Investigating Radiotherapy Response in a Novel Syngeneic Model of Prostate Cancer

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
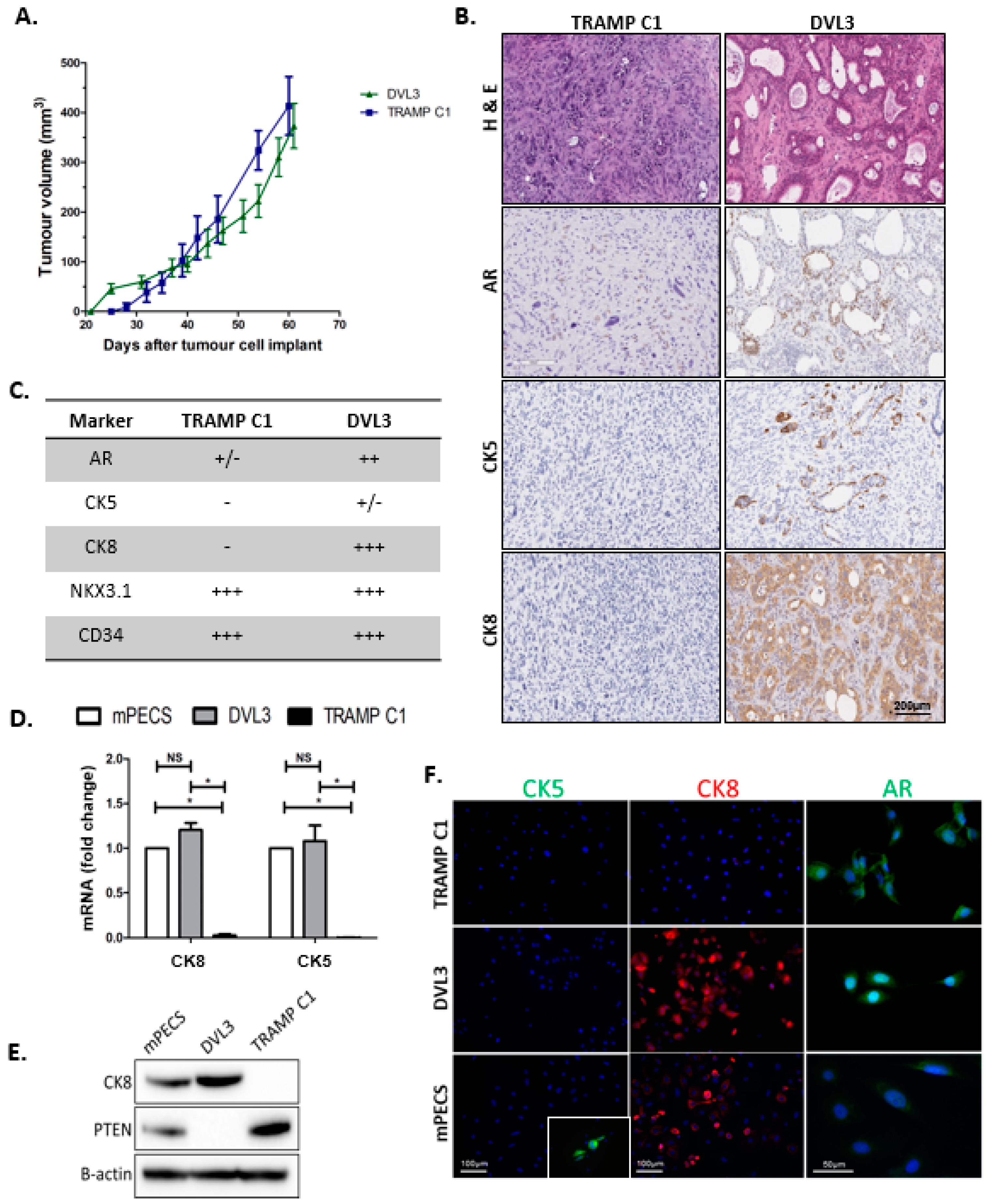
2.1. DVL3 Cell Engraftment in Immunocompetent Mice Results in Tumour Formation, which Accurately Models Human Prostate Adenocarcinoma
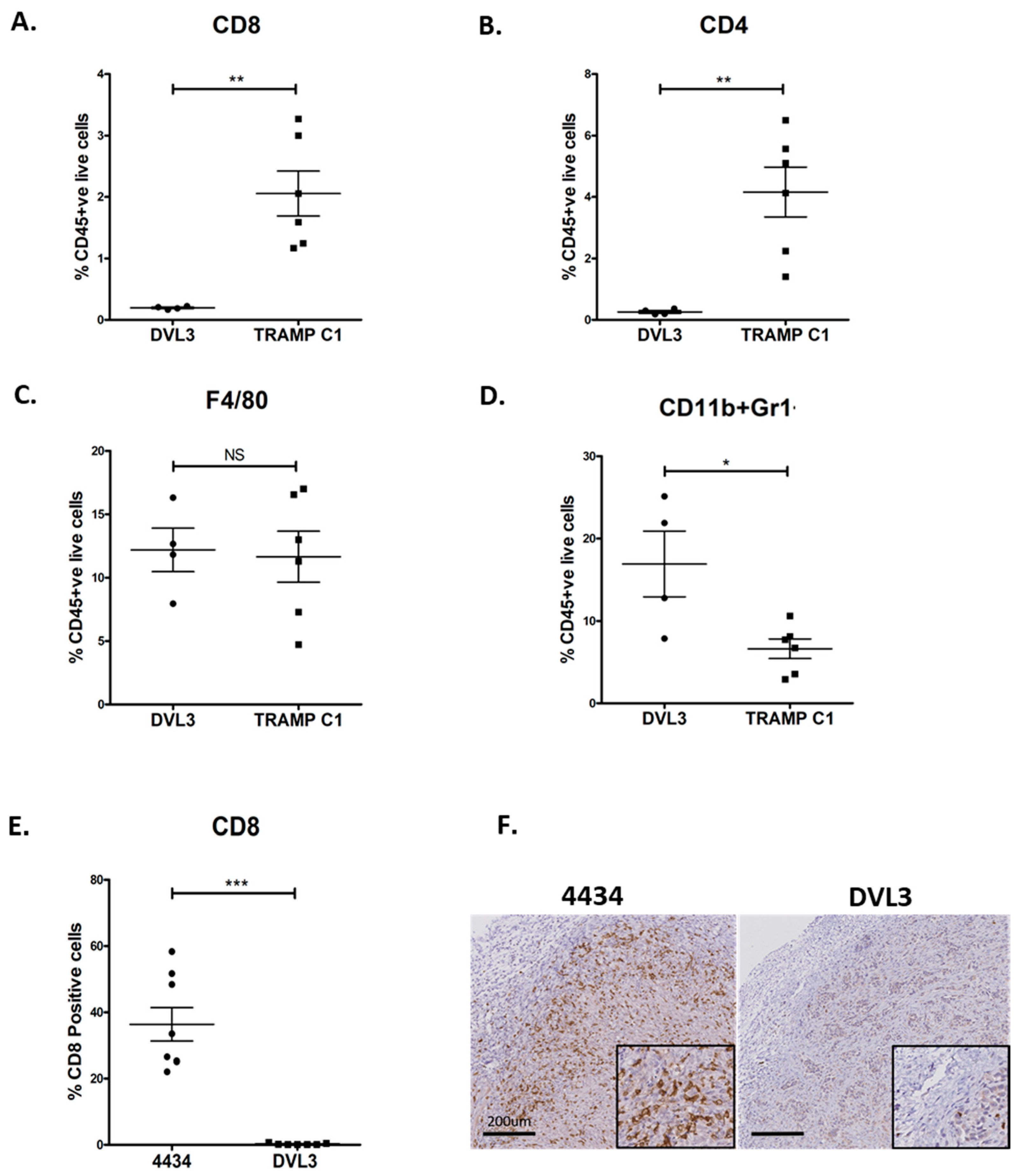
2.2. DVL3 Tumours Have Immunosuppressive Microenvironment Similar to Human Prostate Adenocarcinomas
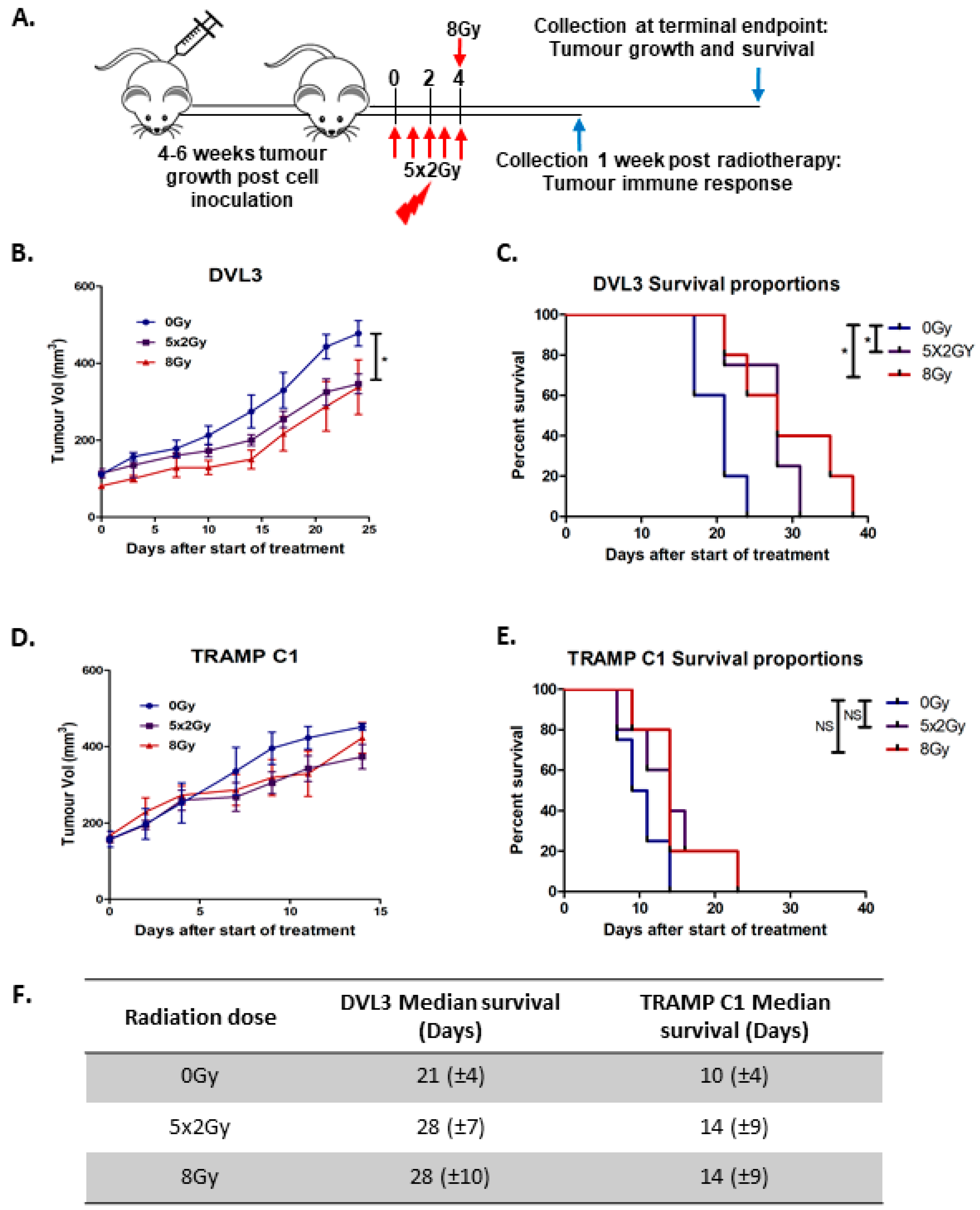
2.3. Fractionated Radiotherapy Leads to Marginal Growth Delay and Alters the Local Tumour Immune Microenvironment
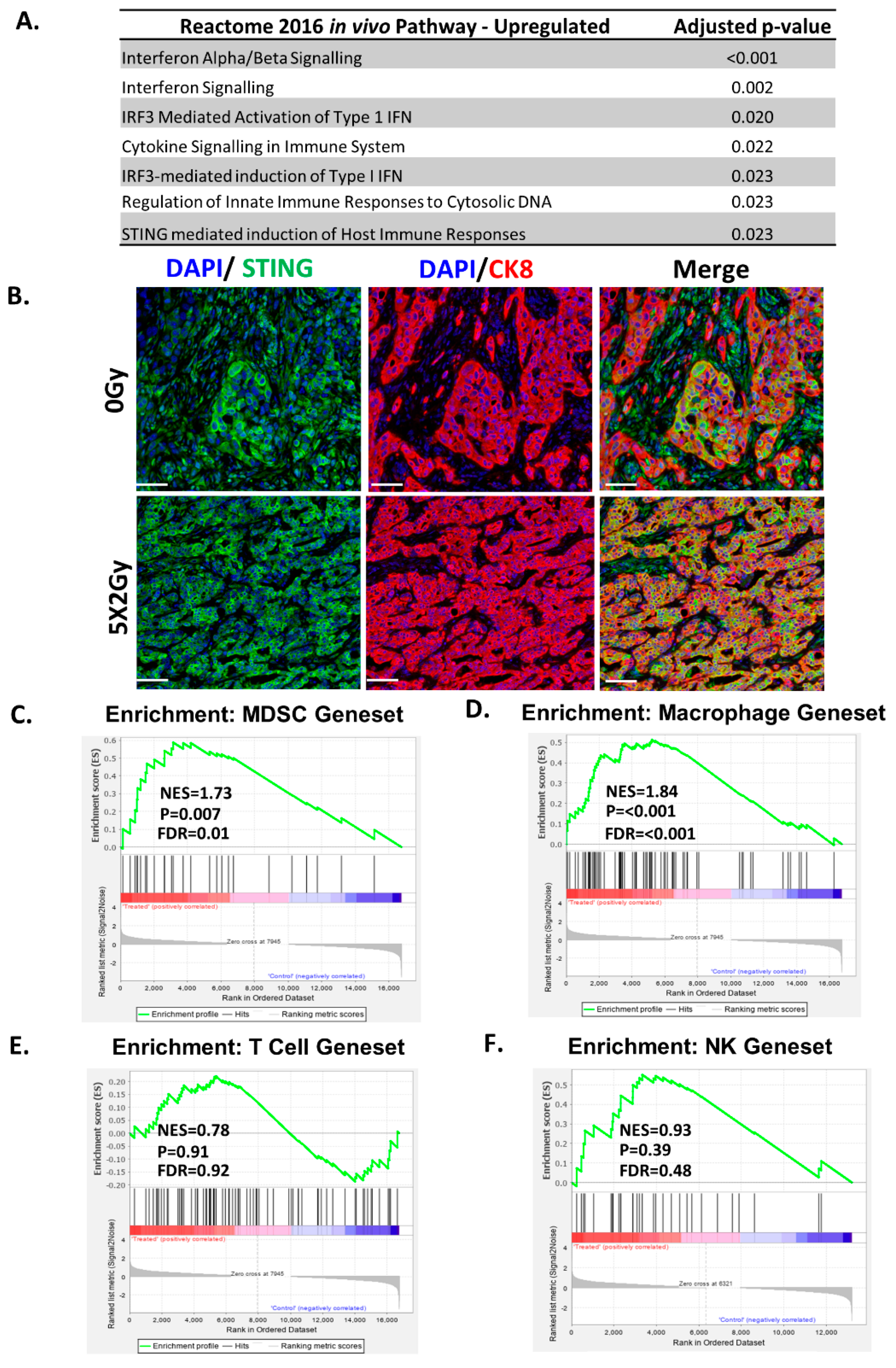
2.4. Fractionated Radiotherapy Differentially Upregulated Genes Associated with STING/ Type-1 Interferon Signalling and Myeloid Signatures
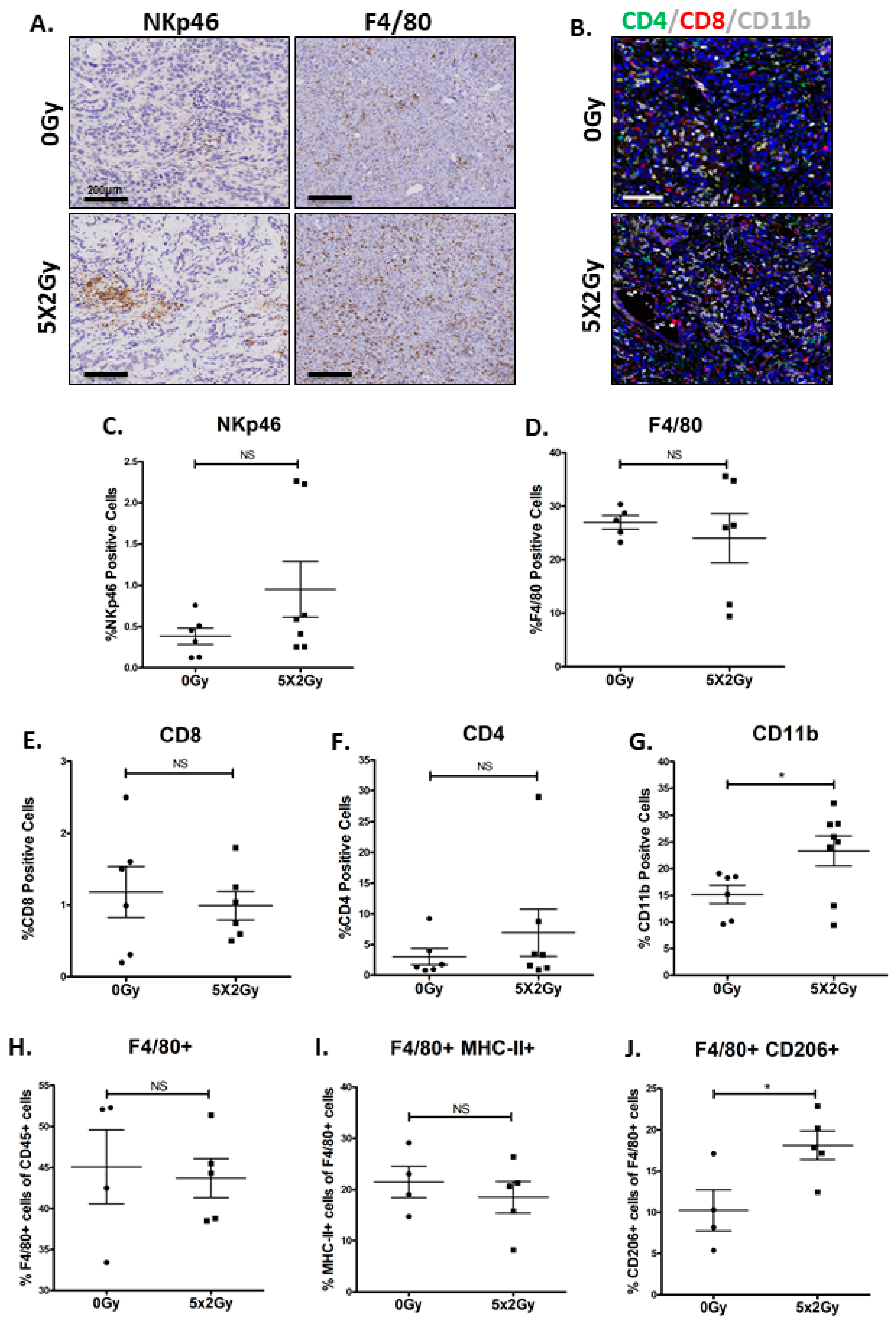
2.5. Fractionated Radiotherapy Resulted in an Increase in Infiltration of Myeloid Cells, but no Changes Were Observed in T-Cell Infiltrates
3. Discussion
4. Materials and Methods
4.1. Cell Line Derivation and Maintenance
4.2. Syngeneic Modelling
4.3. Sample Preparation
4.4. Immunohistochemistry
4.5. Immunocytochemistry
4.6. RNA Isolation and cDNA Synthesis
4.7. qRT-PCR
4.8. Western Blotting
4.9. Flow Cytometry on Cell Lines
4.10. Radiotherapy
4.11. RNA Extraction for RNA Seq Analysis
4.12. RNA Library Preparation, Sequencing and Analysis
4.13. Flow Cytometry on Tumour Tissue
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethical Approval
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef]
