Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Altered Glucose Metabolism in Different Subtypes of BC
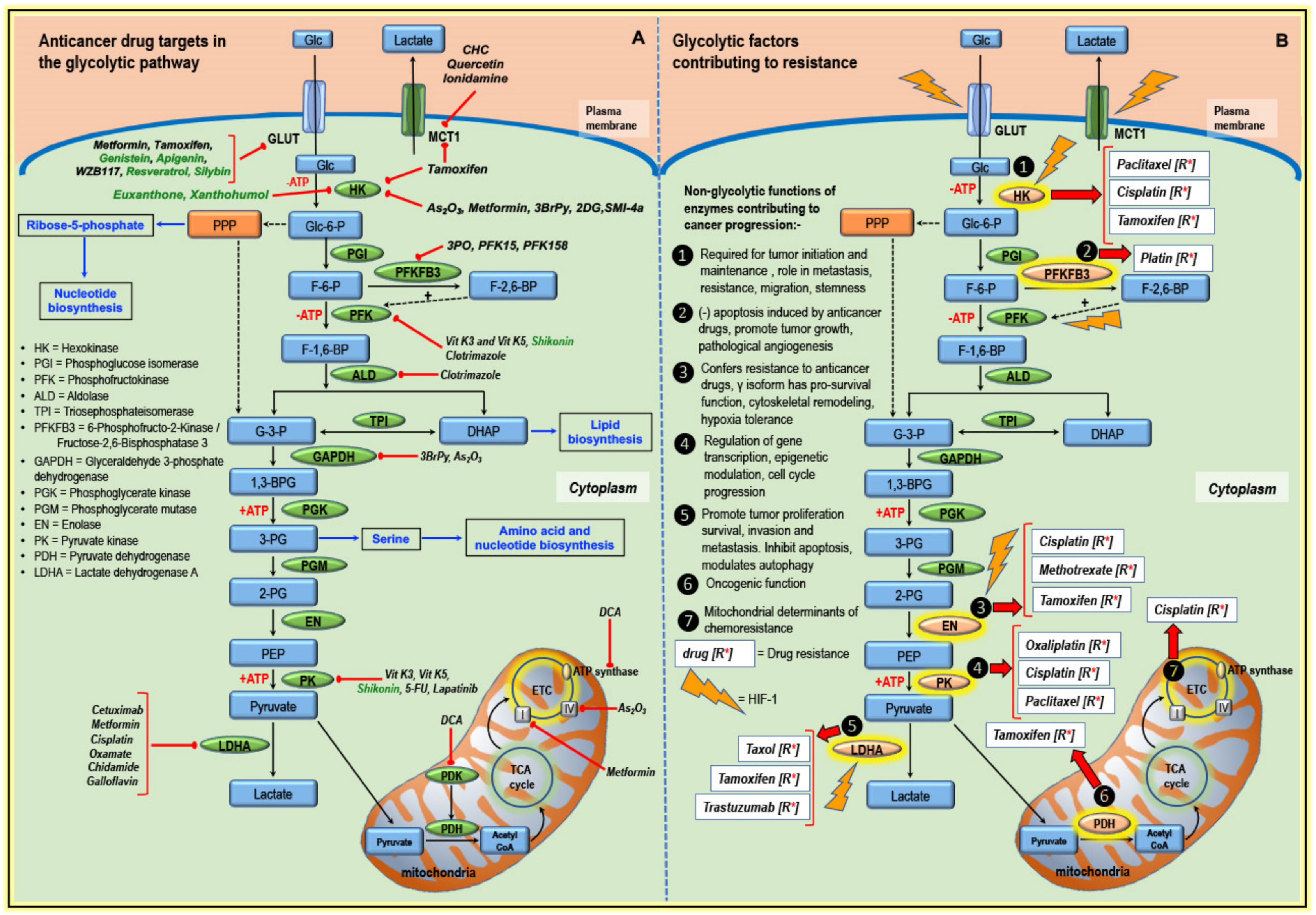
3. Glycolytic Pathway Targeted by Classical Anticancer Drugs and Other Compounds
3.1. Targeting Transporters
3.2. Targeting Enzymes of the Glycolytic Pathway
3.3. Targeting Components of Mitochondria
4. Altered Glucose Metabolism and Anticancer Drug Resistance
4.1. Hexokinase and Drug Resistance
4.2. PFKFB3 and Drug Resistance
4.3. Enolase and Drug Resistance
4.4. Pyruvate Kinase and Drug Resistance
4.5. LDHA and Drug Resistance
4.6. PDH/PDK and Drug Resistance
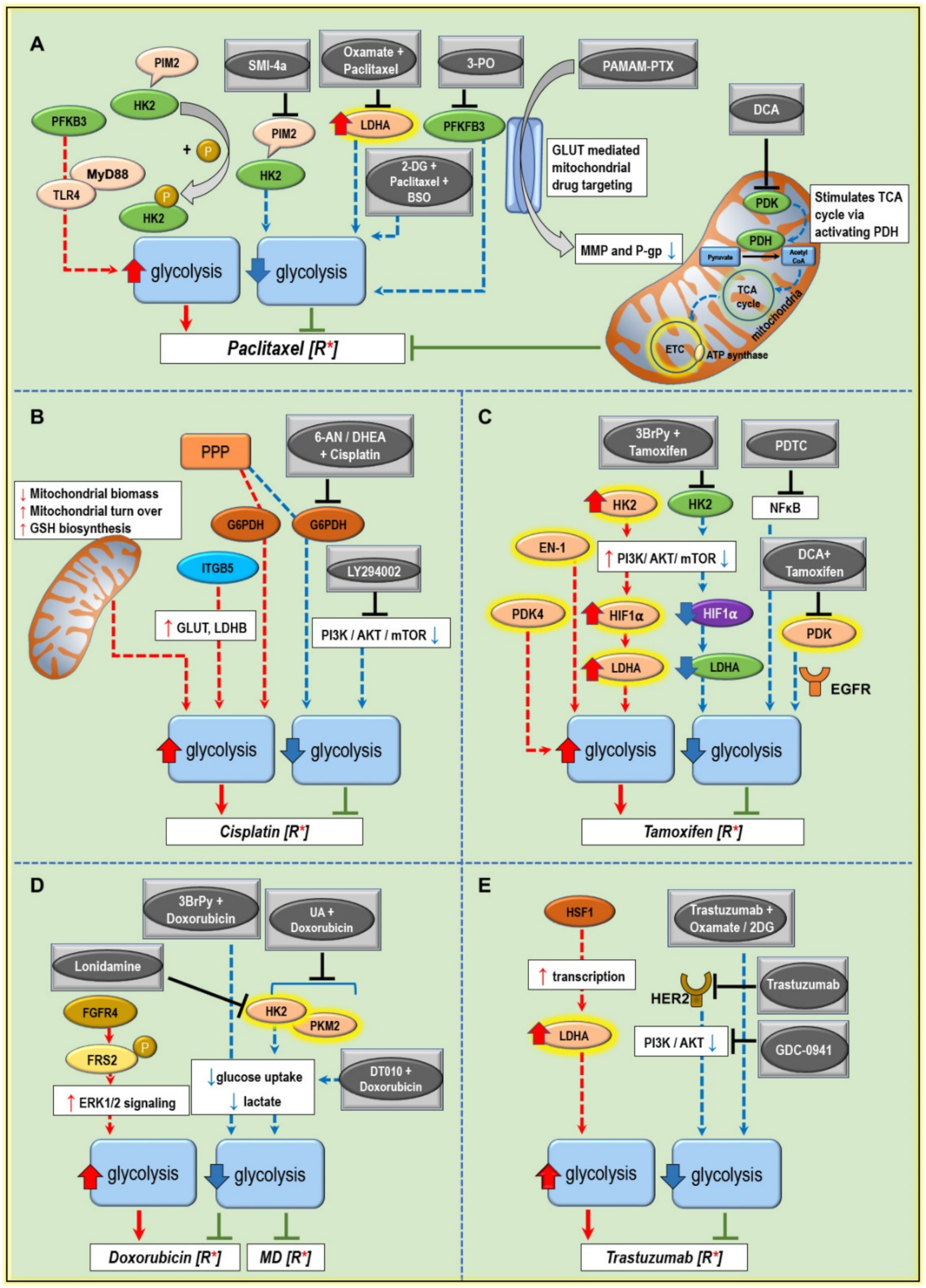
5. Modulating Glucose Metabolism to Increase the Anticancer Drug Efficiency and Combat Treatment Resistance
5.1. Paclitaxel Resistance
5.2. Cisplatin Resistance
5.3. Tamoxifen Resistance
5.4. Doxorubicin Resistance
5.5. Trastuzumab Resistance
5.6. Palbociclib Resistance
6. The Scope of Targeting Warburg’s Effect for Resensitizing BC Chemotherapy
6.1. PI3K/AKT Pathway is a Crucial Signal Contributing to Therapy Resistance
6.2. Targeting Glycolytic Enzymes Prove to Be Beneficial In Both In Vitro and In Vivo Cancer Models
6.3. Novel Methods to Improve Anticancer Drug Efficacy by Modulating the Tumor Glucose Metabolism
6.4. The Power of Combinations for Enhanced Efficacy of Chemotherapy in BC
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-DG | 2-Deoxy-D-glucose |
| 3-BrPy | 3-bromopyruvate |
| 3-PO | 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one |
| 6-AN | 6-aminonicotinamide |
| AKT | Protein kinase B |
| AMPK | 5’ adenosine monophosphate-activated protein kinase |
| ASCT2 | Alanine-, serine-, cysteine transporter 2 |
| ATO | Arsenic trioxide |
| ATP | Adenosine triphosphate |
| BC | Breast cancer |
| BSO | Buthionine sulfoximine |
| CHC | α-cyano-4-hydroxycinnamate |
| DCA | Dichloroacetate |
| ECM | Extracellular matrix |
| EGFR | Epidermal growth factor receptor |
| EN | Enolase |
| ER | Estrogen receptor |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GLUT | Glucose transporter |
| HIF-1α | Hypoxia inducible factor alpha |
| HK | Hexokinase |
| HSF1 | Heat shock factor 1 |
| IC50 | Half maximal inhibitory concentration |
| IGFR | Insulin-like growth factor receptor |
| LDHA | Lactate Dehydrogenase A |
| MCP | Mitochondrial pyruvate carrier |
| MCT1 | Monocarboxylate transporter 1 |
| MDR | Multidrug resistance |
| MET | Mesenchymal-epithelial transition factor |
| OCT4 | Octamer binding transcription factor/protein 4 |
| P-gp | Permeability glycoprotein |
| PAMAM-PTX | Paclitaxel-conjugated polyamidoamine |
| PDTC | Pyrrolidine dithiocarbamate |
| PFK | Phosphofructokinase |
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 3 |
| PI3K | Phosphatidylinositol 3-kinase |
| PK | Pyruvate kinase |
| PPP | Pentose phosphate pathway |
| Ras | Rat sarcoma viral proto-oncogene |
| ROS | Reactive oxygen species |
| RTK | Receptor tyrosine kinase |
| SOX2 | Sex determining region Y Box 2 |
| TCA | Tricarboxylic acid cycle |
| TNBC | Triple negative breast cancer |
| UA | Ursolic acid |
| VEGFα | Vascular endothelial growth factor alpha |
| WT | Wildtype |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef]
- American Cancer Society. Breast Cancer Facts & Figures 2019–2020; American Cancer Society, Inc.: Atlanta, GA, USA, 2019. [Google Scholar]
- DeMichele, A.; Yee, D.; Esserman, L. Mechanisms of Resistance to Neoadjuvant Chemotherapy in Breast Cancer. N. Engl. J. Med. 2017, 377, 2287–2289. [Google Scholar] [CrossRef]
- Mayor, S. Side-effects of cancer drugs are under-reported in trials. Lancet Oncol. 2015, 16, e107. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Calaf, G.M.; Zepeda, A.B.; Castillo, R.L.; Figueroa, C.A.; Arias, C.; Figueroa, E.; Farias, J.G. Molecular aspects of breast cancer resistance to drugs (Review). Int. J. Oncol. 2015, 47, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Lyon, R.C.; Cohen, J.S.; Faustino, P.J.; Megnin, F.; Myers, C.E. Glucose metabolism in drug-sensitive and drug-resistant human breast cancer cells monitored by magnetic resonance spectroscopy. Cancer Res. 1988, 48, 870–877. [Google Scholar]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.M.; Desai, K.V. Pathways to Endocrine Therapy Resistance in Breast Cancer. Front. Endocrinol. 2019, 10, 573. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zong, X. Metabolic Symbiosis in Chemoresistance: Refocusing the Role of Aerobic Glycolysis. Front. Oncol. 2020, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, E.; Samuel, S.M.; Abotaleb, M.; Cheema, S.; Mamtani, R.; Busselberg, D. The “Yin and Yang” of Natural Compounds in Anticancer Therapy of Triple-Negative Breast Cancers. Cancers 2018, 10, 346. [Google Scholar] [CrossRef] [Green Version]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbiah, S.; Gopu, G.; Senthilkumar, P.; Muniasamy, P. Molecular subtypes as a predictor of response to neoadjuvant chemotherapy in breast cancer patients. Indian J. Cancer 2017, 54, 652–657. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramson, V.G.; Mayer, I.A. Molecular Heterogeneity of Triple Negative Breast Cancer. Curr. Breast Cancer Rep. 2014, 6, 154–158. [Google Scholar] [CrossRef] [Green Version]
- Cappelletti, V.; Iorio, E.; Miodini, P.; Silvestri, M.; Dugo, M.; Daidone, M.G. Metabolic Footprints and Molecular Subtypes in Breast Cancer. Dis. Markers 2017, 2017, 7687851. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Xia, L.; Liang, J.; Han, Y.; Wang, H.; Oyang, L.; Tan, S.; Tian, Y.; Rao, S.; Chen, X.; et al. The roles of glucose metabolic reprogramming in chemo- and radio-resistance. J. Exp. Clin. Cancer Res. 2019, 38, 218. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Esechie, A.; Du, G. Increased lipogenesis in cancer. Commun. Integr. Biol. 2009, 2, 545–548. [Google Scholar] [CrossRef]
- Villa, E.; Ali, E.S.; Sahu, U.; Ben-Sahra, I. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers 2019, 11, 688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, S.M.; Varghese, E.; Varghese, S.; Büsselberg, D. Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treat. Rev. 2018, 70, 98–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, L.C.; Franklin, R.B. ‘Why do tumour cells glycolyse?’: From glycolysis through citrate to lipogenesis. Mol. Cell. Biochem. 2005, 280, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Moreadith, R.W.; Lehninger, A.L. The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. Role of mitochondrial NAD(P)+-dependent malic enzyme. J. Biol. Chem. 1984, 259, 6215–6221. [Google Scholar]
- Zhou, Z.; Ibekwe, E.; Chornenkyy, Y. Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change. Antioxidants 2018, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef]
