LncRNAs in Cancer: From garbage to Junk





1
Institute of Genetics and Biophysics “Adriano Buzzati-Traverso”, CNR, 80131 Naples, Italy
2
Laboratory for RNA Cancer Biology, Department of Oncology, KULeuven, LKI, Herestraat 49, 3000 Leuven, Belgium
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this paper.
‡
These authors contributed equally to this paper.
Cancers 2020, 12(11), 3220; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113220
Submission received: 10 October 2020
/
Revised: 28 October 2020
/
Accepted: 29 October 2020
/
Published: 31 October 2020
(This article belongs to the Collection Regulatory and Non-Coding RNAs in Cancer Epigenetic Mechanisms)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Long non-coding RNAs (lncRNAs) are generally defined as transcripts longer than 200 nucleotides and not coding for proteins. This review provides an overview of the main functions of lncRNAs in the cells and their role in tumorigenesis. Additionally, key examples of oncogenic and/or tumor suppressive lncRNAs with a well-established role in tumor development and progression are discussed. Finally, the currently available technological approaches to target lncRNAs (e.g., by modifying their expression, stability, processing and/or binding capacity), with a specific focus on the pros and cons of their use as therapeutic targets are considered.
Abstract
Sequencing-based transcriptomics has significantly redefined the concept of genome complexity, leading to the identification of thousands of lncRNA genes identification of thousands of lncRNA genes whose products possess transcriptional and/or post-transcriptional regulatory functions that help to shape cell functionality and fate. Indeed, it is well-established now that lncRNAs play a key role in the regulation of gene expression through epigenetic and posttranscriptional mechanims. The rapid increase of studies reporting lncRNAs alteration in cancers has also highlighted their relevance for tumorigenesis. Herein we describe the most prominent examples of well-established lncRNAs having oncogenic and/or tumor suppressive activity. We also discuss how technical advances have provided new therapeutic strategies based on their targeting, and also report the challenges towards their use in the clinical settings.
1. Introduction
In the human genome, the protein-coding genes represent less than 2%, whereas a large fraction is constituted by regions—often transcribed on both DNA strands—whose products lack any significant translational potential [1]. In the last two decades, some gaps in the knowledge of the genomic complexity has been filled in by the identification of several families of long RNAs. In particular, long non-coding RNAs (lncRNAs)—some of which already had attributed gene-specific regulatory functions as early as the 1990s (e.g., H19 and XIST)—are arbitrarily defined as RNAs longer than 200 nucleotides lacking coding potential. They constitute a highly heterogeneous family of ncRNAs transcribed by RNA polymerase II and arise from intergenic (gene deserts), as well as intronic or gene-dense regions [2]. Generally, lncRNAs are 5’ capped and 3’ polyadenylated and often undergo splicing similarly to mRNAs [3,4]. Sequencing-based transcriptomics approaches—and especially RNA sequencing—have completely redefined the picture of human genome transcription, enabling the identification of a very large number of lncRNA genes [5,6,7]. Indeed, more than 48,600 transcripts from about 18,000 genes in the human transcriptome are annotated as bona fide lncRNAs (evidence-based annotation of the human genome release GRCh38, GENCODE version 35).
Despite the rapid advancement in their identification and expression analysis, the functional characterization of lncRNA is still lagging behind, possibly because, unlike other ncRNAs, they can employ a wide array of functions. They can act as decoys, guides, and scaffolds, exerting transcriptional and/or post-transcriptional regulatory activities both in the nucleus and in the cytoplasm by direct or indirect interactions with chromatin, proteins, and other RNAs [8]. Therefore, lncRNAs participate virtually in all relevant cellular processes (e.g., maintenance of stemness, proliferation, angiogenesis, apoptosis etc.), both in physiological and pathological conditions and, not surprisingly, alterations in lncRNA expression have been frequently associated with cancer onset and progression [9].
Among the large-scale sequencing projects aiming to characterize cancer genomes, the Cancer Genome Atlas (TCGA) Consortium has provided a wide molecular characterization of about 11,000 primary cancers, uncovering a substantial fraction of new somatic alterations (i.e., point mutations and/or small indels, gene rearrangements, and copy-number alterations). Khurana and colleagues reported that ~99% of somatic SNVs in tumors occur in noncoding regions, which include transcription factor binding sites (TFBS), ncRNAs, and pseudogenes [10]. A recent study based on TCGA and lncRNA expression data from TANRIC [11] reveals that mutational frequencies in lncRNAs whose expression is affected by somatic alterations (MutLncs) are low and that, to a certain extent, their alteration tends to be cancer type-specific [12]. Moreover, a large proportion of mutations are located within lncRNA TFBS, especially of ESR1, TRPS1, ERG, and RUNX1.
A growing number of studies reveals a previously unrecognized role for lncRNAs as conditional and constitutive oncogenes and tumor suppressors by means of their ability to regulate each and every cancer hallmark (e.g., uncontrolled proliferation, metastasis, immune escape, etc.) [13,14,15,16,17]. These findings, and especially the cancer-specific expression of most of them, established the rationale for assessing lncRNAs as possible biomarkers and/or therapeutic targets, as their silencing would not cause side-effects on other tissues.
This review provides an overview of the main regulatory functions of lncRNAs in tumorigenesis, their alteration in different cancer types and the main strategies to target them. Furthermore, we discuss future directions in cancer research based on the emerging lncRNA properties.
2. Epigenetic Regulation and Translational Reprogramming
Compartmentalization in time and space [18,19,20] offered the first indication that lncRNAs may not be mere transcriptional noise, but they could be implicated in virtually all fundamental cellular processes. Indeed, disproving the notion that sequence conservation is essential to postulate a function, the number of lncRNAs with attributed regulatory roles is exponentially increasing [21,22]
With a few exceptions, lncRNAs are generally poorly expressed [18,20] and dispensable for organismal viability in physiological conditions [23,24,25]; however, they have been implicated in all known stress response pathways [26,27]. As such, they play crucial roles in the epigenetic and post-transcriptional regulation of gene expression.
Nuclear lncRNAs have broadened our knowledge of transcriptional regulation from a transcription factor-centric to a more complex view that integrates protein- and RNA-based regulation. The latter controls histone and DNA modifications and the transcriptional machinery by recruiting specific factors to the DNA or sponging them away from it. In this way they contribute both to the regulation of a large number of genes or of specific loci [28].
Cytoplasmic lncRNAs, which are proportionally more abundant than nuclear ones [29], have emerged in the past decade as modulators of RNA stability, localization, and translation. Apart from interaction with the translational machinery, which was revealed by ribosome profiling and translating ribosome affinity purification (TRAP) approaches [30,31], cytoplasmic lncRNAs can also sponge proteins or other RNA molecules, serving as competing-endogenous RNAs (ceRNAs). The discovery of lncRNAs has therefore added a layer of complexity to cytoplasmic post-transcriptional and translational control, dominated before by proteins and miRNAs [32].
2.1. LncRNAs in Epigenetic and Transcriptional Regulation (DNA Methylation, Nucleosome Positioning, and Histone Modifications)
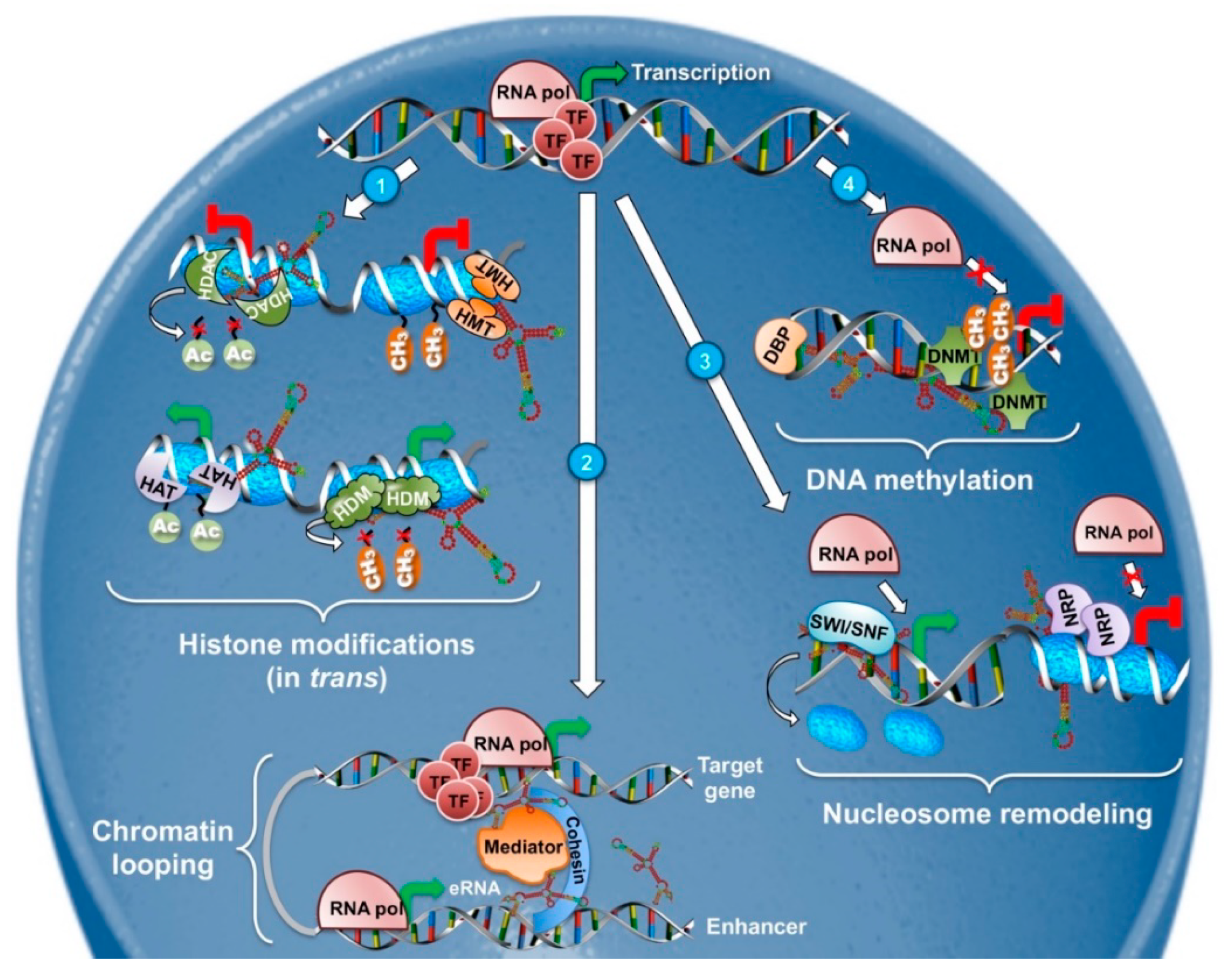
Historically, lncRNA implication in nuclear activities such as epigenetics and transcriptional regulation was the first to be recognized and it is probably the best characterized. LncRNAs can regulate the expression of protein-coding genes in close proximity (cis-acting) or even influence distant transcriptional activators and repressors (trans-acting) (Figure 1) [33]. Their mechanisms of action extend from chromatin remodeling, to binding and regulating transcription factors and epigenetic regulators and in some instances, the sole act of their transcription is sufficient to exert a regulatory function [34,35].
2.1.1. DNA Methylation
DNA methylation is a fundamental epigenetic process regulating gene expression and is orchestrated by several DNA methyltransferases. Changes in DNA methylation patterns are crucial for normal development [36,37] but also for cancer. Aberrant DNA hyper- or hypomethylation regulates the expression of key oncogenes and tumor suppressors and affects genome stability, thus directly participating in cancer development and progression [38]. Since the discovery of lncRNAs, increasing evidence has been linking them to the regulation of DNA methylation. For instance, ecCEBP lncRNA was shown to directly interact with DNA-methyltransferases (DNMTs) preventing DNA methylation at the CENPA locus [39].
Later on, 148 other lncRNAs were identified as interactors of DNMT1 in colon cancer cells by RIP-seq [40]. One of them, DACOR1 (DNMT1-associated colon cancer repressed lncRNA 1) is highly expressed in normal colon but is suppressed in various colon tumors and patient-derived cancer cell lines. Expression of DACOR1 is sufficient to restore the DNA methylation status at thousands of CpG sites hypomethylated in colorectal cancer [41]. Furthermore, DACOR1 induction reduces colon cancer clonogenic ability, thus supporting a tumor-suppressor function for this lncRNA [40].
The emerging widespread indications that lncRNAs can influence DNA-methylation have recently been gathered in the Lnc2Meth database. This web tool aims to close the gap on the regulatory relationships between DNA-methylation and lncRNAs with experimentally verified information [42].
2.1.2. Histone Modifications
Apart from DNA methylation, chromatin structure -and thus transcription- can be influenced by post-translational modifications of histone proteins. Histones are subject to numerous modifications catalyzed by specific histone-modifying enzymes, including acetylation and methylation of lysine, methylation of arginine, phosphorylation of serine and threonine, ubiquitylation of lysine residues, glycosylation, sumoylation, adenosine diphosphate ribosylation, and carbonylation [43]. Demethylation and histone acetylation, for example, mediated by histone demethylases (HDMs) and histone acetyltransferases (HATs), respectively, favors chromatin decondensation, whereas trimethylation of lysine 27 on histone 3 by the polycomb system, as well as deacetylation by histone deacetylases (HDACs), are implicated in repressive chromatin (schematized in Figure 1). Specific patterns of local and global histone modifications are required for the maintenance of the cells identity and alterations have been linked to cancer formation [44,45,46]. Apart from the modifications themselves, deregulation of the enzymes responsible for them have also been documented in many cancers [47,48], highlighting the importance of this epigenetic mechanism.
Although lncRNAs have been only recently associated with histone modifying enzymes [49,50,51] a paradigm of this category has long been known. X-inactive–specific transcript (XIST) RNA was one of the first long non-coding RNAs to be discovered in the early 1990s [52,53,54]. As its name dictates, XIST is a master regulator of the X chromosome inactivation (XCI) in the female mammal, a process meant to balance X-chromosome gene expression between the two sexes [55,56]. The initiation of XCI depends on XIST, which tethers polycomb-repressive complexes to the X chromosome in cis, in order to trimethylate histone H3 on lysine 27 (H3K27me3) resulting in transcriptional silencing. The tremendous effects of XIST on chromatin formation result in the conversion of an entire chromosome into a unique heterochromatic entity known as the Barr body [57]. After the completion of XCI, XIST is no longer needed but continues to be expressed, albeit at low levels, throughout female life [58]. X chromosomes’ aneuploidies have been associated with human cancers for a long time [59,60] and Lee’s lab proved the role of XIST in this process. Deletion of Xist was shown to be sufficient to induce a female-specific, aggressive and ultimately lethal blood cancer, possibly due to upregulation of X-linked genes [58].
Another example of lncRNAs exerting their regulatory functions through local chromatin remodeling is ANRIL (antisense non-coding RNA in the INK4 locus). ANRIL is a 3834 bp long transcript in the INK4A-ARF-INK4B gene cluster. Visel and colleagues provided the first genetic evidence of a negative regulation of p16INK4A and p15INK4B by non-coding RNAs residing in this genomic region [61]. Later on, it was demonstrated that ANRIL recruits the Polycomb repressive complex 2 (PRC2), well-known for its histone methyltransferase (HMT) activity on histone 3 lysine 27, to the p15INK4B locus [62]. ANRIL acts as a molecular scaffold which physically interacts with SUZ2, a PRC2 component, forcing the complex to occupy and thus regulate the p15INK4B locus. As one of the most important tumor suppressor loci, the INK4A-ARF-INK4B gene cluster is frequently deleted or silenced in many human cancers, making ANRIL one of the most altered lncRNAs [63,64,65,66].
Epigenetic silencing mediated by the interaction of lncRNAs with PRC2 has been proposed as a general mechanism for numerous nuclear lncRNAs [49,67,68]. However, several studies showing PRC2 promiscuous interaction with more RNA species [69,70,71], challenge the specificity and functional importance of lncRNA-PRC2 interactions. Indeed, Portoso and colleagues demonstrated that HOTAIR (Hox transcript antisense intergenic RNA) lncRNA, previously shown to interact with PRC2 in order to exert its transcriptional repressive functions on the HOXD locus [72], does not depend on PRC2 for the deposition of H3K27 trimethylation marks. On the contrary, the expression and correct localization of HOTAIR are necessary for silencing [73]. These results support the notion that even if a lncRNA-PRC2 complex forms, it might not, itself, be directly implicated in gene regulation and call for critical evaluation of the functionality of such complexes.
Finally, although less studied, a few lncRNAs have been associated with regulation of other histone marks. An example is lncPRESS1 which is a p53 target that disrupts SIRT6-mediated H3K56 deacethylation. Although the role of lncPRESS1 in cancer has not been investigated yet, it is very likely considering that it is associated with the guardian of the genome [74].