- James, N.D.; Spears, M.R.; Clarke, N.W.; Dearnaley, D.P.; De Bono, J.S.; Gale, J.; Hetherington, J.; Hoskin, P.J.; Jones, R.; Laing, R.; et al. Survival with Newly Diagnosed Metastatic Prostate Cancer in the “Docetaxel Era”: Data from 917 Patients in the Control Arm of the STAMPEDE Trial (MRC PR08, CRUK/06/019). Eur. Urol. 2015, 67, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Donovan, J.L.; Hamdy, F.C.; Lane, J.A.; Mason, M.; Metcalfe, C.; Walsh, E.; Blazeby, J.; Peters, T.J.; Holding, P.; Bonnington, S.; et al. Patient-Reported Outcomes after Monitoring, Surgery, or Radiotherapy for Prostate Cancer. N. Engl. J. Med. 2016, 375, 1425–1437. [Google Scholar] [CrossRef] [Green Version]
- Foster, B.A.; Gingrich, J.R.; Kwon, E.D.; Madias, C.; Greenberg, N.M. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997, 57, 3325–3330. [Google Scholar] [PubMed]
- Berman-Booty, L.D.; Knudsen, K.E. Models of neuroendocrine prostate cancer. Endocr.-Relat. Cancer 2014, 22, R33–R49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafarana, G.; Ishkanian, A.S.; Malloff, C.A.; Locke, J.A.; Sykes, J.; Thoms, J.; Lam, W.L.; Squire, J.A.; Yoshimoto, M.; Ramnarine, V.R.; et al. Copy number alterations of c-MYC and PTEN are prognostic factors for relapse after prostate cancer radiotherapy. Cancer 2012, 118, 4053–4062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geybels, M.S.; Fang, M.; Wright, J.L.; Qu, X.; Bibikova, M.; Klotzle, B.; Fan, J.-B.; Feng, Z.; Ostrander, E.A.; Nelson, P.S.; et al. PTEN loss is associated with prostate cancer recurrence and alterations in tumor DNA methylation profiles. Oncotarget 2017, 8, 84338–84348. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.; Liu, Y.-N.; Pierce, R.; Abou-Kheir, W.; Casey, O.; Seng, V.; Camacho, D.; Simpson, R.M.; Kelly, K. Prostate Epithelial Pten/TP53 Loss Leads to Transformation of Multipotential Progenitors and Epithelial to Mesenchymal Transition. Am. J. Pathol. 2011, 179, 422–435. [Google Scholar] [CrossRef]