- Zhang, F.; Du, G. Dysregulated lipid metabolism in cancer. World J. Biol. Chem. 2012, 3, 167–174. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, D.H.; Jung, W.H.; Koo, J.S. Succinate dehydrogenase expression in breast cancer. Springerplus 2013, 2, 299. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.-O.; Li, C.-W.; Xia, W.; Lee, H.-H.; Chang, S.-S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H.; et al. EGFR Signaling Enhances Aerobic Glycolysis in Triple-Negative Breast Cancer Cells to Promote Tumor Growth and Immune Escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef] [Green Version]
- Varghese, S.; Samuel, S.M.; Varghese, E.; Kubatka, P.; Busselberg, D. High Glucose Represses the Anti-Proliferative and Pro-Apoptotic Effect of Metformin in Triple Negative Breast Cancer Cells. Biomolecules 2019, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Miao, W.; Huang, M.; Li, L.; Dai, X.; Wang, Y. Elevated Hexokinase II Expression Confers Acquired Resistance to 4-Hydroxytamoxifen in Breast Cancer Cells. Mol. Cell. Proteom. 2019, 18, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.-H.; Chang, C.-C.; Chen, C.-S.; Tam, K.-W.; Wang, Y.-J.; Lee, C.-H.; Lin, H.-W.; Cheng, T.-C.; Huang, C.-S.; Chu, J.-S.; et al. Increased expression of enolase α in human breast cancer confers tamoxifen resistance in human breast cancer cells. Breast Cancer Res. Treat. 2010, 121, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Sato-Tadano, A.; Suzuki, T.; Amari, M.; Takagi, K.; Miki, Y.; Tamaki, K.; Watanabe, M.; Ishida, T.; Sasano, H.; Ohuchi, N. Hexokinase II in breast carcinoma: A potent prognostic factor associated with hypoxia-inducible factor-1a and Ki-67. Cancer Sci. 2013, 104, 1380–1388. [Google Scholar] [CrossRef] [Green Version]
- Zancan, P.; Sola-Penna, M.; Furtado, C.M.; Da Silva, D. Differential expression of phosphofructokinase-1 isoforms correlates with the glycolytic efficiency of breast cancer cells. Mol. Genet. Metab. 2010, 100, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, M.; Wang, M.; Yu, X.; Guo, J.; Sun, T.; Li, X.; Yao, L.; Dong, H.; Xu, Y. Metabolic Reprogramming in Triple-Negative Breast Cancer. Front. Oncol. 2020, 10, 428. [Google Scholar] [CrossRef]
- Pucci-Minafra, I.; Fontana, S.; Cancemi, P.; Alaimo, G.; Minafra, S. Proteomic Patterns of Cultured Breast Cancer Cells and Epithelial Mammary Cells. Ann. N. Y. Acad. Sci. 2002, 963, 122–139. [Google Scholar] [CrossRef] [Green Version]
- Altenberg, B.; Greulich, K.O. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef]
- Cerella, C.; Radogna, F.; Dicato, M.; Diederich, M. Natural compounds as regulators of the cancer cell metabolism. Int. J. Cell Biol. 2013, 2013, 639401. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.-L.; Chen, Y.-G. Natural compounds regulate glycolysis in hypoxic tumor microenvironment. BioMed Res. Int. 2015, 2015, 354143. [Google Scholar] [CrossRef]
- Rivenzon-Segal, D.; Boldin-Adamsky, S.; Seger, D.; Seger, R.; Degani, H. Glycolysis and glucose transporter 1 as markers of response to hormonal therapy in breast cancer. Int. J. Cancer 2003, 107, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Samuel, S.M.; Varghese, E.; Kubatka, P.; Triggle, C.R.; Büsselberg, D. Metformin: The Answer to Cancer in a Flower? Current Knowledge and Future Prospects of Metformin as an Anti-Cancer Agent in Breast Cancer. Biomolecules 2019, 9, 846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahdan-Alaswad, R.S.; Edgerton, S.M.; Salem, H.S.; Thor, A.D. Metformin Targets Glucose Metabolism in Triple Negative Breast Cancer. J. Oncol. Transl. Res. 2018, 4. [Google Scholar] [CrossRef]
- Yang, Y.; Wolfram, J.; Boom, K.; Fang, X.; Shen, H.; Ferrari, M. Hesperetin impairs glucose uptake and inhibits proliferation of breast cancer cells. Cell Biochem. Funct. 2013, 31, 374–379. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, C.; Correia-Branco, A.; Araújo, J.R.; Guimarães, J.T.; Keating, E.; Martel, F. The chemopreventive effect of the dietary compound kaempferol on the MCF-7 human breast cancer cell line is dependent on inhibition of glucose cellular uptake. Nutr. Cancer 2015, 67, 504–513. [Google Scholar] [CrossRef]
- Wei, R.; Mao, L.; Xu, P.; Zheng, X.; Hackman, R.M.; Mackenzie, G.G.; Wang, Y. Suppressing glucose metabolism with epigallocatechin-3-gallate (EGCG) reduces breast cancer cell growth in preclinical models. Food Funct. 2018, 9, 5682–5696. [Google Scholar] [CrossRef]
- Granchi, C.; Fortunato, S.; Minutolo, F. Anticancer agents interacting with membrane glucose transporters. MedChemComm 2016, 7, 1716–1729. [Google Scholar] [CrossRef] [Green Version]
- Catanzaro, D.; Gabbia, D.; Cocetta, V.; Biagi, M.; Ragazzi, E.; Montopoli, M.; Carrara, M. Silybin counteracts doxorubicin resistance by inhibiting GLUT1 expression. Fitoterapia 2018, 124, 42–48. [Google Scholar] [CrossRef]
- Levenson, A.S.; Thurn, T.E.; Simons, L.A.; Osipo, C.; Jordan, V.C.; Satcher, R.L.; Gartenhaus, R.B. Overexpression of MCT1 oncogene contributes to increased tumorigenicity of MCF7 breast cancer cells. Cancer Res. 2005, 65, 662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipa, M.-S.; Vera, M.-G.; Sílvia, P.; André, F.V.; Joana, P.; Fernando, C.S.; Fátima, B.; Céline, P. Differential sensitivities to lactate transport inhibitors of breast cancer cell lines. Endocr. Relat. Cancer 2014, 21, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Marini, C.; Salani, B.; Massollo, M.; Amaro, A.; Esposito, A.I.; Maria Orengo, A.; Capitanio, S.; Emionite, L.; Riondato, M.; Bottoni, G.; et al. Direct inhibition of hexokinase activity by metformin at least partially impairs glucose metabolism and tumor growth in experimental breast cancer. Cell Cycle 2013, 12, 3490–3499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aft, R.L.; Zhang, F.W.; Gius, D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: Mechanism of cell death. Br. J. Cancer 2002, 87, 805–812. [Google Scholar] [CrossRef] [Green Version]
- O’Neal, J.; Clem, A.; Reynolds, L.; Dougherty, S.; Imbert-Fernandez, Y.; Telang, S.; Chesney, J.; Clem, B.F. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res. Treat. 2016, 160, 29–40. [Google Scholar] [CrossRef]
- Shi, L.; Pan, H.; Liu, Z.; Xie, J.; Han, W. Roles of PFKFB3 in cancer. Signal Transduct. Target. Ther. 2017, 2, 17044. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Kunjithapatham, R.; Geschwind, J.F. Anticancer efficacy of the metabolic blocker 3-bromopyruvate: Specific molecular targeting. Anticancer Res. 2013, 33, 13–20. [Google Scholar]
- Fan, T.; Sun, G.; Sun, X.; Zhao, L.; Zhong, R.; Peng, Y. Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment. Cancers 2019, 11, 317. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Hu, X.; Cui, J. Shikonin, vitamin K3 and vitamin K5 inhibit multiple glycolytic enzymes in MCF-7 cells. Oncol. Lett. 2018, 15, 7423–7432. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Liu, J.; Jackson, K.; Shi, R.; Zhao, Y. Sensitizing the therapeutic efficacy of taxol with shikonin in human breast cancer cells. PLoS ONE 2014, 9, e94079. [Google Scholar] [CrossRef]
