Inflammasome Sensor NLRP1 Confers Acquired Drug Resistance to Temozolomide in Human Melanoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
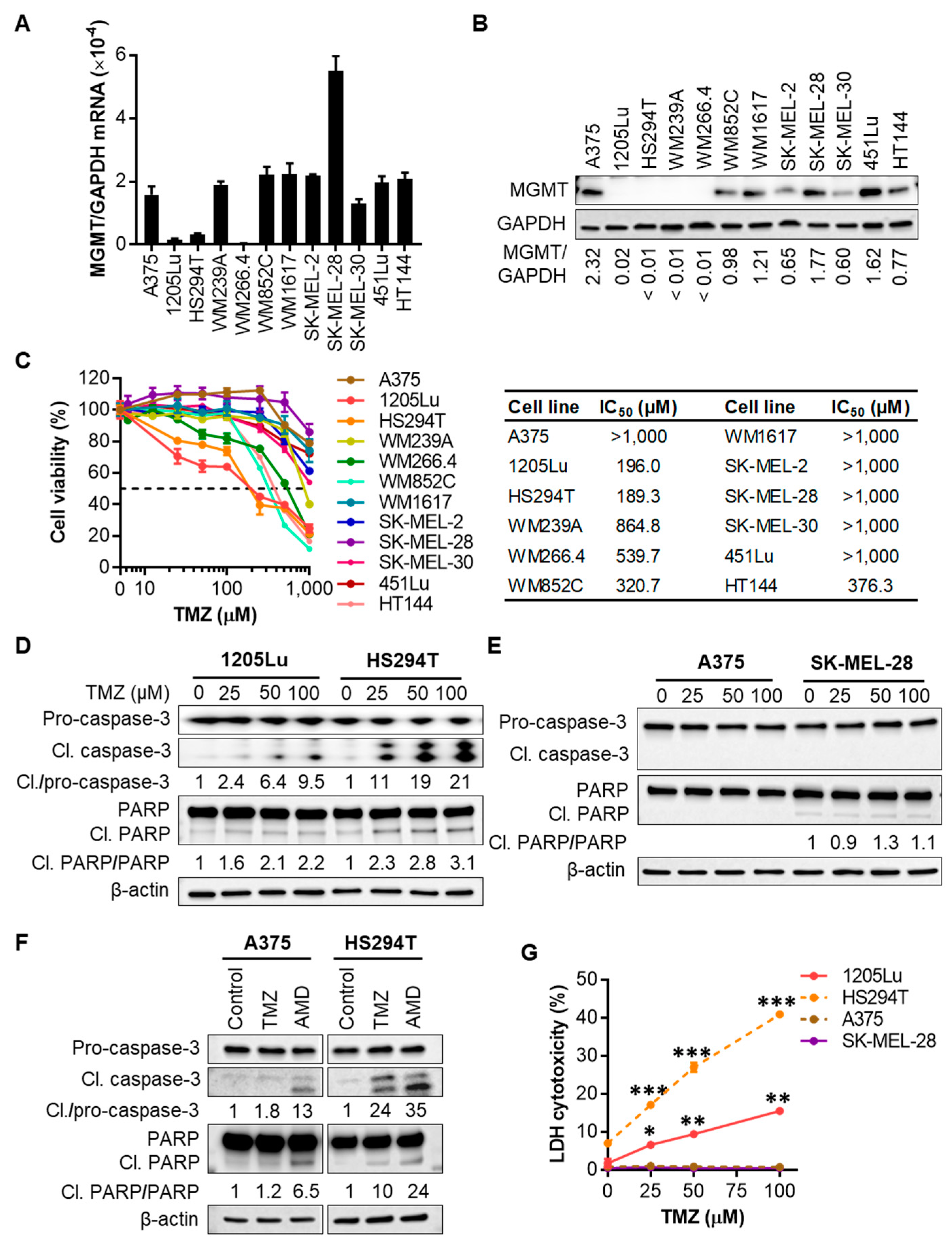
2.1. Human Melanoma Cell Lines That Express Low Levels of MGMT Are Initially Sensitive to TMZ
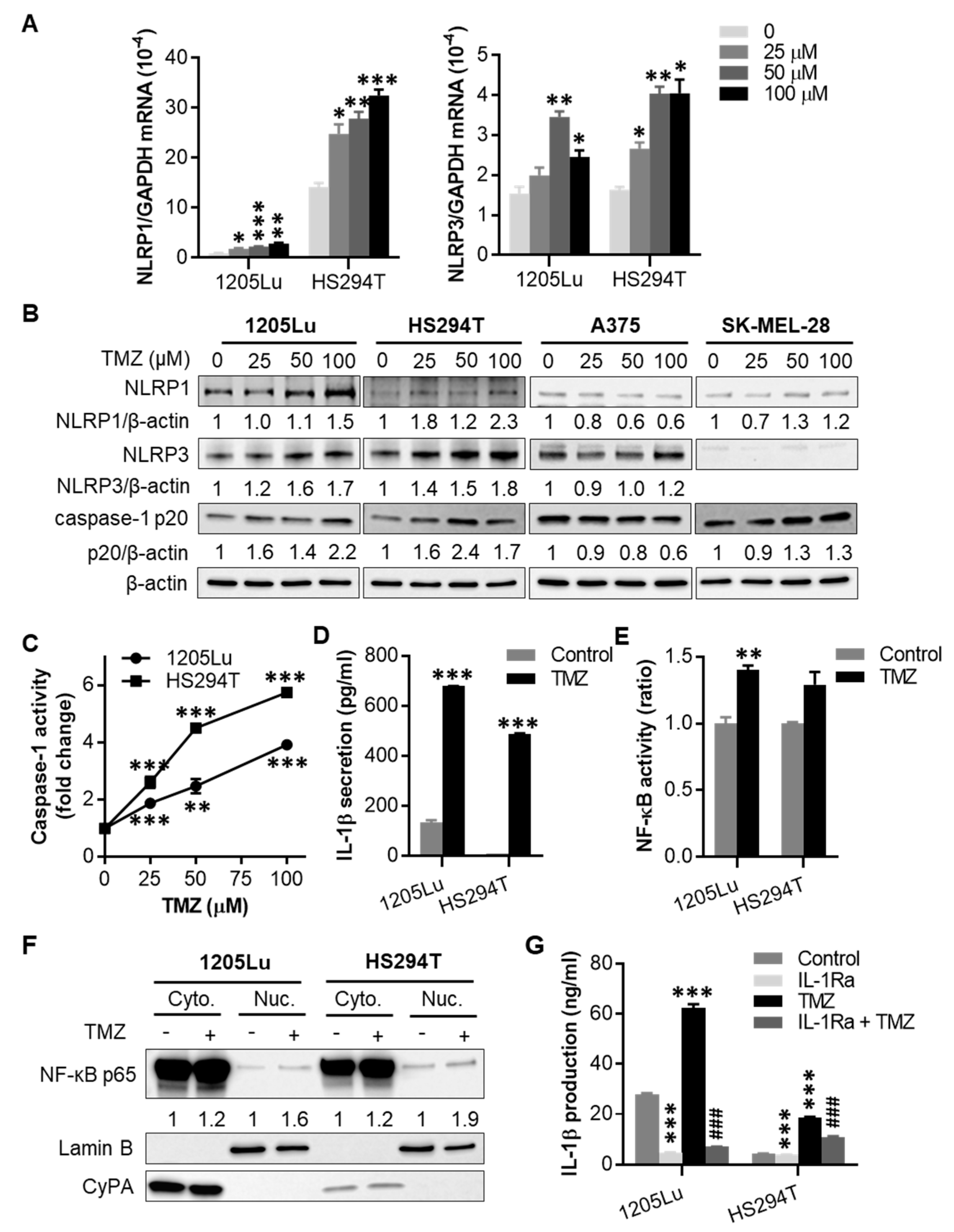
2.2. TMZ Enhances NLRP1 and NLRP3 Expression, Activates NLRP Inflammasomes, and Induces IL-1β Secretion in MGMT-Low Human Melanoma Cells
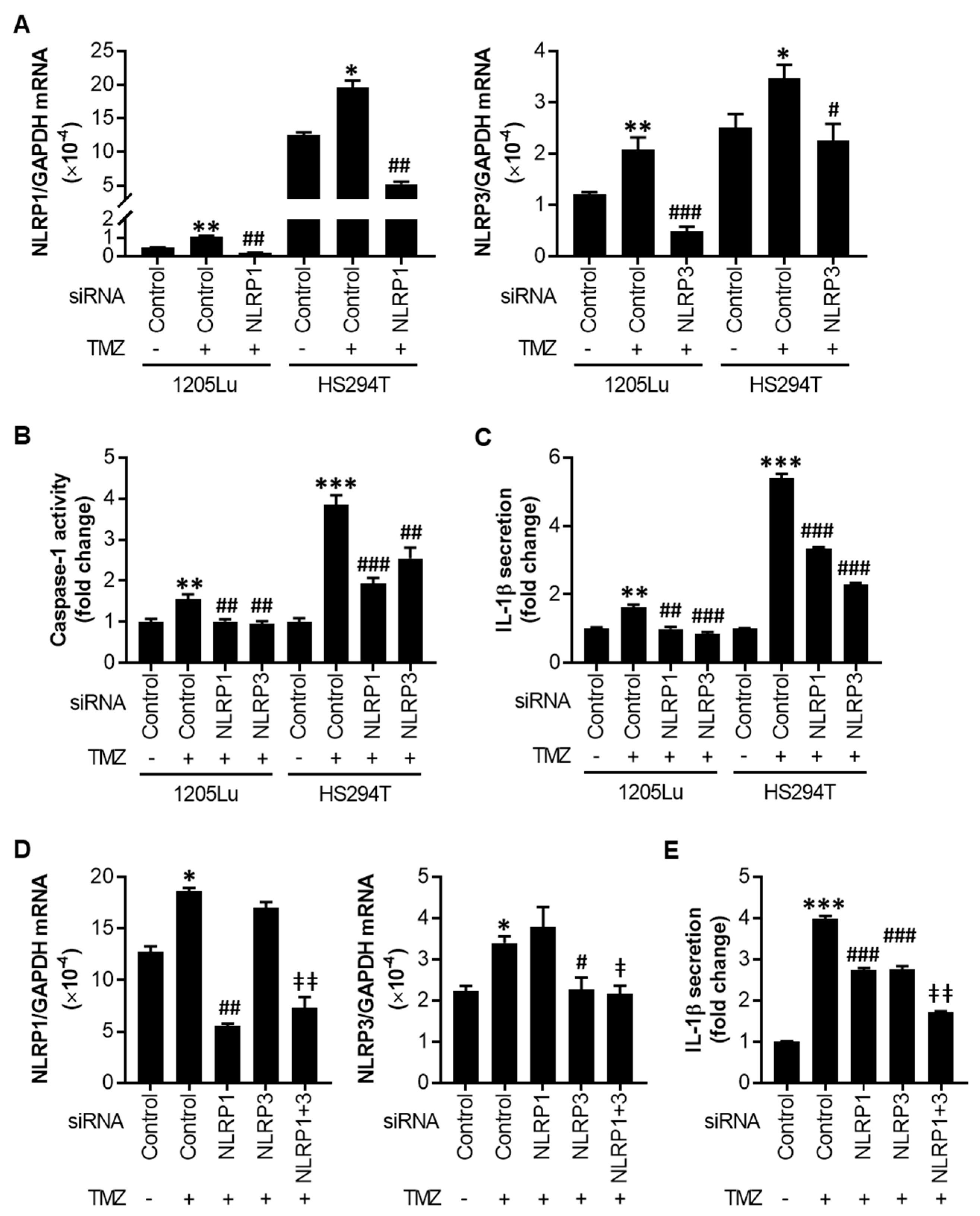
2.3. TMZ-Mediated Initial Inflammasome Activation Is NLRP1- and NLRP3-Dependent
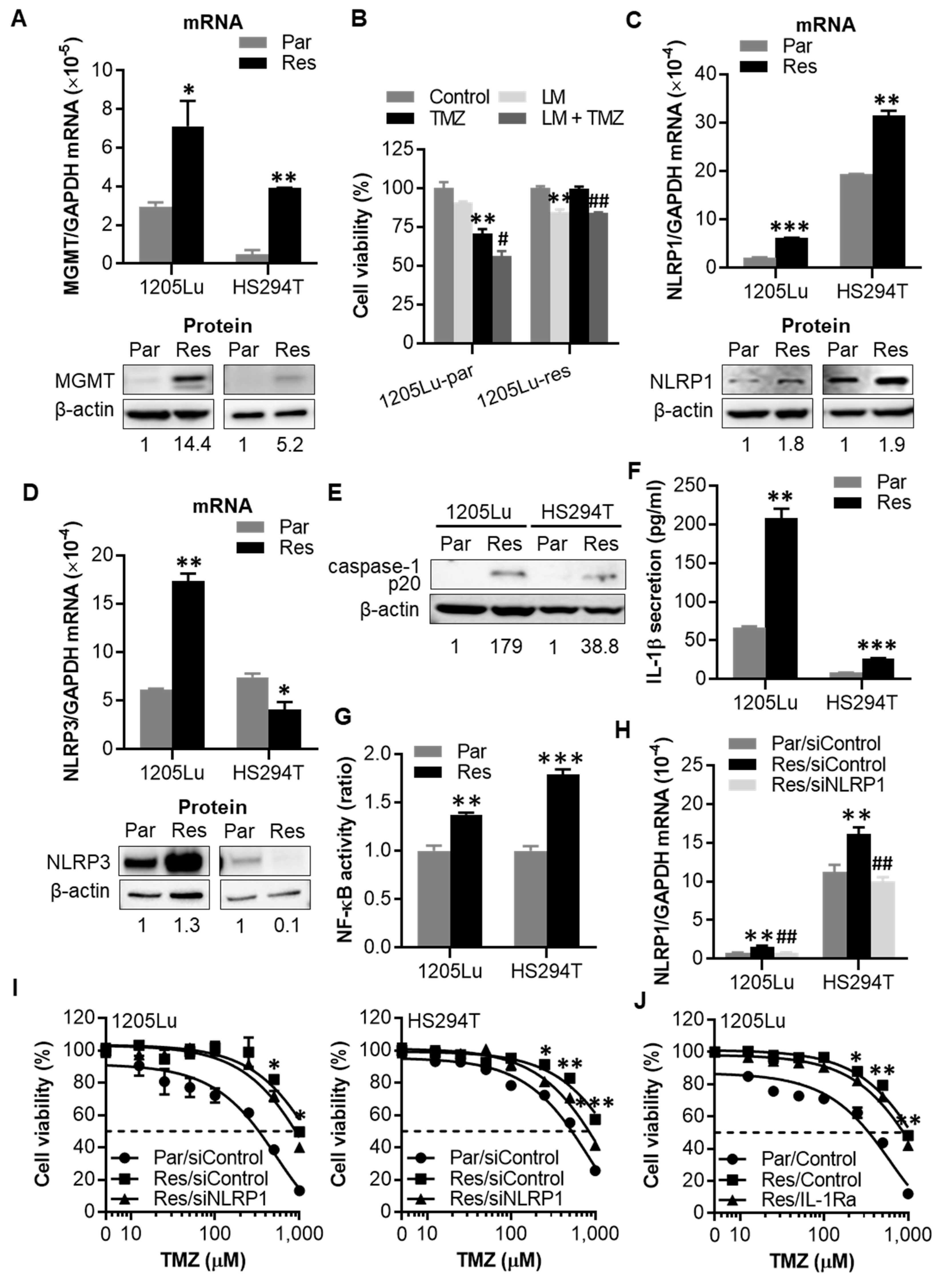
2.4. After Chronic Exposure to TMZ, Human Melanoma Cells Display Acquired Resistance to TMZ through Activation of NLRP1 Inflammasomes and IL-1β Secretion
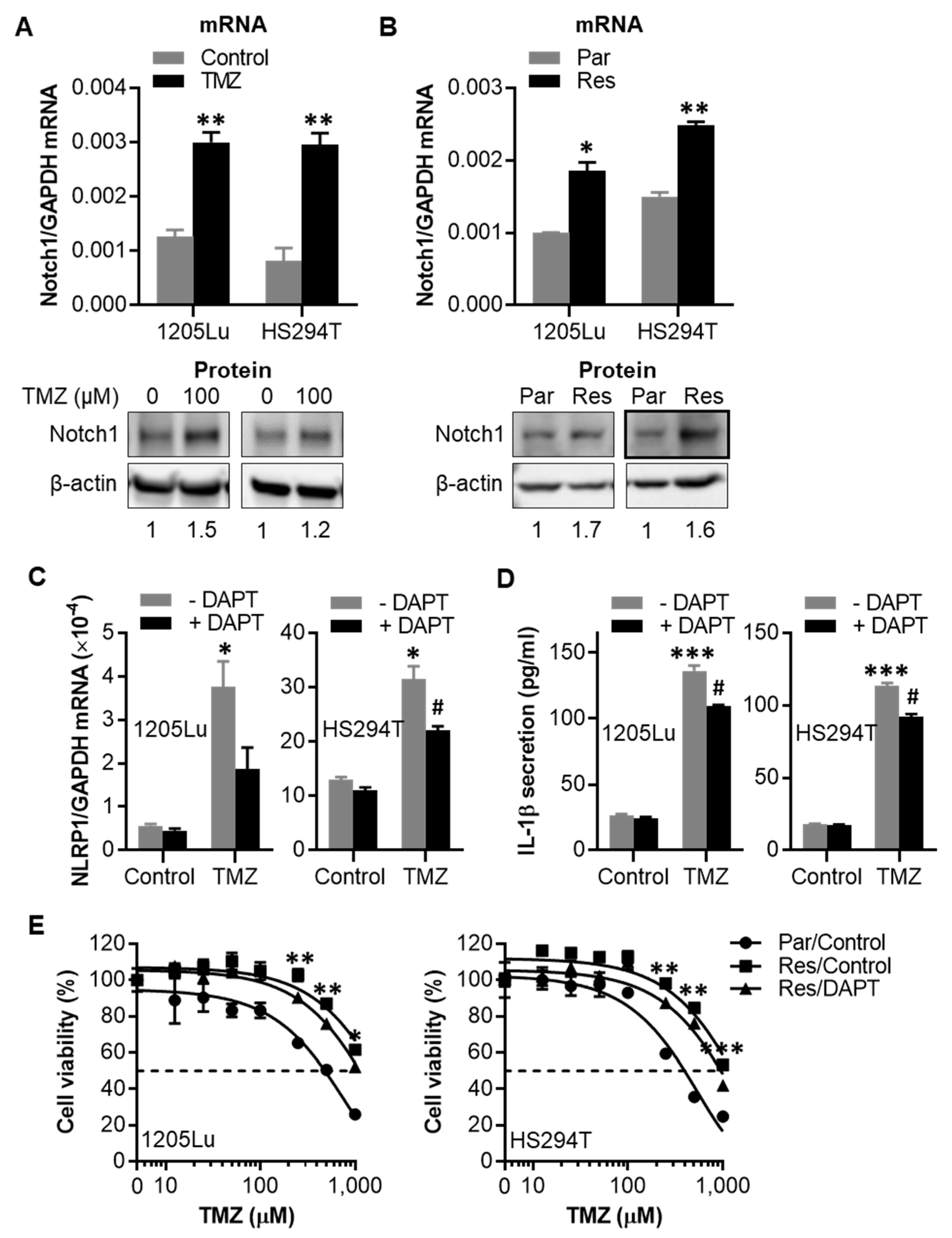
2.5. TMZ-Induced NLRP1 Inflammasome Activation Functions Downstream of Notch Signaling
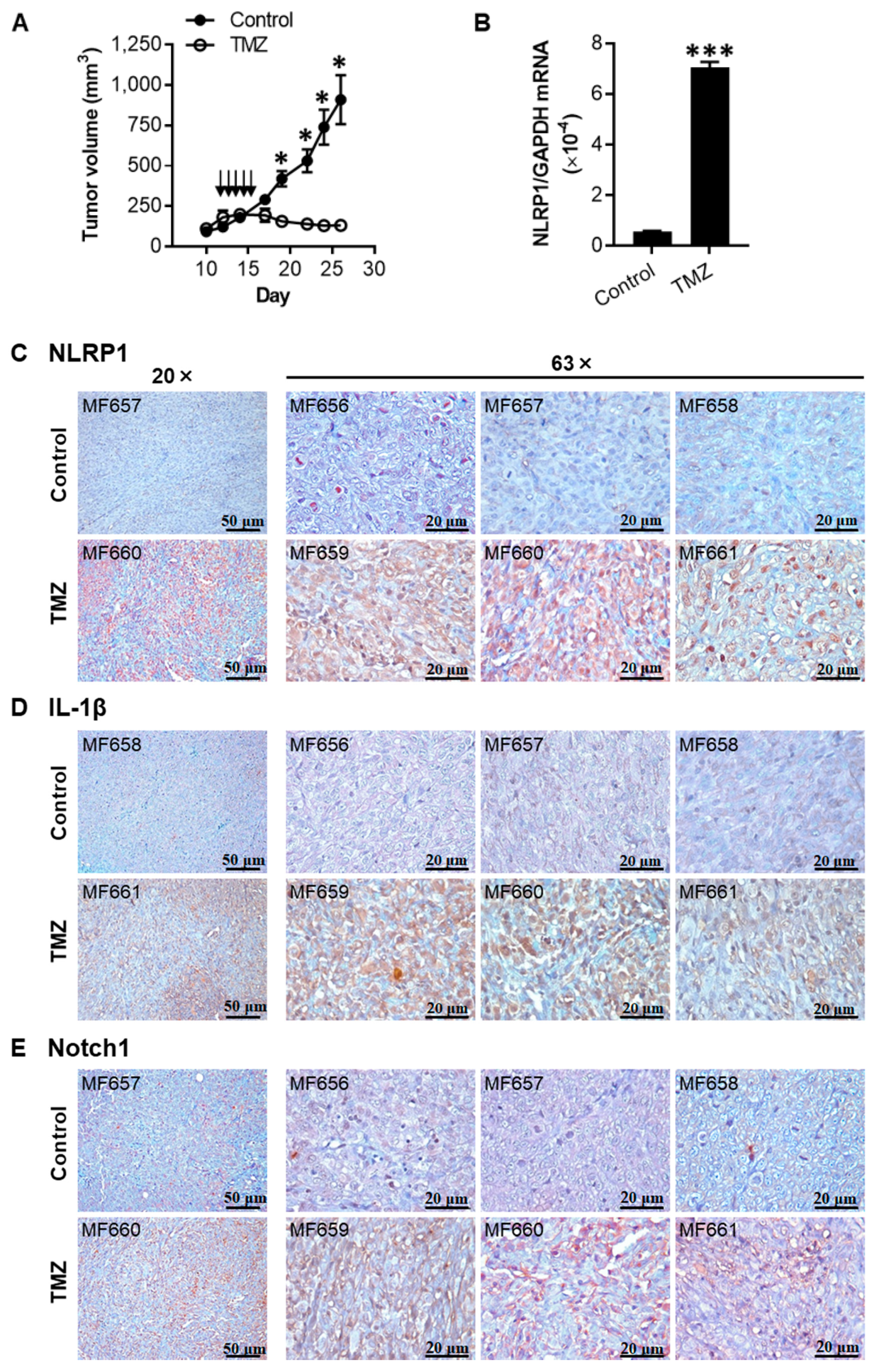
2.6. TMZ Enhances the Expression of NLRP1, IL-1β, and Notch1 in 1205Lu Tumors
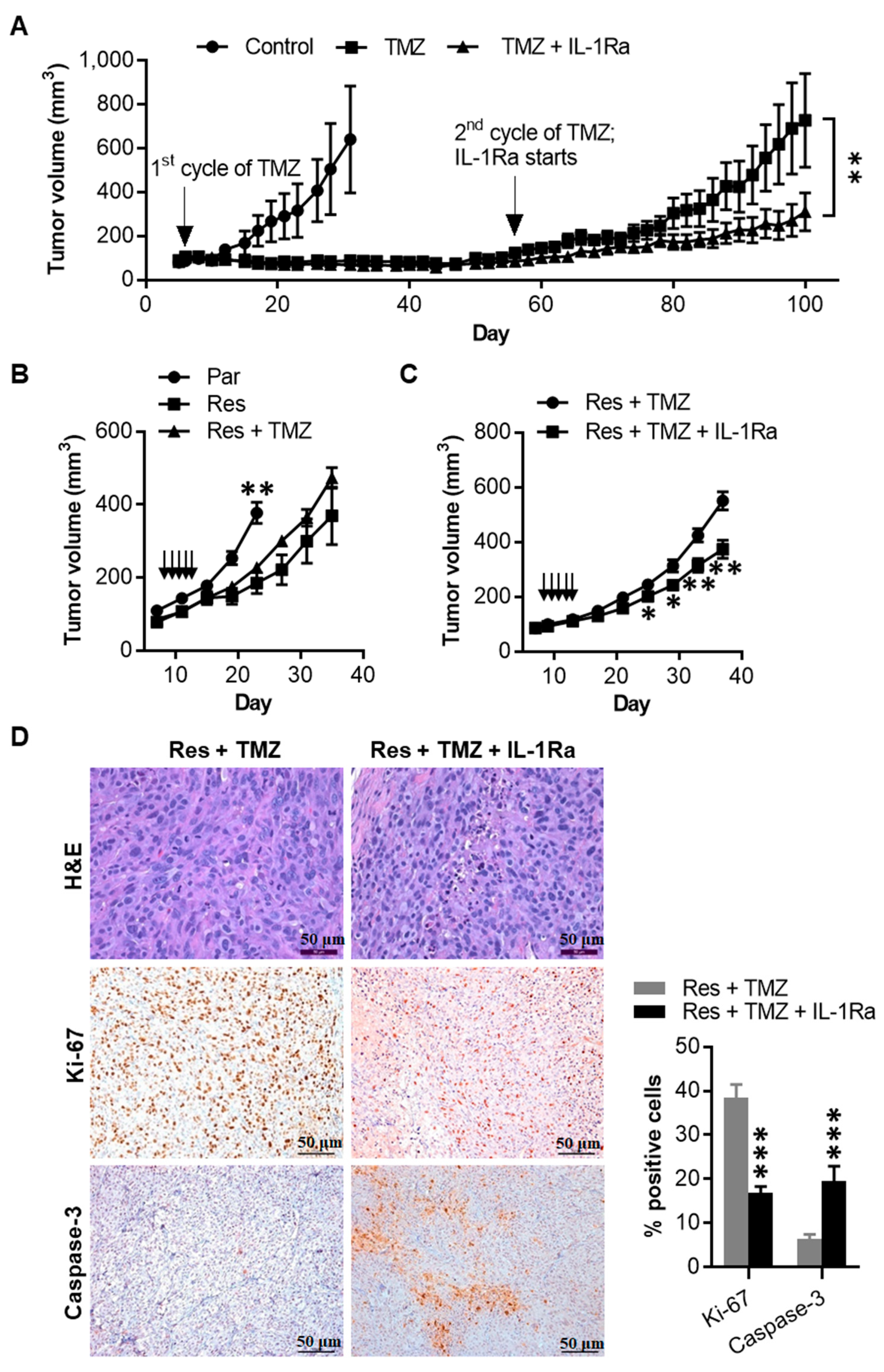
2.7. IL-1Ra Inhibits Tumor Growth of Melanoma Cells That Have Acquired TMZ Resistance
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Growth Inhibition
4.4. Generation of Acquired Resistance to TMZ
4.5. NLRP1 and NLRP3 Knockdown
4.6. Quantitative RT-PCR
4.7. Western Blot
4.8. Enzyme-Linked Immunosorbent Assay (ELISA)
4.9. Caspase-1 Activity Assay
4.10. Lactate Dehydrogenase (LDH) Assay
4.11. NF-κB Activity Assay
4.12. Tumor Formation In Vivo
4.13. Immunohistochemistry
4.14. Statistical analysis
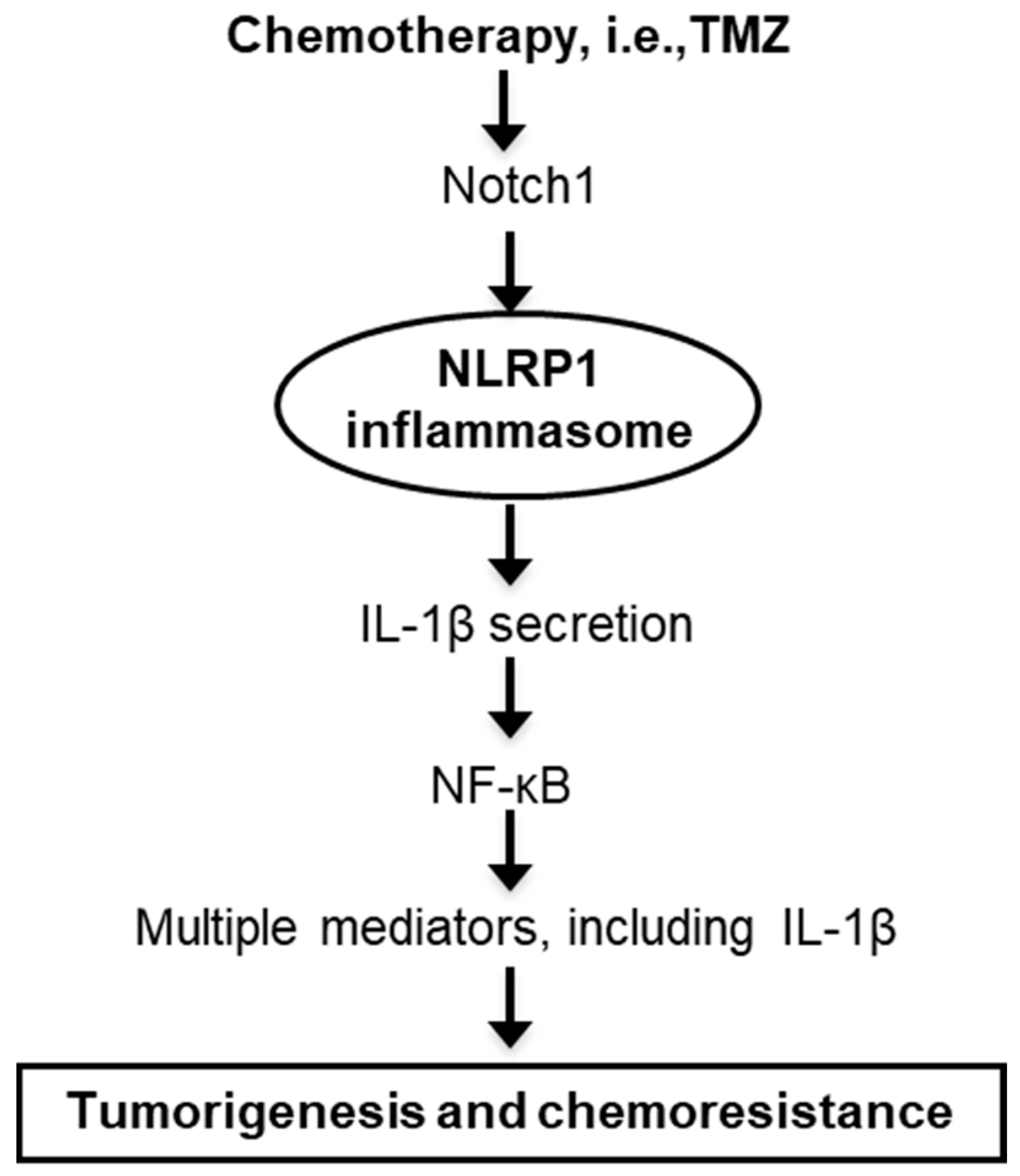
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treatment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Sun, Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016, 380, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, D.; Laput, G.; Vyas, A.K. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. Oncol. Targets Ther. 2014, 7, 1015–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Muerkoster, S.S.; Lust, J.; Arlt, A.; Hasler, R.; Witt, M.; Sebens, T.; Schreiber, S.; Folsch, U.R.; Schafer, H. Acquired chemoresistance in pancreatic carcinoma cells: Induced secretion of IL-1beta and NO lead to inactivation of caspases. Oncogene 2006, 25, 3973–3981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J. Biol. Chem. 2010, 285, 6477–6488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Kolb, R.; Liu, G.H.; Janowski, A.M.; Sutterwala, F.S.; Zhang, W. Inflammasomes in cancer: A double-edged sword. Protein Cell 2014, 5, 12–20. [Google Scholar] [CrossRef]
- Zhai, Z.; Liu, W.; Kaur, M.; Luo, Y.; Domenico, J.; Samson, J.M.; Shellman, Y.G.; Norris, D.A.; Dinarello, C.A.; Spritz, R.A.; et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 2017, 36, 3820–3830. [Google Scholar] [CrossRef] [Green Version]
- Zhong, F.L.; Mamai, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverenyi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016, 167, 187–202. [Google Scholar] [CrossRef] [Green Version]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlt, A.; Vorndamm, J.; Muerkoster, S.; Yu, H.; Schmidt, W.E.; Folsch, U.R.; Schafer, H. Autocrine production of interleukin 1beta confers constitutive nuclear factor kappaB activity and chemoresistance in pancreatic carcinoma cell lines. Cancer Res. 2002, 62, 910–916. [Google Scholar] [PubMed]
- Caporali, S.; Levati, L.; Graziani, G.; Muzi, A.; Atzori, M.G.; Bonmassar, E.; Palmieri, G.; Ascierto, P.A.; D’Atri, S. NF-kappaB is activated in response to temozolomide in an AKT-dependent manner and confers protection against the growth suppressive effect of the drug. J. Transl. Med. 2012, 10, 252. [Google Scholar] [CrossRef] [Green Version]
- Augustine, C.K.; Yoo, J.S.; Potti, A.; Yoshimoto, Y.; Zipfel, P.A.; Friedman, H.S.; Nevins, J.R.; Ali-Osman, F.; Tyler, D.S. Genomic and molecular profiling predicts response to temozolomide in melanoma. Clin. Cancer Res. 2009, 15, 502–510. [Google Scholar] [PubMed] [Green Version]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharm. 2012, 5, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat Res. Rev. Mutat Res. 2016, 769, 19–35. