Mitochondrial Metabolism, Contact Sites and Cellular Calcium Signaling: Implications for Tumorigenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
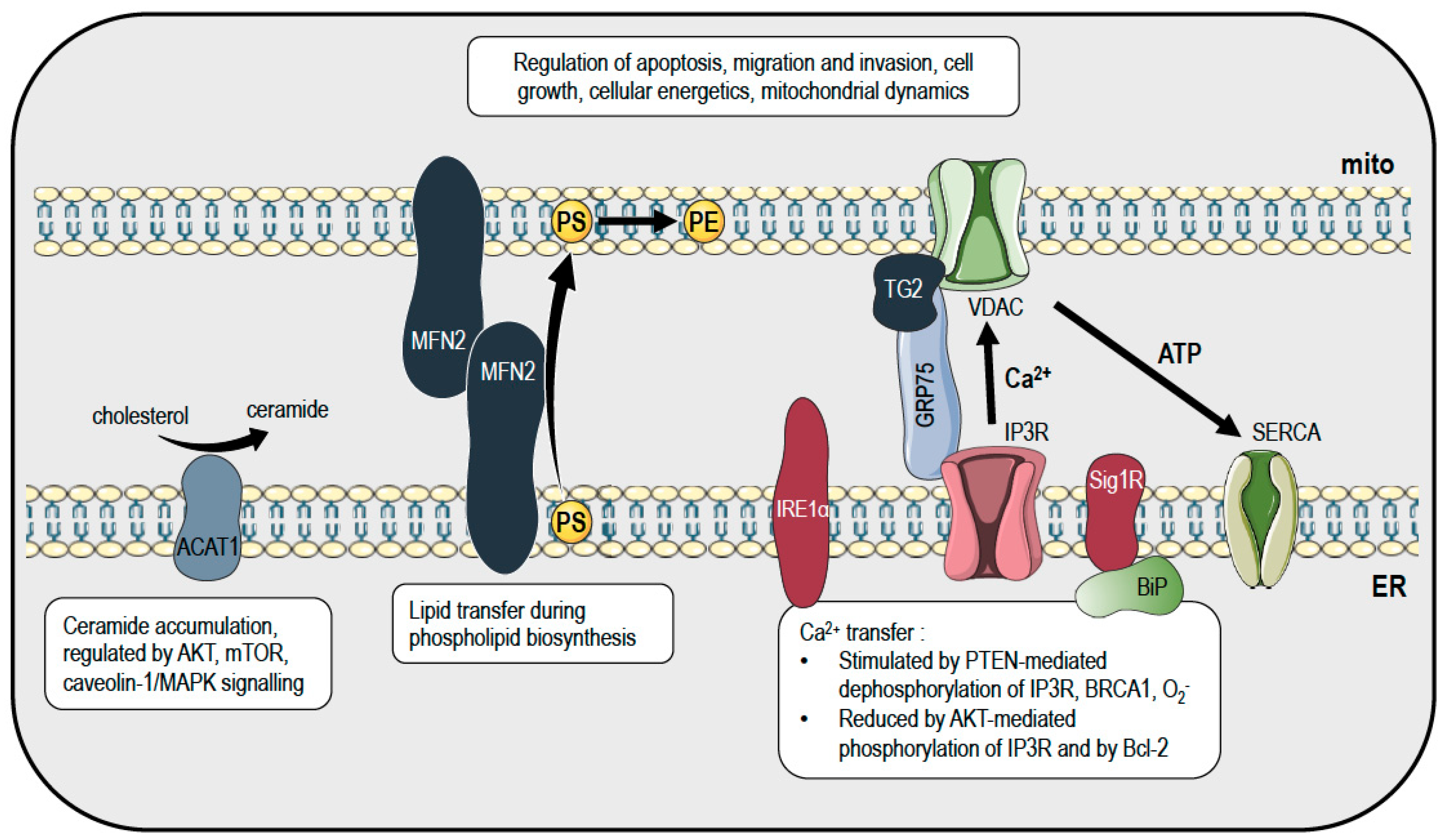
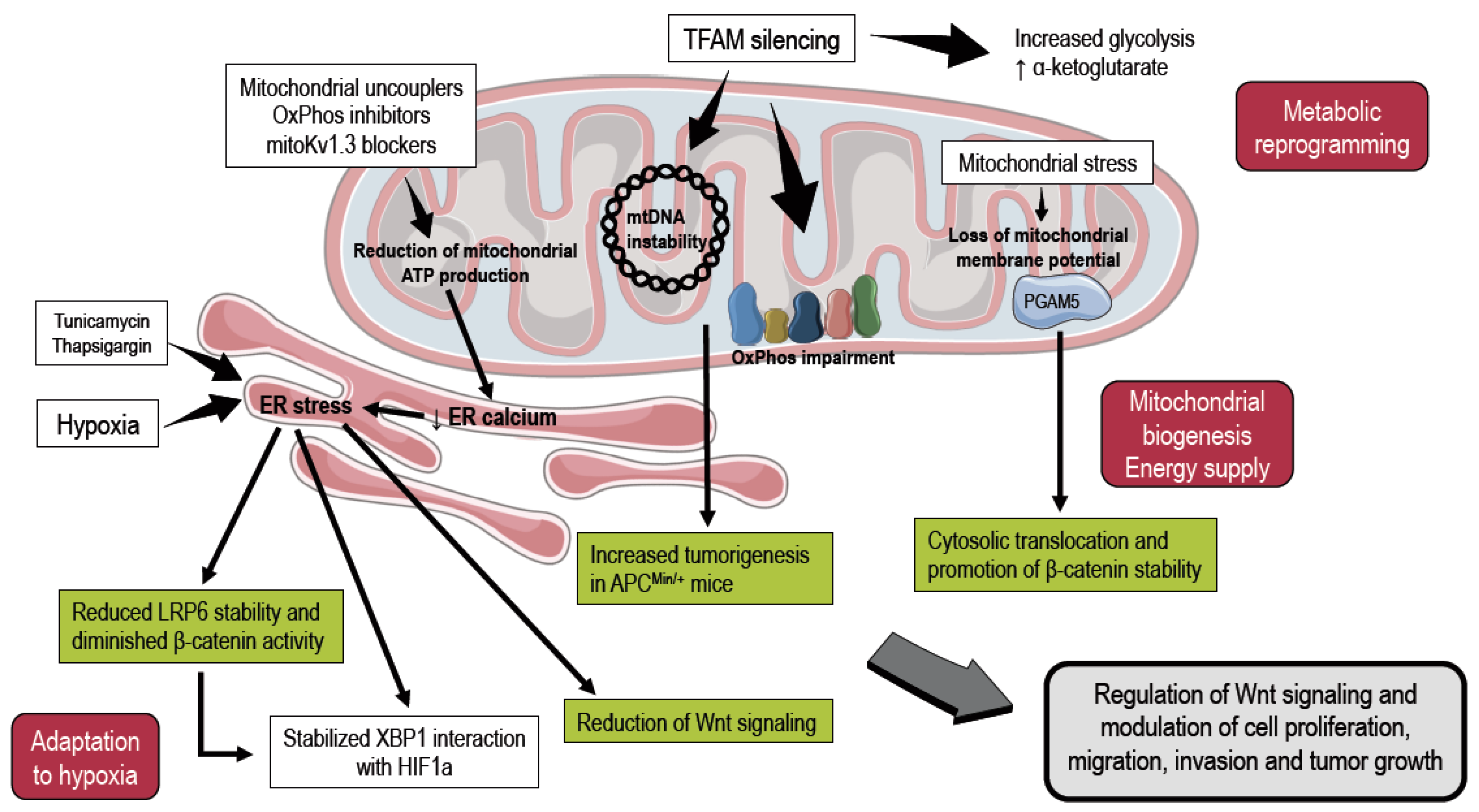
2. Mitochondria and Endoplasmic Reticulum
3. Mitochondria and Lysosomes
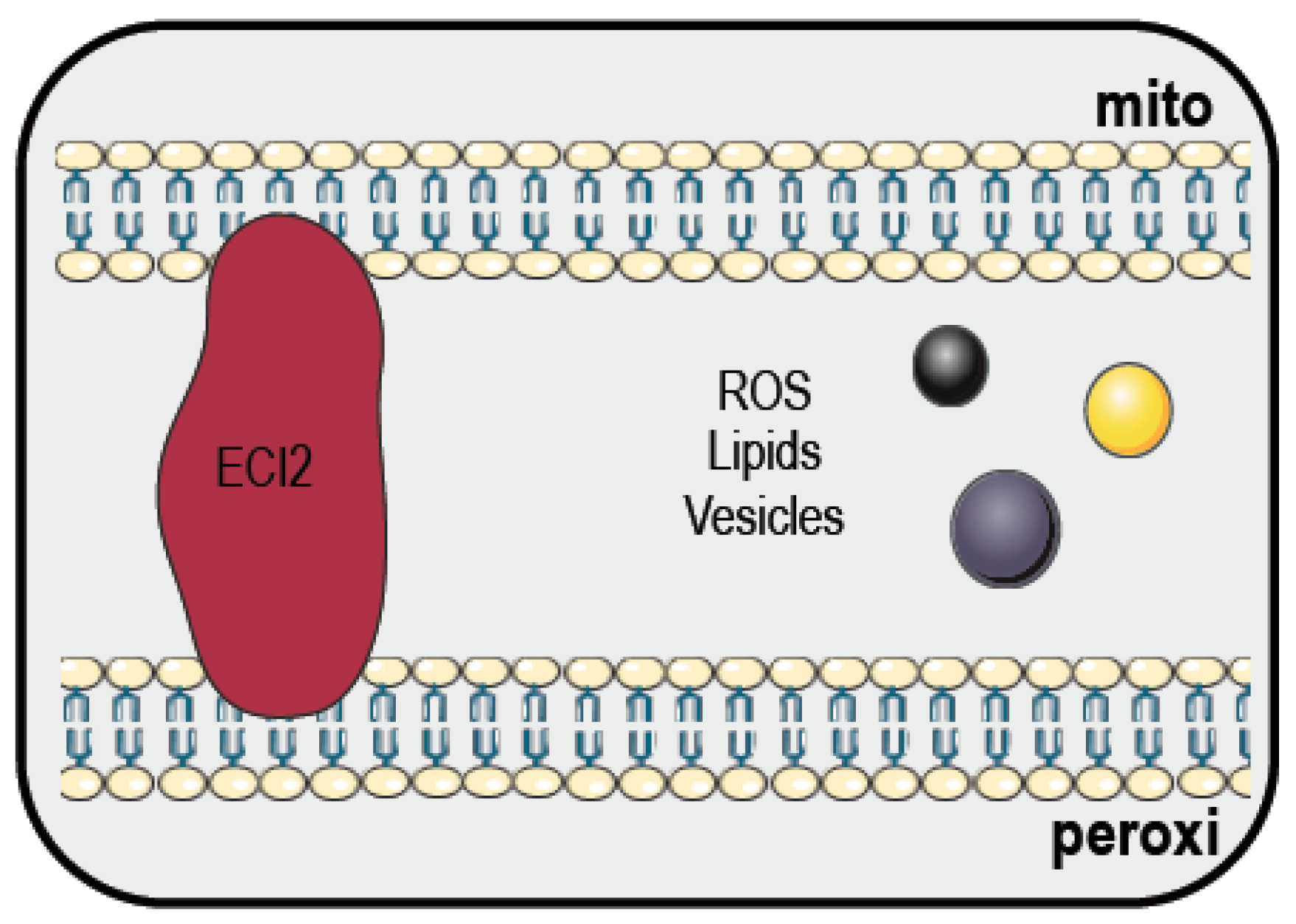
4. Mitochondria and Peroxisomes
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dyall, S.D.; Brown, M.T.; Johnson, P.J. Ancient Invasions: From Endosymbionts to Organelles. Science 2004, 304, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmis, J.N.; Ayliff, M.A.; Huang, C.Y.; Martin, W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004, 5, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Peruzzo, R.; Bachmann, M.; Montà, G.D.; Vicario, M.; Santinon, G.; Mattarei, A.; Moro, E.; Quintana-Cabrera, R.; Scorrano, L.; et al. Impaired Mitochondrial ATP Production Downregulates Wnt Signaling via ER Stress Induction. Cell Rep. 2019, 28, 1949–1960.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhola, P.D.; Letai, A. Mitochondria-Judges and Executioners of Cell Death Sentences. Mol. Cell 2016, 61, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J. Mitochondria—Striking a balance between host and endosymbiont. Science 2019, 365, eaaw9855. [Google Scholar] [CrossRef] [PubMed]
- Merry, T.L.; Ristow, M. Mitohormesis in exercise training. Free Radic. Biol. Med. 2016, 98, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Suaga, P.; Paillusson, S.; Miller, C.C.J. ER-mitochondria signaling regulates autophagy. Autophagy 2017, 13, 1250–1251. [Google Scholar] [CrossRef] [Green Version]
- Gordaliza-Alaguero, I.; Cantó, C.; Zorzano, A. Metabolic implications of organelle–mitochondria communication. EMBO Rep. 2019, 20, e47928. [Google Scholar] [CrossRef]
- Gómez-Suaga, P.; Bravo-San Pedro, J.M.; González-Polo, R.A.; Fuentes, J.M.; Niso-Santano, M. ER-mitochondria signaling in Parkinson’s disease review-article. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Naia, L.; Ferreira, I.L.; Ferreiro, E.; Rego, A.C. Mitochondrial Ca2+ handling in Huntington’s and Alzheimer’s diseases—Role of ER-mitochondria crosstalk. Biochem. Biophys. Res. Commun. 2017, 483, 1069–1077. [Google Scholar] [CrossRef]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef]
- Rieusset, J. Role of endoplasmic reticulum-mitochondria communication in type 2 diabetes. In Advances in Experimental Medicine and Biology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Simmen, T.; Herrera-Cruz, M.S. Plastic mitochondria-endoplasmic reticulum (ER) contacts use chaperones and tethers to mould their structure and signaling. Curr. Opin. Cell Biol. 2018, 53, 61–69. [Google Scholar] [CrossRef]
- Morciano, G.; Marchi, S.; Morganti, C.; Sbano, L.; Bittremieux, M.; Kerkhofs, M.; Corricelli, M.; Danese, A.; Karkucinska-Wieckowska, A.; Wieckowski, M.R.; et al. Role of Mitochondria-Associated ER Membranes in Calcium Regulation in Cancer-Specific Settings. Neoplasia 2018, 20, 510–523. [Google Scholar] [CrossRef]
- Xia, M.F.; Zhang, Y.Z.; Jin, K.; Lu, Z.T.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci. 2019, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Eisenhut, M.; Hocken, N.; Weber, A.P.M. Plastidial metabolite transporters integrate photorespiration with carbon, nitrogen, and sulfur metabolism. Cell Calcium 2015, 58, 98–104. [Google Scholar] [CrossRef]
