heterozygosity or telomeric allelic imbalance, are utilized in commercial assays, such as the Myriad myChoice HRD assay, and also appear to correlate with PARPi response [29]. Whether level of PARP1 expression correlates with response to PARPi remains under investigation. One study evaluating primary ovarian cancer samples found no correlation [34], whereas a radiotracer-PARP1 study found a significant correlation with response to two PARPi [35].
6. PARP Inhibitors in Ovarian Cancer
The clinical use of PARPi in the treatment of EOC has expanded dramatically. Olaparib, rucaparib, and niraparib were initially approved for use in the recurrence setting as monotherapy [36,37,38] agnostic of sensitivity to platinum, followed by approval as post-chemotherapy maintenance for platinum sensitive disease [39]. PARPi are now FDA approved as frontline maintenance. Olaparib obtained FDA approval in 2018 as maintenance following response to frontline platinum-based therapy for patients with germline or somatic BRCA-mutated EOC [40]. In April 2020, niraparib received FDA approval as maintenance following response to frontline platinum regardless of HR status [41], and the combination of olaparib/bevacizumab received FDA approval in May 2020 as maintenance for patients with HRD EOC [42].
Analysis of the frontline PARPi maintenance studies may yield the best indicators of baseline rates and kinetics of de novo and acquired PARPi resistance, as this population is the least affected by prior lines of treatment. The randomized placebo-controlled phase III SOLO-1 trial studied maintenance olaparib following partial or complete responses to platinum-based frontline chemotherapy in patients with germline or somatic BRCA-mutated advanced HGSOC or high grade endometrioid EOC [43]. Olaparib maintenance was estimated to improve median PFS by approximately 36 months, as median PFS had not yet been reached at the time of data reporting. However, the continued negative slope of the Kaplan–Meier survival curve for patients receiving olaparib highlights disease recurrence despite PARPi treatment. Similarly, the randomized placebo-controlled phase III PRIMA/ENGOT-Ov26 trial studied niraparib in the frontline maintenance setting in patients with or without a known BRCA mutation or evidence of HRD by the Myriad myChoice assay [44]. Median PFS improved from 8.2 months to 13.8 months with niraparib maintenance in the overall population, and prespecified molecular subgroup analysis demonstrated that the benefit was greatest in those with BRCA mutations (median PFS 22.1 versus 10.9 months), followed by those with non-BRCA HR deficiency (19.6 versus 8.2 months). Regardless of this susceptibility, however, disease recurrence substantiates the development of PARPi resistance.
7. Mechanisms of PARP Inhibitor Resistance
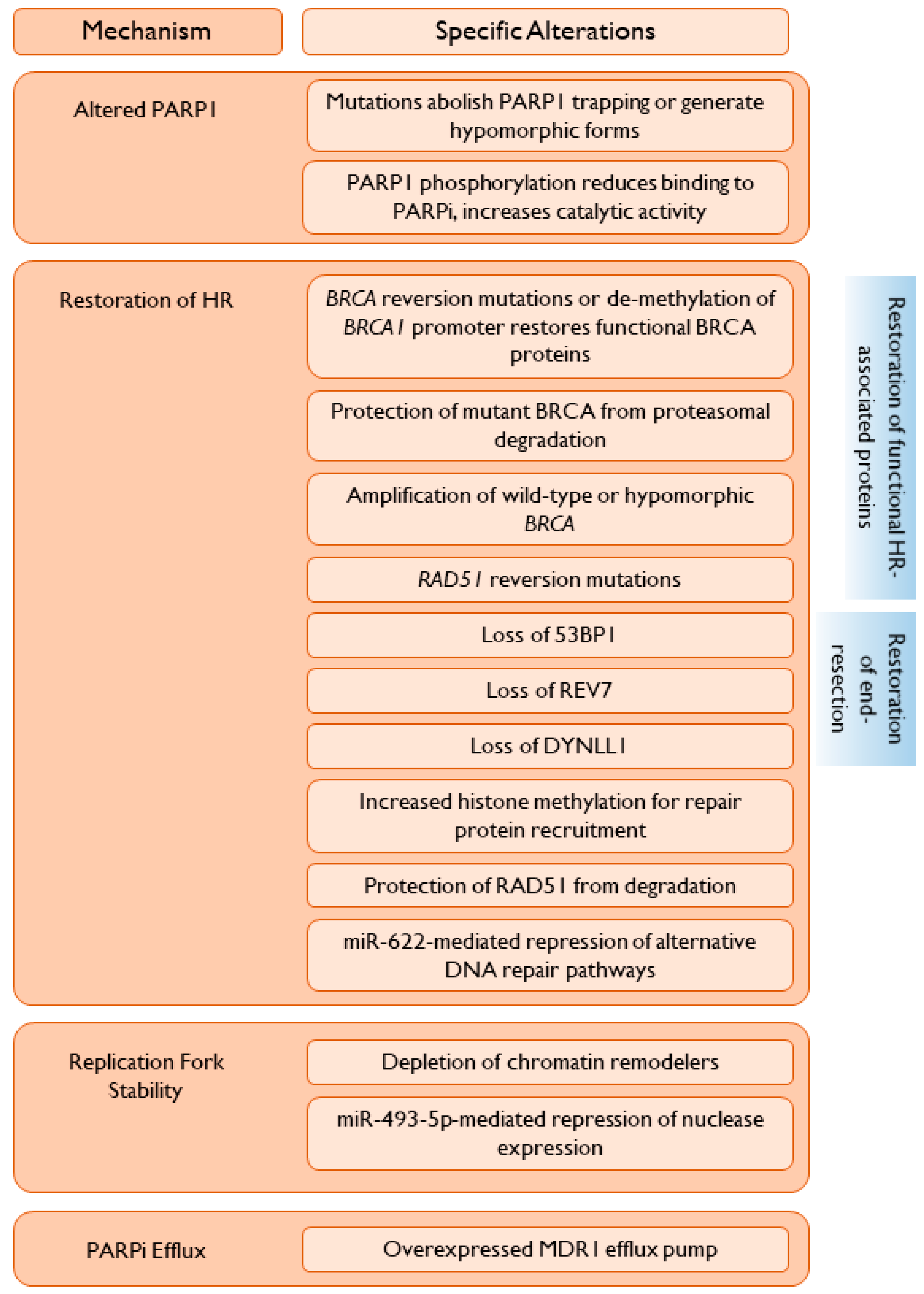
The increase in PARPi use will be paralleled by an increasing number of women who are found to have de novo or acquired resistance to PARPi. As seen in many trials evaluating PARPi as post-chemotherapy maintenance or as monotherapy, a substantial proportion of women progress on treatment, highlighting an urgent need to determine appropriate post-progression treatment, the choice of which may be influenced by the mechanism(s) of PARPi resistance [45]. Select resistance mechanisms (summarized in Figure 1) are discussed in detail below.
7.1. Alterations in PARP1
7.1.1. PARP1 Mutations
Mutations in PARP1 can diminish the binding of PARPi or allow PARP1 to maintain endogenous functions. A large-scale Crispr-cas9 mutagenesis screen with in vitro clonal selection following PARPi selection pressure identified in-frame mutations occurring within the DNA-binding zinc-finger domain of PARP1 [46]. Mutations were frequently seen involving residues K119 and S120 and the surrounding region, impacting the ability of PARP1 to bind sites of DNA damage. This suggests that the abolishment of PARP1 trapping underlies those clones’ resistance to PARPi. A mutation arising in a region predicted to contribute to the DNA-binding interface abolished PARP1 trapping, as did a separate unique mutation located in the PARP1 regulatory region. A catalytic domain mutation yielded a mutant PARP1 that had retained but diminished recruitment to sites of DNA damage, PARylation, and DNA binding capacity. In response to PARPi, there was only transient PARP trapping. These preclinical data highlight that PARP1 mutations altering PARP trapping may serve as a mechanism of PARPi resistance. This hypothesis is supported by a reported case of a patient with EOC and de novo resistance to olaparib, who was subsequently found to have a PARP1 mutation affecting a region critical for the communication between the DNA-binding and catalytic domains [46]. The resulting PARP1 protein retained DNA-binding capacity but was unable to become trapped in response to PARPi.
7.1.2. Post-Translational Modifications of PARP1
Post-translational modifications of PARP1 may also impact its function and confer PARPi resistance. Preclinical studies in models of triple negative breast cancer found that MET, activated in response to oxidative DNA damage, was able to phosphorylate PARP1 [47]. This modification resulted in reduced binding by PARPi, an increase in PARP1 enzymatic activity, and resistance to PARPi. Cells were re-sensitized to PARPi with the use of a MET inhibitor, and dual MET and PARP inhibition led to synergistic anti-tumor activity in breast and lung cancer xenografts. In contrast, in a separate model of hepatocellular carcinoma, PARP1 phosphorylation was dependent on the nuclear translocation of EGFR and formation of an EGFR/MET heterodimer causing MET activation [48]. Whether PARP1 phosphorylation in EOC relies on MET alone or requires another receptor tyrosine kinase, such as EGFR, is unknown.
7.2. Restoration of Homologous Recombination
A common mechanism of resistance to PARPi is the restoration of at least partial HR capabilities, such that DSBs can be repaired, decreasing genomic instability and replication stress. This can occur through the restoration of a deficient HR-related protein to a functional capacity, or by the alteration of inhibitory proteins such that HR can proceed (Figure 1).