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Kitange, G.J.; James, C.D.; Plummer, R.; Calvert, H.; Weller, M.; Wick, W. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin. Cancer Res. 2008, 14, 2900–2908. [Google Scholar] [CrossRef] [Green Version]
- Agnihotri, S.; Burrell, K.; Buczkowicz, P.; Remke, M.; Golbourn, B.; Chornenkyy, Y.; Gajadhar, A.; Fernandez, N.A.; Clarke, I.D.; Barszczyk, M.S.; et al. ATM regulates 3-methylpurine-DNA glycosylase and promotes therapeutic resistance to alkylating agents. Cancer Discov. 2014, 4, 1198–1213. [Google Scholar] [CrossRef] [Green Version]
- Raineri, A.; Fasoli, S.; Campagnari, R.; Gotte, G.; Menegazzi, M. Onconase Restores Cytotoxicity in Dabrafenib-Resistant A375 Human Melanoma Cells and Affects Cell Migration, Invasion and Colony Formation Capability. Int. J. Mol. Sci. 2019, 20, 5980. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Luo, Q.; Zhang, H.; Wang, H.; Chen, W.; Meng, G.; Chen, F. The role of NLRP3 inflammasome in 5-fluorouracil resistance of oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 81. [Google Scholar] [CrossRef] [Green Version]
- Goldman, A.; Khiste, S.; Freinkman, E.; Dhawan, A.; Majumder, B.; Mondal, J.; Pinkerton, A.B.; Eton, E.; Medhi, R.; Chandrasekar, V.; et al. Targeting tumor phenotypic plasticity and metabolic remodeling in adaptive cross-drug tolerance. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar] [PubMed]
- Naumann, S.C.; Roos, W.P.; Jost, E.; Belohlavek, C.; Lennerz, V.; Schmidt, C.W.; Christmann, M.; Kaina, B. Temozolomide- and fotemustine-induced apoptosis in human malignant melanoma cells: Response related to MGMT, MMR, DSBs, and p53. Br. J. Cancer 2009, 100, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makita, K.; Hara, H.; Sano, E.; Okamoto, Y.; Ochiai, Y.; Harada, T.; Ueda, T.; Nakayama, T.; Aizawa, S.; Yoshino, A. Interferon-beta sensitizes human malignant melanoma cells to temozolomide-induced apoptosis and autophagy. Int. J. Oncol. 2019, 54, 1864–1874. [Google Scholar] [PubMed]
