Intrinsic Balance between ZEB Family Members Is Important for Melanocyte Homeostasis and Melanoma Progression

Abstract
:1. Introduction
2. The ZEB Transcription Factors Family
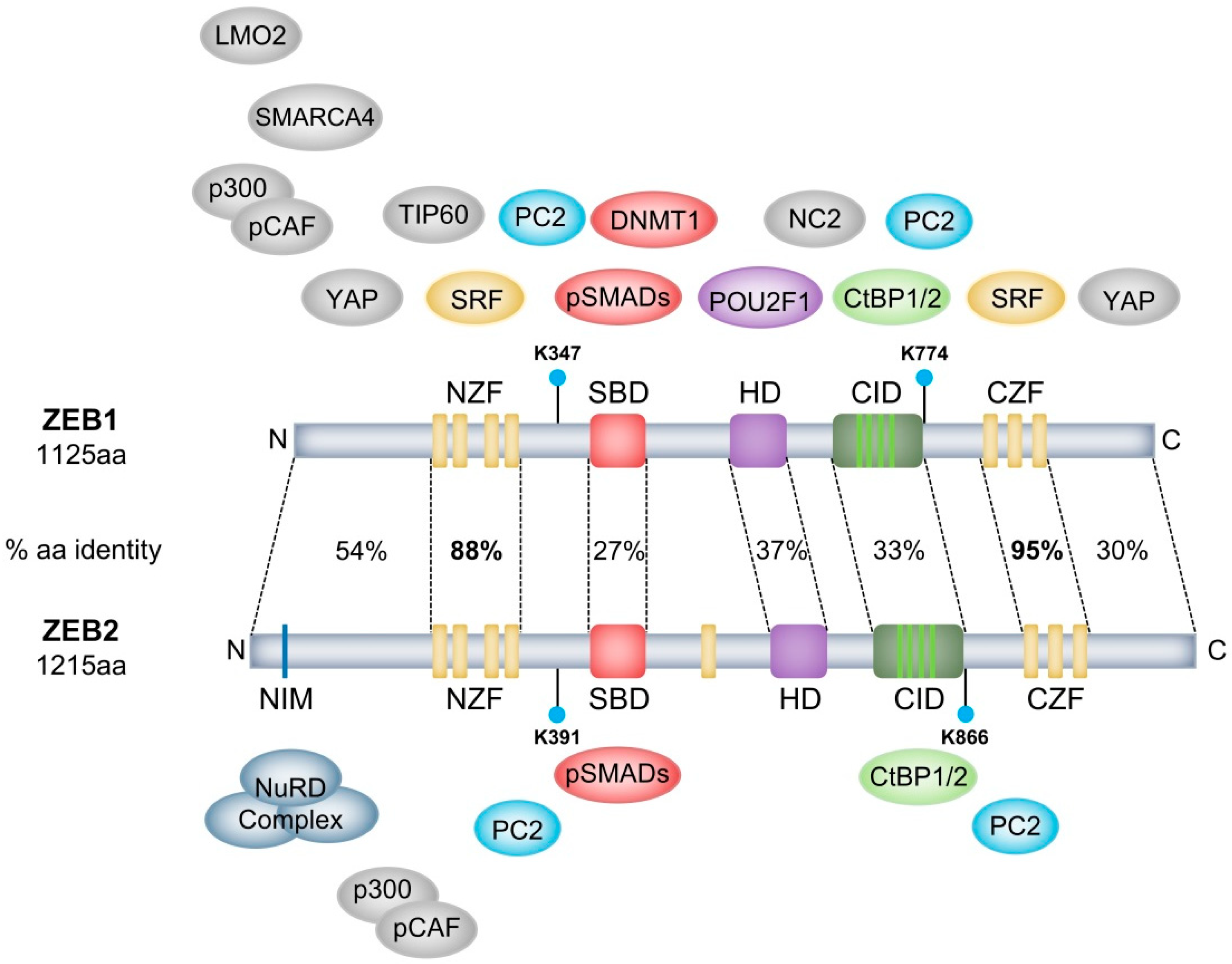
2.1. ZEB1 and ZEB2 Homology and Diversification
2.2. ZEB1 and ZEB2 Interact with the CtBP1/2 Co-Repressor Complex
2.3. CtBP1/2-Independent Transcriptional Repression via ZEB1 and ZEB2
2.4. ZEB1 and ZEB2 as Transcriptional Activators
2.5. ZEB1 and ZEB2 Functional Versatility
3. ZEB1 and ZEB2 as Modulators of TGF-β/BMP Signals
3.1. TGF-β/BMP Signalling Pathways
3.2. ZEB1 and ZEB2 Interact with SMAD Proteins
4. ZEBs during Specification from Neural Crest Cells towards the Melanocyte Lineage and in Adult Melanocyte Homeostasis
4.1. ZEB1 and ZEB2 in Neural Crest Cells
4.2. Neural Crest Cells Migration and Differentiation in the Melanocyte Lineage
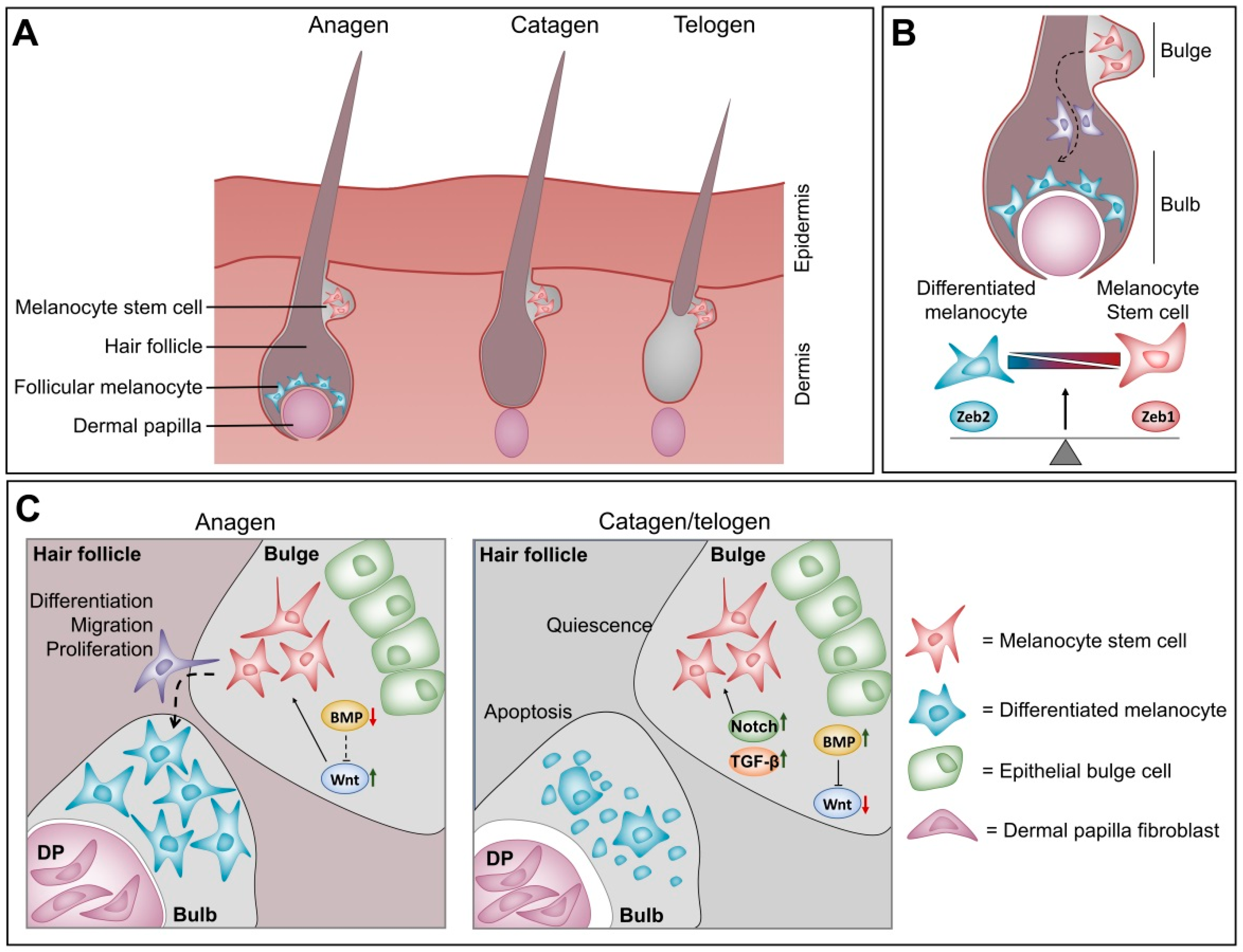
4.3. Balance between ZEB1 and ZEB2 in Melanocyte Homeostasis at the Hair Follicle
5. ZEB1 and ZEB2 Induce EMT-Driven Cellular Plasticity during Carcinoma Progression
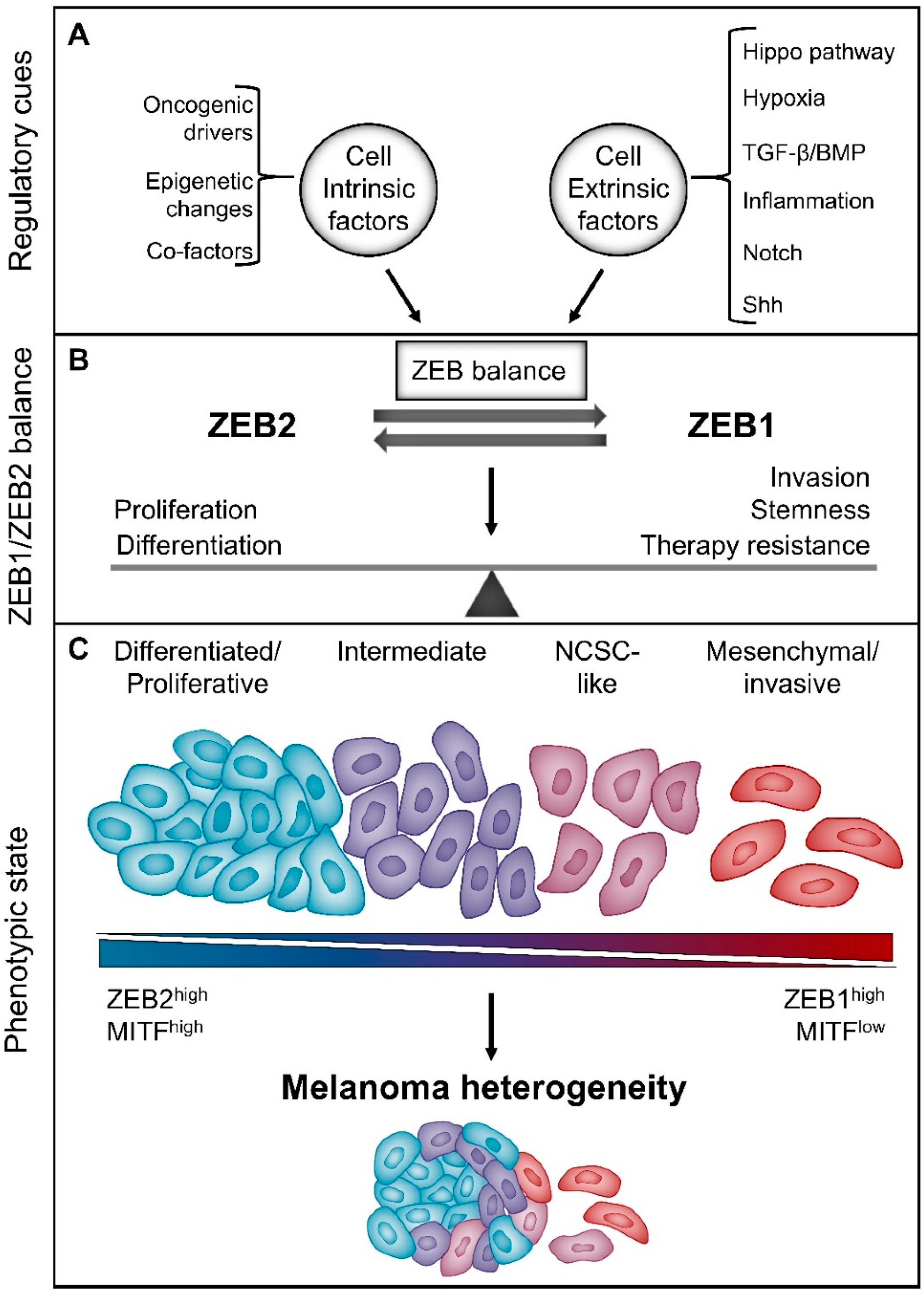
6. The “Phenotype-Switching” Model for Melanoma Heterogeneity
6.1. The Reversible “Phenotype-Switching” Model
6.2. Implications of “Intermediate” Melanoma Cell States for Therapy Resistance
7. ZEB1 and ZEB2 in Melanoma Phenotype Switching
7.1. ZEBs and Melanoma Differentiation/Proliferation vs Invasion
7.2. ZEBs and Melanoma Stemness
7.3. ZEBs and Melanoma Therapy Resistance
8. Micro-Environmental Cues Regulate the Reversible Balance between ZEB1 and ZEB2
8.1. TGF-β/Shh Signalling
8.2. Hypoxia
8.3. Hippo Signalling Pathway
8.4. EMT-Associated Transcription Factors
9. Conclusions and Perspectives: Targeting ZEB1 and ZEB2 to Modulate Melanoma Phenotype Switching
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Yuan, L.; Alanazi, S.; Garrett, J.T. Current advances in the treatment of braf-mutant melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schummer, P.; Schilling, B.; Gesierich, A. Long-term outcomes in braf-mutated melanoma treated with combined targeted therapy or immune checkpoint blockade: Are we approaching a true cure? Am. J. Clin. Dermatol. 2020, 21, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Vandewalle, C.; Van Roy, F.; Berx, G. The role of the zeb family of transcription factors in development and disease. Cell. Mol. Life Sci. 2009, 66, 773–787. [Google Scholar] [CrossRef]
- Remacle, J.E.; Kraft, H.; Lerchner, W.; Wuytens, G.; Collart, C.; Verschueren, K.; Smith, J.C.; Huylebroeck, D. New mode of DNA binding of multi-zinc finger transcription factors: Deltaef1 family members bind with two hands to two target sites. EMBO J. 1999, 18, 5073–5084. [Google Scholar] [CrossRef] [Green Version]
- Verschueren, K.; Remacle, J.E.; Collart, C.; Kraft, H.; Baker, B.S.; Tylzanowski, P.; Nelles, L.; Wuytens, G.; Su, M.T.; Bodmer, R.; et al. Sip1, a novel zinc finger/homeodomain repressor, interacts with smad proteins and binds to 5’-cacct sequences in candidate target genes. J. Biol. Chem. 1999, 274, 20489–20498. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.E.; Darling, D.S. Combination of a zinc finger and homeodomain required for protein-interaction. Mol. Biol. Rep. 2003, 30, 199–206. [Google Scholar] [CrossRef]
- Nelles, L.; Van de Putte, T.; van Grunsven, L.; Huylebroeck, D.; Verschueren, K. Organization of the mouse zfhx1b gene encoding the two-handed zinc finger repressor smad-interacting protein-1. Genomics 2003, 82, 460–469. [Google Scholar] [CrossRef]
- Beltran, M.; Puig, I.; Pena, C.; Garcia, J.M.; Alvarez, A.B.; Pena, R.; Bonilla, F.; de Herreros, A.G. A natural antisense transcript regulates zeb2/sip1 gene expression during snail1-induced epithelial-mesenchymal transition. Genes Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef] [Green Version]
- Funahashi, J.; Sekido, R.; Murai, K.; Kamachi, Y.; Kondoh, H. Delta-crystallin enhancer binding protein delta ef1 is a zinc finger-homeodomain protein implicated in postgastrulation embryogenesis. Developement 1993, 119, 433–446. [Google Scholar]