7.2.1. Restoration of Functional HR-Associated Proteins
Reversion Mutations
Somatic BRCA reversion mutations are a well described mechanism of resistance to PARPi, occurring in approximately 20% of cases of EOC [49,50]. As pathogenic BRCA1/2 mutations may lead to frameshifts, protein truncation, and the generation of a hypomorphic or non-functional protein, reversion mutations can be insertions or deletions that restore the open reading frame, remove the original deleterious mutation to restore a nearly full-length wild-type sequence, or cause a synonymous mutation, restoring wild-type amino acid sequences [51,52]. This results in the restoration of a functional BRCA protein [50,51,53,54,55,56,57,58,59]. Micro-homology associated with reversion alterations suggest that they arise as a result of the error-prone altNHEJ or SSA pathways of DNA repair, utilized in the setting of HR deficiency [53,54,60]. Multiple concurrent reversion mutations may evolve following PARPi treatment, representing multiple subclones [49,54,56,59,61,62] that may contribute to clinically heterogenous intra-patient progression of disease. In one case, 12 separate reversion mutations were found within one patient who developed resistance to rucaparib [61].
BRCA1 Promoter Alterations
The de-methylation of BRCA1 leading to the re-expression of protein and restored HR was demonstrated in patient-derived xenograft (PDX) models of PARPi-resistant breast cancer [60]. This was reported clinically, with BRCA1 de-methylation occurring at the time of relapse in a patient with PARPi resistant EOC [56] and in two patients following neoadjuvant platinum-based chemotherapy [65]. The zygosity of BRCA1 methylation appears to be important for PARPi sensitivity or resistance, with heterozygous methylation correlating with PARPi resistance in HGSOC PDX models [65]. Additionally, BRCA1 promoter methylation can be subverted by intrachromosomal rearrangements that place BRCA1 expression under the control of a different promoter, as was found in PARPi-resistant PDX tumors [60].
Generation of Hypomorphic BRCA Proteins
Alternative splicing of BRCA1 can generate a hypomorphic protein with residual function. The expression of the BRCA1Δ11q splice variant, with partial skipping and exclusion of most of the exon 11 nucleotides, was associated with in vitro and in vivo PARPi resistance [66]. Mutations in the highly conserved RING domain of BRCA1, crucial for interaction with BARD1, yielded a protein that demonstrated increased stability independent of BARD1, with retained function and PARPi resistance [67]. Furthermore, RING domain deficient BRCA1 was detected in patient derived specimens with germline BRCA1 mutations, confirming this as a clinically relevant mechanism of resistance. Similarly, the generation of hypomorphic BRCA1 can result from mutations occurring in close proximity to Alu elements in intron 15 of BRCA1 [68]. These mutant BRCA1 isoforms were able to avoid proteasomal degradation, retained partial RAD51-loading capability despite loss of the BRCT domain, and promoted PARPi resistance [68]. Additionally, the authors determined that BRCA1 gene rearrangements led to the translation of intron 15. This BRCA1 isoform was functional, retained RAD51 loading ability, and also generated PARPi resistance in vitro [68].
Decreased Proteasomal Degradation
Mutations within the BRCT domain itself can create a misfolded protein, which typically undergoes proteasomal degradation [69,70]. However, under PARPi selection pressure in vitro, PARPi resistant cells were found to have increased expression of the mutant BRCA1 due to Hsp90-mediated stabilization [71]. These BRCT-mutated proteins were functional and able to interact with PALB2-BRCA2 for RAD51 foci formation, thereby engendering PARPi resistance. Analysis of specimens obtained from platinum-treated recurrent BRCA1-mutated EOC patients found that, of four patients with BRCT domain mutations, two patients had increased BRCA1 protein expression in the absence of reversion mutations. Analogous to the in vitro data, this was likely due to Hsp90-mediated stabilization [71].
Amplification of Wild-Type BRCA
Copy number gain or upregulation of the wild-type, functional BRCA allele was found to underlie the PARPi resistance in a cohort of patients with HGSOC [72]. In one patient, the primary tumor had single-copy loss of the majority of chromosome 17q. However, at the time of progression, there was single-copy gain of the remaining wild-type allele, resulting in two copies of the non-mutated BRCA1 allele. A second patient developed upregulation of the wild-type BRCA2 allele at time of progression. However, in vitro analysis of BRCA2-mutant PARPi resistant clones found that amplification and copy number gain of even the mutant BRCA2 allele was sufficient for the restoration of HR and PARPi resistance. Therefore, this may be a mechanism of resistance in situations of a hypomorphic BRCA mutation [73].
7.2.2. Restoration of End-Resection
A crucial step in HR is end-resection, initiated by the MRN complex and extended by additional nucleases, including EXO-1 and DNA2. Alterations in proteins associated with end-resection may restore HR capability in previously HR-deficient cancers, leading to PARPi resistance (Figure 1).
Loss of 53BP1, a repressor of end-resection, was shown in vitro to partially restore HR despite BRCA1 deficiency, conferring PARPi resistance [74,75]. The loss of 53BP1 was seen in 20% of PARPi-resistant breast cancer PDXs [76]. One patient with PARPi-resistant BRCA-mutant breast cancer, identified through genomic analysis of a breast cancer cohort, was found to have biallelic inactivation of TP53BP1 [77]. How the loss of 53BP1 restores HR is likely manifold. The loss of 53BP1-mediated suppression of ATM-dependent RPA phosphorylation rescues RPA loading onto ssDNA. Concomitantly, 53BP1 loss appears to abrogate ATM-mediated cell cycle checkpoint arrest, allowing the restoration of cell proliferation [78,79]. Lastly, if BRCA1 is deficient and cannot facilitate PALB2 recruitment, the loss of 53BP1 allows the exposure of the nucleosome region required for PALB2 self-localization and direct DNA binding [75].
The restoration of HR due to the loss of 53BP1 appears specific to BRCA1 but not BRCA2 deficiency [78], with the type of BRCA1 mutation dictating the extent to which HR is restored. For example, in cells with 53BP1 loss and BRCA1 mutations not affecting the coiled-coil domain (required for interacting with PALB2), HR was restored to a greater degree compared to BRCA1 with disrupted coiled-coil domains [80,81,82]. Similarly, the depletion of REV7, a component of the shieldin complex, was found in vitro to restore HR through CtIP-mediated end-resection, leading to PARPi resistance. This was seen in BRCA1-, but not BRCA2-, deficient cells, highlighting the different steps in HR in which each BRCA protein acts [83].
DYNLL1 suppresses the activity of several components of the end-resection machinery involved in HR, including the MRN complex, thereby limiting end resection. The loss of DYNLL1 permitted the uninhibited recruitment of end-resection proteins to areas of DNA damage, allowing end resection and HR to proceed [84]. Even in BRCA1 mutant cells, DYNLL1 loss enhanced end-resection, restored RAD51 foci formation, and restored HR-mediated DSB repair. This was correlated clinically in a cohort of HGSOC specimens in which BRCA1-deficient cancers with low expression of DYNLL1 were associated with fewer chromosomal abnormalities [84]. In vitro, concurrent DYNLL1 loss and BRCA1 deficiency resulted in resistance to PARPi. In effect, the loss of DYNLL1 compensated for BRCA1 deficiency.
7.2.3. Promotion of Repair Protein Recruitment
Preclinical data suggest that PARPi resistance may arise from adaptive epigenetic changes. For example, the overexpression of the histone methyltransferases EHMT1/2 was found in PARPi resistant HGSOC [85]. EHMT1/2 promote the recruitment of DNA damage repair proteins [86,87], including those related to HR, and have been implicated in replication fork stability [88], suggesting two downstream mechanisms of resistance. The inhibition of EHMT1/2 ablated markers of HR and NHEJ in PARPi resistant but not sensitive cells, suggests that PARPi resistant cells developed a reliance on EHMT1/2-facilitated repair [85]. In both BRCA-deficient and -proficient cells, the inhibition of EHMT1/2 re-sensitized previously resistant cells to olaparib [85].
In PARPi resistant cells, RAD51 levels are elevated due to the downregulation of EMI1, a mitotic regulator that assembles a ubiquitin ligase complex involved in RAD51 degradation [89]. Therefore, EMI1 downregulation allows the accumulation of RAD51, which may facilitate HR and PARPi resistance [90].
7.2.4. Repression of Alternative DNA Repair Pathways
If alternative DNA repair pathways are suppressed, HR may be preferentially employed instead. This was demonstrated with microRNA-622 (miR-622), which clinically is associated with worse overall and disease-free survival in BRCA1 mutated cases of EOC [91]. This association was not seen in cases with wild-type BRCA. Mechanistically, miR-622 targeted and decreased the expression of Ku80, suppressing NHEJ. In parallel, this allowed the unfettered recruitment of the MRN complex to DSBs and rescue of HR in BRCA1 mutant cancers, leading to PARPi resistance [91]. The upregulation of Wnt/β-catenin signaling, which may occur secondary to the methylation of FZD10, a receptor in the Wnt pathway [92], also induced PARPi resistance in vitro [93]. Functional assays demonstrated that HR was primarily enhanced, however NHEJ was also promoted. The inhibition of β-catenin significantly impeded HR and NHEJ and restored sensitivity of cells to PARPi [92,94]. This may occur through the modulation of LIG4 by β-catenin, which is involved in NHEJ and upregulation of MRE11 [95,96].