- Zheng, M.; Bocangel, D.; Ramesh, R.; Ekmekcioglu, S.; Poindexter, N.; Grimm, E.A.; Chada, S. Interleukin-24 overcomes temozolomide resistance and enhances cell death by down-regulation of O6-methylguanine-DNA methyltransferase in human melanoma cells. Mol. Cancer Ther. 2008, 7, 3842–3851. [Google Scholar] [PubMed] [Green Version]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef] [Green Version]
- Decker, P.; Muller, S. Modulating poly (ADP-ribose) polymerase activity: Potential for the prevention and therapy of pathogenic situations involving DNA damage and oxidative stress. Curr. Pharm. Biotechnol. 2002, 3, 275–283. [Google Scholar] [CrossRef]
- Wang, W.; Mani, A.M.; Wu, Z.H. DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression. J. Cancer Metastasis Treat. 2017, 3, 45–59. [Google Scholar] [CrossRef]
- Erice, O.; Smith, M.P.; White, R.; Goicoechea, I.; Barriuso, J.; Jones, C.; Margison, G.P.; Acosta, J.C.; Wellbrock, C.; Arozarena, I. MGMT Expression Predicts PARP-Mediated Resistance to Temozolomide. Mol. Cancer 2015, 14, 1236–1246. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Liu, M.; Gonzalez-Perez, R.R. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim. Biophys. Acta 2011, 1815, 197–213. [Google Scholar] [CrossRef] [Green Version]
- Capaccione, K.M.; Pine, S.R. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis 2013, 34, 1420–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordaro, F.G.; De Presbiteris, A.L.; Camerlingo, R.; Mozzillo, N.; Pirozzi, G.; Cavalcanti, E.; Manca, A.; Palmieri, G.; Cossu, A.; Ciliberto, G.; et al. Phenotype characterization of human melanoma cells resistant to dabrafenib. Oncol. Rep. 2017, 38, 2741–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–732. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jia, L.; Jin, X.; Liu, Q.; Cao, W.; Gao, X.; Yang, M.; Sun, B. NF-kappaB inhibitor reverses temozolomide resistance in human glioma TR/U251 cells. Oncol. Lett. 2015, 9, 2586–2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, Y.T.; McDonald, K.L.; Grewal, T.; Munoz, L. Interleukins in glioblastoma pathophysiology: Implications for therapy. Br. J. Pharm. 2013, 168, 591–606. [Google Scholar] [CrossRef] [Green Version]