- Zhou, M.; Zhao, Y.; Ding, Y.; Liu, H.; Liu, Z.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.; Owen, L.B.; et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol. Cancer 2010, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Xie, J.; Li, J.; Liu, F.; Wu, X.; Wang, Z. Cisplatin suppresses the growth and proliferation of breast and cervical cancer cell lines by inhibiting integrin beta5-mediated glycolysis. Am. J. Cancer Res. 2016, 6, 1108–1117. [Google Scholar] [PubMed]
- Bai, X.; Jiang, H.; Han, G.; He, Q. Chidamide suppresses the glycolysis of triple negative breast cancer cells partially by targeting the miR-33a-5p-LDHA axis. Mol. Med. Rep. 2019, 20, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Li, X.; Luo, Z.; Liu, J.; Fan, Z. Cetuximab reverses the Warburg effect by inhibiting HIF-1-regulated LDH-A. Mol. Cancer Ther. 2013, 12, 2187–2199. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.C.; Board, P.G.; Blackburn, A.C. Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells. Mol. Cancer 2011, 10, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrzejewski, S.; Gravel, S.P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.C.; Fadia, M.; Dahlstrom, J.E.; Parish, C.R.; Board, P.G.; Blackburn, A.C. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res. Treat. 2010, 120, 253–260. [Google Scholar] [CrossRef]
- Verma, A.; Lam, Y.M.; Leung, Y.C.; Hu, X.; Chen, X.; Cheung, E.; Tam, K.Y. Combined use of arginase and dichloroacetate exhibits anti-proliferative effects in triple negative breast cancer cells. J. Pharm. Pharmacol. 2019, 71, 306–315. [Google Scholar] [CrossRef]
- Mobasheri, A.; Richardson, S.; Mobasheri, R.; Shakibaei, M.; Hoyland, J.A. Hypoxia inducible factor-1 and facilitative glucose transporters GLUT1 and GLUT3: Putative molecular components of the oxygen and glucose sensing apparatus in articular chondrocytes. Histol. Histopathol. 2005, 20, 1327–1338. [Google Scholar] [CrossRef]
- Krzeslak, A.; Wojcik-Krowiranda, K.; Forma, E.; Jozwiak, P.; Romanowicz, H.; Bienkiewicz, A.; Brys, M. Expression of GLUT1 and GLUT3 Glucose Transporters in Endometrial and Breast Cancers. Pathol. Oncol. Res. 2012, 18, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Robey, I.F.; Lien, A.D.; Welsh, S.J.; Baggett, B.K.; Gillies, R.J. Hypoxia-inducible factor-1alpha and the glycolytic phenotype in tumors. Neoplasia 2005, 7, 324–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, R.; van Der Hoeven, J.J.; van Der Wall, E.; van Der Groep, P.; van Diest, P.J.; Comans, E.F.; Joshi, U.; Semenza, G.L.; Hoekstra, O.S.; Lammertsma, A.A.; et al. Biologic correlates of (18)fluorodeoxyglucose uptake in human breast cancer measured by positron emission tomography. J. Clin. Oncol. 2002, 20, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, K.E.; Rekman, J.F.; Gunnink, L.K.; Busscher, B.M.; Scott, J.L.; Tidball, A.M.; Stehouwer, N.R.; Johnecheck, G.N.; Looyenga, B.D.; Louters, L.L. Quercetin inhibits glucose transport by binding to an exofacial site on GLUT1. Biochimie 2018, 151, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Pérez, A.; Ojeda, P.; Ojeda, L.; Salas, M.; Rivas, C.I.; Vera, J.C.; Reyes, A.M. Hexose transporter GLUT1 harbors several distinct regulatory binding sites for flavones and tyrphostins. Biochemistry 2011, 50, 8834–8845. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef] [Green Version]
- Zambrano, A.; Molt, M.; Uribe, E.; Salas, M. Glut 1 in Cancer Cells and the Inhibitory Action of Resveratrol as A Potential Therapeutic Strategy. Int. J. Mol. Sci. 2019, 20, 3374. [Google Scholar] [CrossRef] [Green Version]
- Sawayama, H.; Ogata, Y.; Ishimoto, T.; Mima, K.; Hiyoshi, Y.; Iwatsuki, M.; Baba, Y.; Miyamoto, Y.; Yoshida, N.; Baba, H. Glucose transporter 1 regulates the proliferation and cisplatin sensitivity of esophageal cancer. Cancer Sci. 2019, 110, 1705–1714. [Google Scholar] [CrossRef] [Green Version]
- Mathupala, S.; Parajuli, P.; Sloan, A. Silencing of monocarboxylate transporters via small interfering ribonucleic acid inhibits glycolysis and induces cell death in malignant glioma: An in vitro study. Neurosurgery 2004, 55, 1410–1419. [Google Scholar] [CrossRef]
- Gallagher, S.; Castorino, J.; Wang, D.; Philp, N. Monocarboxylate transporter 4 regulates maturation and trafficking of CD147 to the plasma membrane in the metastatic breast cancer cell line MDA-MB-231. Cancer Res. 2007, 67, 4182–4189. [Google Scholar] [CrossRef] [Green Version]
- Baltazar, F.; Pinheiro, C.; Morais-Santos, F.; Azevedo-Silva, J.; Queirós, O.; Preto, A.; Casal, M. Monocarboxylate transporters as targets and mediators in cancer therapy response. Histol. Histopathol. 2014, 29, 1511–1524. [Google Scholar] [CrossRef]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, C.S.; Graham, N.A.; Gu, W.; Espindola Camacho, C.; Mah, V.; Maresh, E.L.; Alavi, M.; Bagryanova, L.; Krotee, P.A.L.; Gardner, B.K.; et al. MCT1 Modulates Cancer Cell Pyruvate Export and Growth of Tumors that Co-express MCT1 and MCT4. Cell Rep. 2016, 14, 1590–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.M.; Cotzia, P.; Fratamico, R.; Mikkilineni, L.; Chen, J.; Colombo, D.; Mollaee, M.; Whitaker-Menezes, D.; Domingo-Vidal, M.; Lin, Z.; et al. MCT1 in Invasive Ductal Carcinoma: Monocarboxylate Metabolism and Aggressive Breast Cancer. Front. Cell Dev. Biol. 2017, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Leung, Y.H.; Belanger, F.; Lu, J.; Turgeon, J.; Michaud, V. Effects of a Series of Acidic Drugs on L-Lactic Acid Transport by the Monocarboxylate Transporters MCT1 and MCT4. Curr. Pharm. Biotechnol. 2017, 18, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ertay, A.; Peng, P.; Li, J.; Liu, D.; Xiong, H.; Zou, Y.; Qiu, H.; Hancock, D.; Yuan, X.; et al. SGLT1 is required for the survival of triple-negative breast cancer cells via potentiation of EGFR activity. Mol. Oncol. 2019, 13, 1874–1886. [Google Scholar] [CrossRef]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.J.; Hwang, H.S.; Park, J.H.; Bang, S.H.; Kang, W.J.; Yun, M.; Lee, J.D. Evaluation of the role of hexokinase type II in cellular proliferation and apoptosis using human hepatocellular carcinoma cell lines. J. Nucl. Med. 2009, 50, 1525–1532. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.; Song, L.; Fan, N.; Feng, T.; Liu, L.; Yang, X.; Wang, M.; Li, Y.; Tian, Y.; Zhao, F.; et al. Combination of metformin and sorafenib suppresses proliferation and induces autophagy of hepatocellular carcinoma via targeting the mTOR pathway. Int. J. Oncol. 2017, 50, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Wick, A.N.; Drury, D.R.; Nakada, H.I.; Wolfe, J.B. Localization of the primary metabolic block produced by 2-deoxyglucose. J. Biol. Chem. 1957, 224, 963–969. [Google Scholar]
