The Metastatic Bone Marrow Niche in Neuroblastoma: Altered Phenotype and Function of Mesenchymal Stromal Cells

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
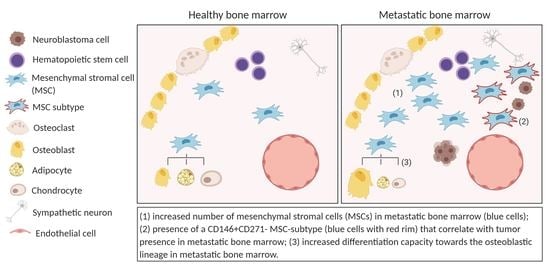
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
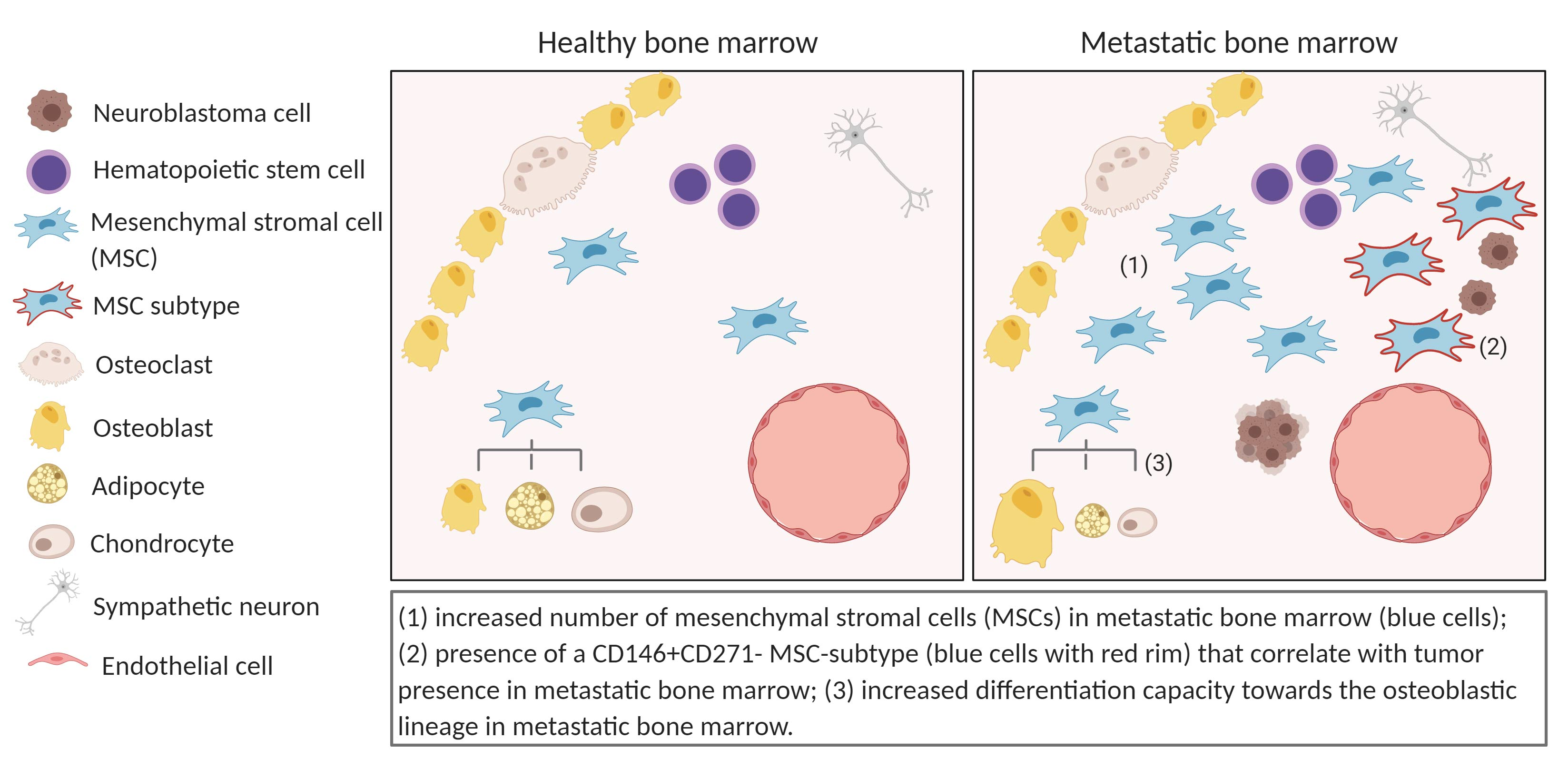
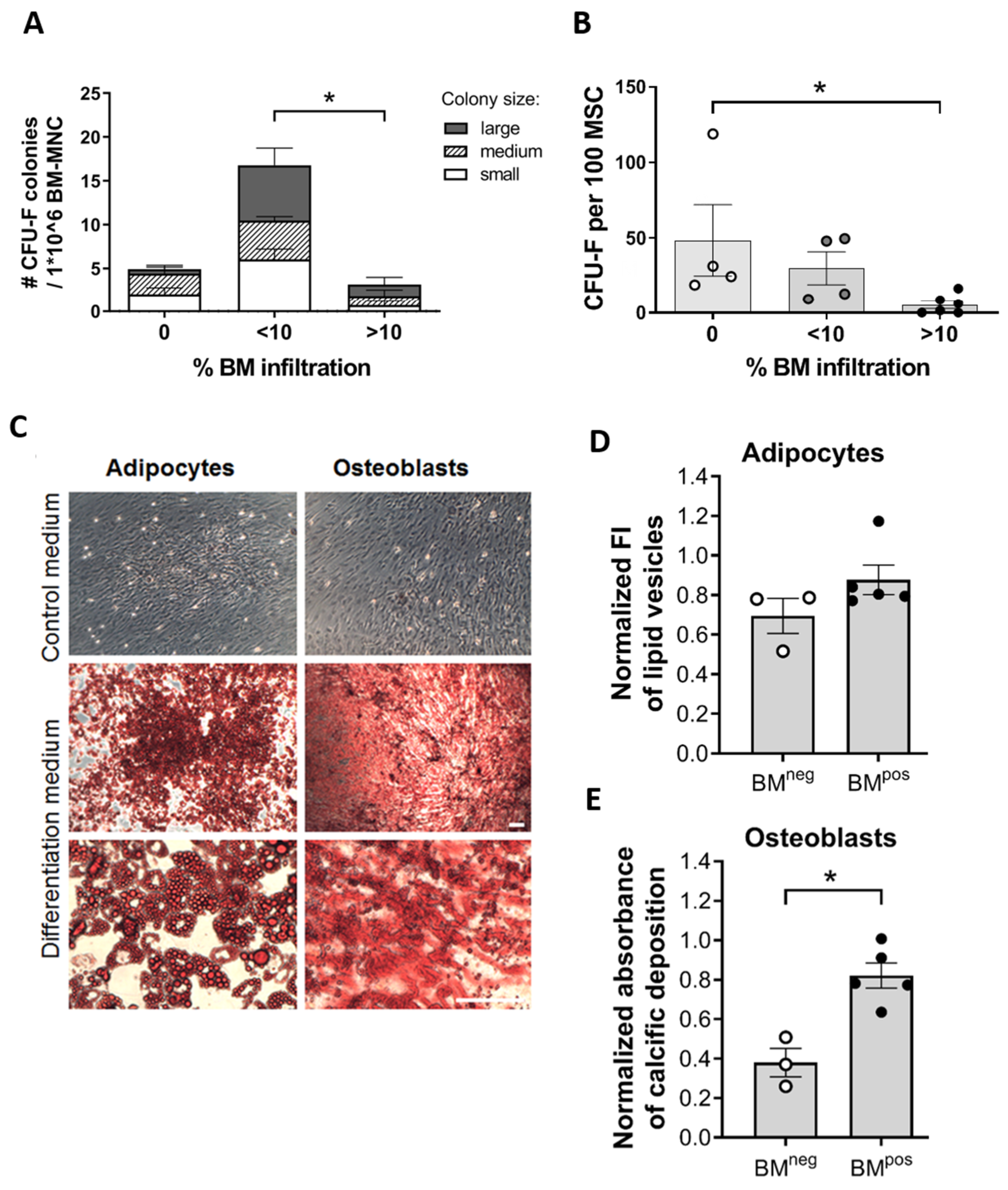
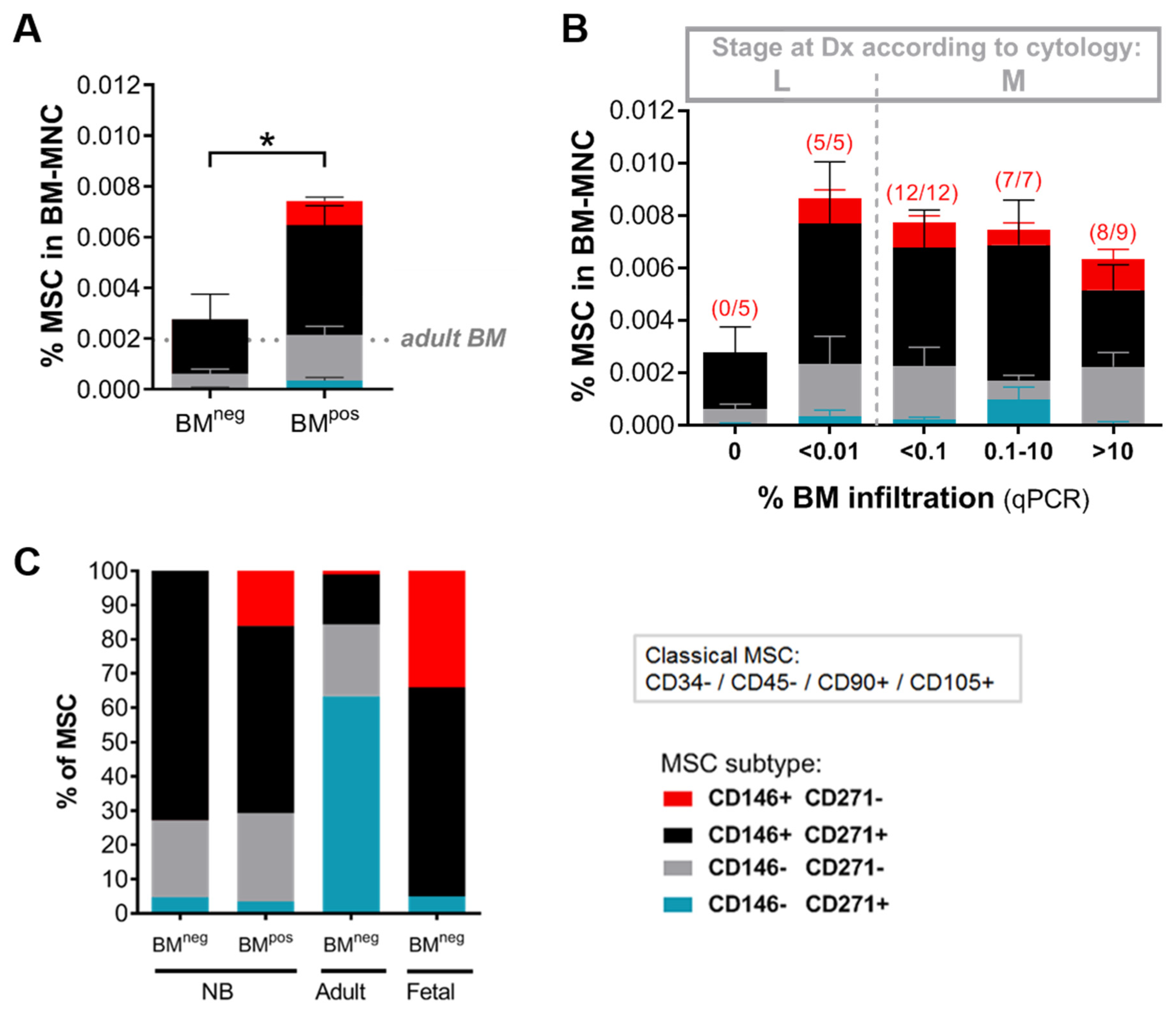