- Koseki, T.; Inohara, N.; Chen, S.; Nunez, G. ARC, an inhibitor of apoptosis expressed in skeletal muscle and heart that interacts selectively with caspases. Proc. Natl. Acad. Sci. USA 1998, 95, 5156–5160. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liang, P.; Jiang, B.; Tang, Y.; Lv, Q.; Hao, H.; Liu, Z.; Xiao, X. CARD9 inhibits mitochondria-dependent apoptosis of cardiomyocytes under oxidative stress via interacting with Apaf-1. Free Radic. Biol. Med. 2019, 141, 172–181. [Google Scholar] [CrossRef]
- Martz, C.A.; Ottina, K.A.; Singleton, K.R.; Jasper, J.S.; Wardell, S.E.; Peraza-Penton, A.; Anderson, G.R.; Winter, P.S.; Wang, T.; Alley, H.M.; et al. Systematic identification of signaling pathways with potential to confer anticancer drug resistance. Sci. Signal. 2014, 7, ra121. [Google Scholar] [CrossRef] [Green Version]
- Nair, J.S.; Sheikh, T.; Ho, A.L.; Schwartz, G.K. PTEN regulates sensitivity of melanoma cells to RO4929097, the gamma-secretase inhibitor. Anticancer Res. 2013, 33, 1307–1316. [Google Scholar]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Wu, H.; Xu, H.; Xiong, H.; Chu, Q.; Yu, S.; Wu, G.S.; Wu, K. Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett. 2015, 369, 20–27. [Google Scholar] [PubMed] [Green Version]
- Shin, H.M.; Minter, L.M.; Cho, O.H.; Gottipati, S.; Fauq, A.H.; Golde, T.E.; Sonenshein, G.E.; Osborne, B.A. Notch1 augments NF-kappaB activity by facilitating its nuclear retention. EMBO J. 2006, 25, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faustin, B.; Lartigue, L.; Bruey, J.M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, J.C. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 2007, 25, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Sinnberg, T.; Lasithiotakis, K.; Niessner, H.; Schittek, B.; Flaherty, K.T.; Kulms, D.; Maczey, E.; Campos, M.; Gogel, J.; Garbe, C.; et al. Inhibition of PI3K-AKT-mTOR signaling sensitizes melanoma cells to cisplatin and temozolomide. J. Investig. Derm. 2009, 129, 1500–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niessner, H.; Kosnopfel, C.; Sinnberg, T.; Beck, D.; Krieg, K.; Wanke, I.; Lasithiotakis, K.; Bonin, M.; Garbe, C.; Meier, F. Combined activity of temozolomide and the mTOR inhibitor temsirolimus in metastatic melanoma involves DKK1. Exp. Derm. 2017, 26, 598–606. [Google Scholar] [CrossRef]
- Ma, X.H.; Piao, S.; Wang, D.; McAfee, Q.W.; Nathanson, K.L.; Lum, J.J.; Li, L.Z.; Amaravadi, R.K. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin. Cancer Res. 2011, 17, 3478–3489. [Google Scholar] [CrossRef] [Green Version]
- Ryabaya, O.O.; Inshakov, A.N.; Egorova, A.V.; Emelyanova, M.A.; Nasedkina, T.V.; Zasedatelev, A.S.; Khochenkov, D.A.; Stepanova, E.V. Autophagy inhibitors chloroquine and LY294002 enhance temozolomide cytotoxicity on cutaneous melanoma cell lines in vitro. Anticancer Drugs 2017, 28, 307–315. [Google Scholar]
- Chang, X.; Zhu, W.; Zhang, H.; Lian, S. Sensitization of melanoma cells to temozolomide by overexpression of microRNA 203 through direct targeting of glutaminase-mediated glutamine metabolism. Clin. Exp. Derm. 2017, 42, 614–621. [Google Scholar] [CrossRef]
- Okada, M.; Sato, A.; Shibuya, K.; Watanabe, E.; Seino, S.; Suzuki, S.; Seino, M.; Narita, Y.; Shibui, S.; Kayama, T.; et al. JNK contributes to temozolomide resistance of stem-like glioblastoma cells via regulation of MGMT expression. Int. J. Oncol. 2014, 44, 591–599. [Google Scholar]
- Khalili, J.S.; Liu, S.; Rodriguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef] [PubMed]
- Weatherbee, J.L.; Kraus, J.L.; Ross, A.H. ER stress in temozolomide-treated glioblastomas interferes with DNA repair and induces apoptosis. Oncotarget 2016, 7, 43820–43834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Su, J.; Lan, B.; Gao, Y.; Zhao, J. Targeting off-target effects: Endoplasmic reticulum stress and