- Postigo, A.A.; Dean, D.C. Zeb represses transcription through interaction with the corepressor ctbp. Proc. Natl. Acad. Sci. USA 1999, 96, 6683–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Grunsven, L.A.; Taelman, V.; Michiels, C.; Verstappen, G.; Souopgui, J.; Nichane, M.; Moens, E.; Opdecamp, K.; Vanhomwegen, J.; Kricha, S.; et al. Xsip1 neuralizing activity involves the co-repressor ctbp and occurs through bmp dependent and independent mechanisms. Dev. Biol. 2007, 306, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sawada, J.; Sui, G.; Affar el, B.; Whetstine, J.R.; Lan, F.; Ogawa, H.; Luke, M.P.; Nakatani, Y.; Shi, Y. Coordinated histone modifications mediated by a ctbp co-repressor complex. Nature 2003, 422, 735–738. [Google Scholar] [CrossRef]
- Wang, J.; Scully, K.; Zhu, X.; Cai, L.; Zhang, J.; Prefontaine, G.G.; Krones, A.; Ohgi, K.A.; Zhu, P.; Garcia-Bassets, I.; et al. Opposing lsd1 complexes function in developmental gene activation and repression programmes. Nature 2007, 446, 882–887. [Google Scholar] [CrossRef]
- Kuppuswamy, M.; Vijayalingam, S.; Zhao, L.J.; Zhou, Y.; Subramanian, T.; Ryerse, J.; Chinnadurai, G. Role of the pldls-binding cleft region of ctbp1 in recruitment of core and auxiliary components of the corepressor complex. Mol. Cell. Biol. 2008, 28, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Grooteclaes, M.L.; Frisch, S.M. Evidence for a function of ctbp in epithelial gene regulation and anoikis. Oncogene 2000, 19, 3823–3828. [Google Scholar] [CrossRef] [Green Version]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed e box binding zinc finger protein sip1 downregulates e-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Pena, C.; Garcia, J.M.; Silva, J.; Garcia, V.; Rodriguez, R.; Alonso, I.; Millan, I.; Salas, C.; de Herreros, A.G.; Munoz, A.; et al. E-cadherin and vitamin d receptor regulation by snail and zeb1 in colon cancer: Clinicopathological correlations. Hum. Mol. Genet. 2005, 14, 3361–3370. [Google Scholar] [CrossRef]
- Goossens, S.; Peirs, S.; Van Loocke, W.; Wang, J.; Takawy, M.; Matthijssens, F.; Sonderegger, S.E.; Haigh, K.; Nguyen, T.; Vandamme, N.; et al. Oncogenic zeb2 activation drives sensitivity toward kdm1a inhibition in t-cell acute lymphoblastic leukemia. Blood 2017, 129, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases hdac1 and hdac2 by the transcriptional repressor zeb1 downregulates e-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef] [PubMed]
- van Grunsven, L.A.; Michiels, C.; Van de Putte, T.; Nelles, L.; Wuytens, G.; Verschueren, K.; Huylebroeck, D. Interaction between smad-interacting protein-1 and the corepressor c-terminal binding protein is dispensable for transcriptional repression of e-cadherin. J. Biol. Chem. 2003, 278, 26135–26145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. Zeb1 represses e-cadherin and induces an emt by recruiting the swi/snf chromatin-remodeling protein brg1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukagawa, A.; Ishii, H.; Miyazawa, K.; Saitoh, M. Deltaef1 associates with dnmt1 and maintains DNA methylation of the e-cadherin promoter in breast cancer cells. Cancer Med. 2015, 4, 125–135. [Google Scholar] [CrossRef]
- Qin, Y.; Tang, B.; Hu, C.J.; Xiao, Y.F.; Xie, R.; Yong, X.; Wu, Y.Y.; Dong, H.; Yang, S.M. An htert/zeb1 complex directly regulates e-cadherin to promote epithelial-to-mesenchymal transition (emt) in colorectal cancer. Oncotarget 2016, 7, 351–361. [Google Scholar] [CrossRef]
- Hannen, R.; Bartsch, J.W. Essential roles of telomerase reverse transcriptase htert in cancer stemness and metastasis. FEBS Lett. 2018, 592, 2023–2031. [Google Scholar] [CrossRef]
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. Sirt1 induces emt by cooperating with emt transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 2012, 31, 4619–4629. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Li, Q.; Yu, Y.; Li, M.; Zhang, D. Set8 induces epithelialmesenchymal transition and enhances prostate cancer cell metastasis by cooperating with zeb1. Mol. Med. Rep. 2016, 13, 1681–1688. [Google Scholar] [CrossRef]