7.3. Alteration of Replication Fork Dynamics
One means of PARPi cytotoxicity occurs through the dysregulation of replication fork reversal and/or restart. Therefore, resistance to PARPi can arise through the stabilization of replication forks [97].
MRE11 is recruited to stalled replication forks and may be involved in limited resection required for fork restart. However, in the setting of BRCA1/2 deficiency it may also precipitate uncontrolled degradation and fork collapse. Fork remodeling is required for MRE11-dependent nascent DNA degradation. This process is facilitated by SMARCAL1, ZRANB3, and HLTF; depletion of these chromatin remodelers prevented strand degradation by MRE11, leading to fork stability, reduced replication stress-induced DNA damage and chromosomal instability, and resistance to olaparib in BRCA1/2 deficient cells [98]. Additionally, the loss of the recruitment protein PTIP protects nascent DNA strands from extensive degradation by MRE11, as can occur when BRCA1/2 is deficient or nonfunctional and cannot recruit protective RAD51 [97]. Comparably, MRE11 inhibition reduced the levels of PARPi-induced chromosomal abnormalities, likely through replication fork protection.
E2F7, a transcription factor induced by DNA damage and involved in facilitating G1/S arrest, also impacts MRE11 activity [99]. E2F7 represses the expression of key HR contributors, including RAD51 and BRCA1 [100]. In vitro depletion of E2F7 led to increased RAD51 levels, restored RAD51-mediated HR repair, and increased stability of stalled replication forks by preventing MRE11-mediated degradation, overall imparting resistance to PARPi.
Similarly, the recruitment of the nuclease MUS81 by EZH2-directed histone methylation facilitates fork restart in BRCA2-deficient cells [101]. Low EZH2 levels reduced MUS81 recruitment and led to fork stabilization. EZH2 inhibition and MUS81 loss each conferred PARPi resistance in BRCA2-deficient cells and breast cancer model.
The nuclear protein SLFN11 detects replication stress, stalls fork progression, and prolongs S phase arrest to allow the repair of DNA lesions prior to the continuation of the cell cycle. However, prolonged fork stalling eventuates replisome disassembly and fork breakage, contrarily predisposing cells further to the genome-destabilizing effects of PARPi. High SLFN11 expression was associated with increased sensitivity to PARPi [102]. Loss of SLFN11 was associated with resistance to PARPi in BRCA1/2-deficient cells [102,103].
In keeping with the evidence above, miR-493-5p induced PARPi resistance by decreasing protein expression of the end-resection proteins EXO1, MRE11, BLM, and CHD4 in BRCA2-mutant cells [104]. These factors, recruited by PTIP, are involved in destabilizing the replication fork with additional roles in HR and single strand annealing [17,18,97,105]. Therefore, miR-493-5p promotes fork stabilization and diminishes the repair of DSBs via the mutagenic SSA pathway. In BRCA2-mutated EOC cell lines, miR-493-5p upregulation was associated with olaparib resistance [104].
7.4. PARPi Efflux
Upregulation of drug efflux pumps is a well described mechanism of PARPi resistance. Gene alterations in ABCB1, encoding the multi-drug efflux pump MDR1 (also known as p-glycoprotein), include intergenic deletions, transcript fusions, and 5′ region mutations, leading to the increased expression of ABCB1 [56,106]. Transcript fusions can place ABCB1 under the control of heterogeneous genes and promoters. In one study of patients with recurrent HGSOC, fusions accounted for 59% of the specimens with the highest MDR1 expression [106]. Approximately 8% of HGSOC specimens, taken at time of post-PARPi recurrence, found the upregulation of ABCB1 via fusions and translocations [56].
Of note, paclitaxel and doxorubicin are also MDR1 substrates. Theoretically, the use of either agent prior to PARPi may similarly induce MDR1 upregulation and indirectly induce PARPi resistance. In one study, prior paclitaxel use was significantly associated with the presence of ABCB1 fusion transcripts [106]. However, prior doxorubicin use was not significantly associated with ABCB1 fusion transcripts. The association of MDR1 cross-resistance between PARPi and paclitaxel has been demonstrated in additional studies [107,108]. Therefore, conversely, MDR1 overexpression occurring as a PARPi resistance mechanism has implications for which agent to use following PARPi resistance.
8. Implications for Post-Progression PARP Inhibitor Combination Therapies
Barring alterations in PARP1 or increased efflux, PARPi resistance generally occurs via the restoration of HR or replication fork stabilization. Therefore, combination therapies may aim to target one or both to re-sensitize resistant cells to PARPi or to induce PARPi sensitivity in EOC that is HRP at baseline. The rationale behind many combination therapies is depicted in Figure 2. However, many combinatorial strategies are thus far only in the preclinical or early-phase trial stages, where issues, such as additive toxicities precluding therapeutic dosing of one or both agents and lack of comparator arms, hinder the ability to robustly evaluate these combinations [109].
8.1. PARPi and Anti-Angiogenic Agents
Anti-angiogenic agents have been shown to inhibit BRCA1/2 expression, which may be beneficial in cases of reversion alterations leading to functional proteins. The inhibition of VEGFR3 in vitro decreased levels of BRCA1 and BRCA2 and inhibited cell growth [110]. In the setting of a BRCA2-mutated clone that developed a resistance mutation and expressed a functionally wild-type protein, VEGFR3 inhibition was sufficient to restore chemosensitivity [110]. The inhibition of VEGF and VEGFR2 in organoid models prevented Akt-mediated DNA repair, thereby preventing HR and leading to aberrant NHEJ [111]. Anti-angiogenics are also hypothesized to induce or exacerbate intratumoral hypoxia, which itself is associated with impaired HR [112,113,114,115]. Therefore, preclinical data suggest that combining anti-angiogenic agents with PARPi may be effective in PARPi resistant disease. A single arm phase II trial treated 34 patients with PARPi-resistant EOC with olaparib and cediranib (oral anti-angiogenic), yielding 4 patients who achieved partial responses and 18 patients with stable disease [116]. That this combination was effective even in PARPi-resistant cases may be due to cediranib-mediated suppression of BRCA1/2 and RAD51 expression, both indirectly through induction of hypoxia, and directly through transcriptional repression [117]. In a similar patient population of germline BRCAwt platinum sensitive recurrent EOC, combination cediranib/olaparib had greater activity compared to olaparib alone in post-hoc analyses of a phase II trial, prolonging median PFS from 5.7 months to 23.7 months (p = 0.002) and median OS from 23.0 months to 37.8 months (p = 0.047) [118]. In prespecified subset analysis in a subsequent phase III trial (GY-004), however, the population with germline BRCAwt platinum sensitive recurrent EOC performed comparably to platinum-based chemotherapy (ORR 64% cediranib/olaparib versus 72% chemotherapy; HR 0.97, 95% CI 0.73–1.30) [119]. Cediranib/olaparib performed better than chemotherapy in those with BRCA-mutated disease (ORR 89% versus 71%, HR 0.55, 95% CI 0.32–0.94). Due to the trial’s hierarchical testing design, cediranib/olaparib did not meet statistical criteria for comparison to olaparib monotherapy. Importantly, the phase II and III trials of cediranib and olaparib did not report the somatic BRCA status of patients. Additionally, the benefit of anti-angiogenic agents may also be influenced by the immunomodulatory effects of anti-VEGF/VEGFR agents [120,121].
8.2. PARPi and Hsp90 Inhibition
Pairing PARPi with inhibitors of Hsp90 (Hsp90i) may be relevant for EOC with BRCA1 mutations affecting the BRCT domain, given the role of Hsp90 in subverting ubiquitin-directed proteasomal degradation and restoring BRCA1 protein function [71]. Hsp90 stabilization of mutant BRCA1 likely extends beyond BRCT domain mutations [122]. In addition to BRCA1, Hsp90 interacts with several other client proteins involved in DNA repair and cell cycle regulation, including CHK1, BRCA2, RAD51, and MRE11 [122,123]. Therefore, Hsp90i may impair HR by several mechanisms and may additionally impair NHEJ [122]. This was corroborated in preclinical studies using the Hsp90 inhibitor ganetespib. Treatment with ganetespib led to reduced expression of BRCA1, BRCA2, CHK1, ATM, RAD51, MRE11, and CDK1 and was associated with abrogated HR [124]. Pairing ganetespib with talazoparib produced anti-tumor synergy even in BRCAwt, HR-proficient HGSOC cells [124]. This suggests that PARPi resistant cancers with restored HR may be susceptible to combination Hsp90i and PARPi.
8.3. PARPi and PI3K Pathway Inhibition
Impairing DNA damage repair through PI3K inhibition (PI3Ki) occurs through the suppression of BRCA1/2 transcription and depletion of the nucleotide pool [125,126,127,128]. A phase I trial of buparlisib (PI3Ki) with olaparib yielded 12 of 46 patients with EOC who achieved a partial response, of which four patients did not have germline or somatic BRCA mutations [129]. Similarly, a phase Ib trial of alpelisib (PI3Ki) with olaparib in patients with EOC found that 35% (n = 6/17) of patients with germline BRCAwt disease achieved partial responses, similar to those with germline BRCA mutations (30%, n = 3/10) [130]. Both cohorts were enriched for HR proficient disease, as 94% and 90% of each group, respectively had platinum resistant or refractory disease. In prespecified analyses, archival tumor specimens were assessed for somatic mutations, and in a combined group of germline and somatic BRCAwt disease, 33% of patients (4/12) achieved a response. These data suggest that PI3Ki was sufficient to induce HR deficiency in cancers with baseline HR proficiency and without evidence of PI3K pathway mutations, thereby sensitizing to the effects of PARPi. Though both phase I trials allowed prior PARPi use for patients in dose escalation, response attributions and whether these patients had PARPi resistance is unclear.