2.1. The Metastatic BM-Niche Contains a Higher Number of MSC
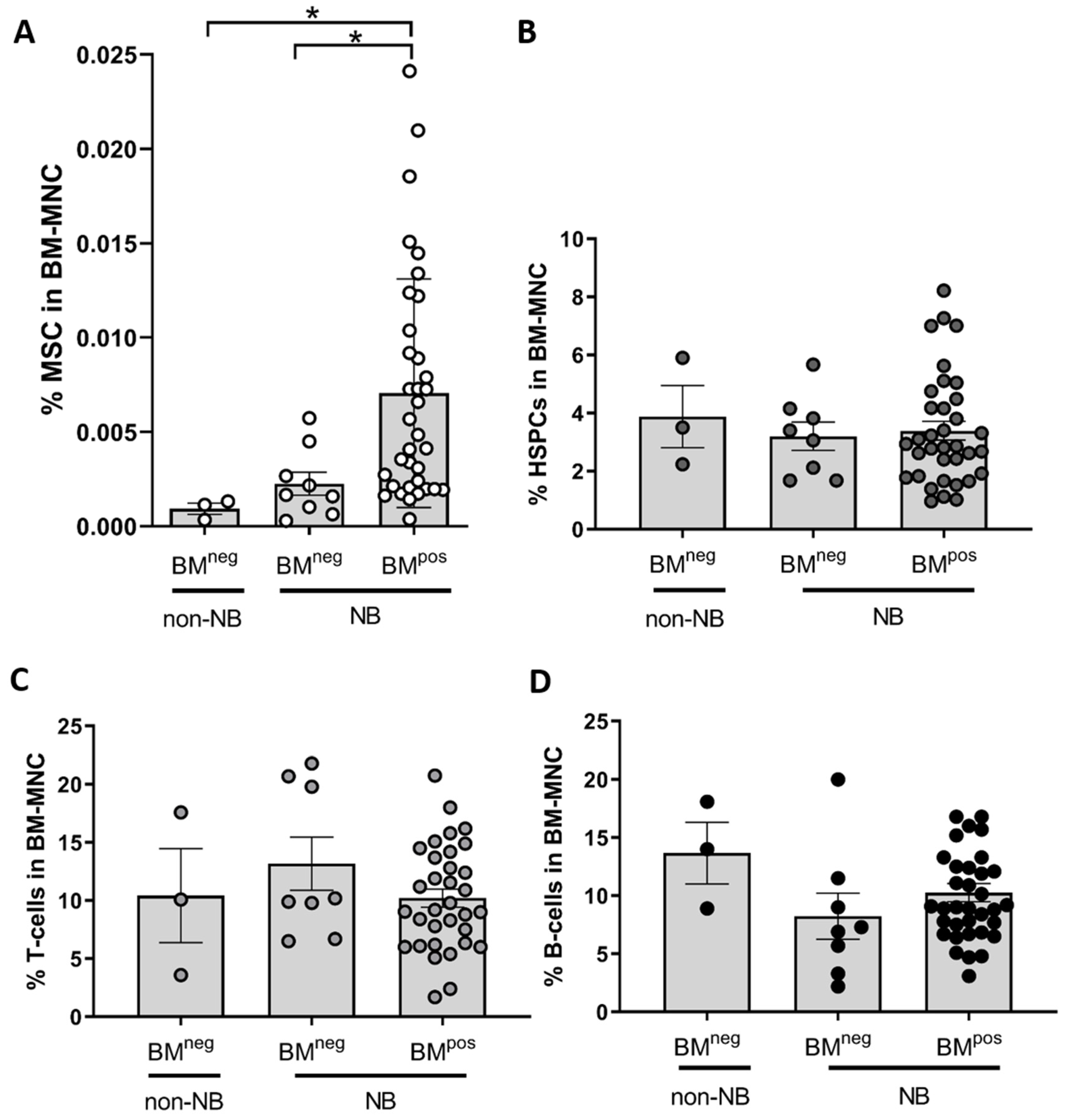
2.2. Exclude Detection of Tumor Cells with MSC-FACS-Panel