- Hlubek, F.; Lohberg, C.; Meiler, J.; Jung, A.; Kirchner, T.; Brabletz, T. Tip60 is a cell-type-specific transcriptional regulator. J. Biochem. 2001, 129, 635–641. [Google Scholar] [CrossRef]
- Ikeda, K.; Halle, J.P.; Stelzer, G.; Meisterernst, M.; Kawakami, K. Involvement of negative cofactor nc2 in active repression by zinc finger-homeodomain transcription factor areb6. Mol. Cell. Biol. 1998, 18, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Verstappen, G.; van Grunsven, L.A.; Michiels, C.; Van de Putte, T.; Souopgui, J.; Van Damme, J.; Bellefroid, E.; Vandekerckhove, J.; Huylebroeck, D. Atypical mowat-wilson patient confirms the importance of the novel association between zfhx1b/sip1 and nurd corepressor complex. Hum. Mol. Genet. 2008, 17, 1175–1183. [Google Scholar] [CrossRef] [Green Version]
- Manshouri, R.; Coyaud, E.; Kundu, S.T.; Peng, D.H.; Stratton, S.A.; Alton, K.; Bajaj, R.; Fradette, J.J.; Minelli, R.; Peoples, M.D.; et al. Zeb1/nurd complex suppresses tbc1d2b to stimulate e-cadherin internalization and promote metastasis in lung cancer. Nat. Commun. 2019, 10, 5125. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, G.; Manabe, I.; Tsushima, K.; Fujiu, K.; Oishi, Y.; Imai, Y.; Maemura, K.; Miyagishi, M.; Higashi, Y.; Kondoh, H.; et al. Deltaef1 mediates tgf-beta signaling in vascular smooth muscle cell differentiation. Dev. Cell. 2006, 11, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Yang, S.; Shen, W.; Li, H.; Gao, Y.; Zhu, T.H. Identification of deltaef1 as a novel target that is negatively regulated by lmo2 in t-cell leukemia. Eur. J. Haematol. 2010, 85, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, W.; Mossmann, D.; Kleemann, J.; Mock, K.; Meisinger, C.; Brummer, T.; Herr, R.; Brabletz, S.; Stemmler, M.P.; Brabletz, T. Zeb1 turns into a transcriptional activator by interacting with yap1 in aggressive cancer types. Nat. Commun. 2016, 7, 10498. [Google Scholar] [CrossRef]
- Long, J.; Zuo, D.; Park, M. Pc2-mediated sumoylation of smad-interacting protein 1 attenuates transcriptional repression of e-cadherin. J. Biol. Chem. 2005, 280, 35477–35489. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, M.; Derynck, R.; Miyazono, K. Tgf-beta and the tgf-beta family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. Tgf-beta family signaling in the control of cell proliferation and survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massague, J. Transforming growth factor-beta signaling in immunity and cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Dituri, F.; Cossu, C.; Mancarella, S.; Giannelli, G. The interactivity between tgfbeta and bmp signaling in organogenesis, fibrosis, and cancer. Cells 2019, 8, 1130. [Google Scholar] [CrossRef] [Green Version]
- Schmierer, B.; Hill, C.S. Tgfbeta-smad signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Gheldof, A.; Hulpiau, P.; van Roy, F.; De Craene, B.; Berx, G. Evolutionary functional analysis and molecular regulation of the zeb transcription factors. Cell. Mol. Life Sci. 2012, 69, 2527–2541. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A. Opposing functions of zeb proteins in the regulation of the tgfbeta/bmp signaling pathway. EMBO J. 2003, 22, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- van Grunsven, L.A.; Taelman, V.; Michiels, C.; Opdecamp, K.; Huylebroeck, D.; Bellefroid, E.J. Deltaef1 and sip1 are differentially expressed and have overlapping activities during xenopus embryogenesis. Dev. Dyn. 2006, 235, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Depp, J.L.; Taylor, J.J.; Kroll, K.L. Regulation of smad signaling through a differential recruitment of coactivators and corepressors by zeb proteins. EMBO J. 2003, 22, 2453–2462. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, S.; Yamazaki, S.; Nakauchi, H.; Morishita, K. Downregulation of zeb1 and overexpression of smad7 contribute to resistance to tgf-beta1-mediated growth suppression in adult t-cell leukemia/lymphoma. Oncogene 2010, 29, 4157–4169. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, A.; Saigou, Y.; Higashi, Y.; Kondoh, H. Regulation of ocular lens development by smad-interacting protein 1 involving foxe3 activation. Development 2005, 132, 4437–4448. [Google Scholar] [CrossRef] [Green Version]