2.1.3. Chromatin Looping
Transcriptional gene regulation can also be achieved at a higher level of DNA compaction through the control of chromatin looping and nucleosome positioning. The effects of chromatin looping on transcription regulation can be probably best described by active enhancers regulating distant genes. Enhancers are tissue-specific DNA elements of low sequence and high functional conservation [75]. They bind transcription factors and mediator complex to recruit Polymerase II and enforce chromatin loops to increase the rate of transcription at distal promoters [76]. Active enhancers are pervasively transcribed giving rise to cell type- and state-specific transcripts called eRNAs [77,78]. eRNAs are non-coding RNAs that can be categorized into short, bidirectional, non-polyadenylated, mono-exonic transcripts or longer, unidirectional, polyadenylated, and spliced, both of which are of low abundance and enriched in the nucleus [78]. Whether eRNAs themselves are needed for a functional enhancer is still under debate; nevertheless, many studies show that eRNAs can modulate chromatin by stabilizing enhancer-promoter looping (Figure 1) [77,79,80]. Under normal conditions active enhancers control the maintenance of different cell types and it is of no surprise that they are deregulated in many human cancers [81]. Along with them, eRNAs are differentially expressed in cancer tissues and were shown to act as oncogenes, promoting the transcriptional activation of oncogenic networks and even inducing chromosome rearrangements and genomic instability [82,83,84].
The eRNA-acting lncRNA Leukemia-induced Non-coding Activator RNA-1 (LUNAR1) was initially identified by RNA-Seq in T-cell leukemia [85]. LUNAR1 is a T-ALL specific, Notch-dependent, nuclear-enriched lncRNA, whose coding neighbor is the insulin-like growth factor receptor 1 (IGF1R) implicated in T-ALL initiation and Notch signaling [86]. Depletion of LUNAR1 leads to downregulation of IGF1R and significant reduction in Mediator Complex and RNA Pol II occupancy at both the IGF1R enhancer and promoter. Hi-C experiments and 3C-qPCR showed that LUNAR1 is physically associated with the Notch-occupied IGF1R enhancer and with LUNAR1 promoter, proving that this lncRNA exploits chromatin looping to reach to its target and then recruits Mediator to activate the target-promoter [85].
2.1.4. Nucleosome Positioning
Nucleosome positioning is a major factor in controlling gene expression since tight interaction of nucleosomes with histone cores can strongly affect DNA accessibility [87]. LncRNAs can regulate nucleosome positioning in various ways ranging from the recruitment of ATP-dependent remodelers to transcription mediated nucleosome stabilization (Figure 1) [88]. The human lncRNA SChLAP1 controls nucleosome positioning via negative regulation of ATP-dependent chromatin remodelers. It physically interacts with the SNF5-subunit of the SWI/SNF chromatin-remodeling complex, responsible for nucleosome positioning, and prevents its binding to the chromatin, thus regulating gene expression. SChLAP1 has high prognostic value for prostate cancer and it is the first documented lncRNA acting as an antagonist of a major epigenetic complex, making it a great example for lncRNAs regulating nucleosome positioning [89].
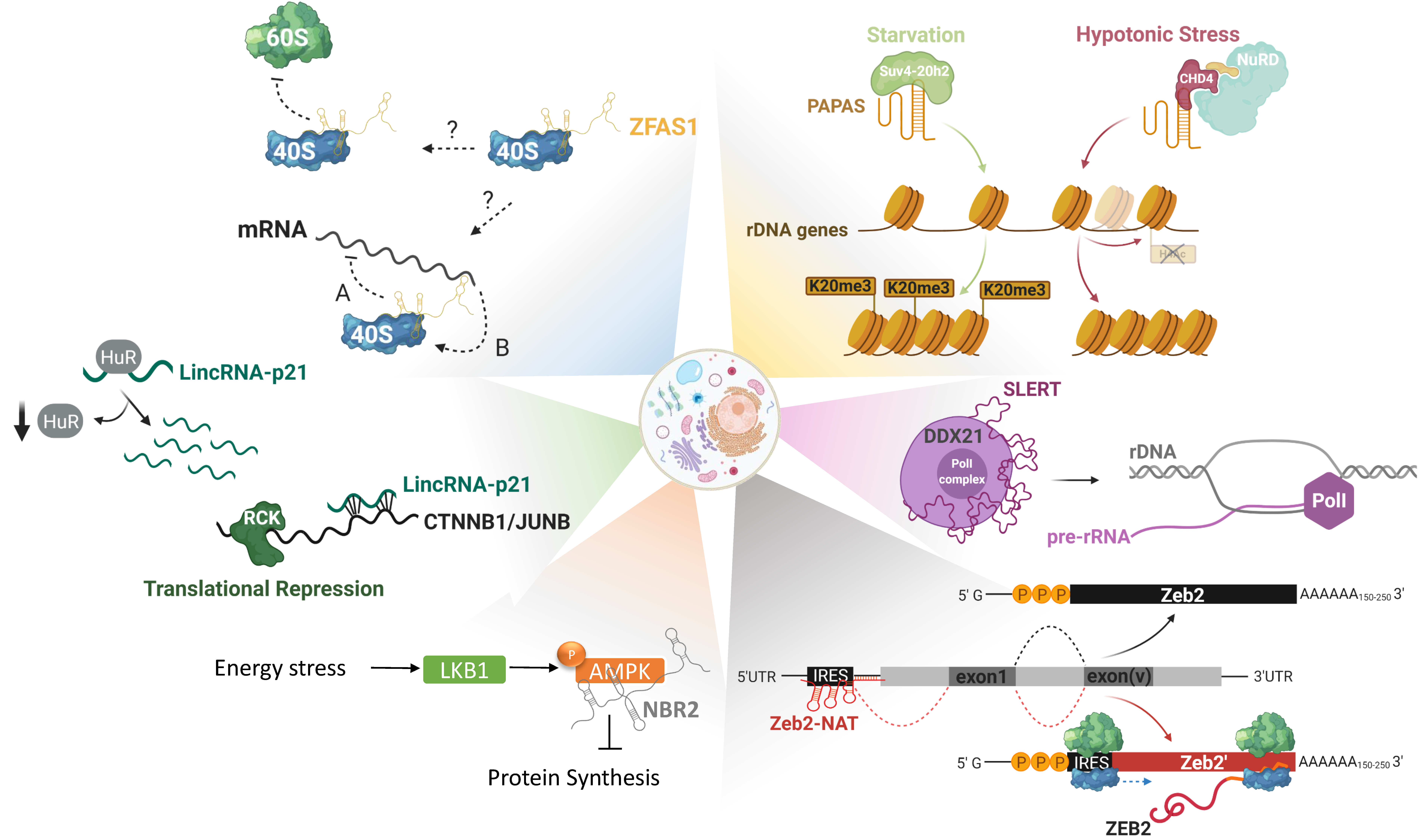
Another lncRNA that exemplifies the complexity of lncRNAs functions in nucleosome positioning is promoter and pre-rRNA antisense PAPAS [90]. PAPAS links histone modifications together with nucleosome remodeling, ribosome biogenesis and by extension protein synthesis. PAPAS is an antisense RNA to pre-rRNA genes which inhibits pre-rRNA synthesis under growth-factor deprivation [91]. This lncRNA is upregulated in starving cells and acts as a scaffold for the histone methyltransferase Suv4-20h2, guiding it to rRNA genes and leading to trimethylation of histone H4 at lysine 20 (H4K20me3) and chromatin compaction. In this way, rRNA promoters become inaccessible to RNA polymerase I, resulting in ribosome biogenesis attenuation [91]. Interestingly enough, upon hypotonic stress PAPAS still suppresses transcription of rRNA genes but through a different mechanism. In these conditions Suv4-20h2 gets ubiquitinylated and degraded, therefore PAPAS interacts instead with CHD4, a subunit of the nucleosome remodeling and deacetylation complex (NuRD). This interaction causes a shift of the promoter-bound nucleosome into a position that blocks transcription initiation [92,93].
Whether this lncRNA is deregulated in cancer is not yet known to our knowledge, however, PAPAS’ functions prove not only that transcriptional control by lncRNAs can be facilitated through many mechanisms but also that a single lncRNA can have different modes of action depending on the cell state.
2.2. LncRNA Acting as ceRNAs
LncRNAs can serve as master regulators at the post-transcriptional level, acting as competing endogenous RNAs (ceRNAs). ceRNAs regulate other (m)RNA targets by competing for the binding of shared miRNAs [94,95]. LncRNA-bound miRNAs can no longer be loaded to the AGO2/RISC and are, thus, unable to repress the translation of their mRNA-targets. Through this miRNA-mediated mode of action, ceRNAs emerge as pluripotent regulators capable of influencing an entire post-transcriptional regulatory network mediated by multiple miRNAs. Their importance is further proved by studies conducted in solid tumors and hematopoietic malignancies where ceRNAs alter the expression of tumor suppressors and oncogenes, in favor of cancer progression [96].
The first example of a non-coding transcript acting as a miRNA-sponge was provided by a processed pseudogene, highly homologous to the PTEN gene, called PTENP1 (phosphatase and tensin homolog pseudogene 1) that harbored several binding sites for PTEN-targeting miRNAs. In prostate cancer, the competition between PTENP1 and PTEN for binding with their shared miRNAs, positively regulates PTEN protein levels, placing PTENP1 in the category of tumor-suppressor lncRNAs [97]. Following their discovery, PTENP1s tumor-suppressor functions were confirmed in many other cancer types [98,99,100] proving beyond doubt that lncRNAs can exert ceRNA-related functions of great impact.
Another paradigm of this function is HULC, a very abundant lncRNA in hepatocellular carcinoma, and many other cancer types, that resembles the mammalian LTR transposon 1A [101]. Acting as an oncogene, it promotes tumor angiogenesis, cell proliferation, and abnormal lipid metabolism, by multiple mechanisms, such as the regulation of sphingosine kinase 1 expression and PI3K/AKT signaling [102,103,104]. The expression levels of HULC in cancer patients have been correlated with their clinical outcome [105]. Furthermore, acting as a ceRNA, HULC ensures its own high expression levels by sponging miR-372 from binding and suppressing translation of PRKACB. PRKACB, in turn induces the phosphorylation of cAMP response element binding protein (CREB) leading to upregulation of HULC transcription [106].
2.3. LncRNAs in Translational Reprogramming
Gene expression regulation is not limited to transcriptional and epigenetic regulatory networks. Protein synthesis is the most energy-demanding process inside the cell and, therefore, it is very tightly regulated [26]. Accordingly, this process is controlled by all the major tumor suppressors and oncogenes at all stages starting from rRNA biosynthesis in the nucleoli and ending with ribosome recycling [107]. Regulation at the translational level offers the advantage to rapidly respond to environmental changes [108].
LncRNAs have been associated with all the steps of protein synthesis, the simplest way being repressing or promoting the translation of specific mRNA-targets. This mode of action can be exemplified by the human lncRNA lincRNA-p21, a cytoplasmic-enriched molecule known to co-localize with ribosomes. In human cervical carcinoma (HeLa) cells lincRNA-p21 was shown to be negatively regulated by the RNA binding protein Hu antigen R (HuR) and, only when HuRs levels decrease, the lncRNA is able to stabilize and interact with its mRNA-targets: mainly CTNNB1 and JUNB. The imperfect base-pairing between lincRNA-p21 at sites in the coding and untranslated regions of its targets, triggers the association of these mRNAs with the translational repressors RCK and FMRP leading to attenuation of their translation [109]. Both CTNNB1 and JUNB are pro-survival proteins [110,111]. Their silencing by lincRNA-p21 dictates its tumor-suppressive functions, which have been elucidated in other cancer types as well [112,113].
Apart from selective translational regulation of mRNA-targets there is accumulating evidence that lncRNAs can have broader effects on translation by regulating ribosome biogenesis in the nucleoli. There, lncRNAs can affect nucleolar structure, rRNA and/or ribosomal protein-transcription and maturation (Figure 2) [26]. Apart from the aforementioned lncRNA PAPAS, there are more lncRNAs implicated in ribosome biogenesis regulation. SLERT (SnoRNA-ended lncRNA enhances pre-ribosomal RNA transcription) is a box H/ACA small nucleolar RNA (snoRNA)-ended lncRNA, expressed in embryonic stem cells but also in several human cancer cell lines. These box H/ACA snoRNAs are present at both ends of the lncRNA and are required for its biogenesis and translocation to the nucleolus. SLERT physically interacts with and sponges the DEAD-box RNA helicase DDX21, a protein that coats polymerase I complexes blocking pre-rRNA transcription. This interaction allows Pol I to freely transcribe pre-rRNAs, making SLERT a positive regulator of ribosome biogenesis [114]. In keeping with this, SLERT inhibition reduces tumorigenic potential both in vitro and in vivo, thus acting as an oncogene.
After ribosome biogenesis in the nucleoli, ribosomal subunits have to be exported to the cytoplasm in order to actively engage in translation. Indications that lncRNAs could bind and modulate the activity of ribosomes themselves are starting to emerge. One such lncRNA could be ZFAS1, initially discovered in murine tissues undergoing mammary gland development and later on shown to be highly expressed in breast cancer, among others. Utilizing polysome profiling, Hansji and colleagues demonstrated that ZFAS1 associates with the 40S small ribosomal subunit [115]. Even though mechanistic insights are still missing, it is intriguing to speculate that lncRNAs could serve as general modulators of protein synthesis initiation, either through modulation of ribosome assembly or via regulation of translation initiation factors (eIFs), elongation, and termination.
All these layers of regulation do not only take place at the steady-state but are actually exacerbated under stress conditions where cells are forced to attenuate global translation in the effort to save energy [116]. Under nutrient-rich conditions, global translation is dictated by the recognition of the 7-methylguanosine (m7Gppp) in the 5’-cap structure of mRNAs by eukaryotic initiation factor 4E (eIF4E) [117]. Translational reprogramming occurs upon environmental cues when the cells cannot meet the energy demand and in effort to survive, they shut-down global translation while still maintaining protein synthesis of essential factors for survival, through alternative mechanisms. The most common ways to activate these alternative routes that are cap-independent, are by recognition of upstream open reading frames (uORFs) and internal ribosome entry sites (IRES) [118]. Although more light still needs to be shed on the contribution of lncRNAs on the regulation of translation rewiring, there are already some indications that they could be implicated in this dynamic process. One lncRNA that could be linked to it is Zeb2-NAT (Zeb2-natural antisense transcript). The protein Zeb2 is a transcriptional repressor of E-cadherin and regulator of epithelial-to-mesenchymal transition (EMT), a process vital during physiological development and cancer progression [119]. Zeb2 contains an IRES sequence in an intron upstream of its first exon and alternative splicing of this intron is actually regulated by Zeb2-NAT. Upon induction of EMT the lncRNA Zeb2-NAT physically binds and masks the splicing site of the IRES, leading to intron retention. Under these conditions, Zeb2 is translated through the IRES, thus leading to downregulation of its target E-cadherin [120]. Zeb2-NAT’s ability to control a fundamental process as EMT has been shown to favor the maintenance and metastasis of some human cancers [121,122].
A last, indirect, method to regulate translation is the modulation of signaling pathways controlling it. The lncRNA NBR2 (neighbor of BRCA1 gene 2) is induced upon energy stress by the LKB1-AMPK pathway, with AMPK being a major sensor of the cellular energy levels. Upon such a stress NBR2 physically interacts with AMPK promoting its kinase activity, signaling towards attenuation protein synthesis and activation of pathways that will restore the energy levels [123]. Depletion of NBR2 leads to unchecked cell cycling, perturbed apoptotic/autophagic responses, and increased tumor development in vivo [124].