autophagy as effective strategies to enhance temozolomide treatment. Onco Targets Ther. 2019, 12, 1857–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Osualdo, A.; Anania, V.G.; Yu, K.; Lill, J.R.; Kaufman, R.J.; Matsuzawa, S.; Reed, J.C. Transcription Factor ATF4 Induces NLRP1 Inflammasome Expression during Endoplasmic Reticulum Stress. PLoS ONE 2015, 10, e0130635. [Google Scholar] [CrossRef]
- Zhai, Z.; Samson, J.M.; Fujita, M. ATF4-dependent regulation of vemurafenib resistance in melanoma by NLRP1. J. Investig. Derm. 2018, 138, S213. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Ribas, A. New Combination Strategies Using Programmed Cell Death 1/Programmed Cell Death Ligand 1 Checkpoint Inhibitors as a Backbone. Cancer J. 2017, 23, 10–22. [Google Scholar] [CrossRef]
- Park, J.; Kim, C.G.; Shim, J.K.; Kim, J.H.; Lee, H.; Lee, J.E.; Kim, M.H.; Haam, K.; Jung, I.; Park, S.H.; et al. Effect of combined anti-PD-1 and temozolomide therapy in glioblastoma. Oncoimmunology 2019, 8, e1525243. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Kim, D.W.; Bassett, R.L.; Cain, S.; Washington, E.; Hwu, W.J.; Kim, K.B.; Papadopoulos, N.E.; Homsi, J.; Hwu, P.; et al. A phase II study of ipilimumab plus temozolomide in patients with metastatic melanoma. Cancer Immunol. Immunother. 2017, 66, 1359–1366. [Google Scholar] [CrossRef]
- Wang, S.; Yao, F.; Lu, X.; Li, Q.; Su, Z.; Lee, J.H.; Wang, C.; Du, L. Temozolomide promotes immune escape of GBM cells via upregulating PD-L1. Am. J. Cancer Res. 2019, 9, 1161–1171. [Google Scholar]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Invest. 2020, 130, 2570–2586. [Google Scholar] [CrossRef] [Green Version]
- Moll, M.; Kuemmerle-Deschner, J.B. Inflammasome and cytokine blocking strategies in autoinflammatory disorders. Clin. Immunol. 2013, 147, 242–275. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, E.; Campbell, M.; Doyle, S.L. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. J. Inflamm. Res. 2015, 8, 15–27. [Google Scholar] [PubMed] [Green Version]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, P.S.; Sandstrom, A.; Vance, R.E. The NLRP1 inflammasome: New mechanistic insights and unresolved mysteries. Curr. Opin. Immunol. 2019, 60, 37–45. [Google Scholar] [CrossRef]