- De Coninck, S.; Berx, G.; Taghon, T.; Van Vlierberghe, P.; Goossens, S. Zeb2 in t-cells and t-all. Adv. Biol. Regul. 2019, 74, 100639. [Google Scholar] [CrossRef]
- Scott, C.L.; Omilusik, K.D. Zebs: Novel players in immune cell development and function. Trends Immunol. 2019, 40, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Epifanova, E.; Babaev, A.; Newman, A.G.; Tarabykin, V. Role of zeb2/sip1 in neuronal development. Brain Res. 2019, 1705, 24–31. [Google Scholar] [CrossRef]
- Vandamme, N.; Berx, G. From neural crest cells to melanocytes: Cellular plasticity during development and beyond. Cell. Mol. Life Sci. 2019, 76, 1919–1934. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Moribe, H.; Kondoh, H.; Higashi, Y. Deltaef1, a zinc finger and homeodomain transcription factor, is required for skeleton patterning in multiple lineages. Development 1998, 125, 21–31. [Google Scholar] [PubMed]
- Van de Putte, T.; Maruhashi, M.; Francis, A.; Nelles, L.; Kondoh, H.; Huylebroeck, D.; Higashi, Y. Mice lacking zfhx1b, the gene that codes for smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of hirschsprung disease-mental retardation syndrome. Am J Hum Genet 2003, 72, 465–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Putte, T.; Francis, A.; Nelles, L.; van Grunsven, L.A.; Huylebroeck, D. Neural crest-specific removal of zfhx1b in mouse leads to a wide range of neurocristopathies reminiscent of mowat-wilson syndrome. Hum Mol Genet 2007, 16, 1423–1436. [Google Scholar] [CrossRef] [Green Version]
- Stryjewska, A.; Dries, R.; Pieters, T.; Verstappen, G.; Conidi, A.; Coddens, K.; Francis, A.; Umans, L.; van, I.W.F.; Berx, G.; et al. Zeb2 regulates cell fate at the exit from epiblast state in mouse embryonic stem cells. Stem Cells 2017, 35, 611–625. [Google Scholar] [CrossRef]
- Kalcheim, C. Epithelial-mesenchymal transitions during neural crest and somite development. J. Clin. Med. 2015, 5, 3261–3275. [Google Scholar] [CrossRef]
- Gouignard, N.; Andrieu, C.; Theveneau, E. Neural crest delamination and migration: Looking forward to the next 150 years. Genesis 2018, 56, e23107. [Google Scholar] [CrossRef]
- Theveneau, E.; Mayor, R. Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 2012, 366, 34–54. [Google Scholar] [CrossRef] [Green Version]
- Adameyko, I.; Lallemend, F.; Aquino, J.B.; Pereira, J.A.; Topilko, P.; Muller, T.; Fritz, N.; Beljajeva, A.; Mochii, M.; Liste, I.; et al. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell 2009, 139, 366–379. [Google Scholar] [CrossRef] [Green Version]
- Adameyko, I.; Lallemend, F. Glial versus melanocyte cell fate choice: Schwann cell precursors as a cellular origin of melanocytes. Cell. Mol. Life Sci. 2010, 67, 3037–3055. [Google Scholar] [CrossRef]
- Dupin, E.; Real, C.; Glavieux-Pardanaud, C.; Vaigot, P.; Le Douarin, N.M. Reversal of developmental restrictions in neural crest lineages: Transition from schwann cells to glial-melanocytic precursors in vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 5229–5233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A. The biology of melanocyte and melanocyte stem cell. Acta Biochim. Biophys. Sin. (Shanghai) 2014, 46, 255–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, A.; Fisher, D.E. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab. Invest 2017, 97, 649–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotto, C.; Abbe, P.; Hemesath, T.J.; Bille, K.; Fisher, D.E.; Ortonne, J.P.; Ballotti, R. Microphthalmia gene product as a signal transducer in camp-induced differentiation of melanocytes. J. Cell Biol. 1998, 142, 827–835. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, A.; Takeda, K.; Ploplis, B.; Tachibana, M. Epistatic relationship between waardenburg syndrome genes mitf and pax3. Nat. Genet. 1998, 18, 283–286. [Google Scholar] [CrossRef]
- Verastegui, C.; Bille, K.; Ortonne, J.P.; Ballotti, R. Regulation of the microphthalmia-associated transcription factor gene by the waardenburg syndrome type 4 gene, sox10. J. Biol. Chem. 2000, 275, 30757–30760. [Google Scholar] [CrossRef] [Green Version]