3. LncRNAs in Tumorigenesis
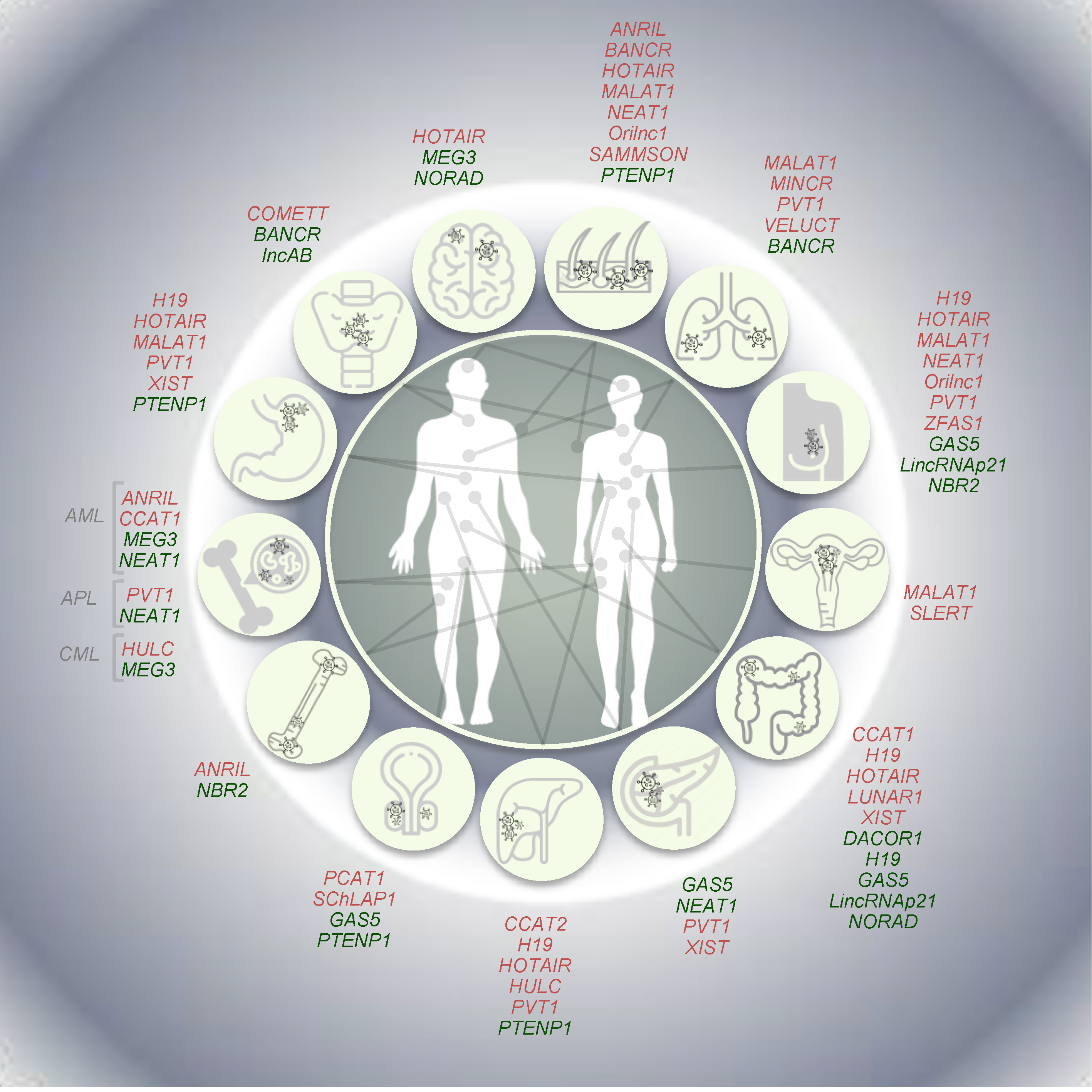
Systematic analyses based on TCGA data ([125] among the others) and the Cancer LncRNA Census (CLC; [126]) have largely contributed to reveal tumor-specific expression signatures of lncRNAs in cancer. Afterwards, different independent studies based on strong functional, genetic, and evolutionary evidence have reported, for a minority of them, a causative role in cancer. Hereafter, we provide—with obvious limitations due to space constraints—a description of the most relevant examples of well-established lncRNAs having oncogenic, tumor suppressive or dual properties, also reporting examples of lncRNAs whose expression is driven by well-known oncogenes (Figure 3).
3.1. Oncogenic LncRNAs
Among several oncogenic lncRNAs, here we focused on MALAT1 and HOTAIR, whose oncogenic potential is mainly related to tissue metastasis, as well as on SAMMSON and VELUCT, especially associated with sustained proliferation and abnormal cell metabolism, respectively. Furthermore, some examples of oncogenic lncRNAs induced downstream of the activation, e.g., by point mutations or rearrangements, of known oncogenes (i.e., BRAF, RET, and MYC) and/or regulating the expression of known oncogenes, such as COMETT, are also described below.
The lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was first identified as aberrantly expressed in metastatic lung cancer [127]. MALAT1, which is one of the most abundant lncRNAs in the cell, localizes to nuclear speckles, where it interacts with serine-rich (SR) proteins, regulating their abundance in the nucleoplasm and their phosphorylation levels, thus affecting alternative splicing [128]. A relevant role in the epigenetic regulation of transcriptional programs has been reported. Indeed, the relocation of growth control genes in the three-dimensional space of the nucleus—from repressive polycomb-enriched bodies (PcGs) to transcription-permissive interchromatin granules (ICGs)—is driven by the binding of MALAT1 and polycomb 2 protein [129]. Mouse xenograft-based studies revealed an oncogenic activity of this lncRNA, as the metastatic properties of injected lung cancer cells were dramatically reduced by its deletion [130]. Despite being initially identified for its oncogenic role in lung, MALAT1 expression was found deregulated in several tumors, including neoplasms of the digestive system [131,132,133] and ovarian cancer [134]. In keeping with this, MALAT1 knockdown has been proposed as a promising strategy to block the metastatic capacity of breast cancer [135]. Stable or transient KD in breast tumor cells resulted in a significant reduction of tumor growth as well as in dramatic impairment of metastasis formation, associated in vivo with increased differentiation state and consequent formation of cystic tumors [135]. Furthermore, somatic mutations in MALAT1 have been so far identified in women with breast cancer (luminal-type) [136] and its depletion in luminal cells from ER+ tumors has been reported to dramatically restrain tumor cells’ proliferation [137]. This evidence supports its KD as a valuable resource, especially in breast cancer treatment.
The human HOX transcript antisense intergenic RNA, known as HOTAIR, is embedded in the HOXC locus and was firstly described as interacting with EZH2 and SUZ12, members of the polycomb repressive complex 2 (PRC2) exerting its function in trans [138]. Interestingly, the cognate mouse Hotair gene (mHotair) is poorly conserved in the mouse genome and has a different exon number with highly variable sequence similarity [139]. In addition, it lacks the binding sites for EZH2, responsible for its methyltransferase activity and typical of the human HOTAIR lncRNA. Indeed, HOTAIR recruits the lysine specific demethylase (LSD1), part of CoREST/REST complex responsible for gene repression by chromatin remodeling. Adopting a secondary structure, human HOTAIR acts as a scaffold between these complexes inducing the formation of a transcription-repressive chromatin state around HOXD locus, which causes its silencing [68,72,140,141]. Conversely, no detectable regulatory effect of mHotair on Hoxd cluster genes was observed in mice, suggesting the presence of redundant mechanisms, cell type-specific functions or a rapid evolution of this lncRNA [139]. An oncogenic role for HOTAIR has been reported in several tumors and, consequently, this lncRNA is considered a potential therapeutic target in different cancer types. Indeed, its activity has been associated with increased invasiveness and metastatic capacity in primary breast and colon tumors, where high levels of HOTAIR positively correlate with metastasis and poor outcome [130,142]. Moreover, it is considered an independent prognostic marker in hepatocarcinoma for recurrence and impaired survival [14,129].
The survival associated mitochondrial melanoma specific oncogenic non-coding RNA (SAMMSON) maps to human chr3p13, a region amplified in 10–15% of human melanomas that also hosts the transcription factor MITF, the master regulator of melanocyte and melanoma biology [143]. Melanoma-specific expression of SAMMSON is thought to increase cancer cell fitness by concertedly enhancing mitochondrial and cytosolic translation via the sequestration of the nuclear RNA-binding protein CARF in the cytoplasm, where it makes aberrant contact with the mitochondrial protein p32 [144]. Of note, the primate-specific lncRNA SAMMSON is detectable in more than 90% of melanoma patients, but not in normal adult tissues, and its silencing with antisense oligonucleotides (ASO) results in the potent induction of cell death and increased sensitivity to MAPK inhibition both in vitro and in patient-derived xenograft models (PDX). Therefore, targeting SAMMSON represents a new promising cancer-specific therapeutic option in melanoma [143].
Then, viability enhancing lung cancer transcript (VELUCT) is a relevant example of extremely low-abundance lncRNAs. Despite this, silencing VELUCT lncRNA in lung cancer cell lines causes a dramatic drop of viability, indicating a potential oncogenic role for this low-copy lncRNA [145]. Novel opportunities for therapeutic strategies are also arising from the identification of lncRNAs whose expression is induced by oncogenes and are able to modulate their activity. For instance, PVT1, CCAT1, CCAT2, PCAT1, and MINCR lncRNAs have been identified among the most promising lncRNAs regulating Myc activity in tumor cells [146,147,148,149,150]. These lncRNAs map in 8q24– in a so-called “gene desert” (due to the lack of protein-coding genes), close to the proto-oncogene MYC. The identification of enhancers physically interacting with MYC and the amplification of this genomic region in different cancer types highlighted the potential role of lncRNAs mapping to 8q24 [151,152]. Among them, the lncRNA plasmacytoma variant translocation 1 (PVT1), discovered as an activator of Myc [153,154], is encoded by the PVT1 gene in humans. This lncRNA is located 51 kb downstream of the MYC locus (RefSeq on hg38 genome release) [146,155] and is co-amplified in a variety of human and animal tumors, which results in Myc stabilization. Furthermore, PVT1 is often involved in DNA rearrangements (e.g., translocations in Burkitt lymphoma, fusion genes with NBEA and WWOX in multiple myeloma, APIP, ATE1, and PPAPDC1A in the human gastric cell line), which could activate Myc by disrupting the gene structure or protein production [146,155,156,157,158,159,160]. Moreover, PVT1 has been demonstrated to be a MYC target and to be part of the same ribonucleo-protein complex [160]. However, although the role of PVT1 in tumorigenesis has been defined as dependent on increases in the MYC copy-number, an independent contribution to tumorigenesis has also been proposed [161]. Indeed, PVT1 inhibition can induce cell apoptosis, even in PVT1-overexpressing cells, whereas MYC silencing does not produce the same effect [162,163], suggesting different mechanisms of PVT1 and MYC cooperation in different cancers. Of note, the over-expression of PVT1 was reported in a wide range of cancers, including breast, gastric, non-small cell lung cancers (NSCLC), pancreatic and hepatocellular carcinomas, and acute promyelocytic leukemia (APL), and serves as a predictor of tumor progression and prognosis [161,163,164,165,166,167].
Likewise, prostate cancer associated transcript 1 (PCAT1) is another lncRNA mapping approximately 710 kb upstream of the MYC gene (RefSeq on hg38 genome release) and responsible for its post-transcriptional regulation. Identified in prostate cancer as a posttranscriptional repressor of BRCA2 [149], PCAT1 also acts as competitive endogenous RNA (ceRNA) by sponging miR34-1 and abrogating its binding to MYC 3’UTR, thus increasing Myc protein levels [168].
Furthermore, the colon cancer-associated transcripts 1 and 2 (CCAT1 and CCAT2, respectively), mapping upstream of the MYC locus at ≈515 kb and 333 kb, respectively (RefSeq on hg38 genome release) have been also reported to regulate Myc [147,169]. Interestingly, their expression correlates with a cancer-associated single nucleotide polymorphism (SNP), the rs6983267, which is located in a super-enhancer around the transcription start site of the MYC gene at 8q24. The silencing of CCAT1 in colon cancer cells reduces proliferation through CDKN1A/p21-mediated cell-cycle arrest in G1 phase and the injection of CCAT1 KD tumor cells delays tumorigenesis in xenograft models [148]. Moreover, CCAT1 is aberrantly upregulated in patients with acute myeloid leukemia (AML) and promotes cell proliferation by regulating miRNA-mediated pathways [170]. Conversely, over-expression of CCAT2 lncRNA, a target of Wnt-signalling, increases the expression of MYC and some of the members of the miR-17-92 cluster through TCF7L2 transcriptional regulation. This event promotes invasive tumor growth in xenograft models, conferring tumor cells with higher metastatic capacity in the liver [147].
Finally, the Myc-induced lncRNA MINCR was identified in Burkitt lymphoma cells as contributing to Myc-mediated regulation of cell cycle genes and having a relevant role in cell cycle progression [150]. Indeed, MINCR silencing causes the downregulation of AURKA and AURKB genes, encoding the Aurora kinase A and B, respectively, and of the chromatin licensing and DNA replication factor 1 (CDT1), determining a dramatic reduction in cell proliferation. Furthermore, MINCR overexpression was assessed in NSCLC patients and cell lines where its silencing induces cell cycle arrest and apoptosis by reducing Myc expression and its downstream effectors (e.g., cyclin A, cyclin D, CDK2, Bcl-2) [171].
However, lncRNAs induced by other oncogenes have also been reported, as the oncogenic RAS-induced lncRNA 1 (Orilnc1)—identified in breast and melanoma cells as being induced by mutated BRAF [172]—and the cytosolic oncogenic antisense to MET transcript (COMETT), over-expressed in BRAF-mutated and RET-rearranged papillary thyroid carcinomas [173]. Zhang and colleagues reported that Orilnc silencing by shRNAs causes a down-regulation of Cyclin E1 inducing G1/S arrest and blocking tumor cell proliferation and growth, both in vitro and in vivo [172]. Similarly, the repression of COMETT markedly reduces the viability and proliferation of tumor cells harboring BRAF mutation or RET oncogene rearrangement, as well as the motility and invasiveness of tumor cells in vitro. Of note, COMETT depletion significantly impairs the expression levels of different MAPK pathway effectors, including—but not limited to—the MET oncogene, also increasing tumor cells’ sensitivity to vemurafenib, a common inhibitor of mutated B-raf [173].
3.2. Tumor Suppressive lncRNAs
An increasing number of studies are reporting lncRNAs acting as tumor suppressors, whereby their inactivation/deletion in tumors has a role in the onset and/or progression toward metastatic advanced cancer stages. Here, we discuss five lncRNAs whose tumor-suppressing activities have been reported in different tumor types.
Among highly expressed lncRNAs, the growth arrest-specific transcript 5 (GAS5) has been reported to be ubiquitously expressed according to serial analysis of gene expression (SAGE) in cancer as well as normal tissue [174]. Well-known for its role in embryogenesis [175], it also plays a key role in other relevant processes, such as p53 signaling [176], growth arrest [177], and apoptosis [178]. One of the reported roles for GAS5 lncRNA is to act as a decoy for the glucocorticoid receptor [179] even though it can also bind to the receptors of androgen and progesterone, having a role in tumors with hormone-dependent induction [175]. GAS5 expression levels are inversely correlated with tumor size, staging, and also metastasis in different tumor types, including breast, bladder, colon, pancreas, and prostate cancer [180,181]. For instance, its overexpression in xenograft mice models of breast cancer significantly reduces tumor growth in vivo through cell-cycle arrest and induction of apoptosis [178].
Discovered as the human ortholog of gene trap locus 2 (Gtl2) in mice, the lncRNA maternally expressed gene 3 (MEG3) is a member of the imprinted genes mapping at 14q32.3 [182,183]. This lncRNA activates p53 and its target genes, showing a tumor suppressor activity related to its ability to inhibit tumor cell proliferation, as shown in various cancer cell lines following in vitro re-expression [184,185,186,187,188]. However, in AML and chronic myeloid leukemia (CML), it has been reported that MEG3 inhibits cell growth also in a p53-indipendent manner [189,190]. According to its tumor-suppressive activity its expression is lost in primary tumors, i.e., neuroblastomas, gliomas, and cell lines, e.g., those derived from brain, bladder, bone marrow, breast, colon, liver and prostate, owing to different mechanisms [184,185,186,187,188,189,190,191,192]. Deletion is the most frequent mechanism for MEG3 silencing in tumor cells. However, its negative epigenetic regulation is very frequent in cancers. MEG3 lncRNA maps in a genomic locus enriched in CpGs, highly sensitive to methylation. In tumors, the hypermethylation of MEG3 promoter and hypomethylation of the surrounding intergenic region have a pronounced effect on its expression levels [185]. The loss of MEG3 expression has also been associated with tumor grade in meningiomas [186]. Independent studies assessed MEG3 ability to inhibit tumor cell proliferation and induce cell apoptosis in different cell lines [184] [187,188]. Furthermore, MEG3 inactivation leads to an increased expression of genes promoting angiogenesis and microvessel formation in the brain [192].
The lncRNA AB074169 (lncAB) is a single-exon lncRNA first identified in a comparative microarray analysis as down-regulated, due to promoter hypermethylation, in papillary thyroid carcinoma samples when compared to adjacent non-tumor counterparts [193]. Accordingly, its knockdown promoted tumor cell proliferation in vitro. Conversely, its forced over-expression resulted in cell-cycle arrest and tumor growth inhibition in vitro and in vivo. Mechanistically, lncAB binds to, and reduces the expression of, KHSRP, increasing p21 levels and simultaneously decreasing CDK2 expression, in turn repressing cell proliferation [193].
The lncRNA Non-coding RNA Activated by DNA damage (NORAD), previously annotated as LINC00657, is conserved and ubiquitously expressed, although RNA-Seq data from normal post-mortem brains collected in GTEx database (https://www.gtexportal.org/home/gene/NORAD) indicate NORAD as highly expressed in frontal cortex. NORAD expression is triggered in a p53-mediated manner by DNA damage [194]. It has been also reported that NORAD acts as decoy in colon cancer for the members 1 and 2 of the RNA-binding proteins belonging to the PUMILIO family [195]. Proteins of this family stimulate deadenylation and decapping of messenger RNAs leading to post-transcriptional repression. In the absence of NORAD, the chromosomal instability is induced by the repression of their targets which, among other things, include many genes encoding proteins crucial for DNA replication and repair processes, affecting in turn the cell cycle and mitosis [194].
Tumor protein P53 pathway corepressor 1 (TP53COR1), commonly known as lincRNA-p21, was identified as highly transcribed upon DNA damage in a p53-mediated manner [196]. LincRNA-p21 is an antisense transcript of CDKN1A, the tumor-suppressor gene known as p21. It is a transcriptional repressor which is induced in p53-dependent transcriptional responses [176,196]. CDKN1A expression is affected by lincRNA-p21 silencing which causes the activation of the protein-coding gene in cis. As reported by Dimitrova and colleagues it causes the disruption of G1/S cell-cycle checkpoint [176]. In addition, lincRNA-p21 expression correlates with tumor staging and invasive phenotype in colon cancer [197]; it interacts with HuR in breast cancer cells causing the transcriptional repression of CTNNB1 and JUNB genes [109]. Finally, lincRNA-p21 is one of the few lncRNAs regulating tumor metabolic rewiring. Indeed, it has been reported that it is transcriptionally induced in hypoxic conditions and its increase is associated with the induction of hypoxia-induced glycolysis in tumor cells [198].