- Yang, Q.; Zhang, T.; Wang, C.; Jiao, J.; Li, J.; Deng, Y. Coencapsulation of epirubicin and metformin in PEGylated liposomes inhibits the recurrence of murine sarcoma S180 existing CD133+ cancer stem-like cells. Eur. J. Pharm. Biopharm. 2014, 88, 737–745. [Google Scholar] [CrossRef]
- Zhang, T.; Zhu, X.; Wu, H.; Jiang, K.; Zhao, G.; Shaukat, A.; Deng, G.; Qiu, C. Targeting the ROS/PI3K/AKT/HIF-1α/HK2 axis of breast cancer cells: Combined administration of Polydatin and 2-Deoxy-d-glucose. J. Cell. Mol. Med. 2019, 23, 3711–3723. [Google Scholar] [CrossRef] [PubMed]
- Bizjak, M.; Malavašič, P.; Dolinar, K.; Pohar, J.; Pirkmajer, S.; Pavlin, M. Combined treatment with Metformin and 2-deoxy glucose induces detachment of viable MDA-MB-231 breast cancer cells in vitro. Sci. Rep. 2017, 7, 1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, F.; Li, Q.; Sun, J.Y.; Luo, Y.; Chen, M.; Bao, Y. PFKFB3 is involved in breast cancer proliferation, migration, invasion and angiogenesis. Int. J. Oncol. 2018, 52, 945–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganapathy-Kanniappan, S. Evolution of GAPDH as a druggable target of tumor glycolysis? Expert Opin. Ther. Targets 2018, 22, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Li, X.; Xie, X.; Ye, F.; Chen, B.; Song, C.; Tang, H.; Xie, X. High expressions of LDHA and AMPK as prognostic biomarkers for breast cancer. Breast 2016, 30, 39–46. [Google Scholar] [CrossRef]
- Pelizzari, G.; Basile, D.; Zago, S.; Lisanti, C.; Bartoletti, M.; Bortot, L.; Vitale, M.G.; Fanotto, V.; Barban, S.; Cinausero, M.; et al. Lactate Dehydrogenase (LDH) Response to First-Line Treatment Predicts Survival in Metastatic Breast Cancer: First Clues for A Cost-Effective and Dynamic Biomarker. Cancers 2019, 11, 1243. [Google Scholar] [CrossRef] [Green Version]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stefano, G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef]
- Jia, L.; Huang, S.; Yin, X.; Zan, Y.; Guo, Y.; Han, L. Quercetin suppresses the mobility of breast cancer by suppressing glycolysis through Akt-mTOR pathway mediated autophagy induction. Life Sci. 2018, 208, 123–130. [Google Scholar] [CrossRef]
- Fan, K.; Fan, Z.; Cheng, H.; Huang, Q.; Yang, C.; Jin, K.; Luo, G.; Yu, X.; Liu, C. Hexokinase 2 dimerization and interaction with voltage-dependent anion channel promoted resistance to cell apoptosis induced by gemcitabine in pancreatic cancer. Cancer Med. 2019, 8, 5903–5915. [Google Scholar] [CrossRef]
- Guo, Y.; Wei, L.; Zhou, Y.; Lu, N.; Tang, X.; Li, Z.; Wang, X. Flavonoid GL-V9 induces apoptosis and inhibits glycolysis of breast cancer via disrupting GSK-3β-modulated mitochondrial binding of HKII. Free Radic. Biol. Med. 2020, 146, 119–129. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, N.; Qiao, C.; Ni, T.; Li, Z.; Yu, B.; Guo, Q.; Wei, L. FV-429 induces apoptosis and inhibits glycolysis by inhibiting Akt-mediated phosphorylation of hexokinase II in MDA-MB-231 cells. Mol. Carcinog. 2016, 55, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Yoon, J.-H. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse Warburg effect and its therapeutic implication. World J. Biol. Chem. 2015, 6, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Büsselberg, D. Arsenic trioxide in environmentally and clinically relevant concentrations interacts with calcium homeostasis and induces cell type specific cell death in tumor and non-tumor cells. Toxicol. Lett. 2008, 179, 34–42. [Google Scholar] [CrossRef]
- Liu, C.; Wang, X.; Zhang, Y. The Roles of HK2 on Tumorigenesis of Cervical Cancer. Technol. Cancer Res. Treat. 2019, 18, 1533033819871306. [Google Scholar] [CrossRef]
- Siu, M.K.Y.; Jiang, Y.-X.; Wang, J.-J.; Leung, T.H.Y.; Han, C.Y.; Tsang, B.K.; Cheung, A.N.Y.; Ngan, H.Y.S.; Chan, K.K.L. Hexokinase 2 Regulates Ovarian Cancer Cell Migration, Invasion and Stemness via FAK/ERK1/2/MMP9/NANOG/SOX9 Signaling Cascades. Cancers 2019, 11, 813. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Ren, C.; Qiao, P.; Han, X.; Wang, L.; Lv, S.; Sun, Y.; Liu, Z.; Du, Y.; Yu, Z. PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene 2018, 37, 5997–6009. [Google Scholar] [CrossRef]
- Atsumi, T.; Chesney, J.; Metz, C.; Leng, L.; Donnelly, S.; Makita, Z.; Mitchell, R.; Bucala, R. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002, 62, 5881–5887. [Google Scholar]
- Klarer, A.C.; O’Neal, J.; Imbert-Fernandez, Y.; Clem, A.; Ellis, S.R.; Clark, J.; Clem, B.; Chesney, J.; Telang, S. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab. 2014, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Roy, D.; Sarkar Bhattacharya, S.; Jin, L.; Jung, D.; Zhang, S.; Kalogera, E.; Staub, J.; Wang, Y.; Xuyang, W.; et al. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int. J. Cancer 2019, 144, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, H.; Dong, X. Diagnostic value of α-enolase expression and serum α-enolase autoantibody levels in lung cancer. J. Bras. Pneumol. 2018, 44, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Cancemi, P.; Buttacavoli, M.; Roz, E.; Feo, S. Expression of Alpha-Enolase (ENO1), Myc Promoter-Binding Protein-1 (MBP-1) and Matrix Metalloproteinases (MMP-2 and MMP-9) Reflect the Nature and Aggressiveness of Breast Tumors. Int. J. Mol. Sci. 2019, 20, 3952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capello, M.; Ferri-Borgogno, S.; Riganti, C.; Chattaragada, M.S.; Principe, M.; Roux, C.; Zhou, W.; Petricoin, E.F.; Cappello, P.; Novelli, F. Targeting the Warburg effect in cancer cells through ENO1 knockdown rescues oxidative phosphorylation and induces growth arrest. Oncotarget 2015, 7, 5598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcinkowska, M.; Stanczyk, M.; Janaszewska, A.; Sobierajska, E.; Chworos, A.; Klajnert-Maculewicz, B. Multicomponent Conjugates of Anticancer Drugs and Monoclonal Antibody with PAMAM Dendrimers to Increase Efficacy of HER-2 Positive Breast Cancer Therapy. Pharm. Res. 2019, 36, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanzaro, D.; Gaude, E.; Orso, G.; Giordano, C.; Guzzo, G.; Rasola, A.; Ragazzi, E.; Caparrotta, L.; Frezza, C.; Montopoli, M. Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget 2015, 6, 30102–30114. [Google Scholar] [CrossRef] [Green Version]
- Woo, Y.M.; Shin, Y.; Lee, E.J.; Lee, S.; Jeong, S.H.; Kong, H.K.; Park, E.Y.; Kim, H.K.; Han, J.; Chang, M.; et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1alpha Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PLoS ONE 2015, 10, e0132285. [Google Scholar] [CrossRef] [Green Version]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.-C.; Hung, W.-C. Pyruvate kinase M2 fuels multiple aspects of cancer cells: From cellular metabolism, transcriptional regulation to extracellular signaling. Mol. Cancer 2018, 17, 35. [Google Scholar] [CrossRef] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Wu, J.; Hu, L.; Chen, M.; Cao, W.; Chen, H.; He, T. Pyruvate kinase M2 overexpression and poor prognosis in solid tumors of digestive system: Evidence from 16 cohort studies. Onco Targets Ther. 2016, 9, 4277–4288. [Google Scholar] [CrossRef] [Green Version]