- Valm, A.M.; Cohen, S.; Legant, W.R.; Melunis, J.; Hershberg, U.; Wait, E.; Cohen, A.R.; Davidson, M.W.; Betzig, E.; Lippincott-Schwartz, J. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 2017, 546, 162–167. [Google Scholar] [CrossRef]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef]
- Lever, J.D.; Chappell, J.B. Mitochondria isolated from rat brown adipose tissue and liver. J. Biophys. Biochem. Cytol. 1958, 4, 287–290. [Google Scholar] [CrossRef] [Green Version]
- Copeland, D.E.; Dalton, A.J. An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J. Biophys. Biochem. Cytol. 1959, 5, 393–396. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Calì, T.; Ottolini, D.; Vicario, M.; Catoni, C.; Vallese, F.; Cieri, D.; Barazzuol, L.; Brini, M. splitGFP Technology Reveals Dose-Dependent ER-Mitochondria Interface Modulation by α-Synuclein A53T and A30P Mutants. Cells 2019, 8, 1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum–Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Simmen, T. Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells 2019, 8, 1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doghman-Bouguerra, M.; Lalli, E. ER-mitochondria interactions: Both strength and weakness within cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 650–662. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Savignac, M.; Simon, M.; Edir, A.; Guibbal, L.; Hovnanian, A. SERCA2 dysfunction in darier disease causes endoplasmic reticulum stress and impaired cell-to-cell adhesion strength: Rescue by miglustat. J. Investig. Dermatol. 2014, 134, 1961–1970. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E. Calcium Signaling. Cell 1995, 80, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Stigler, J.; Rief, M. Calcium-dependent folding of single calmodulin molecules. Proc. Natl. Acad. Sci. USA 2012, 109, 17814–17819. [Google Scholar] [CrossRef] [Green Version]
- Cardenas, C.; Pinton, P.; Bultynck, G. Editorial: Inter-organelle calcium communication in cancer. Front. Oncol. 2018, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Ramirez, F.G.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. P53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. MTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [Green Version]
- Rimessi, A.; Marchi, S.; Patergnani, S.; Pinton, P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene 2013, 33, 2329–2340. [Google Scholar] [CrossRef]
- Raffaello, A.; de Stefani, D.; Rizzuto, R. The mitochondrial Ca2+ uniporter. Cell Calcium 2012, 52, 16–21. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabó, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. Mitochondrial Ca2+ transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Dev. Neurosci. 1993, 15, 165–173. [Google Scholar] [CrossRef]
- Hansford, R.G.; Zorov, D. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol. Cell. Biochem. 1998, 184, 359–369. [Google Scholar] [CrossRef]
- Mammucari, C.; Gherardi, G.; Rizzuto, R. Structure, activity regulation, and role of the mitochondrial calcium uniporter in health and disease. Front. Oncol. 2017, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Das, A.M.; Harris, D.A. Control of mitochondrial ATP synthase in heart cells: Inactive to active transitions caused by beating or positive inotropic agents. Cardiovasc. Res. 1990, 24, 411–417. [Google Scholar] [CrossRef]