In a phase I trial of olaparib and the AKT inhibitor capivasertib, 11 of 25 patients with EOC achieved clinical benefit (CR + PR + SD ≥ 4 months) [131]. Of these 11 patients, four were PARPi resistant. Despite this, one patient achieved a PR, and two patients achieved prolonged SD of 56 and 115 weeks, respectively. These data suggest that combination therapy was able to re-induce sensitivity to PARPi. Of additional interest is correlative cfDNA analysis that showed BRCA1/2 reversion mutations developing at the time of progression, demonstrating that PARPi resistance can develop despite combination therapy [131]. A separate analysis of patients with endometrial, ovarian, and triple-negative breast cancers treated with olaparib/capivasertib determined that markers of DNA damage checkpoint activation (high phospho-Chk1, -Wee1, -CDC2) and decreased mTOR activity were associated with response, whereas resistance to the combination was associated with high receptor tyrosine kinase activity levels and mTOR activation [132]. Though it is unclear whether markers differed between malignancies or whether any patients included in the analysis had received prior PARPi, these data provide some indication of a molecularly-defined population who may be more likely to respond, and provide greater insight to additional resistance mechanisms.
8.4. PARPi and MEK Inhibition
RAS-mutated cell lines were found to be HRP and PARPi resistant, and acquired PARPi resistance was associated with the upregulation of the RAS/MAPK pathway [133]. This suggested that the MAPK pathway may be a target for re-sensitization to PARPi. MEK inhibition (MEKi) was found to decrease cellular capacity for HR by decreasing the expression of MRE11, RAD50, NBN, and BRCA1/2 [133,134], predisposing to effects of PARPi. Combining MEKi and PARPi induced greater DNA damage and apoptosis in vitro, with synergistic anti-tumor activity in vivo [133,134]. An ongoing phase I/II trial of olaparib and selumetinib (MEKi) (NCT03162627) includes an expansion cohort of PARPi-resistant EOC.
8.5. PARPi and Inhibition of ATR, Chk1, and Wee1
Inhibitors of the ATR/Chk1/Wee1 axis affect both HR and replication fork stability, promoting re-sensitization to PARPi in settings of both BRCA1 and BRCA2 deficiency. In BRCA1-deficient cells, ATR inhibitors (ATRi) disrupted the acquired ATR-dependent recruitment of PALB2-BRCA2 and RAD51 loading [135,136], thereby reestablishing HR deficiency and overcoming the RAD51-related protection of stalled forks. The synergistic anti-tumor effect of combination PARPi and ATRi was demonstrated in PARPi-resistant BRCA1-mutant EOC models [137] and breast cancer models [138]. Furthermore, the role of ATR in coupling DNA damage repair with cell cycle regulation enhances the synergism of combination ATRi and PARPi, particularly in p53-mutated EOC [139]. In BRCA2-mutant EOC PDX models, ATRi released G2/M arrest, leading to premature mitosis with unrepaired PARPi-induced DNA damage [140]. This corresponded with increased tumor suppression in murine models.
The inhibition of Chk1 (Chk1i), a downstream effector protein activated by ATR, produced similar results when administered with PARPi, in both BRCA mutant and wild-type EOC models [137,140,141]. Prexasertib, a Chk1 inhibitor, has demonstrated preclinical sensitization to PARPi [141,142] and early evidence of clinical effectiveness in a phase I trial [143]. Interestingly, two patients with PARPi-resistant, BRCA1-mutant HGSOC achieved partial responses with combination olaparib and prexasertib [143]. Effects of Chk1i may result from its known role interacting with Cdc25a and Cdc25c, possibly from effects on the transcription factor E2F7 [99], and by preventing RAD51 foci formation [141].
Acting downstream of Chk1, Wee1 inhibition synergizes with PARPi in preclinical data across several cancer types [144,145,146]. The safety of combination olaparib and the Wee1 inhibitor adavosertib was demonstrated in a phase 1b trial of refractory solid tumors [147], and this regimen is under investigation in a phase II trial specifically in PARPi-resistant EOC (NCT03579316). Interestingly, the sequential administration of PARPi and adavosertib was as effective as a concurrent administration in anti-tumor efficacy in vivo, but was better tolerated with fewer hematopoietic effects [146]. This alternate dosing schedule could be considered in future trials.
8.6. PARPi and BET/BRD4 Inhibition
Bromodomain containing 4 (BRD4) is a member of the BET protein family with roles in epigenetic gene regulation. The re-sensitization of PARPi-resistant cells using BRD4 inhibition (BRD4i) or broader BET inhibition (BETi) appears to be through the repression of HR-associated genes, including BRCA1, RAD51, and CtIP, thereby generating a state of HR deficiency [148,149,150]. Combined PARPi/BRD4i demonstrated antitumor synergy in vitro and in vivo [148,149,150]. This combination was effective in cell lineages that were BRCAwt (mimicking BRCA reversion mutations), 53BP1 deficient (mimicking TP53BP1 mutations and/or 53BP1 loss), and PARP1 deficient (mimicking PARP1 mutations or post-translational modifications), suggesting effectiveness across several mechanisms of PARPi resistance [148]. A phase I trial evaluating the combination of olaparib and AZD5153 (BETi) (NCT03205176) in advanced solid tumors, including EOC, allows prior PARPi exposure.
8.7. PARPi and CDK12 Inhibition
Cyclin-dependent kinase 12 (CDK12) modulates transcription by acting on RNA polymerase II. In vitro, CDK12 loss-of-function mutations and CDK12 inhibition (CDK12i) reduced the expression of HR-related genes, including BRCA1, ATR, and Fanconi-anemia pathway genes, due to premature cleavage and polyadenylation [151,152,153], leading to reduced capacity for HR repair [154,155,156]. This mechanism of re-inducing HR underlies the synthetic lethality between CDK12i and PARPi, seen in models of HGSOC [157] and breast cancer [156] and may be effective for PARPi-resistant disease in which HR is restored. There are thus far no clinical data for this combination.
8.8. PARPi and Immune Checkpoint Inhibition
Pairing PARPi and immune checkpoint inhibition (ICI) exploits two main premises for synergy. The first centers on the ability of PARPi to propagate DNA damage and generate cytosolic DNA. This activates the cGAS/STING pathway [158,159,160,161,162,163], with the downstream expression of type 1 interferons, T-cell-recruiting cytokines, and paracrine stimulation of dendritic cells [158,164]. However, this may be compromised if the mechanism of PARPi resistance is the restoration of HR proficiency. For example, downstream markers of cGAS/STING activation were decreased in BRCA proficient cells compared to BRCA deficient cells [158,165]. The second premise arises from the multiple immunomodulatory effects of PARPi, including on T cell differentiation, macrophage polarization [166,167,168], increased susceptibility to NK cell-mediated death [169,170], and PD-L1 upregulation [171,172]. These effects may be compromised if acquired alterations affect PARPi binding or allow the persistence of PARP1 function. The combination PARPi/ICI seeks to capitalize on the immunostimulatory effects of PARPi while negating the effects of PD-L1 upregulation.
The combination of PARPi/ICI may still be effective in the setting of restored HR proficiency. In the phase I/II TOPACIO/KEYNOTE-162 trial, the combination of niraparib and pembrolizumab was evaluated in patients with platinum-resistant EOC, of whom the majority were BRCAwt (79%) or HRP (53%) [173]. Overall response rates (ORR) were similar regardless of HR status; an ORR of 19% was seen in those patients with HRP disease, compared to an ORR of 14% in patients with HR deficient disease. This was comparable to the ORR of 14% (5/35) seen in a phase II trial of olaparib and durvalumab in a predominantly platinum-resistant (86%), BRCAwt (77%) patient population [174]. Of the five responders, two were BRCAwt and HRP. Paired pre- and on-treatment specimens showed that olaparib/durvalumab promoted an immunologically-inflamed environment, with increased IFNγ and TNFα production, increased tumor-infiltrating lymphocytes, and increased PD-L1 expression [174]. Therefore, though the clinical effectiveness of PARPi/ICI is modest in patients with measured or surrogate markers of HR proficiency, this combination may still be beneficial in select patients.