- Kos, R.; Reedy, M.V.; Johnson, R.L.; Erickson, C.A. The winged-helix transcription factor foxd3 is important for establishing the neural crest lineage and repressing melanogenesis in avian embryos. Development 2001, 128, 1467–1479. [Google Scholar]
- Denecker, G.; Vandamme, N.; Akay, O.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a zeb2-mitf-zeb1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014, 21, 1250–1261. [Google Scholar] [CrossRef] [Green Version]
- Osawa, M.; Egawa, G.; Mak, S.S.; Moriyama, M.; Freter, R.; Yonetani, S.; Beermann, F.; Nishikawa, S. Molecular characterization of melanocyte stem cells in their niche. Development 2005, 132, 5589–5599. [Google Scholar] [CrossRef] [Green Version]
- Alonso, L.; Fuchs, E. The hair cycle. J. Cell Sci. 2006, 119, 391–393. [Google Scholar] [CrossRef]
- Moriyama, M.; Osawa, M.; Mak, S.S.; Ohtsuka, T.; Yamamoto, N.; Han, H.; Delmas, V.; Kageyama, R.; Beermann, F.; Larue, L.; et al. Notch signaling via hes1 transcription factor maintains survival of melanoblasts and melanocyte stem cells. J. Cell Biol. 2006, 173, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Kumano, K.; Masuda, S.; Sata, M.; Saito, T.; Lee, S.Y.; Sakata-Yanagimoto, M.; Tomita, T.; Iwatsubo, T.; Natsugari, H.; Kurokawa, M.; et al. Both notch1 and notch2 contribute to the regulation of melanocyte homeostasis. Pigment. Cell Melanoma Res. 2008, 21, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, E.K.; Suzuki, M.; Igras, V.; Du, J.; Lonning, S.; Miyachi, Y.; Roes, J.; Beermann, F.; Fisher, D.E. Key roles for transforming growth factor beta in melanocyte stem cell maintenance. Cell Stem Cell 2010, 6, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandyba, E.; Leung, Y.; Chen, Y.B.; Widelitz, R.; Chuong, C.M.; Kobielak, K. Competitive balance of intrabulge bmp/wnt signaling reveals a robust gene network ruling stem cell homeostasis and cyclic activation. Proc. Natl. Acad. Sci. USA 2013, 110, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, P.; Takeo, M.; Chou, W.; Myung, P.; Bosenberg, M.; Chin, L.; Taketo, M.M.; Ito, M. Coordinated activation of wnt in epithelial and melanocyte stem cells initiates pigmented hair regeneration. Cell 2011, 145, 941–955. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Yang, K.; Carpenter, A.; Lang, R.A.; Andl, T.; Zhang, Y. Cd133-positive dermal papilla-derived wnt ligands regulate postnatal hair growth. Biochem. J. 2016, 473, 3291–3305. [Google Scholar] [CrossRef] [Green Version]
- Goossens, S.; Vandamme, N.; Van Vlierberghe, P.; Berx, G. Emt transcription factors in cancer development re-evaluated: Beyond emt and met. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 584–591. [Google Scholar] [CrossRef]
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-mesenchymal transition: Epigenetic reprogramming driving cellular plasticity. Trends Genet. 2017, 33, 943–959. [Google Scholar] [CrossRef]
- Chin, V.L.; Lim, C.L. Epithelial-mesenchymal plasticity-engaging stemness in an interplay of phenotypes. Stem Cell Investig. 2019, 6, 25. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of emt transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.T.; Yang, M.H. Hybrid epithelial/mesenchymal state in cancer metastasis: Clinical significance and regulatory mechanisms. Cells 2020, 9, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, zeb and bhlh factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining emt during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment. Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef]
- Hoek, K.S.; Schlegel, N.C.; Brafford, P.; Sucker, A.; Ugurel, S.; Kumar, R.; Weber, B.L.; Nathanson, K.L.; Phillips, D.J.; Herlyn, M.; et al. Metastatic potential of melanomas defined by specific gene expression profiles with no braf signature. Pigment. Cell Res. 2006, 19, 290–302. [Google Scholar] [CrossRef]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Dobbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef] [Green Version]