3.3. LncRNAs with Dual Activity
A few cancer-associated lncRNAs have been reported to play divergent roles in different cancer types. These contrasting functions could be explained, at least in part, by the wide genetic and phenotypic heterogeneity of tumors. Thus, tumor- or microenvironment-specific factors could determine oncogenic or tumor suppressor activities of a given lncRNA. However, the use of distinct experimental systems/approaches (e.g., in vivo models vs. in vitro studies) could also contribute to the generation of confounding results. Some clear and well-known examples of lncRNAs with apparently divergent roles in tumors are provided here.
The lncRNA H19 has been discovered for its high expression along mouse embryogenesis. It is a paternally imprinted gene mapping at chr11p15.5. As such, it is repressed at birth in most tissues. H19 expression is induced in cancer cells due to a loss of imprinting at its locus [199,200], especially in breast, colon, and hepatic carcinomas [201,202]. Its transcriptional regulation is exerted by the oncogenes p53, Myc, and hypoxia-induced factor 1a (HIF-1a) which are frequently deleted (p53) or over-expressed/amplified (Myc and HIF-1a) [203,204,205]. RNA-Seq data collected in the Cancer Genome Atlas portal report H19 lncRNA to be over-expressed in colon and gastric cancers [206]. However, a tumor-suppressive role has been reported in colon cancer mouse models as well as in human rhabdoid tumors [207,208,209]. The genetic heterogeneity of tumors and the intrinsic inter-species differences may account for discrepant results. H19 exerts its activity on tumor progression by different mechanisms. It can act as ceRNA for a large subset of microRNAs and it can simultaneously be a precursor of miRNAs. Its activity as a miRNA "sponge", in line with its main localization in the cytosol, has been reported for let-7 and miR-200 family members [210]. However, H19 is also a miR-675 precursor [208]. In colon cancers, the over-expression of H19 is paralleled by miR-675 increase. This miRNA directly binds and inhibits the tumor-suppressor retinoblastoma (RB) protein, promoting cell proliferation [211].
The MENepsilon/beta on chromosome 11 is a direct target of p53 [17] and gives rise to two transcriptional variants of the nuclear enriched abundant transcript 1 (NEAT1) non-coding RNA, a short polyadenylated (3.7 kb) and a long non-polyadenylated (27 kb). The last one is essential for the formation of paraspeckles, i.e., nuclear membraneless bodies [212]. The switch between the two isoforms was recently ascribed to the integrator complex that favors the production of the short form over the long [27]. Furthermore, NEAT1 long form—and, thus, the paraspeckles—promote skin tumorigenesis by increasing survival of tumor cells expressing mutated KRAS in genetically engineered mouse models [17]. In addition, its knockdown synergizes with several genotoxic chemotherapeutics, suggesting NEAT1 levels as good a predictor of responses to chemotherapy [17]. In keeping with its oncogenic role, mutations in CpG islands within NEAT1 promoter have been reported in breast cancer [213], where increased levels of NEAT1—and paraspeckles—correlate with staging in HER+ patients [214]. Nevertheless, the Attardi’s lab reported instead a tumor suppressor role for NEAT1 in p53-/- mutant KRAS mouse model of pancreatic ductal adenocarcinoma [215]. Accordingly, reduced NEAT1 expression was reported in AML and APL [216,217]. However, whether the tumor suppressive function of NEAT1 stems from the long or the short transcript needs to be further investigated. Due to the nature of paraspeckles, which are able to sequester other biomolecules in the nucleus, and to the difference in the germ layer originating from the affected tissue (ectoderm for skin and endoderm for pancreas), a context-specific function for this lncRNA it is very likely.
The BRAF-activated lncRNA (BANCR) has been reported either for its oncogenic or tumor suppressive activity. First identified as an oncogene in melanoma for its ability to activate MAPK pathway and induce tumor cell migration and proliferation [218,219], BANCR has also been reported as tumor suppressor in NSCLC for its role in epithelial-mesenchymal transition (EMT) [220]. In line with this finding, down-regulation of BANCR is associated with increased metastatic potential of tumor cells and poor prognosis [220]. Likewise, its down-regulation has been positively associated with tumor progression in papillary thyroid and clear cell renal cell carcinomas [221], indicating—at least in these cancer types—a tumor suppressive role.
4. Strategies to Target lncRNAs
Technical advances in molecular therapies provide new opportunities for exploring the clinical relevance of lncRNAs. The identification of lncRNAs involved in tumorigenesis provided useful insights in cancer biology, also paving the way to the adoption of new therapeutic strategies based on their targeting—e.g., CRISPR/dCas9-based approaches [222]—and to their evaluation as novel diagnostic/prognostic markers. Especially for lncRNAs with direct or indirect oncogenic activity, different targeting strategies have been adopted by modulation at the (i) genome-level (i.e., affecting their transcription levels), (ii) post-transcriptional RNA-level (i.e., regulating lncRNAs stability and/or processing) and (iii) interaction-level (i.e., modulating their binding capacity to obtain steric inhibition of specific lncRNA-protein interactions).
4.1. Genomic Modulation of LncRNAs by CRISPR-Based Systems
Targeting strategies aimed to modulate lncRNAs activity by genomic regulation benefitted from recent advances in genome editing approaches based on CRISPR-based systems. Particularly, among the systems of the so-called class 2–which use single effectors—the type II CRISPR-system, in its simplest form, consists of a nuclease (Cas9) and a single guide RNA (sgRNA), which leads Cas9 to target precise sites of the DNA [223]. Its main applications consist of the knockout of target genes by the generation of double-stranded breaks (DSB) in the open reading frame and the following induction of the non-homologous end joining (NHEJ) repair system (systems termed CRISPRn mutagenesis), as well as gene corrections or overexpression by inducing homology-directed repair (HDR; CRISPRn HR) or genomic deletions following multiple DSB (CRISPRn excision) [223,224,225]. However, the intricate genomic architecture of lncRNAs, the incomplete knowledge of their promoters and/or interaction motifs, as well as the complex mechanisms of their multiple activities (in cis and in trans) limit the use of standard CRISPR-based approaches. Of note, Goyal and colleagues [224] characterized about 62% of the total lncRNAs as “non-CRISPRable”- due to the presence of internal (35%) or bidirectional (20%) promoters–and only 38% of lncRNAs as specifically targetable.. Indeed, classical systems based on induction of frameshift mutations (e.g., the CRISPRn method) could be applied for modulating lncRNAs activity only if the sequence of the recognized motif is completely characterized and additional mechanisms of action are excluded. Additionally, targeting lncRNA promoters (e.g., by CRISPRn or HDR-based CRISPR approaches) needs a complete knowledge of the nucleotide sequences and could however affect the expression of other genes due to potential overlap and interactions between the promoters, or due to their proximity [226,227,228]. Furthermore, promoter targeting may also be insufficient if other, unknown, internal promoters contribute to the regulation of their expression. Similarly, the total or partial excision (e.g., by CRISPRn system) could be unfeasible if the lncRNA sequence overlaps with other functional regions or could lead to the generation of DNA artifacts as newly combined elements [225,227,228].
Interestingly, innovative CRISPR approaches have provided powerful tools to overcome classical editing allowing to specifically and reliably modulate lncRNAs expression (reviewed in [225,229]). Among the most prominent methods are the double excision CRISPR knockout (DECKO) system and the combination of CRISPR interference (CRISPRi) and "dead"-Cas9 (dCas9) [225,228,230,231,232]. More in detail, dCas9 lacks the nuclease activity but still preserves its RNA-dependent DNA-binding activity. Its fusion with specific effector domains has allowed the development of additional systems for gene expression modulation [224,230]. For instance, dCas9 fusion to the KRAB (Krüppel-associated box) domain of ZNF10 allows a more robust transcription inhibition (CRISPRi), whereas targeted gene expression in cis is induced by its combination with activation domains, such as VP64, p65, or Rta (CRISPR activation, CRISPRa) [224,230]. Of note, the CRISPRi system was employed for targeting promoters of more than 16000 lncRNAs and revealed that about 500 of them have causal roles in cancer cell growth [232,233]. Furthermore, the silencing of different lncRNAs (e.g., MALAT1) was achieved by the CRISPR/Cas9 system and the insertion of an inhibitory signal [234,235]. Even the combination of CRISPR-dCas9 to epigenetic effector proteins (such as DNA methyltransferase or histone modifiers) [236] has been successfully used for epigenetic silencing of lncRNAs. For example, the epigenetic silencing of MALAT1 was reached by excising active histone marks associated to its TSS [237]. Then, a good applicability for targeting lncRNAs has also been showed for CRISPR-based targeted insertion utilizing the NHEJ pathway—given the absence of a homologous donor sequence [238,239]—as well as for the DECKO method, which applies a two-step cloning strategy to generate lentiviral vectors expressing simultaneously two guide RNAs (gRNAs) [228].
The manipulation of lncRNAs expression has been reported also by the CRISPR-mediated tagging and regulation of lncRNAs (CTRL) method, consisting of a modified gene trap vector and a plasmid with Cas9 and 2 sgRNA sequences [240]. This approach—successfully applied for upregulating HOTAIR, DICER1-AS1 and PTENP1, among others—could represent a valuable tool for future screening of several lncRNAs [225,240].
Furthermore, novel Cas enzymes are starting to be utilized, such as the CRISPR/Cas13 system (class 2, type VI). It has showing remarkable potential for the development of basic and clinical approaches in cancer, as well as fortargeting lncRNAs [241,242,243]. Particularly, type VI enzymes possess RNA recognition and cleavage activities and, following the formation of guide-target RNA duplexes, it binds and cleaves targeted complementary sequences producing efficient gene knockdown without genome manipulation [241,244]. Thus, instead of the DNase activity of Cas9, Cas13 holds RNase activity and the distinct subtypes (i.e., Cas13a, Cas13b, Cas13c, and Cas13d), although different in sequence and size, share as common features the presence of higher eukaryotes and prokaryotes nucleotide-binding (HEPN) domains, which account for the RNA-targeted nucleolytic activity [245]. CRISPR/Cas13 systems have been successfully employed for multiple applications (e.g., RNA knockdown, imaging, tracking, and editing). Notably, its high sensitivity and specificity has been also assessed for lncRNA knockdown in a high-throughput phenotypic assay based on survival challenge in anticancer drugs response and the targeting of a subclass of nuclear lncRNAs—i.e., very long intergenic non-coding (vlinc) RNAs—by Cas13a from Leptotrichia wadei (LwCas13a) [243]. Furthermore, the catalytically “dead” Cas13 system (dCas13) has been recently used for labeling NEAT1 lncRNA and to explore the dynamics of associated paraspeckles without inducing genome alterations [242].
However, despite the remarkable advance in the development of innovative CRISP-based systems, the accurate analysis of lncRNA loci (for designing specific RNA guides), the evaluation of expression of neighboring genes and the validation of results by other approaches (e.g., RNAi, ASOs) should be implemented when targeting lncRNAs by CRISPR editing approaches. Future studies will be instrumental for assessing the translational potential of CRISPR-based targeting strategies of lncRNAs in cancer therapies.
4.2. Post-Transcriptional Targeting of LncRNAs
Oligonucleotide-based approaches which depend on RNA-RNA or RNA-DNA duplex formation have been developed as a strategy for the successful post-transcriptional targeting of long non-coding RNAs [246,247]. Double-stranded small-interfering RNAs, i.e., dssiRNAs, represent the most commonly used approach for lncRNA silencing. It is based on Dicer and the RNA-induced silencing complex (RISC) degradation pathway and involves the Argonaute 2 (Ago2) protein [248]. This approach has proven extremely successful in targeting lncRNAs for degradation in many cancer cell lines, although in vivo experiments using siRNAs have been even more challenging. However, their limited bioavailability and susceptibility to nucleases has largely restricted their use for in vivo purposes. However, to prevent siRNAs from enzymatic degradation, some chemical modifications (e.g., 2′-O methyl sugar residues and phosphorothioate bonds at the 3′-end) have been adopted to improve their pharmacokinetic properties [249]. This is the case of patisiran (Alnylam Pharmaceuticals) - a double-stranded siRNA comprised of a 3’-end DNA, RNA, and 2’-O-MOE RNA moieties, used for inhibiting the hepatic synthesis of transthyretin and approved by FDA for the polyneuropathy of hereditary transthyretin amyloidosis [250].
An alternative approach developed more than three decades ago is represented by the antisense oligonucleotides (ASOs) [251]. These were designed for the first time as short (13-mer) single strand DNA (ssDNA) oligos in order to target the Rous sarcoma virus RNA. Since their first use, ASOs have been widely adopted in many studies to successfully modulate lncRNA levels. ASOs show some differences compared to siRNAs. Indeed, the former are single-stranded DNA oligonucleotides (13–15 bp) which bind, via standard Watson-Crick base-pairing, target nucleotides in the RNA of interest. The formation of DNA-RNA duplex causes the degradation of the target RNA by RNAse H, even though specific ASO can act by inhibiting or modifying the expression of their target via steric hindrance and/or they can modulate splicing. However, the most useful feature of ASOs is the capacity to specifically target nuclear RNAs and, due to RNase H enrichment in the nucleus, cause its degradation. For this reason, ASOs are currently employed to achieve significant knockdown of lncRNAs [252]. As described for siRNAs, new generations of ASOs, consisting of longer (15–20 nt) oligos with phosphorothioate modifications, have been developed. These improvements have significantly increased ASO stability, making these oligonucleotides even more resistant to endonuclease-mediated degradation [253,254]. However, in the last two decades new chemical modifications, including 2′-O-methoxyethylation, have been used to maximize the RNAse H-mediated degradation of target RNAs and the duplex stability as well as the resistance of free ASOs to endonuclease-mediated degradation. These modifications have also largely reduced the so-called “off-target” effects, conferring to ASOs with new drug-like features [255]. This new class of chimeric ASOs—commercialy known as Gapmers—consists of RNA-DNA hybrids carrying a 2′-O-methoxyethyl-modified sugar backbone. Other chemical modifications, such as the S-constrained ethyl (cEt) and the locked nucleic acids (LNAs), have finally improved ASO potency revealing new pharmacokinetic properties, which make these oligo-based approaches the most useful tools to target lncRNA both in vitro and in vivo.
4.3. Steric Inhibition of LncRNA-Protein Interactions
Different experimental evidence has demonstrated that lncRNAs can act as scaffolds for single or for multi-protein complexes due to their peculiar folding. As these ncRNAs mediate the interaction of regulatory DNA regions, RNAs, and binding proteins, interfering with their binding can provide new therapeutic solutions. Targeting lncRNA unique structural regions by ASOs represents one of the most reliable approaches. In particular, the adoption of modified ASOs, unable to trigger RNAse H-dependent degradation of RNAs (e.g., morpholinos), as well as of small molecules able to block contact interface/s between lncRNAs and proteins, can result in loss-of-function [256]. Indeed, extending the aforementioned chemical modifications (e.g., cEt and LNAs) to the entire sequence of the ASO would impede RNAse H-dependent degradation of the lncRNA, causing only the alteration of the splicing, as described for splice-switching oligos (SSOs). Indeed, the oligos act by blocking the binding of splicing regulatory elements, such as enhancers or silencers (by steric hindrance), and in turn modulate pre-mRNA splicing [257]. To date, new therapies based on splice-switching oligos have been approved by the FDA for the spinal [258] and Duchenne muscular atrophy [259].
Similarly to SSOs, morpholinos are non-ionic DNA analogs of about 25 oligonucleotides initially developed to block mRNA translation or promote splicing switch, very similarly to LNAs. These molecules, first used to study zebrafish development [260,261], received their first approval in the clinics as DMD splicing modulators in patients affected by Duchenne muscular dystrophy [262,263,264]. In morpholinos sugars are replaced with methylene morpholine rings whereas anionic phosphates of DNA/RNAs are substituted by non-ionic phosphorodiamidate bonds [261,265,266]. Due to these chemical modifications, the morpholinos abolish, or largely suppress, lncRNAprotein binding, also impeding lncRNA binding to gene regulatory regions of the DNA.
LncRNA ability to form stable secondary and tertiary structures with DNA, RNA and proteins has provided new opportunities to target them [267]. New assays, such as selective 2′-hydroxyl acylation by primer extension (SHAPE; [268]) and the psoralen analysis of RNA interactions and structures (PARIS; [269]), have been developed to specifically map lncRNA secondary/tertiary structures. Bacterial/viral riboswitches, having structural elements similar to cancer-associated lncRNAs, have been successfully targeted with small-molecule inhibitors [270]. Hence, it is likely that a similar approach—coupling large scale experimental assays as SHAPE and PARIS to the screening of small-molecule inhibitors—may be used to target cancer-associated lncRNAs.