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP synthase forms a Ca2+-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Cao, H.; Zhan, L.; Sun, X.; Wang, G.; Li, J.; Guo, X.; Ren, T.; Wang, Z.; Lyu, Y.; et al. Mitochondrial fission forms a positive feedback loop with cytosolic calcium signaling pathway to promote autophagy in hepatocellular carcinoma cells. Cancer Lett. 2017, 403, 108–118. [Google Scholar] [CrossRef]
- Bartok, A.; Weaver, D.; Golenár, T.; Nichtova, Z.; Katona, M.; Bánsághi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Simoes, I.C.M.; Morciano, G.; Lebiedzinska-Arciszewska, M.; Aguiari, G.; Pinton, P.; Potes, Y.; Wieckowski, M.R. The mystery of mitochondria-ER contact sites in physiology and pathology: A cancer perspective. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165834. [Google Scholar] [CrossRef]
- Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Poletti, F.; Giorgi, C.; Pandolfi, P.P.; Pinton, P. Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 2013, 20, 1631–1643. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.T.; Wagner, L.; Yule, D.I.; Bhanumathy, C.; Joseph, S.K. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 2005, 281, 3731–3737. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Marinello, M.; Bononi, A.; Bonora, M.; Giorgi, C.; Rimessi, A.; Pinton, P. Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis. Cell Death Dis. 2012, 3, e304. [Google Scholar] [CrossRef] [Green Version]
- Szado, T.; Vanderheyden, V.; Parys, J.B.; De Smedt, H.; Rietdorf, K.; Kotelevets, L.; Chastre, E.; Khan, F.; Landegren, U.; Söderberg, O.; et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2427–2432. [Google Scholar] [CrossRef] [Green Version]
- Hedgepeth, S.C.; Garcia, M.I.; Wagner, L.E.; Rodriguez, A.M.; Chintapalli, S.V.; Snyder, R.R.; Hankins, G.D.V.; Henderson, B.R.; Brodie, K.M.; Yule, D.I.; et al. The BRCA1 tumor suppressor binds to inositol 1,4,5-trisphosphate receptors to stimulate apoptotic calcium release. J. Biol. Chem. 2015, 290, 7304–7313. [Google Scholar] [CrossRef] [Green Version]
- Monaco, G.; Decrock, E.; Akl, H.; Ponsaerts, R.; Vervliet, T.; Luyten, T.; De Maeyer, M.; Missiaen, L.; Distelhorst, C.W.; De Smedt, H.; et al. Selective regulation of IP 3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2011, 19, 295–309. [Google Scholar] [CrossRef]
- Xu, L.; Xie, Q.; Qi, L.; Wang, C.; Xu, N.; Liu, W.; Yu, Y.; Li, S.; Xu, Y. Bcl-2 overexpression reduces cisplatin cytotoxicity by decreasing ER-mitochondrial Ca2+ signaling in SKOV3 cells. Oncol. Rep. 2018, 39, 985–992. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- D’Eletto, M.; Rossin, F.; Occhigrossi, L.; Farrace, M.G.; Faccenda, D.; Desai, R.; Marchi, S.; Refolo, G.; Falasca, L.; Antonioli, M.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573–3581.e4. [Google Scholar] [CrossRef] [Green Version]
- Mangala, L.S.; Fok, J.Y.; Zorrilla-Calancha, I.R.; Verma, A.; Mehta, K. Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene 2007, 26, 2459–2470. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T.P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Gueguinou, M.; Crottès, D.; Chantôme, A.; Rapetti-Mauss, R.; Potier-Cartereau, M.; Clarysse, L.; Girault, A.; Fourbon, Y.; Jézéquel, P.; Guérin-Charbonnel, C.; et al. The SigmaR1 chaperone drives breast and colorectal cancer cell migration by tuning SK3-dependent Ca2+ homeostasis. Oncogene 2017, 36, 3640–3647. [Google Scholar] [CrossRef]
- Aydar, E.; Onganer, P.; Perrett, R.; Djamgoz, M.B.; Palmer, C.P. The expression and functional characterization of sigma (σ) 1 receptors in breast cancer cell lines. Cancer Lett. 2006, 242, 245–257. [Google Scholar] [CrossRef]