- Rzechonek, A.; Kaminska, A.; Mamczur, P.; Drapiewski, A.; Budzynski, W. Limited Clinical Significance of Dimeric Form of Pyruvate Kinase as a Diagnostic and Prognostic Biomarker in Non-small Cell Lung Cancer. Adv. Exp. Med. Biol. 2017, 955, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, F.; Fan, Y.; Qian, X.; Lang, R.; Gu, F.; Gu, J.; Fu, L. Both high expression of pyruvate kinase M2 and vascular endothelial growth factor-C predicts poorer prognosis in human breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 8028–8037. [Google Scholar] [PubMed]
- Castagnoli, L.; Iorio, E.; Dugo, M.; Koschorke, A.; Faraci, S.; Canese, R.; Casalini, P.; Nanni, P.; Vernieri, C.; Di Nicola, M.; et al. Intratumor lactate levels reflect HER2 addiction status in HER2-positive breast cancer. J. Cell. Physiol. 2019, 234, 1768–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Wu, K.; Liu, Y.; Shi, L.; Tao, K.; Wang, G. Prognostic significance of metabolic enzyme pyruvate kinase M2 in breast cancer: A meta-analysis. Medicine 2017, 96, e8690. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lv, F.; Liu, F.; Guo, X.; Fan, Y.; Gu, F.; Gu, J.; Fu, L. High Expression of Pyruvate Kinase M2 is Associated with Chemosensitivity to Epirubicin and 5-Fluorouracil in Breast Cancer. J. Cancer 2015, 6, 1130–1139. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Xie, G.; Tong, J.; Peng, Y.; Huang, H.; Li, J.; Wang, N.; Liang, H. Overexpression of microRNA-122 re-sensitizes 5-FU-resistant colon cancer cells to 5-FU through the inhibition of PKM2 in vitro and in vivo. Cell Biochem. Biophys. 2014, 70, 1343–1350. [Google Scholar] [CrossRef]
- Su, Q.; Luo, S.; Tan, Q.; Deng, J.; Zhou, S.; Peng, M.; Tao, T.; Yang, X. The role of pyruvate kinase M2 in anticancer therapeutic treatments. Oncol. Lett. 2019, 18, 5663–5672. [Google Scholar] [CrossRef]
- Qian, Y.; Bi, L.; Yang, Y.; Wang, D. Effect of pyruvate kinase M2-regulating aerobic glycolysis on chemotherapy resistance of estrogen receptor-positive breast cancer. Anticancer Drugs 2018, 29, 616–627. [Google Scholar] [CrossRef]
- Yoo, B.C.; Ku, J.L.; Hong, S.H.; Shin, Y.K.; Park, S.Y.; Kim, H.K.; Park, J.G. Decreased pyruvate kinase M2 activity linked to cisplatin resistance in human gastric carcinoma cell lines. Int. J. Cancer 2004, 108, 532–539. [Google Scholar] [CrossRef]
- Hou, X.M.; Yuan, S.Q.; Zhao, D.; Liu, X.J.; Wu, X.A. LDH-A promotes malignant behavior via activation of epithelial-to-mesenchymal transition in lung adenocarcinoma. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Han, R.L.; Wang, F.P.; Zhang, P.A.; Zhou, X.Y.; Li, Y. miR-383 inhibits ovarian cancer cell proliferation, invasion and aerobic glycolysis by targeting LDHA. Neoplasma 2017, 64, 244–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizwan, A.; Serganova, I.; Khanin, R.; Karabeber, H.; Ni, X.; Thakur, S.; Zakian, K.L.; Blasberg, R.; Koutcher, J.A. Relationships between LDH-A, lactate, and metastases in 4T1 breast tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 5158–5169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Chun, J.; Pan, C.; Alesi, G.N.; Li, D.; Magliocca, K.R.; Kang, Y.; Chen, Z.G.; Shin, D.M.; Khuri, F.R.; et al. Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis. Oncogene 2017, 36, 3797–3806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, C.K.; Parekh, A.; Parida, P.K.; Bhutia, S.K.; Mandal, M. Lactate dehydrogenase A regulates autophagy and tamoxifen resistance in breast cancer. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1004–1018. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.E.; Im, S.A.; Gelmon, K.A.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.L.; et al. PALOMA-2: Primary results from a phase III trial of palbociclib (P) with letrozole (L) compared with letrozole alone in postmenopausal women with ER+/HER2– advanced breast cancer (ABC). J. Clin. Oncol. 2017, 34. [Google Scholar] [CrossRef]
- Dennison, J.B.; Molina, J.R.; Mitra, S.; González-Angulo, A.M.; Balko, J.M.; Kuba, M.G.; Sanders, M.E.; Pinto, J.A.; Gómez, H.L.; Arteaga, C.L.; et al. Lactate dehydrogenase B: A metabolic marker of response to neoadjuvant chemotherapy in breast cancer. Clin. Cancer Res. 2013, 19, 3703–3713. [Google Scholar] [CrossRef] [Green Version]
- Guda, M.R.; Asuthkar, S.; Labak, C.M.; Tsung, A.J.; Alexandrov, I.; Mackenzie, M.J.; Prasad, D.V.; Velpula, K.K. Targeting PDK4 inhibits breast cancer metabolism. Am. J. Cancer Res. 2018, 8, 1725–1738. [Google Scholar]
- Pelley, J.W. 6-Glycolysis and Pyruvate Oxidation. In Elsevier’s Integrated Biochemistry; Pelley, J.W., Ed.; Mosby: Philadelphia, PA, USA, 2007; pp. 47–53. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Rajendran, G.; Harris, R.A.; Taylor, J.A. Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase:Pyruvate Dehydrogenase Axis. Mol. Cancer Ther. 2019, 18, 1673. [Google Scholar] [CrossRef] [Green Version]
- Sradhanjali, S.; Reddy, M.M. Inhibition of Pyruvate Dehydrogenase Kinase as a Therapeutic Strategy against Cancer. Curr. Top Med. Chem. 2018, 18, 444–453. [Google Scholar] [CrossRef]
- Peng, F.; Wang, J.H.; Fan, W.J.; Meng, Y.T.; Li, M.M.; Li, T.T.; Cui, B.; Wang, H.F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef] [Green Version]
- Walter, W.; Thomalla, J.; Bruhn, J.; Fagan, D.H.; Zehowski, C.; Yee, D.; Skildum, A. Altered regulation of PDK4 expression promotes antiestrogen resistance in human breast cancer cells. Springerplus 2015, 4, 689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.-W.; Lin, S.-C.; Chien, C.-W.; Lin, S.-C.; Lee, C.-T.; Lin, B.-W.; Lee, J.-C.; Tsai, S.-J. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am. J. Pathol. 2011, 179, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Saunier, E.; Benelli, C.; Bortoli, S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int. J. Cancer 2016, 138, 809–817. [Google Scholar] [CrossRef]
- Iorns, E.; Lord, C.J.; Ashworth, A. Parallel RNAi and compound screens identify the PDK1 pathway as a target for tamoxifen sensitization. Biochem. J. 2009, 417, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhang, S.L.; Hu, X.; Tam, K.Y. Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target? Int. J. Biol. Sci. 2015, 11, 1390–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbet, C.; Bastien, E.; Draoui, N.; Doix, B.; Mignion, L.; Jordan, B.F.; Marchand, A.; Vanherck, J.-C.; Chaltin, P.; Schakman, O.; et al. Interruption of lactate uptake by inhibiting mitochondrial pyruvate transport unravels direct antitumor and radiosensitizing effects. Nat. Commun. 2018, 9, 1208. [Google Scholar] [CrossRef] [PubMed]