The use of oligo-based approaches to target lncRNAs in cancer has provided interesting results. Nonetheless, new challenges need to be faced and technical caveats need to be addressed. Indeed, despite the fact that chemical modifications have improved oligo stability and largely reduced, if not abolished, their endonuclease-mediated degradation, one the most limiting factors is represented by their capacity to activate the innate immune response. The intrinsic characteristic of innate immunity protecting cells from exogenous RNAs by Toll-like receptors (TLRs) has largely limited oligo bioavailability [271]. First-generation oligos, including short interfering RNAs, have “CpG” motifs which are recognized by TLR9 and activate a TLR-dependent pro-inflammatory response [272]. Despite ASOs having specific chemical modifications and nucleotide sequences (CpG-poor), which minimize the activiation of these intracellular responses, they can still trigger a TLR-independent mechanism, such as the retinoic acid-inducible gene I-like (RIG-I) RNA helicases (RLHs), which elicit a pro-inflammatory response in the cytosol [272,273]. Upon activation of these pro-inflammatory pathways ASOs are entrapped into endosomes and their bioavailability—and in turn their capacity to target lncRNAs—is dramatically impaired [274]. In this regard, new oligo coniugating strategies need to be adopted and well-tolerated sequences need to be identified [275,276].
In addition, new strategies to avoid off-target effects need to be explored, but most importantly rigorous studies about oligo safety in humans need to be conducted before applying these approaches for cancer therapies.
Nine clinical trials (active, recruiting, or completed; https://clinicaltrials.gov) are currently testing signatures of lncRNAs in breast, lung, thyroid, or stomach tumors or single lncRNAs (XIST, CCAT1 and HOTAIR, in AML, colorectal, and thyroid carcinomas, respectively) as possible prognostic/diagnostic biomarkers. One of them is further validating prognostic and predictive mRNA-lncRNA signatures for triple-negative breast cancer which could be used to classify patients into high- or low-risk of recurrence. This trial is also evaluating these signatures as predictors of chemotherapy efficacy (e.g., doxorubicin, cyclophosphamide, gemcitabine, and cisplatin). However, to the best of our knowledge, no clinical trials targeting lncRNAs in cancer are currently underway.
5. Conclusions
A growing body of evidence has largely established that lncRNAs play a crucial role in tumor onset, progression, and drug responses. The rapid advancement of sequencing-based technologies, paralleled by consolidated functional in vitro approaches, as well as animal and human-derived tumor models, is offering an unprecedented opportunity in oncology, revolutionizing the way cancer is diagnosed and treated. Due to the high specificity in their expression—often restricted to tumor cells—many lncRNAs are currently under evaluation as biomarkers or direct therapeutic targets in clinical trials, whereas others have been proposed as modulators of tumor response to therapy. In this regard, despite RNA-based strategies specifically targeting lncRNAs in vitro and in vivo (e.g., in cancer mouse models) having provided encouraging results, lncRNA-based therapeutic approaches have not yet reached the clinical stage and different challenges are still open. Among them, the level of expression of lncRNAs is a key factor both for diagnostics and therapeutics. Identifying lncRNA candidates whose expression can be easily quantified and monitored is a relevant point to develop easy-to-use diagnostic tests. In addition, detecting lncRNAs through non-invasive approaches (e.g., in blood, urine, or saliva) would significantly boost the use of lncRNAs in daily clinical practice. It is reasonable to speculate that future decision trees in oncology will be revised by integrating patient-specific omics data, such as whole-genome/targeted resequencing (for somatic and germline mutations/rearrangements) and RNA-Seq (for profiling coding and ncRNAs). Hence, lncRNA detection for diagnostic and prognostic purposes or their therapeutic targeting will become a daily practice for clinicians, boosting the perspectives of personalized medicine, and especially of precision oncology.
Author Contributions
All authors participated to manuscript writing, preparation of figures, editing, and critical review. All authors have read and agreed to the published version of the manuscript.
Funding
This work was carried out with the financial support of My First AIRC Grant (MFAG) Programme (grant no. 23453) and Project SATIN—POR Campania FESR 2014/2020 to V.C; M.A.’s research contract is funded by the European Foundation for the Study of Diabetes (EFSD)/BoehringerIngelheim European Research Programme; V.K. is a recipient of FWO PhD fellowship 1S47519N; E.L. is funded by the Melanoma Research Alliance (MRA) Amanda and Jonathan Eilian Young Investigator Award and by the Belgian Federation for Cancer (Stichting Tegen Kanker).
Acknowledgments
We apologize to many colleagues whose relevant work could not be included in this review due to space limitations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Euskirchen, G.; Bertone, P.; Martone, R.; Luscombe, N.M.; Hartman, S.; Harrison, P.M.; Nelson, F.K.; Miller, P.; Gerstein, M.; et al. The transcriptional activity of human Chromosome 22. Genes Dev. 2003, 17, 529–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 457, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Bertone, P.; Stolc, V.; Royce, T.E.; Rozowsky, J.S.; Urban, A.E.; Zhu, X.; Rinn, J.L.; Tongprasit, W.; Samanta, M.; Weissman, S.; et al. Global identification of human transcribed sequences with genome tiling arrays. Science 2004, 306, 2242–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [PubMed] [Green Version]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [Green Version]
- Moran, V.A.; Perera, R.J.; Khalil, A.M. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 2012, 40, 6391–6400. [Google Scholar] [CrossRef]
- Fang, Y.; Fullwood, M.J. Roles, Functions, and Mechanisms of Long Non-coding RNAs in Cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Khurana, E.; Fu, Y.; Colonna, V.; Mu, X.J.; Kang, H.M.; Lappalainen, T.; Sboner, A.; Lochovsky, L.; Chen, J.; Harmanci, A.; et al. Integrative annotation of variants from 1092 humans: Application to cancer genomics. Science 2013, 342, 1235587. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Han, L.; Roebuck, P.; Diao, L.; Liu, L.; Yuan, Y.; Weinstein, J.N.; Liang, H. TANRIC: An Interactive Open Platform to Explore the Function of lncRNAs in Cancer. Cancer Res. 2015, 75, 3728–3737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Li, X.; Zhi, H.; Zhang, Y.; Wang, P.; Wang, Y.; Shang, S.; Fang, Y.; Shen, W.; Ning, S.; et al. Comprehensive Characterization of Somatic Mutations Impacting lncRNA Expression for Pan-Cancer. Mol. Nucleic. Acids 2019, 18, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.J.; Xie, S.L.; Li, Q.; Ma, J.; Wang, G.Y. Large intervening non-coding RNA HOTAIR is associated with hepatocellular carcinoma progression. J. Int. Med. Res. 2011, 39, 2119–2128. [Google Scholar] [CrossRef] [Green Version]
- Niland, C.N.; Merry, C.R.; Khalil, A.M. Emerging Roles for Long Non-Coding RNAs in Cancer and Neurological Disorders. Front. Genet. 2012, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Adriaens, C.; Standaert, L.; Barra, J.; Latil, M.; Verfaillie, A.; Kalev, P.; Boeckx, B.; Wijnhoven, P.W.G.; Radaelli, E.; Vermi, W.; et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat. Med. 2016, 22, 861–868. [Google Scholar] [CrossRef]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [Green Version]
- Mattick, J.S. The central role of RNA in human development and cognition. Febs Lett. 2011, 585, 1600–1616. [Google Scholar] [CrossRef]
- Derrien, T. The long non-coding RNAs: A new (p)layer in the “dark matter”. Front. Genet. 2012, 2, 107. [Google Scholar] [CrossRef] [Green Version]
- Wilson, K.D.; Ameen, M.; Guo, H.; Abilez, O.J.; Tian, L.; Mumbach, M.R.; Diecke, S.; Qin, X.; Liu, Y.; Yang, H.; et al. Endogenous Retrovirus-Derived lncRNA BANCR Promotes Cardiomyocyte Migration in Humans and Non-human Primates. Dev. Cell 2020, 54, 694–709.e9. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.-V.; Brown, E.L.; DeSouza, T.; Jespersen, N.Z.; Nandrup-Bus, C.; Yang, Q.; Yang, Z.; Desai, A.; Min, S.Y.; Rojas-Rodriguez, R.; et al. Human thermogenic adipocyte regulation by the long noncoding RNA LINC00473. Nat. Metab. 2020, 2, 397–412. [Google Scholar] [CrossRef]
- Nakagawa, S.; Naganuma, T.; Shioi, G.; Hirose, T. Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J. Cell Biol. 2011, 193, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Arun, G.; Mao, Y.S.; Lazar, Z.; Hung, G.; Bhattacharjee, G.; Xiao, X.; Booth, C.J.; Wu, J.; Zhang, C.; et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012, 2, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, S.; Horvat, F.; Drutovic, D.; Efenberkova, M.; Pinkas, D.; Jindrova, A.; Pasulka, J.; Iyyappan, R.; Malik, R.; Susor, A.; et al. The most abundant maternal lncRNA Sirena1 acts post-transcriptionally and impacts mitochondrial distribution. Nucleic Acids Res. 2020, 48, 3211–3227. [Google Scholar] [CrossRef]
- Verheyden, Y.; Goedert, L.; Leucci, E. Control of nucleolar stress and translational reprogramming by lncRNAs. Cell Stress 2018, 3, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Barra, J.; Gaidosh, G.S.; Blumenthal, E.; Beckedorff, F.; Tayari, M.M.; Kirstein, N.; Karakach, T.K.; Jensen, T.H.; Impens, F.; Gevaert, K.; et al. Integrator restrains paraspeckles assembly by promoting isoform switching of the lncRNA NEAT1. Sci. Adv. 2020, 6, eaaz9072. [Google Scholar] [CrossRef]
- Geisler, S.; Coller, J. RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013, 14, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Brar, G.A.; Stern-Ginossar, N.; Harris, M.S.; Talhouarne, G.J.S.; Jackson, S.E.; Wills, M.R.; Weissman, J.S. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014, 8, 1365–1379. [Google Scholar] [CrossRef] [Green Version]
- Van Heesch, S.; van Iterson, M.; Jacobi, J.; Boymans, S.; Essers, P.B.; de Bruijn, E.; Hao, W.; MacInnes, A.W.; Cuppen, E.; Simonis, M. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol. 2014, 15, R6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, F.; Shah, A.; Shan, G. Long Non-coding RNAs in the Cytoplasm. Genom. Proteom. Bioinform. 2016, 14, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagano, T.; Fraser, P. No-nonsense functions for long noncoding RNAs. Cell 2011, 145, 178–181. [Google Scholar] [CrossRef] [Green Version]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Begolli, R.; Sideris, N.; Giakountis, A. LncRNAs as Chromatin Regulators in Cancer: From Molecular Function to Clinical Potential. Cancers 2019, 11, 1524. [Google Scholar] [CrossRef] [Green Version]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 2013, 153, 1134–1148. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [Green Version]
- Di Ruscio, A.; Ebralidze, A.K.; Benoukraf, T.; Amabile, G.; Goff, L.A.; Terragni, J.; Figueroa, M.E.; De Figueiredo Pontes, L.L.; Alberich-Jorda, M.; Zhang, P.; et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013, 503, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Merry, C.R.; Forrest, M.E.; Sabers, J.N.; Beard, L.; Gao, X.-H.; Hatzoglou, M.; Jackson, M.W.; Wang, Z.; Markowitz, S.D.; Khalil, A.M. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum. Mol. Genet. 2015, 24, 6240–6253. [Google Scholar] [CrossRef]
- Somasundaram, S.; Forrest, M.E.; Moinova, H.; Cohen, A.; Varadan, V.; LaFramboise, T.; Markowitz, S.; Khalil, A.M. The DNMT1-associated lincRNA DACOR1 reprograms genome-wide DNA methylation in colon cancer. Clin. Epigenetics 2018, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhi, H.; Li, X.; Wang, P.; Gao, Y.; Gao, B.; Zhou, D.; Zhang, Y.; Guo, M.; Yue, M.; Shen, W.; et al. Lnc2Meth: A manually curated database of regulatory relationships between long non-coding RNAs and DNA methylation associated with human disease. Nucleic Acids Res. 2018, 46, D133–D138. [Google Scholar] [CrossRef] [PubMed]
- Khorasanizadeh, S. The nucleosome: From genomic organization to genomic regulation. Cell 2004, 116, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef]
- Fuks, F. DNA methylation and histone modifications: Teaming up to silence genes. Curr. Opin. Genet. Dev. 2005, 15, 490–495. [Google Scholar] [CrossRef]
- Ozdağ, H.; Teschendorff, A.E.; Ahmed, A.A.; Hyland, S.J.; Blenkiron, C.; Bobrow, L.; Veerakumarasivam, A.; Burtt, G.; Subkhankulova, T.; Arends, M.J.; et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genom. 2006, 7, 90. [Google Scholar] [CrossRef] [Green Version]
- Füllgrabe, J.; Kavanagh, E.; Joseph, B. Histone onco-modifications. Nat. Publ. Group 2011, 30, 3391–3403. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-Dinardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef]
- Wagschal, A.; Sutherland, H.G.; Woodfine, K.; Henckel, A.; Chebli, K.; Schulz, R.; Oakey, R.J.; Bickmore, W.A.; Feil, R. G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol. Cell. Biol. 2008, 28, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Xing, Y.; Wen, X.; Jia, R.; Ni, H.; He, J.; Ding, X.; Pan, H.; Qian, G.; Ge, S.; et al. Long non-coding RNA ROR decoys gene-specific histone methylation to promote tumorigenesis. Genome Biol. 2015, 16, 1–17. [Google Scholar]
- Borsani, G.; Tonlorenzi, R.; Simmler, M.C.; Dandolo, L.; Arnaud, D.; Capra, V.; Grompe, M.; Pizzuti, A.; Muzny, D.; Lawrence, C.; et al. Characterization of a murine gene expressed from the inactive X chromosome. Nature 1991, 351, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; McCabe, V.M.; Norris, D.P.; Cooper, P.J.; Swift, S.; Rastan, S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 1992, 71, 515–526. [Google Scholar] [CrossRef]
- Brown, C.J.; Hendrich, B.D.; Rupert, J.L.; Lafrenière, R.G.; Xing, Y.; Lawrence, J.; Willard, H.F. The human XIST gene: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992, 71, 527–542. [Google Scholar] [CrossRef]
- Payer, B.; Lee, J.T. X chromosome dosage compensation: How mammals keep the balance. Annu. Rev. Genet. 2008, 42, 733–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wutz, A. Gene silencing in X-chromosome inactivation: Advances in understanding facultative heterochromatin formation. Nat. Rev. Genet. 2011, 12, 542–553. [Google Scholar] [CrossRef]
- Barr, M.L.; Bertram, E.G. A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. Nature 1949, 163, 676–677. [Google Scholar] [CrossRef]
- Yildirim, E.; Kirby, J.E.; Brown, D.E.; Mercier, F.E.; Sadreyev, R.I.; Scadden, D.T.; Lee, J.T. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 2013, 152, 727–742. [Google Scholar] [CrossRef] [Green Version]
- Liao, D.J.; Du, Q.-Q.; Yu, B.W.; Grignon, D.; Sarkar, F.H. Novel perspective: Focusing on the X chromosome in reproductive cancers. Cancer Investig. 2003, 21, 641–658. [Google Scholar] [CrossRef]
- Pageau, G.J.; Hall, L.L.; Ganesan, S.; Livingston, D.M.; Lawrence, J.B. The disappearing Barr body in breast and ovarian cancers. Nat. Rev. Cancer 2007, 7, 628–633. [Google Scholar] [CrossRef]
- Visel, A.; Zhu, Y.; May, D.; Afzal, V.; Gong, E.; Attanasio, C.; Blow, M.J.; Cohen, J.C.; Rubin, E.M.; Pennacchio, L.A. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 2010, 464, 409–412. [Google Scholar] [CrossRef]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Nat. Publ. Group 2011, 30, 1956–1962. [Google Scholar]