- Dinarello, C.A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Group, C.T. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Uphoff, C.C.; Drexler, H.G. Detecting Mycoplasma contamination in cell cultures by polymerase chain reaction. Methods Mol. Med. 2004, 88, 319–326. [Google Scholar]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Stevens, M.F.; Laughton, C.A.; Madhusudan, S.; Bradshaw, T.D. Acquired resistance to temozolomide in glioma cell lines: Molecular mechanisms and potential translational applications. Oncology 2010, 78, 103–114. [Google Scholar] [CrossRef]
- Luo, Y.; Ellis, L.Z.; Dallaglio, K.; Takeda, M.; Robinson, W.A.; Robinson, S.E.; Liu, W.; Lewis, K.D.; McCarter, M.D.; Gonzalez, R.; et al. Side population cells from human melanoma tumors reveal diverse mechanisms for chemoresistance. J. Investig. Derm. 2012, 132, 2440–2450. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, Z.; Samson, J.M.; Yamauchi, T.; Vaddi, P.K.; Matsumoto, Y.; Dinarello, C.A.; Ravindran Menon, D.; Fujita, M. Inflammasome Sensor NLRP1 Confers Acquired Drug Resistance to Temozolomide in Human Melanoma. Cancers 2020, 12, 2518. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092518
Zhai Z, Samson JM, Yamauchi T, Vaddi PK, Matsumoto Y, Dinarello CA, Ravindran Menon D, Fujita M. Inflammasome Sensor NLRP1 Confers Acquired Drug Resistance to Temozolomide in Human Melanoma. Cancers. 2020; 12(9):2518. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092518
Chicago/Turabian StyleZhai, Zili, Jenny Mae Samson, Takeshi Yamauchi, Prasanna K. Vaddi, Yuko Matsumoto, Charles A. Dinarello, Dinoop Ravindran Menon, and Mayumi Fujita. 2020. "Inflammasome Sensor NLRP1 Confers Acquired Drug Resistance to Temozolomide in Human Melanoma" Cancers 12, no. 9: 2518. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092518