9. Conclusions and Future Perspectives
There are multiple mechanisms for PARPi resistance, and though many described mechanisms require clinical validation, the above data highlight several key issues. First, multiple resistance mechanisms may arise within one individual. This is well illustrated in the case of BRCA reversion mutations, in which several studies found numerous reversion mutations within individuals. The choice of subsequent PARPi combination therapy would need to consider this heterogeneity. Second, it is imperative to reassess the molecular characteristics of disease at each point of progression, given the dynamic nature of treatment response and resistance. “Liquid biopsy”, evaluating cell-free DNA (cfDNA), circulating tumors cells, circulating miRNA, or exosomes, may be more feasible than repeated biopsies [175]. For example, BRCA reversion mutations are readily detected using cfDNA [49,50,59,62]. Importantly, genetic alterations may have unanticipated consequences on the overall restoration of DNA repair or synthesis. In this regard, functional assays, such as those for the detection of HR deficiency/proficiency or replication fork stability, may be more informative than identifying the genetic changes in isolation. This is an area of unmet need, as surrogate measures of HR and replication fork dynamics, such as detection of γH2AX and Rad51 foci, are not yet clinically validated. Finally, PARPi combination therapies have great promise, but few trials currently allow patients with PARPi resistance. This is detrimental to the development of effective treatment strategies, particularly as the number of PARPi-resistant patients will rise with the growing use of PARPi in general. Ideally, post-progression PARPi combination regimens should complement the resistance mechanism(s) and functional status of the tumor at the time of treatment. To do so will require establishing the predictive value of both to optimize the pairing of patient and regimen. Expanding the benefit of PARPi through combination therapies will critically rely on appropriate dose escalation or alternative dose-schedule strategies. Optimizing target modulation while minimizing possible overlapping toxicities will prevent the premature termination of a promising combination. In conclusion, understanding the mechanisms of PARPi resistance, detecting them in real-time, such as through regular sampling by liquid biopsy, and optimizing targeted combinations, are critically needed.
Author Contributions
E.K.L. and U.A.M. both contributed to the conceptualization and preparation of this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This review received no external funding. U.A.M. is supported by the Breast Cancer Research Foundation.
Conflicts of Interest
E.K.L. declares no conflict of interest. U.A.M. reports consulting fees for Merck.
Abbreviations
| altNHEJ | Alternative non-homologous end-joining |
| BER | Base excision repair |
| cfDNA | Cell-free DNA |
| cNHEJ | Classical non-homologous end-joining |
| DSB | Double strand break |
| EOC | Epithelial ovarian cancer |
| HGSOC | High grade serous ovarian cancer |
| HR | Homologous recombination |
| HRD | Homologous recombination deficient |
| HRP | Homologous recombination proficient |
| ICI | Immune checkpoint inhibition |
| NER | Nucleotide excision repair |
| NHEJ | Non-homologous end-joining |
| PARP | Poly(ADP-ribose) polymerase |
| PARPi | PARP inhibitor |
| PDX | Patient-derived xenograft |
| SSA | Single strand annealing |
| SSB | Single strand break |
| ssDNA | Single stranded DNA |
| VEGF/R | Vascular endothelial growth factor/receptor |
References
- Jachimowicz, R.D.; Goergens, J.; Reinhardt, H.C. DNA double-strand break repair pathway choice - from basic biology to clinical exploitation. Cell Cycle 2019, 18, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol 2018, 2, 313–349. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7–shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef]
- Findlay, S.; Heath, J.; Luo, V.M.; Malina, A.; Morin, T.; Coulombe, Y.; Djerir, B.; Li, Z.; Samiei, A.; Simo-Cheyou, E.; et al. SHLD 2/FAM 35A co-operates with REV 7 to coordinate DNA double-strand break repair pathway choice. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e23. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single Strand Annealing and its role in genome maintenance Chromosomal break repair by the Single Strand Annealing (SSA) pathway HHS Public Access. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantzer, F.; Schreiber, V.; Niedergang, C.; Trucco, C.; Flatter, E.; Rubia, G.D.L.; Oliver, J.; Rolli, V.; Ménissier-de Murcia, J.; De Murcia, G. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie 1999, 81, 69–75. [Google Scholar] [CrossRef]
- Vodenicharov, M.D. Base excision repair is efficient in cells lacking poly(ADP-ribose) polymerase 1. Nucleic Acids Res. 2000, 28, 3887–3896. [Google Scholar] [CrossRef] [PubMed]
- Pines, A.; Vrouwe, M.G.; Marteijn, J.A.; Typas, D.; Luijsterburg, M.S.; Cansoy, M.; Hensbergen, P.; Deelder, A.; de Groot, A.; Matsumoto, S.; et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 2012, 199, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Messner, S.; Altmeyer, M.; Zhao, H.; Pozivil, A.; Roschitzki, B.; Gehrig, P.; Rutishauser, D.; Huang, D.; Caflisch, A.; Hottiger, M.O. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 2010, 38, 6350–6362. [Google Scholar] [CrossRef] [Green Version]
- Poirier, G.G.; de Murcia, G.; Jongstra-Bilen, J.; Niedergang, C.; Mandel, P. Poly (ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. USA 1982, 79, 3423–3427. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Chaudhuri, A.R.; Callen, E.; Pang, Y.; Biswas, K.; Klarmann, K.D.; Martin, B.K.; Burkett, S.; Cleveland, L.; Stauffer, S.; et al. Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, A.; Drew, Y.; Matheson, E.; Salehan, M.; Gentles, L.; Pachter, J.A.; Curtin, N.J. Evaluating the potential of kinase inhibitors to suppress DNA repair and sensitise ovarian cancer cells to PARP inhibitors. Biochem. Pharmacol. 2019, 125–132. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Cancer, O.; Group, S.; Sharma, R.; Stewart, C.; Fereday, S.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurman, R.J. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann. Oncol. 2013, 24. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the {cBioPortal.}. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst). 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Stover, E.H.; Konstantinopoulos, P.A.; Matulonis, U.A.; Swisher, E.M. Biomarkers of response and resistance to DNA repair targeted therapies. Clin. Cancer Res. 2016, 22, 5651–5660. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Karlan, B.Y.; Taniguchi, T.; Fountzilas, E.; Francoeur, N.; Levine, D.A.; Cannistra, S.A. Gene Expression Profile of BRCAness That Correlates With Responsiveness to Chemotherapy and With Outcome in Patients With Epithelial Ovarian Cancer. J. Clin. Oncol. 2010, 28, 3555–3561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulonis, U.A.; Penson, R.T.; Domchek, S.M.; Kaufman, B.; Shapira-Frommer, R.; Audeh, M.W.; Kaye, S.; Molife, L.R.; Gelmon, K.A.; Robertson, J.D.; et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: A multistudy analysis of response rates and safety. Ann. Oncol. 2016, 27, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Hjortkjær, M.; Waldstrøm, M.; Jakobsen, A.; Kanstrup, H.; Søgaard-Andersen, E.; Steffensen, K.D. The prognostic value of BRCA1 and PARP expression in epithelial ovarian carcinoma: Immunohistochemical detection. Int. J. Gynecol. Pathol. 2017, 36, 180–189. [Google Scholar] [CrossRef]
- Makvandi, M.; Xu, K.; Lieberman, B.P.; Anderson, R.C.; Effron, S.S.; Winters, H.D.; Zeng, C.; McDonald, E.S.; Pryma, D.A.; Greenberg, R.A.; et al. A radiotracer strategy to quantify PARP-1 expression in vivo provides a biomarker that can enable patient selection for PARP inhibitor therapy. Cancer Res. 2016, 76, 4516–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [Green Version]
- Balasubramaniam, S.; Beaver, J.A.; Horton, S.; Fernandes, L.L.; Tang, S.; Horne, H.N.; Liu, J.; Liu, C.; Schrieber, S.J.; Yu, J.; et al. FDA approval summary: Rucaparib for the treatment of patients with deleterious BRCA mutation–associated advanced ovarian cancer. Clin. Cancer Res. 2017, 23, 7165–7170. [Google Scholar] [CrossRef] [Green Version]
- FDA Approves Niraparib for HRD-Positive Advanced Ovarian Cancer | FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-niraparib-hrd-positive-advanced-ovarian-cancer (accessed on 11 February 2020).
- Ison, G.; Howie, L.J.; Amiri-Kordestani, L.; Zhang, L.; Tang, S.; Sridhara, R.; Pierre, V.; Charlab, R.; Ramamoorthy, A.; Song, P.; et al. FDA approval summary: Niraparib for the maintenance treatment of patients with recurrent ovarian cancer in response to platinum-based chemotherapy. Clin. Cancer Res. 2018, 24, 4066–4071. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food & Drug Administration FDA Approved Olaparib (LYNPARZA) for the Maintenance Treatment of Adult Patients with Deleterious or Suspected Deleterious Germline or Somatic BRCA-Mutated (gBRCAm or sBRCAm) Advanced Epithelial Ovarian, Fallopian Tube or Primary Peritoneal Cancer. Available online: https://www.fda.gov/drugs/fda-approved-olaparib-lynparza-astrazeneca-pharmaceuticals-lp-maintenance-treatment-adult-patients (accessed on 17 December 2019).
- FDA Approves Niraparib for First-Line Maintenance of Advanced Ovarian Cancer | FDA. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-niraparib-first-line-maintenance-advanced-ovarian-cancer (accessed on 5 May 2020).
- FDA Approves Olaparib Plus Bevacizumab as Maintenance Treatment for Ovarian, Fallopian Tube, or Primary Peritoneal Cancers | FDA. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-olaparib-plus-bevacizumab-maintenance-treatment-ovarian-fallopian-tube-or-primary (accessed on 11 May 2020).