- Oliveira, D.S.M.; Dzinic, S.; Bonfil, A.I.; Saliganan, A.D.; Sheng, S.; Bonfil, R.D. The mouse prostate: A basic anatomical and histological guideline. Bosn. J. Basic Med. Sci. 2016, 16, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy-Burman, P.; Wu, H.; Powell, W.C.; Hagenkord, J.; Cohen, M.B. Genetically defined mouse models that mimic natural aspects of human prostate cancer development. Endocr.-Relat. Cancer 2004, 11, 225–254. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.J.; Ruscetti, M.; Arenzana, T.L.; Tran, L.M.; Bianci-Frias, D.; Sybert, E.; Priceman, S.J.; Wu, L.; Nelson, P.S.; Smale, S.T.; et al. Pten Null Prostate Epithelium Promotes Localized Myeloid-Derived Suppressor Cell Expansion and Immune Suppression during Tumor Initiation and Progression. Mol. Cell. Biol. 2014, 34, 2017–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidotto, T.; Saggioro, F.P.; Jamaspishvili, T.; Chesca, D.L.; De Albuquerque, C.G.P.; Reis, R.B.; Graham, C.H.; Berman, D.M.; Siemens, D.R.; Squire, J.A.; et al. PTEN-deficient prostate cancer is associated with an immunosuppressive tumor microenvironment mediated by increased expression of IDO1 and infiltrating FoxP3+ T regulatory cells. Prostate 2019, 79, 969–979. [Google Scholar] [CrossRef]
- Wang, Y.; Hayward, S.W.; Cao, M.; Thayer, K.A.; Cunha, G.R. Cell differentiation lineage in the prostate. Differentiation 2001, 68, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Bhatia-Gaur, R.; Donjacour, A.A.; Sciavolino, P.J.; Kim, M.; Desai, N.; Young, P.; Norton, C.R.; Gridley, T.; Cardiff, R.D.; Cunha, G.R.; et al. Roles for Nkx3.1 in prostate development and cancer. Genes Dev. 1999, 13, 966–977. [Google Scholar] [CrossRef] [Green Version]