- Eichhoff, O.M.; Weeraratna, A.; Zipser, M.C.; Denat, L.; Widmer, D.S.; Xu, M.; Kriegl, L.; Kirchner, T.; Larue, L.; Dummer, R.; et al. Differential lef1 and tcf4 expression is involved in melanoma cell phenotype switching. Pigment. Cell Melanoma Res. 2011, 24, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic emt-inducers drives the development of malignant melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, J.; Stas, M.; Govaere, O.; Barrette, K.; Dudek, A.; Vankelecom, H.; Haydu, L.E.; Thompson, J.F.; Scolyer, R.A.; van den Oord, J.J. A novel hypoxia-associated subset of fn1 high mitf low melanoma cells: Identification, characterization, and prognostic value. Mod. Pathol. 2014, 27, 1088–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riesenberg, S.; Groetchen, A.; Siddaway, R.; Bald, T.; Reinhardt, J.; Smorra, D.; Kohlmeyer, J.; Renn, M.; Phung, B.; Aymans, P.; et al. Mitf and c-jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat. Commun. 2015, 6, 8755. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals teads as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [Green Version]
- Zipser, M.C.; Eichhoff, O.M.; Widmer, D.S.; Schlegel, N.C.; Schoenewolf, N.L.; Stuart, D.; Liu, W.; Gardner, H.; Smith, P.D.; Nuciforo, P.; et al. A proliferative melanoma cell phenotype is responsive to raf/mek inhibition independent of braf mutation status. Pigment. Cell Melanoma Res. 2011, 24, 326–333. [Google Scholar] [CrossRef]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wolfel, T.; Holzel, M.; et al. Melanomas resist t-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to mapk pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Muller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low mitf/axl ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to braf(v600e) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and transcriptomic features of response to anti-pd-1 therapy in metastatic melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falletta, P.; Sanchez-Del-Campo, L.; Chauhan, J.; Effern, M.; Kenyon, A.; Kershaw, C.J.; Siddaway, R.; Lisle, R.; Freter, R.; Daniels, M.J.; et al. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev. 2017, 31, 18–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ennen, M.; Keime, C.; Kobi, D.; Mengus, G.; Lipsker, D.; Thibault-Carpentier, C.; Davidson, I. Single-cell gene expression signatures reveal melanoma cell heterogeneity. Oncogene 2015, 34, 3251–3263. [Google Scholar] [CrossRef]
- Ennen, M.; Keime, C.; Gambi, G.; Kieny, A.; Coassolo, S.; Thibault-Carpentier, C.; Margerin-Schaller, F.; Davidson, G.; Vagne, C.; Lipsker, D.; et al. Mitf-high and mitf-low cells and a novel subpopulation expressing genes of both cell states contribute to intra- and intertumoral heterogeneity of primary melanoma. Clin. Cancer Res. 2017, 23, 7097–7107. [Google Scholar] [CrossRef] [Green Version]
- Wouters, J.; Kalender-Atak, Z.; Minnoye, L.; Spanier, K.I.; De Waegeneer, M.; González-Blas, C.B.; Mauduit, D.; Davie, K.; Hulselmans, G.; Najem, A.; et al. Single-cell gene regulatory network analysis reveals new melanoma cell states and transition trajectories during phenotype switching. bioRxiv 2019. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward minimal residual disease-directed therapy in melanoma. Cell 2018, 174, 843–855.e819. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell 2018, 33, 890–904. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, N.; Denecker, G.; Bruneel, K.; Blancke, G.; Akay, Ö.; Taminau, J.; De Coninck, J.; De Smedt, E.; Skrypek, N.; Van Loocke, W.; et al. The emt transcription factor zeb2 promotes proliferation of primary and metastatic melanoma while suppressing an invasive, mesenchymal-like phenotype. Cancer Res. 2020, 80, 2983–2995. [Google Scholar] [CrossRef]
- Dou, J.; He, X.; Liu, Y.; Wang, Y.; Zhao, F.; Wang, X.; Chen, D.; Shi, F.; Wang, J. Effect of downregulation of zeb1 on vimentin expression, tumour migration and tumourigenicity of melanoma b16f10 cells and cscs. Cell Biol. Int. 2014, 38, 452–461. [Google Scholar] [CrossRef]