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019. [Google Scholar] [CrossRef] [Green Version]
- Wakefield, M.J.; Nesic, K.; Kondrashova, O.; Scott, C.L. Diverse mechanisms of PARP inhibitor resistance in ovarian cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1872. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Yamaguchi, H.; Wei, Y.; Hsu, J.L.; Wang, H.L.; Hsu, Y.H.; Lin, W.C.; Yu, W.H.; Leonard, P.G.; Lee, G.R.; et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med. 2016, 22, 194–201. [Google Scholar] [CrossRef]
- Dong, Q.; Du, Y.; Li, H.; Liu, C.; Wei, Y.; Chen, M.K.; Zhao, X.; Chu, Y.Y.; Qiu, Y.; Qin, L.; et al. EGFR and c-Met cooperate to enhance resistance to PARP inhibitors in hepatocellular carcinoma. Cancer Res. 2019, 79, 819–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigelt, B.; Comino-Méndez, I.; De Bruijn, I.; Tian, L.; Meisel, J.L.; García-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 reversion mutations in circulating cell-free DNA of therapy-resistant breast or ovarian cancer. Clin. Cancer Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef] [Green Version]
- Mayor, P.; Gay, L.M.; Gornstein, E.; Morley, S.; Frampton, G.M.; Heilmann, A.; Sun, J.; Chung, J.; Daniel, S.; Ramkissoon, S.; et al. BRCA1/2 reversion mutations revealed in breast and gynecologic cancers sequenced during routine clinical care using tissue or liquid biopsy. J. Clin. Oncol. 2017, 35, 5551. [Google Scholar] [CrossRef]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov. 2017, 7, 1006–1017. [Google Scholar] [CrossRef] [Green Version]
- Gornstein, E.L.; Sandefur, S.; Chung, J.H.; Gay, L.M.; Holmes, O.; Erlich, R.L.; Soman, S.; Martin, L.K.; Rose, A.V.; Stephens, P.J.; et al. BRCA2 Reversion Mutation Associated With Acquired Resistance to Olaparib in Estrogen Receptor-positive Breast Cancer Detected by Genomic Profiling of Tissue and Liquid Biopsy. Clin. Breast Cancer 2018, 18, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef] [Green Version]
- Quigley, D.; Alumkal, J.J.; Wyatt, A.W.; Kothari, V.; Foye, A.; Lloyd, P.; Aggarwal, R.; Kim, W.; Lu, E.; Schwartzman, J.; et al. Analysis of Circulating Cell-Free DNA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discov. 2017, 7, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Ter Brugge, P.; Kristel, P.; Van Der Burg, E.; Boon, U.; De Maaker, M.; Lips, E.; Mulder, L.; De Ruiter, J.; Moutinho, C.; Gevensleben, H.; et al. Mechanisms of therapy resistance in patient-derived xenograft models of brca1-deficient breast cancer. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Simmons, A.D.; Nguyen, M.; Pintus, E. Polyclonal BRCA2 mutations following carboplatin treatment confer resistance to the PARP inhibitor rucaparib in a patient with mCRPC: A case report. BMC Cancer 2020, 20. [Google Scholar] [CrossRef]
- Christie, E.L.; Fereday, S.; Doig, K.; Pattnaik, S.; Dawson, S.J.; Bowtell, D.D.L. Reversion of BRCA1/2 germline mutations detected in circulating tumor DNA from patients with high-grade serous ovarian cancer. J. Clin. Oncol. 2017, 35, 1274–1280. [Google Scholar] [CrossRef]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 mediates resistance of cancer stem cells to PARP inhibition in triple-negative breast cancer. Clin. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; Van Der Gulden, H.; Van Der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Δ11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Krais, J.J.; Bernhardy, A.J.; Nicolas, E.; Cai, K.Q.; Harrell, M.I.; Kim, H.H.; George, E.; Swisher, E.M.; Simpkins, F.; et al. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J. Clin. Invest. 2016, 126, 3145–3157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bernhardy, A.J.; Nacson, J.; Krais, J.J.; Tan, Y.F.; Nicolas, E.; Radke, M.R.; Handorf, E.; Llop-Guevara, A.; Balmaña, J.; et al. BRCA1 intronic Alu elements drive gene rearrangements and PARP inhibitor resistance. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.S.; Glover, J.N.M. Structural consequences of a cancer-causing BRCA1-BRCT missense mutation. J. Biol. Chem. 2003, 278, 2630–2635. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Green, R.; Marsillac, S.M.; Coquelle, N.; Williams, R.S.; Yeung, T.; Foo, D.; Hau, D.D.; Hui, B.; Monteiro, A.N.A.; et al. Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer Res. 2010, 70, 4880–4890. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.C.; Choi, Y.E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [Green Version]
- Lheureux, S.; Bruce, J.P.; Burnier, J.V.; Karakasis, K.; Shaw, P.A.; Clarke, B.A.; Yang, S.Y.C.; Quevedo, R.; Li, T.; Dowar, M.; et al. Somatic BRCA1/2 recovery as a resistance mechanism after exceptional response to poly (ADP-ribose) polymerase inhibition. J. Clin. Oncol. 2017, 35, 1240–1249. [Google Scholar] [CrossRef]
- Park, P.H.; Yamamoto, T.M.; Li, H.; Alcivar, A.L.; Xia, B.; Wang, Y.; Bernhardy, A.J.; Turner, K.M.; Kossenkov, A.V.; Watson, Z.L.; et al. Amplification of the Mutation-Carrying BRCA2 Allele Promotes RAD51 Loading and PARP Inhibitor Resistance in the Absence of Reversion Mutations. Mol. Cancer Ther. 2020, 19, 602–613. [Google Scholar] [CrossRef] [Green Version]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Belotserkovskaya, R.; Raga Gil, E.; Lawrence, N.; Butler, R.; Clifford, G.; Wilson, M.D.; Jackson, S.P. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Cohen, O.; Kochupurakkal, B.; Kim, D.; Dunn, C.E.; Buendia Buendia, J.; Wander, S.; Helvie, K.; Lloyd, M.R.; Marini, L.; et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; Van Der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Publ. Gr. 2010, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Xu, X.; Bunting, S.F.; Liu, J.; Wang, R.H.; Cao, L.L.; Wu, J.J.; Peng, T.N.; Chen, J.; Nussenzweig, A.; et al. A Selective Requirement for 53BP1 in the Biological Response to Genomic Instability Induced by Brca1 Deficiency. Mol. Cell 2009, 35, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; Van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in BRCA1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Nacson, J.; Krais, J.J.; Bernhardy, A.J.; Clausen, E.; Feng, W.; Wang, Y.; Nicolas, E.; Cai, K.Q.; Tricarico, R.; Hua, X.; et al. BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell Rep. 2018, 24, 3513–3527.e7. [Google Scholar] [CrossRef] [Green Version]
- Duarte, A.A.; Gogola, E.; Sachs, N.; Barazas, M.; Annunziato, S.; De Ruiter, J.R.; Velds, A.; Blatter, S.; Houthuijzen, J.M.; Van De Ven, M.; et al. BrcA-deficient mouse mammary tumor organoids to study cancer-drug resistance. Articles 2018, 134. [Google Scholar] [CrossRef]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [Green Version]
- He, Y.J.; Meghani, K.; Caron, M.C.; Yang, C.; Ronato, D.A.; Bian, J.; Sharma, A.; Moore, J.; Niraj, J.; Detappe, A.; et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 2018, 563, 522–526. [Google Scholar] [CrossRef]
- Watson, Z.L.; Yamamoto, T.M.; McMellen, A.; Kim, H.; Hughes, C.J.; Wheeler, L.J.; Post, M.D.; Behbakht, K.; Bitler, B.G. Histone methyltransferases EHMT1 and EHMT2 (GLP/G9A) maintain PARP inhibitor resistance in high-grade serous ovarian carcinoma. Clin. Epigenetics 2019, 11. [Google Scholar] [CrossRef]
- Yang, Q.; Zhu, Q.; Lu, X.; Du, Y.; Cao, L.; Shen, C.; Hou, T.; Li, M.; Li, Z.; Liu, C.; et al. G9a coordinates with the RPA complex to promote DNA damage repair and cell survival. Proc. Natl. Acad. Sci. USA 2017, 114, E6054–E6063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginjala, V.; Rodriguez-Colon, L.; Ganguly, B.; Gangidi, P.; Gallina, P.; Al-Hraishawi, H.; Kulkarni, A.; Tang, J.; Gheeya, J.; Simhadri, S.; et al. Protein-lysine methyltransferases G9a and GLP1 promote responses to DNA damage. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 2015, 59, 998–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzio, A.; Puccini, J.; Kwon, Y.; Sung, P.; Bar-Sagi, D.; Correspondence, M.P. The F-Box Domain-Dependent Activity of EMI1 Regulates PARPi Sensitivity in Triple-Negative Breast Cancers. Mol. Cell 2019, 73, 224–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamovich, A.I.; Toland, A.E.; Parvin, J.D. F-Box Protein-Mediated Resistance to PARP Inhibitor Therapy. Mol. Cell 2019, 73, 195–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.E.; Meghani, K.; Brault, M.E.; Leclerc, L.; He, Y.J.; Day, T.A.; Elias, K.M.; Drapkin, R.; Weinstock, D.M.; Dao, F.; et al. Platinum and PARP Inhibitor Resistance Due to Overexpression of MicroRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell Rep. 2016, 14, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Fukumoto, T.; Zhu, H.; Nacarelli, T.; Karakashev, S.; Fatkhutdinov, N.; Wu, S.; Liu, P.; Kossenkov, A.V.; Showe, L.C.; Jean, S.; et al. N6-methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Cancer Res. 2019, 79, 2812–2820. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.M.; McMellen, A.; Watson, Z.L.; Aguilera, J.; Ferguson, R.; Nurmemmedov, E.; Thakar, T.; Moldovan, G.L.; Kim, H.; Cittelly, D.M.; et al. Activation of Wnt signaling promotes olaparib resistant ovarian cancer. Mol. Carcinog. 2019, 58, 1770–1782. [Google Scholar] [CrossRef]