- Shete, S.; Hosking, F.J.; Robertson, L.B.; Dobbins, S.E.; Sanson, M.; Malmer, B.; Simon, M.; Marie, Y.; Boisselier, B.; Delattre, J.-Y.; et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat. Genet. 2009, 41, 899–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacey, S.N.; Sulem, P.; Masson, G.; Gudjonsson, S.A.; Thorleifsson, G.; Jakobsdottir, M.; Sigurdsson, A.; Gudbjartsson, D.F.; Sigurgeirsson, B.; Benediktsdottir, K.R.; et al. New common variants affecting susceptibility to basal cell carcinoma. Nat. Genet. 2009, 41, 909–914. [Google Scholar] [CrossRef]
- Turnbull, C.; Ahmed, S.; Morrison, J.; Pernet, D.; Renwick, A.; Maranian, M.; Seal, S.; Ghoussaini, M.; Hines, S.; Healey, C.S.; et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet. 2010, 42, 504–507. [Google Scholar] [CrossRef] [Green Version]
- Pasmant, E.; Sabbagh, A.; Vidaud, M.; Bièche, I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. Faseb J. 2011, 25, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Davidovich, C.; Zheng, L.; Goodrich, K.J.; Cech, T.R. Promiscuous RNA binding by Polycomb repressive complex 2. Nat. Struct. Mol. Biol. 2013, 20, 1250–1257. [Google Scholar] [CrossRef] [Green Version]
- Kretz, M.; Meister, G. RNA binding of PRC2: Promiscuous or well ordered? Mol. Cell 2014, 55, 157–158. [Google Scholar] [CrossRef] [Green Version]
- Davidovich, C.; Wang, X.; Cifuentes-Rojas, C.; Goodrich, K.J.; Gooding, A.R.; Lee, J.T.; Cech, T.R. Toward a consensus on the binding specificity and promiscuity of PRC2 for RNA. Mol. Cell 2015, 57, 552–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portoso, M.; Ragazzini, R.; Brenčič, Ž.; Moiani, A.; Michaud, A.; Vassilev, I.; Wassef, M.; Servant, N.; Sargueil, B.; Margueron, R. PRC2 is dispensable for HOTAIR-mediated transcriptional repression. Embo J. 2017, 36, 981–994. [Google Scholar] [CrossRef]
- Jain, A.K.; Xi, Y.; McCarthy, R.; Allton, K.; Akdemir, K.C.; Patel, L.R.; Aronow, B.; Lin, C.; Li, W.; Yang, L.; et al. LncPRESS1 Is a p53-Regulated LncRNA that Safeguards Pluripotency by Disrupting SIRT6-Mediated De-acetylation of Histone H3K56. Mol. Cell 2016, 64, 967–981. [Google Scholar] [CrossRef] [Green Version]
- Plank, J.L.; Dean, A. Enhancer function: Mechanistic and genome-wide insights come together. Mol. Cell 2014, 55, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Long, H.K.; Prescott, S.L.; Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 2016, 167, 1170–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santa, F.; Barozzi, I.; Mietton, F.; Ghisletti, S.; Polletti, S.; Tusi, B.K.; Muller, H.; Ragoussis, J.; Wei, C.-L.; Natoli, G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010, 8, e1000384. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Ørom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar] [CrossRef] [Green Version]
- Hah, N.; Danko, C.G.; Core, L.; Waterfall, J.J.; Siepel, A.; Lis, J.T.; Kraus, W.L. A Rapid, Extensive, and Transient Transcriptional Response to Estrogen Signaling in Breast Cancer Cells. Cell 2011, 145, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Wang, L.; Ren, S.; Wang, L.; Blackburn, P.R.; McNulty, M.S.; Gao, X.; Qiao, M.; Vessella, R.L.; Kohli, M.; et al. Activation of P-TEFb by Androgen Receptor-Regulated Enhancer RNAs in Castration-Resistant Prostate Cancer. Cell Rep. 2016, 15, 599–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.H.; Leong, W.Z.; Ngoc, P.C.T.; Tan, T.K.; Bertulfo, F.C.; Lim, M.C.; An, O.; Li, Z.; Yeoh, A.E.J.; Fullwood, M.J.; et al. The enhancer RNA ARIEL activates the oncogenic transcriptional program in T-cell acute lymphoblastic leukemia. Blood 2019, 134, 239–251. [Google Scholar] [CrossRef]
- Trimarchi, T.; Bilal, E.; Ntziachristos, P.; Fabbri, G.; Dalla-Favera, R.; Tsirigos, A.; Aifantis, I. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell 2014, 158, 593–606. [Google Scholar] [CrossRef] [Green Version]
- Medyouf, H.; Gusscott, S.; Wang, H.; Tseng, J.-C.; Wai, C.; Nemirovsky, O.; Trumpp, A.; Pflumio, F.; Carboni, J.; Gottardis, M.; et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J. Exp. Med. 2011, 208, 1809–1822. [Google Scholar] [CrossRef] [Green Version]
- Narlikar, G.J.; Sundaramoorthy, R.; Owen-Hughes, T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 2013, 154, 490–503. [Google Scholar] [CrossRef] [Green Version]
- Böhmdorfer, G.; Wierzbicki, A.T. Control of Chromatin Structure by Long Noncoding RNA. Trends Cell Biol. 2015, 25, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Prensner, J.R.; Iyer, M.K.; Sahu, A.; Asangani, I.A.; Cao, Q.; Patel, L.; Vergara, I.A.; Davicioni, E.; Erho, N.; Ghadessi, M.; et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat. Genet. 2013, 45, 1392–1398. [Google Scholar] [CrossRef] [Green Version]
- Vydzhak, O.; Luke, B.; Schindler, N. Non-coding RNAs at the Eukaryotic rDNA Locus: RNA-DNA Hybrids and Beyond. J. Mol. Biol. 2020, 432, 4287–4304. [Google Scholar] [CrossRef]
- Bierhoff, H.; Dammert, M.A.; Brocks, D.; Dambacher, S.; Schotta, G.; Grummt, I. Quiescence-induced LncRNAs trigger H4K20 trimethylation and transcriptional silencing. Mol. Cell 2014, 54, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Dammert, M.A.; Grummt, I.; Bierhoff, H. lncRNA-Induced Nucleosome Repositioning Reinforces Transcriptional Repression of rRNA Genes upon Hypotonic Stress. Cell Rep. 2016, 14, 1876–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Sentürk, N.; Song, C.; Grummt, I. lncRNA PAPAS tethered to the rDNA enhancer recruits hypophosphorylated CHD4/NuRD to repress rRNA synthesis at elevated temperatures. Genes Dev. 2018, 32, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Karreth, F.A.; Tay, Y.; Perna, D.; Ala, U.; Tan, S.M.; Rust, A.G.; DeNicola, G.; Webster, K.A.; Weiss, D.; Perez-Mancera, P.A.; et al. In Vivo Identification of Tumor- Suppressive PTEN ceRNAs in an Oncogenic BRAF-Induced Mouse Model of Melanoma. Cell 2011, 147, 382–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumazin, P.; Yang, X.; Chiu, H.-S.; Chung, W.-J.; Iyer, A.; Llobet-Navas, D.; Rajbhandari, P.; Bansal, M.; Guarnieri, P.; Silva, J.; et al. An Extensive MicroRNA-Mediated Network of RNA-RNA Interactions Regulates Established Oncogenic Pathways in Glioblastoma. Cell 2011, 147, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hou, J.; He, D.; Sun, M.; Zhang, P.; Yu, Y.; Chen, Y. The Emerging Function and Mechanism of ceRNAs in Cancer. Trends Genet. 2016, 32, 211–224. [Google Scholar] [CrossRef] [Green Version]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [Green Version]
- Li, R.-K.; Gao, J.; Guo, L.-H.; Huang, G.-Q.; Luo, W.-H. PTENP1 acts as a ceRNA to regulate PTEN by sponging miR-19b and explores the biological role of PTENP1 in breast cancer. Cancer Gene 2017, 24, 309–315. [Google Scholar] [CrossRef]
- Gao, L.; Ren, W.; Zhang, L.; Li, S.; Kong, X.; Zhang, H.; Dong, J.; Cai, G.; Jin, C.; Zheng, D.; et al. PTENp1, a natural sponge of miR-21, mediates PTEN expression to inhibit the proliferation of oral squamous cell carcinoma. Mol. Carcinog. 2017, 56, 1322–1334. [Google Scholar] [CrossRef]
- Poliseno, L.; Haimovic, A.; Christos, P.J.; Vega Y Saenz de Miera, E.C.; Shapiro, R.; Pavlick, A.; Berman, R.S.; Darvishian, F.; Osman, I. Deletion of PTENP1 pseudogene in human melanoma. J. Investig. Derm. 2011, 131, 2497–2500. [Google Scholar] [CrossRef] [Green Version]
- Panzitt, K.; Tschernatsch, M.M.O.; Guelly, C.; Moustafa, T.; Stradner, M.; Strohmaier, H.M.; Buck, C.R.; Denk, H.; Schroeder, R.; Trauner, M.; et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 2007, 132, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Xiao, Z.; Wang, Y.; Zheng, M.; Song, T.; Cai, X.; Sun, B.; Ye, L.; Zhang, X. Long noncoding RNA HULC modulates abnormal lipid metabolism in hepatoma cells through an miR-9-mediated RXRA signaling pathway. Cancer Res. 2015, 75, 846–857. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Xiao, Z.; Liu, F.; Cui, M.; Li, W.; Yang, Z.; Li, J.; Ye, L.; Zhang, X. Long non-coding RNA HULC promotes tumor angiogenesis in liver cancer by up-regulating sphingosine kinase 1 (SPHK1). Oncotarget 2016, 7, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Li, Y.; Chai, X.; Kang, Q.; Zhao, P.; Xiong, J.; Wang, J. Long noncoding RNA HULC promotes cell proliferation by regulating PI3K/AKT signaling pathway in chronic myeloid leukemia. Gene 2017, 607, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zheng, H.; Chan, M.T.V.; Wu, W.K.K. HULC: An oncogenic long non-coding RNA in human cancer. J. Cell Mol. Med. 2017, 21, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Wu, H.; Ni, P.; Gu, Z.; Qiao, Y.; Chen, N.; Sun, F.; Fan, Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010, 38, 5366–5383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R.J. Translational Control in Cancer. Cold Spring Harb. Perspect Biol. 2019, 11, a032896. [Google Scholar] [CrossRef] [Green Version]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.-H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhan, M.; Becker, K.G.; Gorospe, M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell 2012, 47, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Shaulian, E. AP-1--The Jun proteins: Oncogenes or tumor suppressors in disguise? Cell Signal. 2010, 22, 894–899. [Google Scholar] [CrossRef]
- Fu, Y.; Zheng, S.; An, N.; Athanasopoulos, T.; Popplewell, L.; Liang, A.; Li, K.; Hu, C.; Zhu, Y. β-catenin as a potential key target for tumor suppression. Int. J. Cancer 2011, 129, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lei, Z.-J.; Guo, Y.; Wang, T.; Qin, Z.-Y.; Xiao, H.-L.; Fan, L.-L.; Chen, D.-F.; Bian, X.-W.; Liu, J.; et al. miRNA-regulated delivery of lincRNA-p21 suppresses β-catenin signaling and tumorigenicity of colorectal cancer stem cells. Oncotarget 2015, 6, 37852–37870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Yang, X.; Li, J.; Yang, W.; Ma, H.; Zhang, Z. p53-targeted lincRNA-p21 acts as a tumor suppressor by inhibiting JAK2/STAT3 signaling pathways in head and neck squamous cell carcinoma. Mol. Cancer 2019, 18, 38. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.-H.; Yao, R.-W.; Zhang, Y.; Guo, C.-J.; Jiang, S.; Xu, G.; Dong, R.; Yang, L.; Chen, L.-L. SLERT Regulates DDX21 Rings Associated with Pol I Transcription. Cell 2017, 169, 664–678.e16. [Google Scholar] [CrossRef]
- Hansji, H.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Cameron-Smith, D.; Figueiredo, V.C.; Askarian-Amiri, M.E. ZFAS1: A long noncoding RNA associated with ribosomes in breast cancer cells. Biol. Direct 2016, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Qian, S.-B. Translational reprogramming in cellular stress response. Wiley Interdiscip. Rev. RNA 2014, 5, 301–315. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppek, K.; Das, R.; Barna, M. Functional 5’ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Beltran, M.; Puig, I.; Peña, C.; García, J.M.; Alvarez, A.B.; Peña, R.; Bonilla, F.; de Herreros, A.G. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef] [Green Version]
- Lan, T.; Chang, L.; Wu, L.; Yuan, Y. Downregulation of ZEB2-AS1 decreased tumor growth and metastasis in hepatocellular carcinoma. Mol. Med. Rep. 2016, 14, 4606–4612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Li, H.; Sun, R.; Li, P.; Yang, Z.; Liu, Y.; Wang, Z.; Yang, Y.; Yin, C. Long non-coding RNA ZEB2-AS1 promotes the proliferation, metastasis and epithelial mesenchymal transition in triple-negative breast cancer by epigenetically activating ZEB2. J. Cell Mol. Med. 2019, 23, 3271–3279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xiao, Z.-D.; Han, L.; Zhang, J.; Lee, S.-W.; Wang, W.; Lee, H.; Zhuang, L.; Chen, J.; Lin, H.-K.; et al. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat. Cell Biol. 2016, 18, 431–442. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Z.; Feng, Y.; Hu, X.; Yuan, J.; Zhao, S.D.; Zhang, Y.; Yang, L.; Shan, W.; He, Q.; et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer Cell 2015, 28, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Carlevaro-Fita, J.; Lanzós, A.; Feuerbach, L.; Hong, C.; Mas-Ponte, D.; Pedersen, J.S.; PCAWG Drivers and Functional Interpretation Group; Johnson, R.; PCAWG Consortium. Cancer LncRNA Census reveals evidence for deep functional conservation of long noncoding RNAs in tumorigenesis. Commun. Biol. 2020, 3, 56. [Google Scholar] [CrossRef]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Zhou, L.; Wu, L.-M.; Lai, M.-C.; Xie, H.-Y.; Zhang, F.; Zheng, S.-S. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann. Surg. Oncol. 2011, 18, 1243–1250. [Google Scholar] [CrossRef]
- Gutschner, T.; Hämmerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef] [Green Version]
- Okugawa, Y.; Toiyama, Y.; Hur, K.; Toden, S.; Saigusa, S.; Tanaka, K.; Inoue, Y.; Mohri, Y.; Kusunoki, M.; Boland, C.R.; et al. Metastasis-associated long non-coding RNA drives gastric cancer development and promotes peritoneal metastasis. Carcinogenesis 2014, 35, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Bai, H.-S.; Deng, Y.; Fan, L. High MALAT1 expression predicts a poor prognosis of cervical cancer and promotes cancer cell growth and invasion. Eur. Rev. Med. Pharm. Sci. 2015, 19, 3187–3193. [Google Scholar]
- Li, C.; Cui, Y.; Liu, L.-F.; Ren, W.-B.; Li, Q.-Q.; Zhou, X.; Li, Y.-L.; Li, Y.; Bai, X.-Y.; Zu, X.-B. High Expression of Long Noncoding RNA MALAT1 Indicates a Poor Prognosis and Promotes Clinical Progression and Metastasis in Bladder Cancer. Clin. Genitourin. Cancer 2017, 15, 570–576. [Google Scholar] [CrossRef]
- Jin, Y.; Feng, S.-J.; Qiu, S.; Shao, N.; Zheng, J.-H. LncRNA MALAT1 promotes proliferation and metastasis in epithelial ovarian cancer via the PI3K-AKT pathway. Eur. Rev. Med. Pharm. Sci. 2017, 21, 3176–3184. [Google Scholar]
- Arun, G.; Diermeier, S.; Akerman, M.; Chang, K.-C.; Wilkinson, J.E.; Hearn, S.; Kim, Y.; MacLeod, A.R.; Krainer, A.R.; Norton, L.; et al. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016, 30, 34–51. [Google Scholar] [CrossRef] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Jadaliha, M.; Zong, X.; Malakar, P.; Ray, T.; Singh, D.K.; Freier, S.M.; Jensen, T.; Prasanth, S.G.; Karni, R.; Ray, P.S.; et al. Functional and prognostic significance of long non-coding RNA MALAT1 as a metastasis driver in ER negative lymph node negative breast cancer. Oncotarget 2016, 7, 40418–40436. [Google Scholar] [CrossRef] [Green Version]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [Green Version]
- Schorderet, P.; Duboule, D. Structural and functional differences in the long non-coding RNA hotair in mouse and human. PLoS Genet. 2011, 7, e1002071. [Google Scholar] [CrossRef]
- Somarowthu, S.; Legiewicz, M.; Chillón, I.; Marcia, M.; Liu, F.; Pyle, A.M. HOTAIR forms an intricate and modular secondary structure. Mol. Cell 2015, 58, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Mandal, P.; Sadhukhan, T.; Roy Chowdhury, R.; Ranjan Mondal, N.; Chakravarty, B.; Chatterjee, T.; Roy, S.; Sengupta, S. Bridging Links between Long Noncoding RNA HOTAIR and HPV Oncoprotein E7 in Cervical Cancer Pathogenesis. Sci. Rep. 2015, 5, 11724. [Google Scholar] [CrossRef] [Green Version]
- Kogo, R.; Shimamura, T.; Mimori, K.; Kawahara, K.; Imoto, S.; Sudo, T.; Tanaka, F.; Shibata, K.; Suzuki, A.; Komune, S.; et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011, 71, 6320–6326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Vendramin, R.; Verheyden, Y.; Ishikawa, H.; Goedert, L.; Nicolas, E.; Saraf, K.; Armaos, A.; Delli Ponti, R.; Izumikawa, K.; Mestdagh, P.; et al. SAMMSON fosters cancer cell fitness by concertedly enhancing mitochondrial and cytosolic translation. Nat. Struct. Mol. Biol. 2018, 25, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Seiler, J.; Breinig, M.; Caudron-Herger, M.; Polycarpou-Schwarz, M.; Boutros, M.; Diederichs, S. The lncRNA VELUCT strongly regulates viability of lung cancer cells despite its extremely low abundance. Nucleic Acids Res. 2017, 45, 5458–5469. [Google Scholar] [CrossRef]