- Zhao, F.; He, X.; Wang, Y.; Shi, F.; Wu, D.; Pan, M.; Li, M.; Wu, S.; Wang, X.; Dou, J. Decrease of zeb1 expression inhibits the b16f10 cancer stem-like properties. Biosci. Trends 2015, 9, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heppt, M.V.; Wang, J.X.; Hristova, D.M.; Wei, Z.; Li, L.; Evans, B.; Beqiri, M.; Zaman, S.; Zhang, J.; Irmler, M.; et al. Msx1-induced neural crest-like reprogramming promotes melanoma progression. J. Invest Dermatol. 2018, 138, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabierowski, S.E.; Baubet, V.; Himes, B.; Li, L.; Fukunaga-Kalabis, M.; Patel, S.; McDaid, R.; Guerra, M.; Gimotty, P.; Dahmane, N.; et al. Direct reprogramming of melanocytes to neural crest stem-like cells by one defined factor. Stem Cells 2011, 29, 1752–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, G.; Dalle, S.; Monet, M.A.; Ligier, M.; Boespflug, A.; Pommier, R.M.; de la Fouchardiere, A.; Perier-Muzet, M.; Depaepe, L.; Barnault, R.; et al. Zeb1-mediated melanoma cell plasticity enhances resistance to mapk inhibitors. EMBO Mol. Med. 2016, 8, 1143–1161. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to map kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- O’Connell, M.P.; Marchbank, K.; Webster, M.R.; Valiga, A.A.; Kaur, A.; Vultur, A.; Li, L.; Herlyn, M.; Villanueva, J.; Liu, Q.; et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ror1 and ror2. Cancer Discov. 2013, 3, 1378–1393. [Google Scholar] [CrossRef] [Green Version]
- Holzel, M.; Tuting, T. Inflammation-induced plasticity in melanoma therapy and metastasis. Trends Immunol. 2016, 37, 364–374. [Google Scholar] [CrossRef]
- Schlegel, N.C.; von Planta, A.; Widmer, D.S.; Dummer, R.; Christofori, G. Pi3k signalling is required for a tgfbeta-induced epithelial-mesenchymal-like transition (emt-like) in human melanoma cells. Exp. Dermatol. 2015, 24, 22–28. [Google Scholar] [CrossRef]
- Perrot, C.Y.; Gilbert, C.; Marsaud, V.; Postigo, A.; Javelaud, D.; Mauviel, A. Gli2 cooperates with zeb1 for transcriptional repression of cdh1 expression in human melanoma cells. Pigment. Cell Melanoma Res. 2013, 26, 861–873. [Google Scholar] [CrossRef]
- Gunarta, I.K.; Li, R.; Nakazato, R.; Suzuki, R.; Boldbaatar, J.; Suzuki, T.; Yoshioka, K. Critical role of glioma-associated oncogene homolog 1 in maintaining invasive and mesenchymal-like properties of melanoma cells. Cancer Sci. 2017, 108, 1602–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheli, Y.; Giuliano, S.; Fenouille, N.; Allegra, M.; Hofman, V.; Hofman, P.; Bahadoran, P.; Lacour, J.P.; Tartare-Deckert, S.; Bertolotto, C.; et al. Hypoxia and mitf control metastatic behaviour in mouse and human melanoma cells. Oncogene 2012, 31, 2461–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widmer, D.S.; Hoek, K.S.; Cheng, P.F.; Eichhoff, O.M.; Biedermann, T.; Raaijmakers, M.I.G.; Hemmi, S.; Dummer, R.; Levesque, M.P. Hypoxia contributes to melanoma heterogeneity by triggering hif1alpha-dependent phenotype switching. J. Invest Dermatol. 2013, 133, 2436–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Li, Y.; Luo, J.; Hou, N. Molecular mechanism of hippo-yap1/taz pathway in heart development, disease, and regeneration. Front. Physiol. 2020, 11, 389. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yang, L.; Szeto, P.; Abali, G.K.; Zhang, Y.; Kulkarni, A.; Amarasinghe, K.; Li, J.; Vergara, I.A.; Molania, R.; et al. The hippo pathway oncoprotein yap promotes melanoma cell invasion and spontaneous metastasis. Oncogene 2020, 134, 123–132. [Google Scholar] [CrossRef]
- Wels, C.; Joshi, S.; Koefinger, P.; Bergler, H.; Schaider, H. Transcriptional activation of zeb1 by slug leads to cooperative regulation of the epithelial-mesenchymal transition-like phenotype in melanoma. J. Invest Dermatol. 2011, 131, 1877–1885. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.B.; Kuperwasser, C.; Brunet, J.P.; Ramaswamy, S.; Kuo, W.L.; Gray, J.W.; Naber, S.P.; Weinberg, R.A. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat. Genet. 2005, 37, 1047–1054. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Protein/Complex | Complex | Region of Interaction | Target Gene | Activity | Regulation | Cellular Context | Refs |