- Yamamoto, K.N.; Hirota, K.; Takeda, S.; Haeno, H. Evolution of pre-existing versus acquired resistance to platinum drugs and PARP inhibitors in BRCA-associated cancers. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Jun, S.; Jung, Y.S.; Suh, H.N.; Wang, W.; Kim, M.J.; Oh, Y.S.; Lien, E.M.; Shen, X.; Matsumoto, Y.; McCrea, P.D.; et al. LIG4 mediates Wnt signalling-induced radioresistance. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, R.; Tang, J.; Ma, J.G.; Chen, S.P.; Xia, L.P.; Zhou, W.J.; Li, D.D.; Feng, G.K.; Zeng, Y.X.; Zhu, X.F. PKB/Akt promotes DSB repair in cancer cells through upregulating Mre11 expression following ionizing radiation. Oncogene 2011, 30, 944–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, K.E.; Thakar, T.; Nicolae, C.M.; Liang, X.; Wang, H.-G.; Moldovan, G.-L. Loss of E2F7 confers resistance to poly-ADP-ribose polymerase (PARP) inhibitors in BRCA2-deficient cells. Nucleic Acids Res. 2018, 46, 8898–8907. [Google Scholar] [CrossRef]
- Westendorp, B.; Mokry, M.; Groot Koerkamp, M.J.A.; Holstege, F.C.P.; Cuppen, E.; De Bruin, A. E2F7 represses a network of oscillating cell cycle genes to control S-phase progression. Nucleic Acids Res. 2012, 40, 3511–3523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; Van De Ven, M.; Van Der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef]
- Murai, J.; Feng, Y.; Yu, G.K.; Ru, Y.; Tang, S.W.; Shen, Y.; Pommier, Y. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016, 7, 76534–76550. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Tang, S.W.; Leo, E.; Baechler, S.A.; Redon, C.E.; Zhang, H.; Al Abo, M.; Rajapakse, V.N.; Nakamura, E.; Jenkins, L.M.M.; et al. SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol. Cell 2018, 69, 371–384.e6. [Google Scholar] [CrossRef] [Green Version]
- Meghani, K.; Fuchs, W.; Detappe, A.; Drané, P.; Gogola, E.; Rottenberg, S.; Jonkers, J.; Matulonis, U.; Swisher, E.M.; Konstantinopoulos, P.A.; et al. Multifaceted Impact of MicroRNA 493-5p on Genome-Stabilizing Pathways Induces Platinum and PARP Inhibitor Resistance in BRCA2-Mutated Carcinomas. Cell Rep. 2018, 23, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Guillemette, S.; Serra, R.W.; Peng, M.; Hayes, J.A.; Konstantinopoulos, P.A.; Green, M.R.; Green, M.R.; Cantor, S.B. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 2015, 29, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard, A.P.; Liu, C.; Armstrong, C.M.; D’Abronzo, L.S.; Lou, W.; Chen, H.; Dall’Era, M.; Ghosh, P.M.; Evans, C.P.; Gao, A.C. Overexpressed ABCB1 Induces Olaparib-Taxane Cross-Resistance in Advanced Prostate Cancer. Transl. Oncol. 2019, 12, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Sundar, R.; Valeri, N.; Harrington, K.J.; Yap, T.A. Combining molecularly targeted agents: Is more always better? Clin. Cancer Res. 2017, 23, 1123–1125. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Yang, K.; Taylor-Harding, B.; Ruprecht Wiedemeyer, W.; Buckanovich, R.J. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia 2014, 16, 343–353.e2. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Roman, N.; Chong, M.Y.; Chahal, S.K.; Caragher, S.P.; Jackson, M.R.; Stevenson, K.H.; Dongre, S.A.; Chalmers, A.J. Radiation Responses of 2D and 3D Glioblastoma Cells: A Novel, 3D-specific Radioprotective Role of VEGF/Akt Signaling through Functional Activation of NHEJ. Mol. Cancer Ther. 2020, 19, 575–589. [Google Scholar] [CrossRef]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Maseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-Regulation of Rad51 and Decreased Homologous Recombination in Hypoxic Cancer Cells. Mol. Cell. Biol. 2004, 24, 8504–8518. [Google Scholar] [CrossRef] [Green Version]
- Chan, N.; Koritzinsky, M.; Zhao, H.; Bindra, R.; Glazer, P.M.; Powell, S.; Belmaaza, A.; Wouters, B.; Bristow, R.G. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res. 2008, 68, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Kumareswaran, R.; Ludkovski, O.; Meng, A.; Sykes, J.; Pintilie, M.; Bristow, R.G. Chronic hypoxia compromises repair of DNA double-strand breaks to drive genetic instability. J. Cell Sci. 2012, 125, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism: Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Oaknin, A.; Garg, S.; Bruce, J.; Dhani, N.C.; Madariaga, A.; Bonilla, L.; Lee, Y.C.; Colombo, I.; Bhat, G.; et al. Evolve: A post PARP inhibitor clinical translational phase II trial of cediranib-olaparib in ovarian cancer—A Princess Margaret Consortium – GCIG Phase II Trial. J. Clin. Oncol. 2019, 37, 5521. [Google Scholar] [CrossRef]
- Kaplan, A.R.; Gueble, S.E.; Liu, Y.; Oeck, S.; Kim, H.; Yun, Z.; Glazer, P.M. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci. Transl. Med. 2019, 11, eaav4508. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.J.; Buss, M.K.; Nattam, S.R.; Hurteau, J.; et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer. Ann. Oncol. 2019, 30, 551–557. [Google Scholar] [CrossRef]
- Liu, J.F.; Brady, M.F.; Matulonis, U.A.; Miller, A.; Kohn, E.C.; Swisher, E.M.; Tew, W.P.; Cloven, N.G.; Muller, C.; Bender, D.; et al. A phase III study comparing single-agent olaparib or the combination of cediranib and olaparib to standard platinum-based chemotherapy in recurrent platinum-sensitive ovarian cancer. J. Clin. Oncol. 2020, 38, 6003. [Google Scholar] [CrossRef]
- Datta, M.; Coussens, L.M.; Nishikawa, H.; Hodi, F.S.; Jain, R.K. Reprogramming the Tumor Microenvironment to Improve Immunotherapy: Emerging Strategies and Combination Therapies. Am. Soc. Clin. Oncol. Educ. B. 2019, 39, 165–174. [Google Scholar] [CrossRef]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Stecklein, S.R.; Kumaraswamy, E.; Behbod, F.; Wang, W.; Chaguturu, V.; Harlan-Williams, L.M.; Jensen, R.A. BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proc. Natl. Acad. Sci. USA 2012, 109, 13650–13655. [Google Scholar] [CrossRef] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Publ. Gr. 2017, 18. [Google Scholar] [CrossRef]
- Gabbasov, R.; Benrubi, I.D.; O’Brien, S.W.; Krais, J.J.; Johnson, N.; Litwin, S.; Connolly, D.C. Targeted blockade of HSP90 impairs DNA-damage response proteins and increases the sensitivity of ovarian carcinoma cells to PARP inhibition. Cancer Biol. Ther. 2019, 20, 1035–1045. [Google Scholar] [CrossRef]
- Juvekar, A.; Hu, H.; Yadegarynia, S.; Lyssiotis, C.A.; Ullas, S.; Lien, E.C.; Bellinger, G.; Son, J.; Hok, R.C.; Seth, P.; et al. Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc. Natl. Acad. Sci. USA 2016, 113, E4338–E4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmañà, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K Inhibitor with a PARP Inhibitor Provides an Effective Therapy for BRCA1-Related Breast Cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, F.L.; Lord, C.J.; Ashworth, A. The Promise of Combining Inhibition of PI3K and PARP as Cancer Therapy. Cancer Discov. 2012, 2, 982–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann. Oncol. 2017, 28, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.; Cadoo, K.A.; Shapiro, G.I.; Mayer, E.L.; O’Cearbhaill, R.E.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: A dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; Lim, J.S.J.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I trial of the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib and AKT inhibitor capivasertib in patients with BRCA1/2 and non-BRCA1/2 mutant cancers. Cancer Discov. 2020, CD-20-0163. [Google Scholar] [CrossRef]
- Labrie, M.; Ju, Z.; Litton, J.K.; Kim, T.-B.; Lee, S.; Chen, K.; Soliman, P.T.; Frumovitz, M.; Meyer, L.A.; Moulder, S.; et al. Abstract 2070: Exploration of markers of synergistic lethality of PARP and PI3K-Akt-mTOR inhibitors in women’s cancers. Cancer Res. 2019, 79 (Suppl. 2070), 13. [Google Scholar] [CrossRef]