- Shtivelman, E.; Henglein, B.; Groitl, P.; Lipp, M.; Bishop, J.M. Identification of a human transcription unit affected by the variant chromosomal translocations 2;8 and 8;22 of Burkitt lymphoma. Proc. Natl. Acad. Sci. USA 1989, 86, 3257–3260. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.; Spizzo, R.; Atlasi, Y.; Nicoloso, M.; Shimizu, M.; Redis, R.S.; Nishida, N.; Gafà, R.; Song, J.; Guo, Z.; et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013, 23, 1446–1461. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.; Cui, R.; Jeon, Y.-J.; Lee, J.-H.; Lee, J.H.; Sim, H.; Park, J.K.; Fadda, P.; Tili, E.; Nakanishi, H.; et al. Long-range interaction and correlation between MYC enhancer and oncogenic long noncoding RNA CARLo-5. Proc. Natl. Acad. Sci. USA 2014, 111, 4173–4178. [Google Scholar] [CrossRef] [Green Version]
- Prensner, J.R.; Chen, W.; Han, S.; Iyer, M.K.; Cao, Q.; Kothari, V.; Evans, J.R.; Knudsen, K.E.; Paulsen, M.T.; Ljungman, M.; et al. The long non-coding RNA PCAT-1 promotes prostate cancer cell proliferation through cMyc. Neoplasia 2014, 16, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Doose, G.; Haake, A.; Bernhart, S.H.; López, C.; Duggimpudi, S.; Wojciech, F.; Bergmann, A.K.; Borkhardt, A.; Burkhardt, B.; Claviez, A.; et al. MINCR is a MYC-induced lncRNA able to modulate MYC’s transcriptional network in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 2015, 112, E5261–E5270. [Google Scholar] [CrossRef] [Green Version]
- Huppi, K.; Pitt, J.J.; Wahlberg, B.M.; Caplen, N.J. The 8q24 gene desert: An oasis of non-coding transcriptional activity. Front. Genet. 2012, 3, 69. [Google Scholar] [CrossRef] [Green Version]
- Iaccarino, I. lncRNAs and MYC: An Intricate Relationship. Int. J. Mol. Sci. 2017, 18, 1497. [Google Scholar] [CrossRef] [Green Version]
- Webb, E.; Adams, J.M.; Cory, S. Variant (6;15) translocation in a murine plasmacytoma occurs near an immunoglobulin kappa gene but far from the myc oncogene. Nature 1984, 312, 777–779. [Google Scholar] [CrossRef]
- Cory, S.; Graham, M.; Webb, E.; Corcoran, L.; Adams, J.M. Variant (6;15) translocations in murine plasmacytomas involve a chromosome 15 locus at least 72 kb from the c-myc oncogene. Embo J. 1985, 4, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Shtivelman, E.; Bishop, J.M. The PVT gene frequently amplifies with MYC in tumor cells. Mol. Cell. Biol. 1989, 9, 1148–1154. [Google Scholar] [CrossRef]
- Graham, M.; Adams, J.M. Chromosome 8 breakpoint far 3” of the c-myc oncogene in a Burkitt”s lymphoma 2;8 variant translocation is equivalent to the murine pvt-1 locus. Embo J. 1986, 5, 2845–2851. [Google Scholar] [CrossRef]
- Siwarski, D.; Müller, U.; Andersson, J.; Notario, V.; Melchers, F.; Rolink, A.; Huppi, K. Structure and expression of the c-Myc/Pvt 1 megagene locus. Curr. Top. Microbiol. Immunol. 1997, 224, 67–72. [Google Scholar]
- Nagoshi, H.; Taki, T.; Hanamura, I.; Nitta, M.; Otsuki, T.; Nishida, K.; Okuda, K.; Sakamoto, N.; Kobayashi, S.; Yamamoto-Sugitani, M.; et al. Frequent PVT1 rearrangement and novel chimeric genes PVT1–NBEA and PVT1–WWOX occur in multiple myeloma with 8q24 abnormality. Cancer Res. 2012, 72, 4954–4962. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Shih, D.J.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef]
- Tseng, Y.-Y.; Moriarity, B.S.; Gong, W.; Akiyama, R.; Tiwari, A.; Kawakami, H.; Ronning, P.; Reuland, B.; Guenther, K.; Beadnell, T.C.; et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 2014, 512, 1–19. [Google Scholar] [CrossRef]
- Cui, M.; You, L.; Ren, X.; Zhao, W.; Liao, Q.; Zhao, Y. Biochemical and Biophysical Research Communications. Biochem. Biophys. Res. Commun. 2016, 471, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Kuo, W.-L.; Stilwell, J.L.; Takano, H.; Lapuk, A.V.; Fridlyand, J.; Mao, J.-H.; Yu, M.; Miller, M.A.; Santos, J.L.; et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin. Cancer Res. 2007, 13, 5745–5755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riquelme, E.; Suraokar, M.B.; Rodriguez, J.; Mino, B.; Lin, H.Y.; Rice, D.C.; Tsao, A.; Wistuba, I.I. Frequent coamplification and cooperation between C-MYC and PVT1 oncogenes promote malignant pleural mesothelioma. J. Thorac. Oncol. 2014, 9, 998–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Li, D.; Gong, M.; Wang, J.; Huang, X.; Wu, T.; Wang, C. Expression and clinical significance of the long non-coding RNA PVT1 in human gastric cancer. Onco Targets 2014, 7, 1625–1630. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-R.; Zang, S.-Z.; Zhong, C.-L.; Li, Y.-X.; Zhao, S.-S.; Feng, X.-J. Increased expression of the lncRNA PVT1 promotes tumorigenesis in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 6929–6935. [Google Scholar]
- Huang, C.; Yu, W.; Wang, Q.; Cui, H.; Wang, Y.; Zhang, L.; Han, F.; Huang, T. Increased expression of the lncRNA PVT1 is associated with poor prognosis in pancreatic cancer patients. Minerva Med. 2015, 106, 143–149. [Google Scholar]
- Zeng, C.; Yu, X.; Lai, J.; Yang, L.; Chen, S.; Li, Y. Overexpression of the long non-coding RNA PVT1 is correlated with leukemic cell proliferation in acute promyelocytic leukemia. J. Hematol. Oncol. 2015, 8, 126. [Google Scholar] [CrossRef] [Green Version]
- Prensner, J.R.; Chen, W.; Iyer, M.K.; Cao, Q.; Ma, T.; Han, S.; Sahu, A.; Malik, R.; Wilder-Romans, K.; Navone, N.; et al. PCAT-1, a long noncoding RNA, regulates BRCA2 and controls homologous recombination in cancer. Cancer Res. 2014, 74, 1651–1660. [Google Scholar] [CrossRef] [Green Version]
- Xiang, J.-F.; Yin, Q.-F.; Chen, T.; Zhang, Y.; Zhang, X.-O.; Wu, Z.; Zhang, S.; Wang, H.-B.; Ge, J.; Lu, X.; et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Nat. Publ. Group 2014, 24, 513–531. [Google Scholar]
- Chen, L.; Wang, W.; Cao, L.; Li, Z.; Wang, X. Long Non-Coding RNA CCAT1 Acts as a Competing Endogenous RNA to Regulate Cell Growth and Differentiation in Acute Myeloid Leukemia. Mol. Cells 2016, 39, 330–336. [Google Scholar]
- Chen, S.; Gu, T.; Lu, Z.; Qiu, L.; Xiao, G.; Zhu, X.; Li, F.; Yu, H.; Li, G.; Liu, H. Roles of MYC-targeting long non-coding RNA MINCR in cell cycle regulation and apoptosis in non-small cell lung Cancer. Respir. Res. 2019, 20, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, G.; Hu, X.; Wu, L.; Feng, Y.; He, S.; Zhang, Y.; Hu, Z.; Yang, L.; Tian, T.; et al. Oncogenic RAS regulates long non-coding RNA Orilnc1 in human cancer. Cancer Res. 2017, 77, 3745–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, R.; Esposito, D.; Pallante, P.; Fusco, A.; Ciccodicola, A.; Costa, V. Oncogenic Properties of the Antisense lncRNA COMETin BRAF- and RET-Driven Papillary Thyroid Carcinomas. Cancer Res. 2019, 79, 2124–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibb, E.A.; Vucic, E.A.; Enfield, K.S.S.; Stewart, G.L.; Lonergan, K.M.; Kennett, J.Y.; Becker-Santos, D.D.; MacAulay, C.E.; Lam, S.; Brown, C.J.; et al. Human cancer long non-coding RNA transcriptomes. PLoS ONE 2011, 6, e25915. [Google Scholar] [CrossRef] [Green Version]
- Hudson, W.H.; Pickard, M.R.; de Vera, I.M.S.; Kuiper, E.G.; Mourtada-Maarabouni, M.; Conn, G.L.; Kojetin, D.J.; Williams, G.T.; Ortlund, E.A. Conserved sequence-specific lincRNA-steroid receptor interactions drive transcriptional repression and direct cell fate. Nat. Commun. 2014, 5, 5395. [Google Scholar] [CrossRef] [Green Version]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; King, R.M.; Philipson, L. Genes specifically expressed at growth arrest of mammalian cells. Cell 1988, 54, 787–793. [Google Scholar] [CrossRef]
- Mourtada-Maarabouni, M.; Pickard, M.R.; Hedge, V.L.; Farzaneh, F.; Williams, G.T. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Nat. Publ. Group 2009, 28, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal. 2010, 3, ra8. [Google Scholar] [CrossRef] [Green Version]
- Pickard, M.R.; Williams, G.T. Molecular and Cellular Mechanisms of Action of Tumour Suppressor GAS5 LncRNA. Genes 2015, 6, 484–499. [Google Scholar] [CrossRef] [Green Version]
- Goustin, A.S.; Thepsuwan, P.; Kosir, M.A.; Lipovich, L. The Growth-Arrest-Specific (GAS)-5 Long Non-Coding RNA: A Fascinating lncRNA Widely Expressed in Cancers. Noncoding RNA 2019, 5, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster-Gossler, K.; Bilinski, P.; Sado, T.; Ferguson-Smith, A.; Gossler, A. The mouse Gtl2 gene is differentially expressed during embryonic development, encodes multiple alternatively spliced transcripts, and may act as an RNA. Dev. Dyn 1998, 212, 214–228. [Google Scholar] [CrossRef]
- Miyoshi, N.; Wagatsuma, H.; Wakana, S.; Shiroishi, T.; Nomura, M.; Aisaka, K.; Kohda, T.; Surani, M.A.; Kaneko-Ishino, T.; Ishino, F. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells 2000, 5, 211–220. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, Y.; Mehta, K.R.; Danila, D.C.; Scolavino, S.; Johnson, S.R.; Klibanski, A. A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocrinol. Metab. 2003, 88, 5119–5126. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhong, Y.; Wang, Y.; Zhang, X.; Batista, D.L.; Gejman, R.; Ansell, P.J.; Zhao, J.; Weng, C.; Klibanski, A. Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 2007, 282, 24731–24742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, D.; Chen, X.; Li, J.; Li, L.; Bian, Z.; Sun, F.; Lu, J.; Yin, Y.; Cai, X.; et al. Secreted Monocytic miR-150 Enhances Targeted Endothelial Cell Migration. Mol. Cell 2010, 39, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Braconi, C.; Kogure, T.; Valeri, N.; Huang, N.; Nuovo, G.; Costinean, S.; Negrini, M.; Miotto, E.; Croce, C.M.; Patel, T. microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Nat. Publ. Group 2011, 30, 4750–4756. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Ren, Z.; Sun, P. Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation. J. Cell. Biochem. 2012, 113, 1868–1874. [Google Scholar] [CrossRef]
- Lyu, Y.; Lou, J.; Yang, Y.; Feng, J.; Hao, Y.; Huang, S.; Yin, L.; Xu, J.; Huang, D.; Ma, B.; et al. Dysfunction of the WT1-MEG3 signaling promotes AML leukemogenesis via p53-dependent and -independent pathways. Leukemia 2017, 31, 2543–2551. [Google Scholar] [CrossRef]
- Li, J.; Zi, Y.; Wang, W.; Li, Y. Long Noncoding RNA MEG3 Inhibits Cell Proliferation and Metastasis in Chronic Myeloid Leukemia via Targeting miR-184. Oncol Res. 2018, 26, 297–305. [Google Scholar] [CrossRef]
- Astuti, D.; Latif, F.; Wagner, K.; Gentle, D.; Cooper, W.N.; Catchpoole, D.; Grundy, R.; Ferguson-Smith, A.C.; Maher, E.R. Epigenetic alteration at the DLK1-GTL2 imprinted domain in human neoplasia: Analysis of neuroblastoma, phaeochromocytoma and Wilms’ tumour. Br. J. Cancer 2005, 92, 1574–1580. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhang, X.; Klibanski, A. MEG3 noncoding RNA: A tumor suppressor. J. Mol. Endocrinol. 2012, 48, R45–R53. [Google Scholar] [CrossRef] [PubMed]
- Gou, Q.; Gao, L.; Nie, X.; Pu, W.; Zhu, J.; Wang, Y.; Liu, X.; Tan, S.; Zhou, J.-K.; Gong, Y.; et al. Long Noncoding RNA AB074169 Inhibits Cell Proliferation via Modulation of KHSRP-Mediated CDKN1a Expression in Papillary Thyroid Carcinoma. Cancer Res. 2018, 78, 4163–4174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kopp, F.; Chang, T.-C.; Sataluri, A.; Chen, B.; Sivakumar, S.; Yu, H.; Xie, Y.; Mendell, J.T. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell 2016, 164, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tichon, A.; Gil, N.; Lubelsky, Y.; Havkin Solomon, T.; Lemze, D.; Itzkovitz, S.; Stern-Ginossar, N.; Ulitsky, I. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat. Commun. 2016, 7, 12209–12210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrova, N.; Zamudio, J.R.; Jong, R.M.; Soukup, D.; Resnick, R.; Sarma, K.; Ward, A.J.; Raj, A.; Lee, J.T.; Sharp, P.A.; et al. LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Mol. Cell 2014, 54, 777–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, H.; Fesler, A.; Schee, K.; Fodstad, O.; Flatmark, K.; Ju, J. Clinical significance of long intergenic noncoding RNA-p21 in colorectal cancer. Clin. Colorectal. Cancer 2013, 12, 261–266. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, H.; Mei, Y.; Wu, M. Reciprocal regulation of HIF-1α and lincRNA-p21 modulates the Warburg effect. Mol. Cell 2014, 53, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Doucrasy, S.; Coll, J.; Barrois, M.; Joubel, A.; Prost, S.; Dozier, C.; Stehelin, D.; Riou, G. Expression of the human fetal bac h19 gene in invasive cancers. Int. J. Oncol. 1993, 2, 753–758. [Google Scholar] [CrossRef]
- Berteaux, N.; Lottin, S.; Monté, D.; Pinte, S.; Quatannens, B.; Coll, J.; Hondermarck, H.; Curgy, J.-J.; Dugimont, T.; Adriaenssens, E. H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J. Biol. Chem. 2005, 280, 29625–29636. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.L.; Weintraub, H.; Lassar, A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987, 51, 987–1000. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Zemel, S.; Tilghman, S.M. Parental imprinting of the mouse H19 gene. Nature 1991, 351, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Dugimont, T.; Montpellier, C.; Adriaenssens, E.; Lottin, S.; Dumont, L.; Iotsova, V.; Lagrou, C.; Stehelin, D.; Coll, J.; Curgy, J.J. The H19 TATA-less promoter is efficiently repressed by wild-type tumor suppressor gene product p53. Oncogene 1998, 16, 2395–2401. [Google Scholar] [CrossRef] [PubMed]
- Barsyte-Lovejoy, D.; Lau, S.K.; Boutros, P.C.; Khosravi, F.; Jurisica, I.; Andrulis, I.L.; Tsao, M.S.; Penn, L.Z. The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 2006, 66, 5330–5337. [Google Scholar] [CrossRef] [Green Version]
- Matouk, I.J.; Mezan, S.; Mizrahi, A.; Ohana, P.; Abu-Lail, R.; Fellig, Y.; Degroot, N.; Galun, E.; Hochberg, A. The oncofetal H19 RNA connection: Hypoxia, p53 and cancer. Biochim. Biophys. Acta 2010, 1803, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Rüger, R. Long Non-coding RNAs and their Role in Metastasis. Cancer Genom. Proteom. 2017, 14, 143–160. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Crenshaw, T.; Moulton, T.; Newcomb, E.; Tycko, B. Tumour-suppressor activity of H19 RNA. Nature 1993, 365, 764–767. [Google Scholar] [CrossRef] [PubMed]
- Yoshimizu, T.; Miroglio, A.; Ripoche, M.-A.; Gabory, A.; Vernucci, M.; Riccio, A.; Colnot, S.; Godard, C.; Terris, B.; Jammes, H.; et al. The H19 locus acts in vivo as a tumor suppressor. Proc. Natl. Acad. Sci. USA 2008, 105, 12417–12422. [Google Scholar] [CrossRef] [Green Version]
- Gabory, A.; Ripoche, M.-A.; Le Digarcher, A.; Watrin, F.; Ziyyat, A.; Forné, T.; Jammes, H.; Ainscough, J.F.X.; Surani, M.A.; Journot, L.; et al. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development 2009, 136, 3413–3421. [Google Scholar] [CrossRef] [Green Version]
- Kallen, A.N.; Zhou, X.-B.; Xu, J.; Qiao, C.; Ma, J.; Yan, L.; Lu, L.; Liu, C.; Yi, J.-S.; Zhang, H.; et al. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol. Cell 2013, 52, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Tsang, W.P.; Ng, E.K.O.; Ng, S.S.M.; Jin, H.; Yu, J.; Sung, J.J.Y.; Kwok, T.T. Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis 2010, 31, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 2009, 33, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Rheinbay, E.; Parasuraman, P.; Grimsby, J.; Tiao, G.; Engreitz, J.M.; Kim, J.; Lawrence, M.S.; Taylor-Weiner, A.; Rodriguez-Cuevas, S.; Rosenberg, M.; et al. Recurrent and functional regulatory mutations in breast cancer. Nature 2017, 547, 55–60. [Google Scholar] [PubMed]