- Gurel, B.; Ali, T.Z.; Montgomery, E.A.; Begum, S.; Hicks, J.; Goggins, M.; Eberhart, C.G.; Clark, D.P.; Bieberich, C.J.; Epstein, J.I.; et al. NKX3.1 as a Marker of Prostatic Origin in Metastatic Tumors. Am. J. Surg. Pathol. 2010, 34, 1097–1105. [Google Scholar] [CrossRef] [Green Version]
- Young, H.L.; Rowling, E.J.; Bugatti, M.; Giurisato, E.; Luheshi, N.; Arozarena, I.; Acosta, J.C.; Kamarashev, J.; Frederick, D.T.; Cooper, Z.; et al. An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition. J. Exp. Med. 2017, 214, 1691–1710. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.-H.; Chiang, C.-S.; Wang, C.-C.; Tsai, C.-S.; Jung, S.-M.; Lee, C.-C.; McBride, W.H.; Hong, J.-H. Radiotherapy decreases vascular density and causes hypoxia with macrophage aggregation in TRAMP-C1 prostate tumors. Clin. Cancer Res. 2009, 15, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Walshaw, R.C.; Honeychurch, J.; Illidge, T.; Choudhury, A. The anti-PD-1 era—An opportunity to enhance radiotherapy for patients with bladder cancer. Nat. Rev. Urol. 2017, 15, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Deng, L.; Hou, Y.; Meng, X.; Huang, X.; Rao, E.; Zheng, W.; Mauceri, H.; Mack, M.; Xu, M.; et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 2017, 8, 1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.-X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Lü, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2015, 6, 80–95. [Google Scholar] [CrossRef] [Green Version]