- Sun, C.; Fang, Y.; Yin, J.; Chen, J.; Ju, Z.; Zhang, D.; Chen, X.; Vellano, C.P.; Jeong, K.J.; Ng, P.K.S.; et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Vena, F.; Jia, R.; Esfandiari, A.; Garcia-Gomez, J.J.; Rodriguez-Justo, M.; Ma, J.; Syed, S.; Crowley, L.; Elenbaas, B.; Goodstal, S.; et al. MEK inhibition leads to BRCA2 downregulation and sensitization to DNA damaging agents in pancreas and ovarian cancer models. Oncotarget 2018, 9, 11592–11603. [Google Scholar] [CrossRef] [Green Version]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Kwan, T.T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.F.; Stanciu, M.; Humphrey, M.R.; Gorham, J.; Wakimoto, H.; Nishihara, R.; Lees, J.; Zou, L.; Martuza, R.L.; Wakimoto, H.; et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Burgess, B.T.; Anderson, A.M.; McCorkle, J.R.; Wu, J.; Ueland, F.R.; Kolesar, J.M. Olaparib combined with an ATR or Chk1 inhibitor as a treatment strategy for acquired olaparib-resistant BRCA1 mutant ovarian cells. Diagnostics 2020, 10, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wengner, A.M.; Siemeister, G.; Lucking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bomer, U.; Moosmayer, D.; Eberspacher, U.; et al. The novel ATR inhibitor BAY 1895344 is efficacious as monotherapy and combined with DNA damage—inducing or repair—compromising therapies in preclinical cancer models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef] [Green Version]
- Schoonen, P.M.; Kok, Y.P.; Wierenga, E.; Bakker, B.; Foijer, F.; Spierings, D.C.J.; van Vugt, M.A.T.M. Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells. Mol. Oncol. 2019, 13, 2422–2440. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [Green Version]
- Brill, E.; Yokoyama, T.; Nair, J.; Yu, M.; Ahn, Y.R.; Lee, J.M. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer. Oncotarget 2017, 8, 111026–111040. [Google Scholar] [CrossRef]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Farkkila, A.; Reznichenko, E.; et al. The CHK1 inhibitor prexasertib exhibits monotherapy activity in high-grade serous ovarian cancer models and sensitizes to PARP inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Do, K.T.; Hill, S.J.; Kochupurakkal, B.; Supko, J.G.; Gannon, C.; Anderson, A.; Muzikansky, A.; Wolanski, A.; Hedglin, J.; Parmar, K.; et al. Abstract CT232: Phase I combination study of the CHK1 inhibitor prexasertib (LY2606368) and olaparib in patients with high-grade serous ovarian cancer and other advanced solid tumors. Cancer Res. 2019, 79 (Suppl. CT232), 13. [Google Scholar] [CrossRef]
- Karnak, D.; Engelke, C.G.; Parsels, L.A.; Kausar, T.; Wei, D.; Robertson, J.R.; Marsh, K.B.; Davis, M.A.; Zhao, L.; Maybaum, J.; et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin. Cancer Res. 2014, 20, 5085–5096. [Google Scholar] [CrossRef] [Green Version]
- Parsels, L.A.; Karnak, D.; Parsels, J.D.; Zhang, Q.; Velez-Padilla, J.; Reichert, Z.R.; Wahl, D.R.; Maybaum, J.; O’Connor, M.J.; Lawrence, T.S.; et al. PARP1 Trapping and DNA Replication Stress Enhance Radiosensitization with Combined WEE1 and PARP Inhibitors. Mol. Cancer Res. 2018, 16, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; McGrail, D.J.; Sun, C.; Labrie, M.; Chen, X.; Zhang, D.; Ju, Z.; Vellano, C.P.; Lu, Y.; Li, Y.; et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy. Cancer Cell 2019, 35, 851–867.e7. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Falchook, G.S.; Wang, J.S.; Fu, S.; Oza, A.; Karen, S.; Imedio, E.R.; Kumar, S.; Ottesen, L.; Mugundu, G.M.; et al. Abstract CT025: Phase Ib study of adavosertib in combination with olaparib in patients with refractory solid tumors: Dose escalation. Cancer Res. 2019, 79, CT025. [Google Scholar] [CrossRef]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.J.; Stubbs, M.; Liu, P.; Ruggeri, B.; Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018, 149, 575–584. [Google Scholar] [CrossRef]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1757. [Google Scholar] [CrossRef] [Green Version]
- Dubbury, S.J.; Boutz, P.L.; Sharp, P.A. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature 2018, 564, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Chirackal Manavalan, A.P.; Pilarova, K.; Kluge, M.; Bartholomeeusen, K.; Rajecky, M.; Oppelt, J.; Khirsariya, P.; Paruch, K.; Krejci, L.; Friedel, C.C.; et al. CDK12 controls G1/S progression by regulating RNAPII processivity at core DNA replication genes. EMBO Rep. 2019, 20, e47592. [Google Scholar] [CrossRef]
- Ekumi, K.M.; Paculova, H.; Lenasi, T.; Pospichalova, V.; Osken, C.A.B.; Rybarikova, J.; Bryja, V.; Geyer, M.; Blazek, D.; Barboric, M. Ovarian carcinoma CDK12 mutations misregulate expression of DNA repair genes via deficient formation and function of the Cdk12/CycK complex. Nucleic Acids Res. 2015, 43, 2575–2589. [Google Scholar] [CrossRef]
- Joshi, P.M.; Sutor, S.L.; Huntoon, C.J.; Karnitz, L.M. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. J. Biol. Chem. 2014, 289, 9247–9253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R.; Duckett, D.R. Therapeutic Targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell 2019, 36, 545–558.e7. [Google Scholar] [CrossRef] [PubMed]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8 + T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Chabanon, R.M.; Lord, C.J.; Postel-Vinay, S. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer The Journal of Clinical Investigation. J Clin Invest 2019, 129. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1-/- murine model of ovarian cancer. Biochem. Biophys. Res. Commun. 2015, 463, 551–556. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.Y.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R.; et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [Green Version]
- Kis-Toth, K.; Szanto, A.; Thai, T.-H.; Tsokos, G.C. Cytosolic DNA-Activated Human Dendritic Cells Are Potent Activators of the Adaptive Immune Response. J. Immunol. 2011, 187, 1222–1234. [Google Scholar] [CrossRef] [Green Version]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Li, L.; Chen, L.; Yuan, W.; Dong, L.; Zhang, Y.; Wu, H.; Wang, C. PARP-1 Mediates LPS-Induced HMGB1 Release by Macrophages through Regulation of HMGB1 Acetylation. J. Immunol. 2014, 193, 6114–6123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditsworth, D.; Zong, W.X.; Thompson, C.B. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J. Biol. Chem. 2007, 282, 17845–17854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.; Porat, A.; He, M.; Suurmond, J.; Santiago-Schwarz, F.; Andersson, U.; Coleman, T.R.; Volpe, B.T.; Tracey, K.J.; Al-Abed, Y.; et al. C1q and HMGB1 reciprocally regulate human macrophage polarization. Blood 2016, 128, 2218–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Liu, L.; Xie, M.; Sun, X.; Yu, Y.; Kang, R.; Yang, L.; Zhu, S.; Cao, L.; Tang, D. Poly-ADP-ribosylation of HMGB1 regulates TNFSF10/TRAIL resistance through autophagy. Autophagy 2015, 11, 214–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.W.; Koh, B.D.; Zhang, J.S.; Flatten, K.S.; Schneider, P.A.; Billadeau, D.D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Kaufmann, S.H. Poly(ADP-ribose) polymerase inhibitors sensitize cancer cells to death receptor-mediated apoptosis by enhancing death receptor expression. J. Biol. Chem. 2014, 289, 20543–20558. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Lampert, E.J.; Zimmer, A.S.; Padget, M.R.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP inhibitor olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: A proof-of-concept phase 2 study. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Asante, D.B.; Calapre, L.; Ziman, M.; Meniawy, T.M.; Gray, E.S. Liquid biopsy in ovarian cancer using circulating tumor DNA and cells: Ready for prime time? Cancer Lett. 2020, 468, 59–71. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Thematic mechanisms of resistance to PARP inhibition.

Figure 2.
Rationale for re-sensitization to PARPi. Almost all HGSOC harbor mutant p53, with cell cycle dysregulation at baseline. Inhibition of ATM or ATR further prevents the appropriate halting of the cell cycle. Using agents that reestablish HR deficiency allows for the accumulation of endogenous and exogenous DNA damage, trapped PARP, and replication stress, ultimately leading to mitotic catastrophe and cell death.
Figure 2.
Rationale for re-sensitization to PARPi. Almost all HGSOC harbor mutant p53, with cell cycle dysregulation at baseline. Inhibition of ATM or ATR further prevents the appropriate halting of the cell cycle. Using agents that reestablish HR deficiency allows for the accumulation of endogenous and exogenous DNA damage, trapped PARP, and replication stress, ultimately leading to mitotic catastrophe and cell death.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, E.K.; Matulonis, U.A. PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies. Cancers 2020, 12, 2054. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082054
AMA Style
Lee EK, Matulonis UA. PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies. Cancers. 2020; 12(8):2054. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082054
Chicago/Turabian StyleLee, Elizabeth K., and Ursula A. Matulonis. 2020. "PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies" Cancers 12, no. 8: 2054. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082054
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.