- Krabbendam, I.E.; Honrath, B.; Culmsee, C.; Dolga, A.M. Mitochondrial Ca(2+)-activated K(+) channels and their role in cell life and death pathways. Cell Calcium 2018, 69, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Carreras-Sureda, A.; Jaña, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernández, E.; Sassano, M.L.; et al. Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Bánsághi, S.; Golenár, T.; Madesh, M.; Csordás, G.; RamachandraRao, S.; Sharma, K.; Yule, D.I.; Joseph, S.K.; Hajnóczky, G. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 2014, 289, 8170–8181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Pennanen, C.; Parra, V.; López-Crisosto, C.; Morales, P.E.; del Campo, A.; Gutierrez, T.; Rivera-Mejías, P.; Kuzmicic, J.; Chiong, M.; Zorzano, A.; et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J. Cell Sci. 2014, 127, 2659–2671. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, Y.; Takata, N.; Suzuki-Karasaki, M.; Yoshida, Y.; Tokuhashi, Y.; Suzuki-Karasaki, Y. Disrupting mitochondrial Ca2+ homeostasis causes tumor-selective TRAIL sensitization through mitochondrial network abnormalities. Int. J. Oncol. 2017, 51, 1146–1158. [Google Scholar] [CrossRef]
- Han, X.J.; Lu, Y.F.; Li, S.A.; Kaitsuka, T.; Sato, Y.; Tomizawa, K.; Nairn, A.C.; Takei, K.; Matsui, H.; Matsushita, M. CaM kinase Iα-induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 2008, 182, 573–585. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 595–609. [Google Scholar] [CrossRef]
- Gizak, A.; Pirog, M.; Rakus, D. Muscle FBPase binds to cardiomyocyte mitochondria under glycogen synthase kinase-3 inhibition or elevation of cellular Ca2+ level. FEBS Lett. 2012, 586, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.-A.; Xiong, X.; Scott, T.; Li, A.T.; Wang, C.; Weiss, H.L.; Tan, L.; Bradford, E.; Fan, T.W.M.; Chandel, N.S.; et al. The mitochondrial retrograde signaling regulates Wnt signaling to promote tumorigenesis in colon cancer. Cell Death Differ. 2019, 26, 1955–1969. [Google Scholar] [CrossRef]
- Delgado-Deida, Y.; Alula, K.M.; Theiss, A.L. The influence of mitochondrial-directed regulation of Wnt signaling on tumorigenesis. Gastroenterol. Rep. 2020, 8, 215–223. [Google Scholar] [CrossRef]
- Tsai, F.C.; Seki, A.; Yang, H.W.; Hayer, A.; Carrasco, S.; Malmersjö, S.; Meyer, T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 2014, 16, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Gar, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer. Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [Green Version]
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.S.; Berecz, T.; Duchen, M.R.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Mol. Med. 2016, 8, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Wang, Y.; Peng, J.; Shen, Q.; Chen, M.; Tang, W.; Li, X.; Cai, C.; Wang, B.; Cai, S.; et al. Mitochondrial calcium uniporter as a target of microRNA-340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget 2017, 8, 83831–83844. [Google Scholar] [CrossRef] [Green Version]
- Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. The Warburg effect and mitochondrial stability in cancer cells. Mol. Aspects Med. 2010, 31, 60–74. [Google Scholar] [CrossRef]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of Neoplastic Tissue. IV. A Study of Lipid Synthesis in Neoplastic Tissue Slices in Vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar]