- Ko, E.C.; Formenti, S.C. Radiotherapy and checkpoint inhibitors: A winning new combination? Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef]
- Dovedi, S.J.; Adlard, A.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired Resistance to Fractionated Radiotherapy Can Be Overcome by Concurrent PD-L1 Blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef] [Green Version]
- Brand, D.H.; Tree, A.C.; Ostler, P.; Van Der Voet, H.; Loblaw, A.; Chu, W.; Ford, D.; Tolan, S.; Jain, S.; Martin, A.; et al. Intensity-modulated fractionated radiotherapy versus stereotactic body radiotherapy for prostate cancer (PACE-B): Acute toxicity findings from an international, randomised, open-label, phase 3, non-inferiority trial. Lancet Oncol. 2019, 20, 1531–1543. [Google Scholar] [CrossRef]
- Dearnaley, D.P.; Syndikus, I.; Mossop, H.; Khoo, V.; Birtle, A.; Bloomfield, D.; Graham, J.D.; Kirkbride, P.; Logue, J.; Malik, Z.; et al. Conventional versus hypofractionated high-dose intensity-modulated radiotherapy for prostate cancer: 5-year outcomes of the randomised, non-inferiority, phase 3 CHHiP trial. Lancet Oncol. 2016, 17, 1047–1060. [Google Scholar] [CrossRef] [Green Version]
- Payne, H.; Mason, M. Androgen deprivation therapy as adjuvant/neoadjuvant to radiotherapy for high-risk localised and locally advanced prostate cancer: Recent developments. Br. J. Cancer 2011, 105, 1628–1634. [Google Scholar] [CrossRef] [Green Version]
- Parker, C.; James, N.D.; Brawley, C.D.; Clarke, N.W.; Hoyle, A.P.; Ali, A.; Ritchie, A.W.S.; Attard, G.; Chowdhury, S.; Cross, W.; et al. Radiotherapy to the primary tumour for newly diagnosed, metastatic prostate cancer (STAMPEDE): A randomised controlled phase 3 trial. Lancet 2018, 392, 2353–2366. [Google Scholar] [CrossRef] [Green Version]