|---|---|---|---|---|---|---|---|
| POU2F1 | Na | HD | Na | Na | Na | Rat ZEB1 | 8 |
| CtBP1/2 | CtBP repressor complex | CID | CDH1 & GH | Repression | Transcriptional & Epigenetic | Breast carcinoma & pituitary differentiation | 12,14,15 |
| HDAC1/2 | Na | Not defined | CDH1 | Repression | Epigenetic | Pancreatic cancer | 21 |
| SMARCA4 | SWI/SNF | N-terminal | CDH1 | Repression | Epigenetic | HEK293T cells | 23 |
| DNMT1 | Na | SBD | CDH1 | Repression | Epigenetic | Mammary gland/Breast cancer | 24 |
| hTERT | Na | Not defined | CDH1 | Repression | Transcriptional | Colorectal cancer | 25 |
| SIRT1 | Na | Not defined | CDH1 | Repression | Epigenetic | Prostate cancer | 27 |
| SET8 | Na | Not defined | CDH1 | Repression | Epigenetic | Prostate cancer | 28 |
| TIP60 | Na | N-terminal half | CD4 | Repression | Epigenetic | Jurkat/Hela cells | 29 |
| NC2 | Na | Between HD & CZF | In vitro reporter constructs | Repression | Transcriptional | Hela cells | 30 |
| NuRD complex | NuRD | Not defined | TBC1D2b | Repression | Epigenetic | Lung cancer | 32 |
| SRF | SMAD3 | NZF & CZF | α-actin | Activation | Transcriptional | Smooth muscle cell differentiation | 33 |
| LMO2 | Na | N-terminal | ZEB1 | Represses ZEB1 transcription/ activity | Transcriptional/ post-translational | T-cell leukemia | 34 |
| YAP | Na | N-terminal & C-terminal | Common ZEB1/YAP target gene set | Activation | Transcriptional | Breast cancer | 35 |
| PC2 | Na | Lys-347 & Lys-774 | ZEB1 | Sumoylates and represses ZEB1 activity | Post-translational | COS-1 cells | 36 |
| pCAF/p300 | SMAD3 | N-terminal | Xbra/Gata2/Msx1 | Activation | Transcriptional | Neurogenesis | 44,45 |
| SMAD1 | Na | SBD | Na | Na | Na | HEK293T cells | 43 |
| SMAD2 | Na | SBD | Na | Na | Na | HEK293T cells | 43 |
| SMAD3 | SRF & Na & pCAF/p300 | SBD | α-actin & BMP- /TGF-β-signalling & Xbra/Gata2/Msx1 | Activation & Repression | Transcriptional | Smooth muscle cell - & Osteoblast differentiation & Neurogenesis | 33,43,44,45 |
| SMAD7 | Na | SBD | SMAD7 | Represses SMAD7 activity | Post-translational | T-cell leukemia/lymphoma model | 46 |
| Protein/Complex | Complex | Region of Interaction | Target Gene | Activity | Regulation | Cellular Context | Refs |
|---|---|---|---|---|---|---|---|
| CtBP1/2 | CtBP repressor complex | CID | BMP4 | Repression | Transcriptional & Epigenetic | Neurulation | 13 |
| LSD1 | Na | Not defined | CD11b | Repression | Epigenetic | T-ALL | 20 |
| NuRD complex | NuRD | NIM | CDH1 | Repression | Epigenetic | HEK293T cells | 31 |
| PC2 | Na | Lys-391 & Lys-866 | ZEB2 | Represses ZEB2 activity | Post-translational | COS-1 cells | 36 |
| pCAF/p300 | SMAD3 | N-terminal | Xbra/Gata2/Msx1 | Activation | Transcriptional | Neurogenesis | 44,45 |
| SMAD1 | Na | SBD | Xbra2 | Repression | Transcriptional | Mesoderm differentiation | 7 |
| SMAD2 | Na | SBD | Xbra2 | Repression | Transcriptional | Mesoderm differentiation | 7 |
| SMAD3 | Na & pCAF/p300 | SBD | BMP/TGF-β-signalling & Xbra/Gata2/Msx1 | Repression & Activation | Transcriptional | Osteoblast differentiation & Neurogenesis | 43,44,45 |
| SMAD5 | Na | SBD | Xbra2 | Repression | Transcriptional | Mesoderm differentiation | 7 |
| SMAD8 | Na | SBD | Foxe3 | Activation | Transcriptional | Ocular lens development | 47 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruneel, K.; Verstappe, J.; Vandamme, N.; Berx, G. Intrinsic Balance between ZEB Family Members Is Important for Melanocyte Homeostasis and Melanoma Progression. Cancers 2020, 12, 2248. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082248
Bruneel K, Verstappe J, Vandamme N, Berx G. Intrinsic Balance between ZEB Family Members Is Important for Melanocyte Homeostasis and Melanoma Progression. Cancers. 2020; 12(8):2248. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082248
Chicago/Turabian StyleBruneel, Kenneth, Jeroen Verstappe, Niels Vandamme, and Geert Berx. 2020. "Intrinsic Balance between ZEB Family Members Is Important for Melanocyte Homeostasis and Melanoma Progression" Cancers 12, no. 8: 2248. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082248