- Rusiñol, A.E.; Cui, Z.; Chen, M.H.; Vance, J.E. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar]
- Kainu, V.; Hermansson, M.; Hänninen, S.; Hokynar, K.; Somerharju, P. Import of phosphatidylserine to and export of phosphatidylethanolamine molecular species from mitochondria. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 429–437. [Google Scholar] [CrossRef]
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernández, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895.e17. [Google Scholar] [CrossRef]
- Guo, D. Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern. Med. Rev. 2017, 3. [Google Scholar] [CrossRef] [Green Version]
- Mulas, M.F.; Abete, C.; Pulisci, D.; Pani, A.; Massidda, B.; Dessì, S.; Mandas, A. Cholesterol esters as growth regulators of lymphocytic leukaemia cells. Cell Prolif. 2011, 44, 360–371. [Google Scholar] [CrossRef]
- Antalis, C.J.; Arnold, T.; Rasool, T.; Lee, B.; Buhman, K.K.; Siddiqui, R.A. High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Res. Treat. 2010, 122, 661–670. [Google Scholar] [CrossRef]
- Gaida, M.M.; Mayer, C.; Dapunt, U.; Stegmaier, S.; Schirmacher, P.; Wabnitz, G.H.; Maria Hänsch, G. Expression of the bitter receptor T2R38 in pancreatic cancer: Localization in lipid droplets and activation by a bacteria-derived quorum-sensing molecule. Oncotarget 2016, 7, 12623–12632. [Google Scholar] [CrossRef] [Green Version]
- Accioly, M.T.; Pacheco, P.; Maya-Monteiro, C.M.; Carrossini, N.; Robbs, B.K.; Oliveira, S.S.; Kaufmann, C.; Morgado-Diaz, J.A.; Bozza, P.T.; Viola, J.P.B. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008, 68, 1732–1740. [Google Scholar] [CrossRef] [Green Version]
- Naon, D.; Scorrano, L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2184–2194. [Google Scholar] [CrossRef]
- Stopsack, K.H.; Gerke, T.A.; Andrén, O.; Andersson, S.O.; Giovannucci, E.L.; Mucci, L.A.; Rider, J.R. Cholesterol uptake and regulation in high-grade and lethal prostate cancers. Carcinogenesis 2017, 38, 806–811. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gu, D.; Lee, S.S.Y.; Song, B.; Bandyopadhyay, S.; Chen, S.; Konieczny, S.F.; Ratliff, T.L.; Liu, X.; Xie, J.; et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 2016, 35, 6378–6388. [Google Scholar] [CrossRef] [Green Version]
- Ohmoto, T.; Nishitsuji, K.; Yoshitani, N.; Mizuguchi, M.; Yanagisawa, Y.; Saito, H.; Sakashita, N. K604, a specific acyl-CoA:cholesterol acyltransferase 1 inhibitor, suppresses proliferation of U251-MG glioblastoma cells. Mol. Med. Rep. 2015, 12, 6037–6042. [Google Scholar] [CrossRef]
- Kim, M.P.; Gallick, G.E. Gemcitabine resistance in pancreatic cancer: Picking the key players. Clin. Cancer Res. 2008, 14, 1284–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liscovitch, M.; Lavie, Y. Multidrug resistance: A role for cholesterol efflux pathways? Trends Biochem. Sci. 2000, 25, 530–534. [Google Scholar] [CrossRef]
- Thoudam, T.; Ha, C.M.; Leem, J.; Chanda, D.; Park, J.S.; Kim, H.J.; Jeon, J.H.; Choi, Y.K.; Liangpunsakul, S.; Huh, Y.H.; et al. PDK4 augments ER–mitochondria contact to dampen skeletal muscle insulin signaling during obesity. Diabetes 2019, 68, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.; Jeyaraju, D.V.; Prudent, J.; Caron, A.; Lemieux, P.; McBride, H.M.; Laplante, M.; Tóth, K.; Pellegrini, L. A Mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc. Natl. Acad. Sci. USA 2014, 111, 16017–16022. [Google Scholar] [CrossRef] [Green Version]