- Weichselbaum, R.R. The 46th David A. Karnofsky Memorial Award Lecture: Oligometastasis—From Conception to Treatment. J. Clin. Oncol. 2018, 36, 3240–3250. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Ellwood-Yen, K.; King, J.C.; Wongvipat, J.; Lebeau, M.M.; Sawyers, C.L. Context-Dependent Hormone-Refractory Progression Revealed through Characterization of a Novel Murine Prostate Cancer Cell Line. Cancer Res. 2005, 65, 11565–11571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, J.; Wang, S.; Qiao, R.; Vivanco, I.; Watson, P.A.; Sawyers, C.L.; Wu, H. Murine Cell Lines Derived from Pten Null Prostate Cancer Show the Critical Role of PTEN in Hormone Refractory Prostate Cancer Development. Cancer Res. 2007, 67, 6083–6091. [Google Scholar] [CrossRef] [Green Version]
- Carm, K.T.; Hoff, A.M.; Bakken, A.C.; Axcrona, U.; Axcrona, K.; Lothe, R.A.; Skotheim, R.I.; Løvf, M. Interfocal heterogeneity challenges the clinical usefulness of molecular classification of primary prostate cancer. Sci. Rep. 2019, 9, 13579–13586. [Google Scholar] [CrossRef] [Green Version]
- Bilusic, M.; Madan, R.A.; Gulley, J.L. Immunotherapy of Prostate Cancer: Facts and Hopes. Clin. Cancer Res. 2017, 23, 6764–6770. [Google Scholar] [CrossRef] [Green Version]
- Vitkin, N.; Nersesian, S.; Siemens, D.R.; Koti, M. The Tumor Immune Contexture of Prostate Cancer. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Di Mitri, D.; Toso, A.; Chen, J.; Sarti, M.; Pinton, S.; Jost, T.R.; D’Antuono, R.; Montani, E.; García-Escudero, R.; Guccini, I.; et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 2014, 515, 134–137. [Google Scholar] [CrossRef]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Horn, L.A.; Ciavattone, N.G. Radiotherapy Both Promotes and Inhibits Myeloid-Derived Suppressor Cell Function: Novel Strategies for Preventing the Tumor-Protective Effects of Radiotherapy. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Vatner, R.E.; Formenti, S.C. Myeloid-Derived Cells in Tumors: Effects of Radiation. Semin. Radiat. Oncol. 2015, 25, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasero, C.; Gravis, G.; Guérin, M.; Granjeaud, S.; Piana, J.T.; Rocchi, P.; Paciencia-Gros, M.; Poizat, F.; Bentobji, M.; Azario-Cheillan, F.; et al. Inherent and Tumor-Driven Immune Tolerance in the Prostate Microenvironment Impairs Natural Killer Cell Antitumor Activity. Cancer Res. 2016, 76, 2153–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanpouille-Box, C.; Formenti, S.C.; Demaria, S. Toward precision radiotherapy for use with immune checkpoint blockers. Clin. Cancer Res. 2018, 24, 259–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, W.; Klement, J.D.; Lu, C.; Ibrahim, M.L.; Liu, K. IFNAR1 Controls Autocrine Type I IFN Regulation of PD-L1 Expression in Myeloid-Derived Suppressor Cells. J. Immunol. 2018, 201, 264–277. [Google Scholar] [CrossRef] [Green Version]
- Grabowska, M.M.; DeGraff, D.J.; Yu, X.; Jin, R.J.; Chen, Z.; Borowsky, A.D.; Matusik, R.J. Mouse models of prostate cancer: Picking the best model for the question. Cancer Metastasis Rev. 2014, 33, 377–397. [Google Scholar] [CrossRef] [PubMed]