- Knutsen, E.; Lellahi, S.M.; Aure, M.R.; Nord, S.; Fismen, S.; Larsen, K.B.; Gabriel, M.T.; Hedberg, A.; Bjørklund, S.S.; Oslo Breast Cancer Research Consortium (OSBREAC); et al. The expression of the long NEAT1_2 isoform is associated with human epidermal growth factor receptor 2-positive breast cancers. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mello, S.S.; Sinow, C.; Raj, N.; Mazur, P.K.; Bieging-Rolett, K.; Broz, D.K.; Imam, J.F.C.; Vogel, H.; Wood, L.D.; Sage, J.; et al. Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes Dev. 2017, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, C.; Xu, Y.; Xu, L.; Yu, X.; Cheng, J.; Yang, L.; Chen, S.; Li, Y. Inhibition of long non-coding RNA NEAT1 impairs myeloid differentiation in acute promyelocytic leukemia cells. BMC Cancer 2014, 14, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Wang, S.; Zhao, Y.; Du, F.; Wang, W.; Lv, P.; Qi, L. Long noncoding RNA NEAT1 modulates cell proliferation and apoptosis by regulating miR-23a-3p/SMC1A in acute myeloid leukemia. J. Cell Physiol. 2019, 234, 6161–6172. [Google Scholar] [CrossRef]
- Flockhart, R.J.; Webster, D.E.; Qu, K.; Mascarenhas, N.; Kovalski, J.; Kretz, M.; Khavari, P.A. BRAFV600E remodels the melanocyte transcriptome and induces BANCR to regulate melanoma cell migration. Genome Res. 2012, 22, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Zhang, L.; Jia, L.; Duan, Y.; Li, Y.; Bao, L.; Sha, N. Long non-coding RNA BANCR promotes proliferation in malignant melanoma by regulating MAPK pathway activation. PLoS ONE 2014, 9, e100893. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Liu, X.-H.; Wang, K.-M.; Nie, F.-Q.; Kong, R.; Yang, J.-S.; Xia, R.; Xu, T.-P.; Jin, F.-Y.; Liu, Z.-J.; et al. Downregulation of BRAF activated non-coding RNA is associated with poor prognosis for non-small cell lung cancer and promotes metastasis by affecting epithelial-mesenchymal transition. Mol. Cancer 2014, 13, 68. [Google Scholar] [CrossRef] [Green Version]
- Xue, S.; Jiang, S.-Q.; Li, Q.-W.; Wang, S.; Li, J.; Yang, S.; Zhang, H.-M.; Xu, Y.-F.; Wang, L.-S.; Zheng, J.-H. Decreased expression of BRAF-activated long non-coding RNA is associated with the proliferation of clear cell renal cell carcinoma. BMC Urol. 2018, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Goyal, A.; Myacheva, K.; Gross, M.; Klingenberg, M.; Duran Arqué, B.; Diederichs, S. Challenges of CRISPR/Cas9 applications for long non-coding RNA genes. Nucleic Acids Res. 2017, 45, e12. [Google Scholar] [CrossRef] [Green Version]
- Awwad, D.A. Beyond classic editing: Innovative CRISPR approaches for functional studies of long non-coding RNA. Biol. Methods Protoc. 2019, 4, bpz017. [Google Scholar] [PubMed]
- Han, J.; Zhang, J.; Chen, L.; Shen, B.; Zhou, J.; Hu, B.; Du, Y.; Tate, P.H.; Huang, X.; Zhang, W. Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol. 2014, 11, 829–835. [Google Scholar] [CrossRef]
- Yin, Y.; Yan, P.; Lu, J.; Song, G.; Zhu, Y.; Li, Z.; Zhao, Y.; Shen, B.; Huang, X.; Zhu, H.; et al. Opposing Roles for the lncRNA Haunt and Its Genomic Locus in Regulating HOXA Gene Activation during Embryonic Stem Cell Differentiation. Cell Stem Cell 2015, 16, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Aparicio-Prat, E.; Arnan, C.; Sala, I.; Bosch, N.; Guigó, R.; Johnson, R. DECKO: Single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs. BMC Genom. 2016, 16, 1–15. [Google Scholar]
- Esposito, R.; Bosch, N.; Lanzós, A.; Polidori, T.; Pulido-Quetglas, C.; Johnson, R. Hacking the Cancer Genome: Profiling Therapeutically Actionable Long Non-coding RNAs Using CRISPR-Cas9 Screening. Cancer Cell 2019, 35, 545–557. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, L. Functional genomics: Screening for lncRNA function. Nat. Rev. Genet. 2017, 18, 70. [Google Scholar]
- Zhu, S.; Li, W.; Liu, J.; Chen, C.-H.; Liao, Q.; Xu, P.; Xu, H.; Xiao, T.; Cao, Z.; Peng, J.; et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat. Biotechnol. 2016, 34, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Han, X.; Yuan, J.; Geng, T.; Chen, S.; Hu, X.; Cui, I.H.; Cui, H. Biallelic insertion of a transcriptional terminator via the CRISPR/Cas9 system efficiently silences expression of protein-coding and non-coding RNA genes. J. Biol. Chem. 2017, 292, 5624–5633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Zhang, G.; Li, J.; Zhang, X.; Huang, S.; Xiang, S.; Hu, X.; Liu, C. CRISPRlnc: A manually curated database of validated sgRNAs for lncRNAs. Nucleic Acids Res. 2019, 47, D63–D68. [Google Scholar] [CrossRef] [Green Version]
- Pulecio, J.; Verma, N.; Mejía-Ramírez, E.; Huangfu, D.; Raya, A. CRISPR/Cas9-Based Engineering of the Epigenome. Cell Stem Cell 2017, 21, 431–447. [Google Scholar] [CrossRef] [Green Version]
- Janga, H.; Aznaourova, M.; Boldt, F.; Damm, K.; Grünweller, A.; Schulte, L.N. Cas9-mediated excision of proximal DNaseI/H3K4me3 signatures confers robust silencing of microRNA and long non-coding RNA genes. PLoS ONE 2018, 13, e0193066. [Google Scholar] [CrossRef] [Green Version]
- Schmid-Burgk, J.L.; Höning, K.; Ebert, T.S.; Hornung, V. CRISPaint allows modular base-specific gene tagging using a ligase-4-dependent mechanism. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Cheng, T.-L.; Qiu, Z. Long non-coding RNA tagging and expression manipulation via CRISPR/Cas9-mediated targeted insertion. Protein Cell 2018, 9, 820–825. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.-Z.; Wang, Y.; Li, S.-Q.; Yao, R.-W.; Luan, P.-F.; Wu, H.; Carmichael, G.G.; Chen, L.-L. Dynamic Imaging of RNA in Living Cells by CRISPR-Cas13 Systems. Mol. Cell 2019, 76, 981–997.e7. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Cai, Y.; Tang, L.; Han, X.; Gao, F.; Cao, H.; Qi, F.; Kapranov, P. A CRISPR/Cas13-based approach demonstrates biological relevance of vlinc class of long non-coding RNAs in anticancer drug response. Sci. Rep. 2020, 10, 1794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; You, Y. CRISPR-Cas13a system: A novel approach to precision oncology. Cancer Biol. Med. 2020, 17, 6–8. [Google Scholar]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Hannon, G.J.; Rossi, J.J. Unlocking the potential of the human genome with RNA interference. Nature 2004, 431, 371–378. [Google Scholar] [CrossRef]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, N.; Gyntelberg, F.; Faber, J. The appraisal of chronic stress and the development of the metabolic syndrome: A systematic review of prospective cohort studies. Endocr. Connect. 2014, 3, R55–R80. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Subasinghe, C.; Shinozuka, K.; Cohen, J.S. Physicochemical properties of phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 1988, 16, 3209–3221. [Google Scholar] [CrossRef]
- Allerson, C.R.; Sioufi, N.; Jarres, R.; Prakash, T.P.; Naik, N.; Berdeja, A.; Wanders, L.; Griffey, R.H.; Swayze, E.E.; Bhat, B. Fully 2’-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem. 2005, 48, 901–904. [Google Scholar] [CrossRef]
- Yoo, B.H.; Bochkareva, E.; Bochkarev, A.; Mou, T.-C.; Gray, D.M. 2’-O-methyl-modified phosphorothioate antisense oligonucleotides have reduced non-specific effects in vitro. Nucleic Acids Res. 2004, 32, 2008–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloosterman, W.P.; Lagendijk, A.K.; Ketting, R.F.; Moulton, J.D.; Plasterk, R.H.A. Targeted inhibition of miRNA maturation with morpholinos reveals a role for miR-375 in pancreatic islet development. PLoS Biol. 2007, 5, e203. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef]
- Geary, R.S.; Baker, B.F.; Crooke, S.T. Clinical and preclinical pharmacokinetics and pharmacodynamics of mipomersen (kynamro(®)): A second-generation antisense oligonucleotide inhibitor of apolipoprotein B. Clin. Pharm. 2015, 54, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Anthony, K.; Feng, L.; Arechavala-Gomeza, V.; Guglieri, M.; Straub, V.; Bushby, K.; Cirak, S.; Morgan, J.; Muntoni, F. Exon skipping quantification by quantitative reverse-transcription polymerase chain reaction in Duchenne muscular dystrophy patients treated with the antisense oligomer eteplirsen. Hum. Gene Methods 2012, 23, 336–345. [Google Scholar] [CrossRef]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene “knockdown” in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef]
- Moulton, J.D. Using Morpholinos to Control Gene Expression. Curr. Protoc. Nucleic Acid Chem. 2017, 68, 4.30.1–4.30.29. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Janson, A.A.M.; Kaman, W.E.; Bremmer-Bout, M.; den Dunnen, J.T.; Baas, F.; van Ommen, G.-J.B.; van Deutekom, J.C.T. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum. Mol. Genet. 2003, 12, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Alter, J.; Lou, F.; Rabinowitz, A.; Yin, H.; Rosenfeld, J.; Wilton, S.D.; Partridge, T.A.; Lu, Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006, 12, 175–177. [Google Scholar] [CrossRef]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Staton, A.A.; Giraldez, A.J. Use of target protector morpholinos to analyze the physiological roles of specific miRNA-mRNA pairs in vivo. Nat. Protoc. 2011, 6, 2035–2049. [Google Scholar] [CrossRef]
- Blum, M.; De Robertis, E.M.; Wallingford, J.B.; Niehrs, C. Morpholinos: Antisense and Sensibility. Dev. Cell 2015, 35, 145–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegueroles, C.; Gabaldón, T. Secondary structure impacts patterns of selection in human lncRNAs. BMC Biol. 2016, 14, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, K.A.; Merino, E.J.; Weeks, K.M. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006, 1, 1610–1616. [Google Scholar] [CrossRef]
- Lu, Z.; Zhang, Q.C.; Lee, B.; Flynn, R.A.; Smith, M.A.; Robinson, J.T.; Davidovich, C.; Gooding, A.R.; Goodrich, K.J.; Mattick, J.S.; et al. RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 2016, 165, 1267–1279. [Google Scholar] [CrossRef] [Green Version]
- Howe, J.A.; Wang, H.; Fischmann, T.O.; Balibar, C.J.; Xiao, L.; Galgoci, A.M.; Malinverni, J.C.; Mayhood, T.; Villafania, A.; Nahvi, A.; et al. Selective small-molecule inhibition of an RNA structural element. Nature 2015, 526, 672–677. [Google Scholar] [CrossRef]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.-H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Senn, J.J.; Burel, S.; Henry, S.P. Non-CpG-containing antisense 2’-methoxyethyl oligonucleotides activate a proinflammatory response independent of Toll-like receptor 9 or myeloid differentiation factor 88. J. Pharm. Exp. 2005, 314, 972–979. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Vollmer, J.; Weeratna, R.D.; Jurk, M.; Samulowitz, U.; McCluskie, M.J.; Payette, P.; Davis, H.L.; Schetter, C.; Krieg, A.M. Oligodeoxynucleotides lacking CpG dinucleotides mediate Toll-like receptor 9 dependent T helper type 2 biased immune stimulation. Immunology 2004, 113, 212–223. [Google Scholar] [CrossRef]
- Kandimalla, E.R.; Bhagat, L.; Wang, D.; Yu, D.; Sullivan, T.; La Monica, N.; Agrawal, S. Design, synthesis and biological evaluation of novel antagonist compounds of Toll-like receptors 7, 8 and 9. Nucleic Acids Res. 2013, 41, 3947–3961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
LncRNAs in epigenetic regulation of gene expression. Schematic models of (1) trans-acting lncRNAs recruiting histone deacetylases (HDACs, upper left), histone methyltransferases (HMTs, upper right) associated to gene repression, or histone acetylases (HATs, lower left) and histone demethylases (HDMs, lower right) associated to transcription-permissive chromatin; (2) enancher RNA (eRNA) recruiting the Mediator complex—with cohesin-based stabilizing contacts—responsible for DNA looping and distal transcriptional activation of target genes; (3) lncRNAs interacting with nucleosome destabilizing proteins, such as the SWI/SNF complex (left), causing nucleosome exclusion and gene activation, or with remodeling proteins (NRPs, right) associated with chromatin condensation and transcriptional repression; and (4) lncRNA acting as a scaffold for DNA methyltransferases (DNMTs) and DNA-binding proteins (DBP), and driving gene promoters’ methylation and their transcriptional repression.
Figure 1.
LncRNAs in epigenetic regulation of gene expression. Schematic models of (1) trans-acting lncRNAs recruiting histone deacetylases (HDACs, upper left), histone methyltransferases (HMTs, upper right) associated to gene repression, or histone acetylases (HATs, lower left) and histone demethylases (HDMs, lower right) associated to transcription-permissive chromatin; (2) enancher RNA (eRNA) recruiting the Mediator complex—with cohesin-based stabilizing contacts—responsible for DNA looping and distal transcriptional activation of target genes; (3) lncRNAs interacting with nucleosome destabilizing proteins, such as the SWI/SNF complex (left), causing nucleosome exclusion and gene activation, or with remodeling proteins (NRPs, right) associated with chromatin condensation and transcriptional repression; and (4) lncRNA acting as a scaffold for DNA methyltransferases (DNMTs) and DNA-binding proteins (DBP), and driving gene promoters’ methylation and their transcriptional repression.

Figure 2.
LncRNAs in translation regulation. In the nucleus, lncRNAs PAPAS and SLERT regulate ribosome biogenesis through regulation of rRNA transcription, while Zeb2-NAT by base-paring to its sense coding gene Zeb2, causes inclusion of an IRES thus leading to its translation. In the cytoplasm, lncRNAs ZFAS1 binds the small ribosomal subunit and may have a role in mRNA translation. NBR2 attenuates global translation by interacting with AMPK and promoting its kinase activity. LincRNA-p21 regulates specific mRNA-targets by promoting their interaction with the translational repressors.
Figure 2.
LncRNAs in translation regulation. In the nucleus, lncRNAs PAPAS and SLERT regulate ribosome biogenesis through regulation of rRNA transcription, while Zeb2-NAT by base-paring to its sense coding gene Zeb2, causes inclusion of an IRES thus leading to its translation. In the cytoplasm, lncRNAs ZFAS1 binds the small ribosomal subunit and may have a role in mRNA translation. NBR2 attenuates global translation by interacting with AMPK and promoting its kinase activity. LincRNA-p21 regulates specific mRNA-targets by promoting their interaction with the translational repressors.

Figure 3.
LncRNAs involved in different cancer types. Main examples of lncRNAs with oncogenic (red) or tumor-suppressive (green) properties reported in different tumors (i.e., clockwise from top left: neuroblastoma, melanoma, lung c., breast c., ovarian c., colon c., pancreatic c., hepatocellular c., prostate c., osteosarcoma, leukemia, gastric c., papillary thyroid c.). AML = acute myeloid leukemia; APL = acute promyelocytic leukemia; CML = chronic myeloid leukemia.
Figure 3.
LncRNAs involved in different cancer types. Main examples of lncRNAs with oncogenic (red) or tumor-suppressive (green) properties reported in different tumors (i.e., clockwise from top left: neuroblastoma, melanoma, lung c., breast c., ovarian c., colon c., pancreatic c., hepatocellular c., prostate c., osteosarcoma, leukemia, gastric c., papillary thyroid c.). AML = acute myeloid leukemia; APL = acute promyelocytic leukemia; CML = chronic myeloid leukemia.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Aprile, M.; Katopodi, V.; Leucci, E.; Costa, V. LncRNAs in Cancer: From garbage to Junk. Cancers 2020, 12, 3220. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113220
AMA Style
Aprile M, Katopodi V, Leucci E, Costa V. LncRNAs in Cancer: From garbage to Junk. Cancers. 2020; 12(11):3220. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113220
Chicago/Turabian StyleAprile, Marianna, Vicky Katopodi, Eleonora Leucci, and Valerio Costa. 2020. "LncRNAs in Cancer: From garbage to Junk" Cancers 12, no. 11: 3220. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113220
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.