- Mustacchi, G.; De Laurentiis, M. The role of taxanes in triple-negative breast cancer: Literature review. Drug Des. Dev. Ther. 2015, 9, 4303–4318. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.Z.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.Y.; Hu, S.S.; Dong, Q.; Cai, J.X.; Zhang, W.P.; Sun, J.Y.; Wang, T.T.; Xie, J.; He, H.R.; Xing, J.F.; et al. Establishment of Paclitaxel-resistant Breast Cancer Cell Line and Nude Mice Models, and Underlying Multidrug Resistance Mechanisms in Vitro and in Vivo. Asian Pac. J. Cancer Prev. 2013, 14, 6135–6140. [Google Scholar] [CrossRef] [Green Version]
- Němcová-Fürstová, V.; Kopperová, D.; Balušíková, K.; Ehrlichová, M.; Brynychová, V.; Václavíková, R.; Daniel, P.; Souček, P.; Kovář, J. Characterization of acquired paclitaxel resistance of breast cancer cells and involvement of ABC transporters. Toxicol. Appl. Pharmacol. 2016, 310, 215–228. [Google Scholar] [CrossRef]
- Zajdel, A.; Wolny, D.; Kalucka-Janik, M.; Wilczok, A. Paclitaxel in breast cancer–drug resistance and strategies to counteract it. Adv. Hyg. Exp. Med. 2019, 73, 508–515. [Google Scholar] [CrossRef]
- Nakano, A.; Tsuji, D.; Miki, H.; Cui, Q.; El Sayed, S.M.; Ikegame, A.; Oda, A.; Amou, H.; Nakamura, S.; Harada, T.; et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PLoS ONE 2011, 6, e27222. [Google Scholar] [CrossRef] [PubMed]
- Zuo, K.Q.; Zhang, X.P.; Zou, J.; Li, D.; Lv, Z.W. Establishment of a Paclitaxel Resistant Human Breast Cancer Cell Strain (MCF-7/Taxol) and Intracellular Paclitaxel Binding Protein Analysis. J. Int. Med Res. 2010, 38, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Schittek, B. The multiple facets of dermcidin in cell survival and host defense. J. Innate Immun. 2012, 4, 349–360. [Google Scholar] [CrossRef]
- Hadzic, T.; Aykin-Burns, N.; Zhu, Y.; Coleman, M.C.; Leick, K.; Jacobson, G.M.; Spitz, D.R. Paclitaxel combined with inhibitors of glucose and hydroperoxide metabolism enhances breast cancer cell killing via H2O2-mediated oxidative stress. Free Radic. Biol. Med. 2010, 48, 1024–1033. [Google Scholar] [CrossRef] [Green Version]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; Dipaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef]
- Mishra, D.; Banerjee, D. Lactate Dehydrogenases as Metabolic Links between Tumor and Stroma in the Tumor Microenvironment. Cancers 2019, 11, 750. [Google Scholar] [CrossRef] [Green Version]
- El-Sisi, A.E.; Sokar, S.S.; Abu-Risha, S.E.; El-Mahrouk, S.R. Oxamate potentiates taxol chemotherapeutic efficacy in experimentally-induced solid ehrlich carcinoma (SEC) in mice. Biomed. Pharmacother. 2017, 95, 1565–1573. [Google Scholar] [CrossRef]
- Ge, X.; Cao, Z.; Gu, Y.; Wang, F.; Li, J.; Han, M.; Xia, W.; Yu, Z.; Lyu, P. PFKFB3 potentially contributes to paclitaxel resistance in breast cancer cells through TLR4 activation by stimulating lactate production. Cell. Mol. Biol. 2016, 62, 119–125. [Google Scholar]
- Cretella, D.; Fumarola, C.; Bonelli, M.; Alfieri, R.; La Monica, S.; Digiacomo, G.; Cavazzoni, A.; Galetti, M.; Generali, D.; Petronini, P.G. Pre-treatment with the CDK4/6 inhibitor palbociclib improves the efficacy of paclitaxel in TNBC cells. Sci. Rep. 2019, 9, 13014. [Google Scholar] [CrossRef]
- El Sayed, R.; El Jamal, L.; El Iskandarani, S.; Kort, J.; Abdel Salam, M.; Assi, H. Endocrine and Targeted Therapy for Hormone-Receptor-Positive, HER2-Negative Advanced Breast Cancer: Insights to Sequencing Treatment and Overcoming Resistance Based on Clinical Trials. Front. Oncol. 2019, 9, 510. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, R.; Yu, Z.; Li, R.; Li, J.; Zhao, X.; Song, S.; Liu, J.; Huang, G. Dichloroacetate restores drug sensitivity in paclitaxel-resistant cells by inducing citric acid accumulation. Mol. Cancer 2015, 14, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuello-Carrión, F.D.; Shortrede, J.E.; Alvarez-Olmedo, D.; Cayado-Gutiérrez, N.; Castro, G.N.; Zoppino, F.C.M.; Guerrero, M.; Martinis, E.; Wuilloud, R.; Gómez, N.N.; et al. HER2 and β-catenin protein location: Importance in the prognosis of breast cancer patients and their correlation when breast cancer cells suffer stressful situations. Clin. Exp. Metastasis 2015, 32, 151–168. [Google Scholar] [CrossRef] [PubMed]
- El-Sahli, S.; Xie, Y.; Wang, L.; Liu, S. Wnt Signaling in Cancer Metabolism and Immunity. Cancers 2019, 11, 904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Gupta, P.; Srivastava, S.K. Penfluridol overcomes paclitaxel resistance in metastatic breast cancer. Sci. Rep. 2019, 9, 5066. [Google Scholar] [CrossRef]
- Xiang, F.; Fan, Y.; Ni, Z.; Liu, Q.; Zhu, Z.; Chen, Z.; Hao, W.; Yue, H.; Wu, R.; Kang, X. Ursolic Acid Reverses the Chemoresistance of Breast Cancer Cells to Paclitaxel by Targeting MiRNA-149-5p/MyD88. Front. Oncol. 2019, 9, 501. [Google Scholar] [CrossRef] [Green Version]
- Guan, D.G.; Chen, H.M.; Liao, S.F.; Zhao, T.Z. Combination of temozolomide and Taxol exerts a synergistic inhibitory effect on Taxolresistant glioma cells via inhibition of glucose metabolism. Mol. Med. Rep. 2015, 12, 7705–7711. [Google Scholar] [CrossRef]
- Garon, E.B.; Christofk, H.R.; Hosmer, W.; Britten, C.D.; Bahng, A.; Crabtree, M.J.; Hong, C.S.; Kamranpour, N.; Pitts, S.; Kabbinavar, F.; et al. Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2014, 140, 443–452. [Google Scholar] [CrossRef] [Green Version]
- García-Castillo, V.; López-Urrutia, E.; Villanueva-Sánchez, O.; Ávila-Rodríguez, M.Á.; Zentella-Dehesa, A.; Cortés-González, C.; López-Camarillo, C.; Jacobo-Herrera, N.J.; Pérez-Plasencia, C. Targeting Metabolic Remodeling in Triple Negative Breast Cancer in a Murine Model. J. Cancer 2017, 8, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Amadori, D.; Frassineti, G.L.; De Matteis, A.; Mustacchi, G.; Santoro, A.; Cariello, S.; Ferrari, M.; Nascimben, O.; Nanni, O.; Lombardi, A.; et al. Modulating effect of lonidamine on response to doxorubicin in metastatic breast cancer patients: Results from a multicenter prospective randomized trial. Breast Cancer Res. Treat. 1998, 49, 209–217. [Google Scholar] [CrossRef]
- Attia, Y.M.; El-Abhar, H.S.; Al Marzabani, M.M.; Shouman, S.A. Targeting glycolysis by 3-bromopyruvate improves tamoxifen cytotoxicity of breast cancer cell lines. BMC Cancer 2015, 15, 838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cufi, S.; Corominas-Faja, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Dorca, J.; Bosch-Barrera, J.; Martin-Castillo, B.; Menendez, J.A. Metformin-induced preferential killing of breast cancer initiating CD44+CD24-/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget 2012, 3, 395–398. [Google Scholar] [CrossRef] [Green Version]