- Theurey, P.; Tubbs, E.; Vial, G.; Jacquemetton, J.; Bendridi, N.; Chauvin, M.A.; Alam, M.R.; Le Romancer, M.; Vidal, H.; Rieusset, J. Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell Biol. 2016, 8, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Carraro, R.S.; Souza, G.F.; Solon, C.; Razolli, D.S.; Chausse, B.; Barbizan, R.; Victorio, S.C.; Velloso, L.A. Hypothalamic mitochondrial abnormalities occur downstream of inflammation in diet-induced obesity. Mol. Cell. Endocrinol. 2018, 460, 238–245. [Google Scholar] [CrossRef]
- Theurey, P.; Rieusset, J. Mitochondria-Associated Membranes Response to Nutrient Availability and Role in Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Horndasch, M.; Lienkamp, S.; Springer, E.; Schmitt, A.; Pavenstädt, H.; Walz, G.; Gloy, J. The C/EBP homologous protein CHOP (GADD153) is an inhibitor of Wnt/TCF signals. Oncogene 2006, 25, 3397–3407. [Google Scholar] [CrossRef]
- Verras, M.; Papandreou, I.; Lim, A.L.; Denko, N.C. Tumor Hypoxia Blocks Wnt Processing and Secretion through the Induction of Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2008, 28, 7212–7224. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.A.L.; Huang, S. Crosstalk of the Wnt/β-catenin pathway with other pathways in cancer cells. Genes Dis. 2016, 3, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.P.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; Van Wichen, D.; De Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC(-/-) colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchartre, Y.; Kim, Y.M.; Kahn, M. The Wnt signaling pathway in cancer. Crit. Rev. Oncol. Hematol. 2016, 99, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Bernkopf, D.B.; Jalal, K.; Brückner, M.; Knaup, K.X.; Gentzel, M.; Schambony, A.; Behrens, J. Pgam5 released from damaged mitochondria induces mitochondrial biogenesis via Wnt signaling. J. Cell Biol. 2018, 217, 1383–1394. [Google Scholar] [CrossRef] [Green Version]
- Bernkopf, D.B.; Behrens, J. Feedback regulation of mitochondrial homeostasis via Wnt/β-catenin signaling. Mol. Cell. Oncol. 2018, 5, e1458015. [Google Scholar] [CrossRef] [Green Version]
- Woo, D.K.; Green, P.D.; Santos, J.H.; D’Souza, A.D.; Walther, Z.; Martin, W.D.; Christian, B.E.; Chandel, N.S.; Shadel, G.S. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC Min/+ mice. Am. J. Pathol. 2012, 180, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Wu, S.; Wei, X.; Liao, Y.; Yi, P.; Liu, Y.; Liu, J.; Liu, J. Hypoxic ER stress suppresses β-catenin expression and promotes cooperation between the transcription factors XBP1 and HIF1α for cell survival. J. Biol. Chem. 2019, 294, 13811–13821. [Google Scholar] [CrossRef]
- Zhang, L.S.; Lum, L. Chemical Modulation of WNT Signaling in Cancer. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Shikata, Y.; Kiga, M.; Futamura, Y.; Aono, H.; Inoue, H.; Kawada, M.; Osada, H.; Imoto, M. Mitochondrial uncoupler exerts a synthetic lethal effect against β-catenin mutant tumor cells. Cancer Sci. 2017, 108, 772–784. [Google Scholar] [CrossRef]
- Ishii, I.; Harada, Y.; Kasahara, T. Reprofiling a classical anthelmintic, pyrvinium pamoate, as an anti-cancer drug targeting mitochondrial respiration. Front. Oncol. 2012, 2, 137. [Google Scholar] [CrossRef] [Green Version]