- Ittmann, M.M.; Huang, J.; Radaelli, E.; Martin, P.; Signoretti, S.; Sullivan, R.; Simons, B.W.; Ward, J.M.; Robinson, B.D.; Chu, G.C.; et al. Animal Models of Human Prostate Cancer: The Consensus Report of the New York Meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013, 73, 2718–2736. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Laurila, E.; Tamayo, P.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirmal, A.J.; Regan, T.; Shih, B.B.; Hume, D.A.; Sims, A.H.; Freeman, T. Immune Cell Gene Signatures for Profiling the Microenvironment of Solid Tumors. Cancer Immunol. Res. 2018, 6, 1388–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cursons, J.; Souza-Fonseca-Guimaraes, F.; Foroutan, M.; Anderson, A.; Hollande, F.; Hediya-Zadeh, S.; Behren, A.; Huntington, N.; Davis, M.J. A gene signature predicting natural killer infiltration an dimproved survival in melanoma patients. Cancer Immunol. Res 2019. [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haughey, C.M.; Mukherjee, D.; Steele, R.E.; Popple, A.; Dura-Perez, L.; Pickard, A.; Patel, M.; Jain, S.; Mullan, P.B.; Williams, R.; et al. Investigating Radiotherapy Response in a Novel Syngeneic Model of Prostate Cancer. Cancers 2020, 12, 2804. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12102804
Haughey CM, Mukherjee D, Steele RE, Popple A, Dura-Perez L, Pickard A, Patel M, Jain S, Mullan PB, Williams R, et al. Investigating Radiotherapy Response in a Novel Syngeneic Model of Prostate Cancer. Cancers. 2020; 12(10):2804. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12102804
Chicago/Turabian StyleHaughey, Charles M., Debayan Mukherjee, Rebecca E. Steele, Amy Popple, Lara Dura-Perez, Adam Pickard, Mehjabin Patel, Suneil Jain, Paul B. Mullan, Rich Williams, and et al. 2020. "Investigating Radiotherapy Response in a Novel Syngeneic Model of Prostate Cancer" Cancers 12, no. 10: 2804. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12102804