- Martin-Castillo, B.; Dorca, J.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Lopez-Bonet, E.; Garcia, M.; Del Barco, S.; Menendez, J.A. Incorporating the antidiabetic drug metformin in HER2-positive breast cancer treated with neo-adjuvant chemotherapy and trastuzumab: An ongoing clinical-translational research experience at the Catalan Institute of Oncology. Ann. Oncol. 2010, 21, 187–189. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaal, E.A.; Berkers, C.R. The Influence of Metabolism on Drug Response in Cancer. Front. Oncol. 2018, 8, 500. [Google Scholar] [CrossRef]
- Egawa-Takata, T.; Endo, H.; Fujita, M.; Ueda, Y.; Miyatake, T.; Okuyama, H.; Yoshino, K.; Kamiura, S.; Enomoto, T.; Kimura, T.; et al. Early reduction of glucose uptake after cisplatin treatment is a marker of cisplatin sensitivity in ovarian cancer. Cancer Sci. 2010, 101, 2171–2178. [Google Scholar] [CrossRef]
- Dhar, S.; Lippard, S.J. Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc. Natl. Acad. Sci. USA 2009, 106, 22199–22204. [Google Scholar] [CrossRef] [Green Version]
- Gang, B.P.; Dilda, P.J.; Hogg, P.J.; Blackburn, A.C. Targeting of two aspects of metabolism in breast cancer treatment. Cancer Biol. Ther. 2014, 15, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.O.; Kang, M.J.; Byun, W.S.; Kim, S.A.; Seo, I.H.; Han, J.A.; Moon, J.W.; Kim, J.H.; Kim, S.J.; Lee, E.J.; et al. Metformin overcomes resistance to cisplatin in triple-negative breast cancer (TNBC) cells by targeting RAD51. Breast Cancer Res. 2019, 21, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criscitiello, C.; Fumagalli, D.; Saini, K.S.; Loi, S. Tamoxifen in early-stage estrogen receptor-positive breast cancer: Overview of clinical use and molecular biomarkers for patient selection. Onco Targets Ther. 2010, 4, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, K.-C. Selective Estrogen Receptor Modulators. Asian Spine J. 2016, 10, 787–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.H.; Seo, S.K.; Park, Y.; Kim, E.K.; Seong, M.K.; Kim, H.A.; Song, J.Y.; Hwang, S.G.; Lee, J.K.; Noh, W.C.; et al. Dichloroacetate potentiates tamoxifen-induced cell death in breast cancer cells via downregulation of the epidermal growth factor receptor. Oncotarget 2016, 7, 59809–59819. [Google Scholar] [CrossRef] [Green Version]
- Manna, S.; Holz, M.K. Tamoxifen Action in ER-Negative Breast Cancer. Signal Transduct. Insights 2016, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Daurio, N.A. Tamoxifen Mediated Metabolic Stress: Molecular Mechanism and Therapeutic Opportunities. Ph.D. Thesis, University of Pennsylvania, Philadelphia, PA, USA, 2016. [Google Scholar]
- Mayer, I. Role of mTOR inhibition in preventing resistance and restoring sensitivity to hormone-targeted and HER2-targeted therapies in breast cancer. Clin. Adv. Hematol. Oncol. 2013, 11, 217–224. [Google Scholar]
- Ferrari, P.; Nicolini, A. Chapter 29-Overcoming Endocrine Resistance in Breast Cancer: mTOR Inhibitors and New Drugs. In Oncogenomics; Dammacco, F., Silvestris, F., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 393–422. [Google Scholar] [CrossRef]
- Lovitt, C.J.; Shelper, T.B.; Avery, V.M. Doxorubicin resistance in breast cancer cells is mediated by extracellular matrix proteins. BMC Cancer 2018, 18, 41. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.; Watson, M.B.; Kane, S.L.; Drew, P.J.; Lind, M.J.; Cawkwell, L. The analysis of doxorubicin resistance in human breast cancer cells using antibody microarrays. Mol. Cancer Ther. 2006, 5, 2115. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.; Yamazaki, M. Clinical Relevance of P-Glycoprotein in Drug Therapy. Drug Metab. Rev. 2003, 35, 417–454. [Google Scholar] [CrossRef]
- Jones, P.M.; George, A.M. The ABC transporter structure and mechanism: Perspectives on recent research. Cell. Mol. Life Sci. CMLS 2004, 61, 682–699. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.F.; Qiu, Y.-Y.; Yu, S.; Clark, S.; Chu, F.; Madonna, M.B. Glycolysis inhibition and its effect in doxorubicin resistance in neuroblastoma. J. Pediatr. Surg. 2014, 49, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK. Cancers 2020, 12, 862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Chan, J.Y.; Zhou, X.; Cui, G.; Yan, Z.; Wang, L.; Yan, R.; Di, L.; Wang, Y.; Hoi, M.P.; et al. A Novel Agent Enhances the Chemotherapeutic Efficacy of Doxorubicin in MCF-7 Breast Cancer Cells. Front. Pharmacol. 2016, 7, 249. [Google Scholar] [CrossRef] [Green Version]
- Zong, L.; Cheng, G.; Liu, S.; Pi, Z.; Liu, Z.; Song, F. Reversal of multidrug resistance in breast cancer cells by a combination of ursolic acid with doxorubicin. J. Pharm. Biomed. Anal. 2019, 165, 268–275. [Google Scholar] [CrossRef]
- Xu, M.; Chen, S.; Yang, W.; Cheng, X.; Ye, Y.; Mao, J.; Wu, X.; Huang, L.; Ji, J. FGFR4 Links Glucose Metabolism and Chemotherapy Resistance in Breast Cancer. Cell. Physiol. Biochem. 2018, 47, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Qu, C.; Zhang, W.; Zheng, G.; Zhang, Z.; Yin, J.; He, Z. Metformin reverses multidrug resistance and epithelial-mesenchymal transition (EMT) via activating AMP-activated protein kinase (AMPK) in human breast cancer cells. Mol. Cell. Biochem. 2014, 386, 63–71. [Google Scholar] [CrossRef]
- Marinello, P.C.; Panis, C.; Silva, T.N.X.; Binato, R.; Abdelhay, E.; Rodrigues, J.A.; Mencalha, A.L.; Lopes, N.M.D.; Luiz, R.C.; Cecchini, R.; et al. Metformin prevention of doxorubicin resistance in MCF-7 and MDA-MB-231 involves oxidative stress generation and modulation of cell adaptation genes. Sci. Rep. 2019, 9, 5864. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, H.; Liu, Z.; Ding, Y.; LeDoux, S.P.; Wilson, G.L.; Voellmy, R.; Lin, Y.; Lin, W.; Nahta, R.; et al. Overcoming Trastuzumab Resistance in Breast Cancer by Targeting Dysregulated Glucose Metabolism. Cancer Res. 2011, 71, 4585. [Google Scholar] [CrossRef] [Green Version]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Lewis Phillips, G.D.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 2009, 15, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoopmann, M.; Warm, M.; Mallmann, P.; Thomas, A.; Göhring, U.J.; Schöndorf, T. Tumor M2 pyruvate kinase—Determination in breast cancer patients receiving trastuzumab therapy. Cancer Lett. 2002, 187, 223–228. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, A.; Henry, N.L. Palbociclib for the Treatment of Estrogen Receptor—Positive, HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2015, 21, 3591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorito, N.; Bacci, M.; Smiriglia, A.; Mannelli, M.; Parri, M.; Comito, G.; Ippolito, L.; Giannoni, E.; Bonechi, M.; Benelli, M.; et al. Glucose Metabolic Reprogramming of ER Breast Cancer in Acquired Resistance to the CDK4/6 Inhibitor Palbociclib+. Cells 2020, 9, 668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickaert, A.; Saiselet, M.; Dom, G.; De Deken, X.; Dumont, J.E.; Feron, O.; Sonveaux, P.; Maenhaut, C. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene 2017, 36, 2637–2642. [Google Scholar] [CrossRef] [Green Version]