- Thorne, C.A.; Hanson, A.J.; Schneider, J.; Tahinci, E.; Orton, D.; Cselenyi, C.S.; Jernigan, K.K.; Meyers, K.C.; Hang, B.I.; Waterson, A.G.; et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol. 2010, 6, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; He, X.; Zou, Y.; Chen, D.; Yang, L.; Rao, J.; Chen, H.; Chan, M.C.W.; Li, L.; Guo, Z.; et al. Mitochondria-targeted platinum(II) complexes: Dual inhibitory activities on tumor cell proliferation and migration/invasion: Via intracellular trafficking of β-catenin. Metallomics 2017, 9, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Nangia-Makker, P.; Yu, Y.; Vasudevan, A.; Farhana, L.; Rajendra, S.G.; Levi, E.; Majumdar, A.P.N. Metformin: A potential therapeutic agent for recurrent colon cancer. PLoS ONE 2014, 9, e84369. [Google Scholar] [CrossRef] [PubMed]
- Fennelly, C.; Amaravadi, R.K. Lysosomal Biology in Cancer. Methods Mol. Biol. 2017, 1594, 293–308. [Google Scholar]
- Guicciardi, M.E.; Leist, M.; Gores, G.J. Lysosomes in cell death. Oncogene 2004, 23, 2881–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nylandsted, J.; Gyrd-Hansen, M.; Danielewicz, A.; Fehrenbacher, N.; Lademann, U.; Høyer-Hansen, M.; Weber, E.; Multhoff, G.; Rohde, M.; Jäättelä, M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 2004, 200, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Kirkegaard, T.; Jäättelä, M. Lysosomal involvement in cell death and cancer. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Madge, L.A.; Li, J.H.; Choi, J.; Pober, J.S. Inhibition of phosphatidylinositol 3-kinase sensitizes vascular endothelial cells to cytokine-initiated cathepsin-dependent apoptosis. J. Biol. Chem. 2003, 278, 21295–21306. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Sterea, A.M.; Almasi, S.; El Hiani, Y. The hidden potential of lysosomal ion channels: A new era of oncogenes. Cell Calcium 2018, 72, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ren, D. Lysosomal Physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Y.; Song, F. Intracellular calcium signaling regulates autophagy via calcineurinmediated TFEB dephosphorylation. Autophagy 2015, 11, 1192–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.J.; Xu, J.; Fu, C.; Zhang, J.; Zheng, Y.G.; Jia, H.; Liu, J.O. Regulation of mTORC1 by lysosomal calcium and calmodulin. Elife 2016, 5, e19360. [Google Scholar] [CrossRef]
- Zhu, M.X. A well-known potassium channel plays a critical role in lysosomes. J. Cell Biol. 2017, 216, 1513–1515. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Wong, Y.C.; Kim, S.; Peng, W.; Krainc, D. Regulation and Function of Mitochondria-Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 2019, 29, 500–513. [Google Scholar] [CrossRef]
- Eisenberg-Bord, M.; Shai, N.; Schuldiner, M.; Bohnert, M. A Tether Is a Tether Is a Tether: Tethering at Membrane Contact Sites. Dev. Cell 2016, 39, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Onoue, K.; Jofuku, A.; Ban-Ishihara, R.; Ishihara, T.; Maeda, M.; Koshiba, T.; Itoh, T.; Fukuda, M.; Otera, H.; Oka, T.; et al. Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. J. Cell Sci. 2013, 126, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end—Directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [Green Version]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Scorrano, L. Keeping mitochondria in shape: A matter of life and death. Eur. J. Clin. Investig. 2013, 43, 886–893. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Lacas-Gervais, S.; Adaixo, R.; Ilc, K.; Rouleau, M.; Notte, A.; Dieu, M.; Michiels, C.; Voeltzel, T.; Maguer-Satta, V.; et al. Local Mitochondrial-Endolysosomal Microfusion Cleaves Voltage-Dependent Anion Channel 1 To Promote Survival in Hypoxia. Mol. Cell. Biol. 2015, 35, 1491–1505. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Wong, Y.C.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial Ca(2+) dynamics via lysosomal TRPML1. Proc. Natl. Acad. Sci. USA 2020, 117, 19266–19275. [Google Scholar] [CrossRef]