- Heaster, T.M.; Landman, B.A.; Skala, M.C. Quantitative Spatial Analysis of Metabolic Heterogeneity Across in vivo and in vitro Tumor Models. Front. Oncol. 2019, 9, 1144. [Google Scholar] [CrossRef] [Green Version]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’Agostino, D.; et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Chen, J.; Yang, L.; Liu, J.; Zhang, X.; Zhang, Y.; Tu, Q.; Yin, D.; Lin, D.; Wong, P.P.; et al. Extracellular vesicle-packaged HIF-1α-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat. Cell Biol. 2019, 21, 498–510. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Schulze, A. Non-canonical functions of enzymes facilitate cross-talk between cell metabolic and regulatory pathways. Exp. Mol. Med. 2018, 50, 34. [Google Scholar] [CrossRef] [Green Version]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Blumenschein, G.R., Jr. Defining biomarkers to predict sensitivity to PI3K/Akt/mTOR pathway inhibitors in breast cancer. Cancer Treat. Rev. 2013, 39, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, E.; Liskova, A.; Kubatka, P.; Mathews Samuel, S.; Busselberg, D. Anti-Angiogenic Effects of Phytochemicals on miRNA Regulating Breast Cancer Progression. Biomolecules 2020, 10, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.-C.; Hung, M.-C. Induction of Akt Activity by Chemotherapy Confers Acquired Resistance. J. Formos. Med Assoc. 2009, 108, 180–194. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, S.; Łuc, M.; Ziółkowski, P.; Agrawal, A.K.; Pielka, E.; Walaszek, K.; Zduniak, K.; Woźniak, M. Insulin-induced enhancement of MCF-7 breast cancer cell response to 5-fluorouracil and cyclophosphamide. Tumor Biol. 2017, 39, 1010428317702901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasalvia-Prisco, E.; Cucchi, S.; Vázquez, J.; Lasalvia-Galante, E.; Golomar, W.; Gordon, W. Insulin-induced enhancement of antitumoral response to methotrexate in breast cancer patients. Cancer Chemother. Pharmacol. 2004, 53, 220–224. [Google Scholar] [CrossRef]
- Zhou, P.; Bogacki, R.; McReynolds, L.; Howley, P.M. Harnessing the ubiquitination machinery to target the degradation of specific cellular proteins. Mol. Cell 2000, 6, 751–756. [Google Scholar] [CrossRef]
- Rodriguez-Gonzalez, A.; Cyrus, K.; Salcius, M.; Kim, K.; Crews, C.M.; Deshaies, R.J.; Sakamoto, K.M. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene 2008, 27, 7201–7211. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, A.M.; Martel, F. Targeting Glucose Transporters for Breast Cancer Therapy: The Effect of Natural and Synthetic Compounds. Cancers 2020, 12, 154. [Google Scholar] [CrossRef] [Green Version]
- Calvaresi, E.C.; Hergenrother, P.J. Glucose conjugation for the specific targeting and treatment of cancer. Chem. Sci. 2013, 4, 2319–2333. [Google Scholar] [CrossRef] [Green Version]
- Miskimins, W.K.; Ahn, H.J.; Kim, J.Y.; Ryu, S.; Jung, Y.-S.; Choi, J.Y. Synergistic Anti-Cancer Effect of Phenformin and Oxamate. PLoS ONE 2014, 9, e85576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Wang, H.; Yu, W.; Qiao, F.; Su, X.; Xu, H. Downregulation of hexokinase 2 improves radiosensitivity of breast cancer. Transl. Cancer Res. 2019, 8, 290–297. [Google Scholar] [CrossRef]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Phan, L.M.; Yeung, S.-C.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Serial Number | Target | Compounds | Model | Reference |
|---|---|---|---|---|
| 1 | GLUT | Apigenin, Genistein, Cisplatin, Metformin, Tamoxifen, EGCG, Hesperetin, Kaempferol, Silybin | MCF-7, ZR-75-1, T-47D, MDA-MB-231, 4T1 | [42,44,45,46,47,48,49,50,51] |
| 2 | MCT | Quercetin, α-cyano-4-hydroxycinnamate and Lonidamine | MDA-MB-231, MDA-MB-468, MCF-7/AZ, SKBr3, Hs 578T, BT-20 | [52,53] |
| 3 | HK | Polydatin, 2-DG, Tamoxifen, Metformin, 3-BrPy, EGCG | MDA-MB-231, 4T1 | [54,55] |
| 4 | PFKFB3 | 3PO, PFK15, PFK158 | SKBR3, BT-474 | [56,57] |
| 5 | GAPDH | 3-BrPy | HepG2, PC-3 | [58,59] |
| 6 | PK | Vit K3, Vit K5, Shikonin, 5-FU, Lapatinib, EGCG, Quercetin | MDA-MB-231 MCF-7, 4T1 | [60,61] |
| 7 | LDHA LDHB | Cetuximab, Metformin, Oxamate, Chidamide, Galloflavin Cisplatin | MBA-MD-231 MCF-7 | [46,62,63,64,65] |
| 8 | Mitochondria | DCA, Metformin, As2O3 | T47D, BT-20, MCF-10A, MDA-MB-468, MDA-MB-231 | [66,67,68,69] |
| Drug | Model | Dose | Observation | Side Effects | Mechanism | Study Method | Ref. |
|---|---|---|---|---|---|---|---|
| DCA + cisplatin | Stage IV BC (1 case) | DCA 6.25 mg/kg | – | Pulmonary embolism, edema | – | NCT01029925 (2014) | [170] |
| DCA | Rat mammary adenocarcinoma | 200 mg/kg/day. (1.5–3mM plasma level) | 50% reduction in lung metastasis | Minimal side effects | Inhibition of PDK | In vivo | [68] |
| Oxamate + paclitaxel | Solid Ehrlich Carcinoma | Oxamate (300 mg/kg) and paclitaxel (10 or 20 mg/kg) | >40% Reduction in volume of SEC, ATP, IL-17 | – | LDH-A inhibition induced apoptosis | In vivo | [160] |
| Doxorubicin + metformin + oxamate | TNBC xenograft | Doxorubicin 1 mg/kg, Metformin 200 mg/kg, and Oxamate 15 mg/kg | Reduced tumor volume | – | LDH-A inhibition induced apoptosis | In vivo | [171] |
| Doxorubicin + lonidamine | Metastatic BC patients | Doxorubicin 75 mg/m2/21day + lonidamine 600 mg/m2/day | Overall response rate is 68% in BC with liver metastasis | Cardiotoxicity, myalgia | – | Randomized clinical trial (1998) | [172] |
| Tamoxifen + 3-BrPy | Solid Ehrlich Carcinoma | Tamoxifen 5 mg/kg and 3-BrPy 10 mg/kg | 80% reduction in tumor volume, increased oxidative stress | – | Decreased MMP-2/9, VEGF | In vivo | [173] |
| Trastuzumab + metformin Paclitaxel + trastuzumab + metformin | Xenograft HER2+ primary BC | Trastuzumab 5 mg/kg/once a week + Metformin 12 cycles of weekly paclitaxel 80 mg/m2 + Metformin 1500 mg/day | Reduced the tumor volume by 4-fold in HER2+, trastuzumab resistant tumor | Metformin reduces the cardiotoxicity induced by trastuzumab | Metformin lowers circulating insulin-like growth factor (IGF). Inhibition of AMPK/mTOR/p70S6K1 pathway | In vivo Phase II trial (2010) | [174,175] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varghese, E.; Samuel, S.M.; Líšková, A.; Samec, M.; Kubatka, P.; Büsselberg, D. Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer. Cancers 2020, 12, 2252. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082252
Varghese E, Samuel SM, Líšková A, Samec M, Kubatka P, Büsselberg D. Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer. Cancers. 2020; 12(8):2252. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082252
Chicago/Turabian StyleVarghese, Elizabeth, Samson Mathews Samuel, Alena Líšková, Marek Samec, Peter Kubatka, and Dietrich Büsselberg. 2020. "Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer" Cancers 12, no. 8: 2252. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082252