- Schrader, M.; Costello, J.; Godinho, L.F.; Islinger, M. Peroxisome-mitochondria interplay and disease. J. Inherit. Metab. Dis. 2015, 38, 681–702. [Google Scholar] [CrossRef]
- Cohen, Y.; Klug, Y.A.; Dimitrov, L.; Erez, Z.; Chuartzman, S.G.; Elinger, D.; Yofe, I.; Soliman, K.; Gärtner, J.; Thoms, S.; et al. Peroxisomes are juxtaposed to strategic sites on mitochondria. Mol. Biosyst. 2014, 10, 1742–1748. [Google Scholar] [CrossRef]
- Hosoi, K.I.; Miyata, N.; Mukai, S.; Furuki, S.; Okumoto, K.; Cheng, E.H.; Fujiki, Y. The VDAC2-BAK axis regulates peroxisomal membrane permeability. J. Cell Biol. 2017, 216, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoms, S.; Grønborg, S.; Gärtner, J. Organelle interplay in peroxisomal disorders. Trends Mol. Med. 2009, 15, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Lismont, C.; Walton, P. The peroxisome-mitochondria connection: How and why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; He, Y.; Zhang, P.; Wang, J.; Fan, C.; Yang, L.; Xiong, F.; Zhang, S.; Gong, Z.; Nie, S.; et al. LncRNAs regulate the cytoskeleton and related Rho/ROCK signaling in cancer metastasis. Mol. Cancer 2018, 17, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, C.L.; Pomatto, L.C.D.; Tripathi, D.N.; Davies, K.J.A. Redox regulation of homeostasis and proteostasis in peroxisomes. Physiol. Rev. 2018, 98, 89–115. [Google Scholar] [CrossRef]
- Wei, F.; Tang, L.; He, Y.; Wu, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Liao, Q.; et al. BPIFB1 (LPLUNC1) inhibits radioresistance in nasopharyngeal carcinoma by inhibiting VTN expression. Cell Death Dis. 2018, 9, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef]
- He, Y.; Jing, Y.; Wei, F.; Tang, Y.; Yang, L.; Luo, J.; Yang, P.; Ni, Q.; Pang, J.; Liao, Q.; et al. Long non-coding RNA PVT1 predicts poor prognosis and induces radioresistance by regulating DNA repair and cell apoptosis in nasopharyngeal carcinoma. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Raza, M.H.; Siraj, S.; Arshad, A.; Waheed, U.; Aldakheel, F.; Alduraywish, S.; Arshad, M. ROS-modulated therapeutic approaches in cancer treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 1789–1809. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Managò, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct Pharmacological Targeting of a Mitochondrial Ion Channel Selectively Kills Tumor Cells In Vivo. Cancer Cell 2017, 31, 516–531.e10. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peruzzo, R.; Costa, R.; Bachmann, M.; Leanza, L.; Szabò, I. Mitochondrial Metabolism, Contact Sites and Cellular Calcium Signaling: Implications for Tumorigenesis. Cancers 2020, 12, 2574. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092574
Peruzzo R, Costa R, Bachmann M, Leanza L, Szabò I. Mitochondrial Metabolism, Contact Sites and Cellular Calcium Signaling: Implications for Tumorigenesis. Cancers. 2020; 12(9):2574. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092574
Chicago/Turabian StylePeruzzo, Roberta, Roberto Costa, Magdalena Bachmann, Luigi Leanza, and Ildikò Szabò. 2020. "Mitochondrial Metabolism, Contact Sites and Cellular Calcium Signaling: Implications for Tumorigenesis" Cancers 12, no. 9: 2574. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092574