2.3. MSC in the Metastatic BM-Niche Differ Functionally and Contain a Specific Subtype
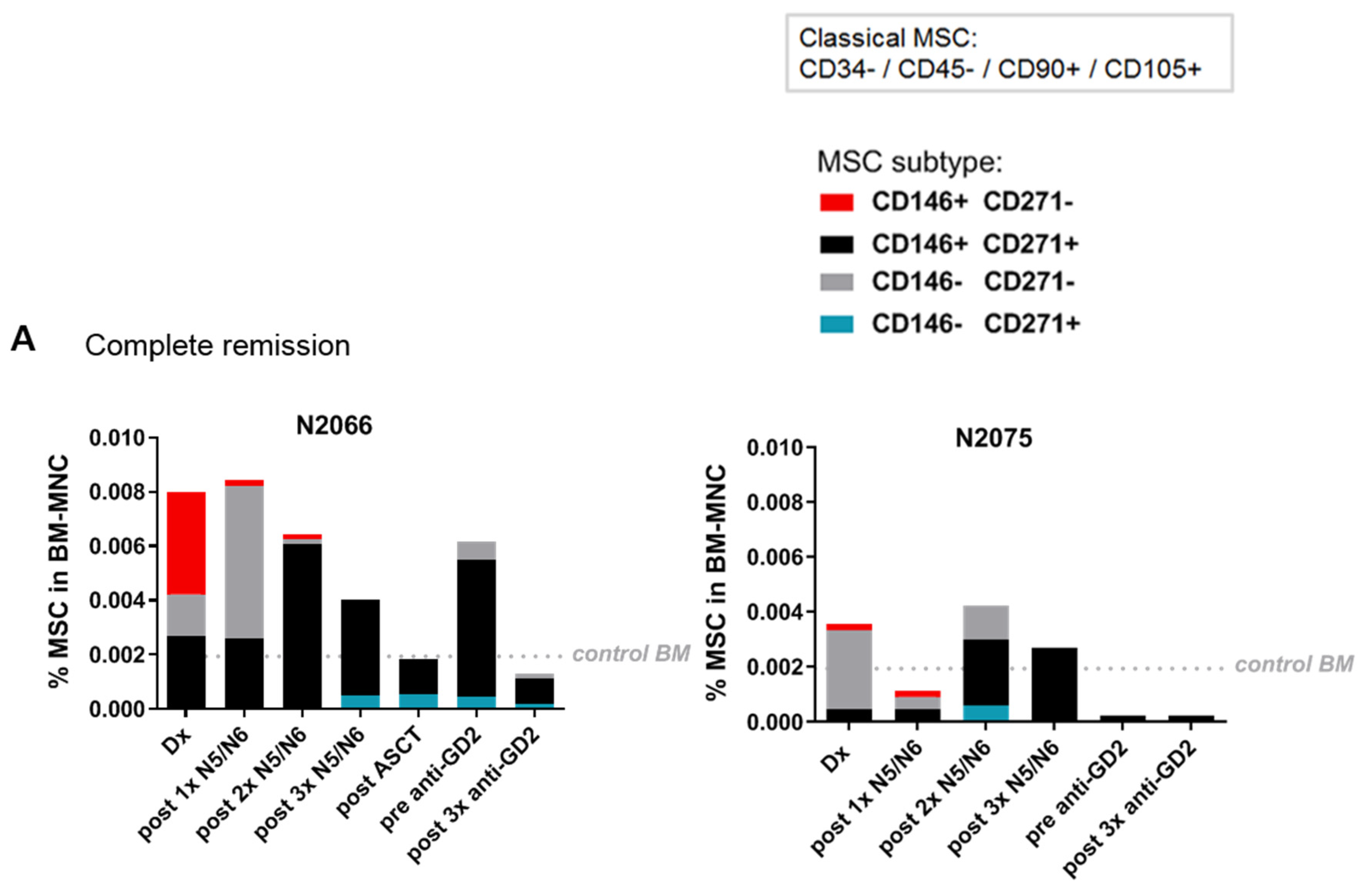
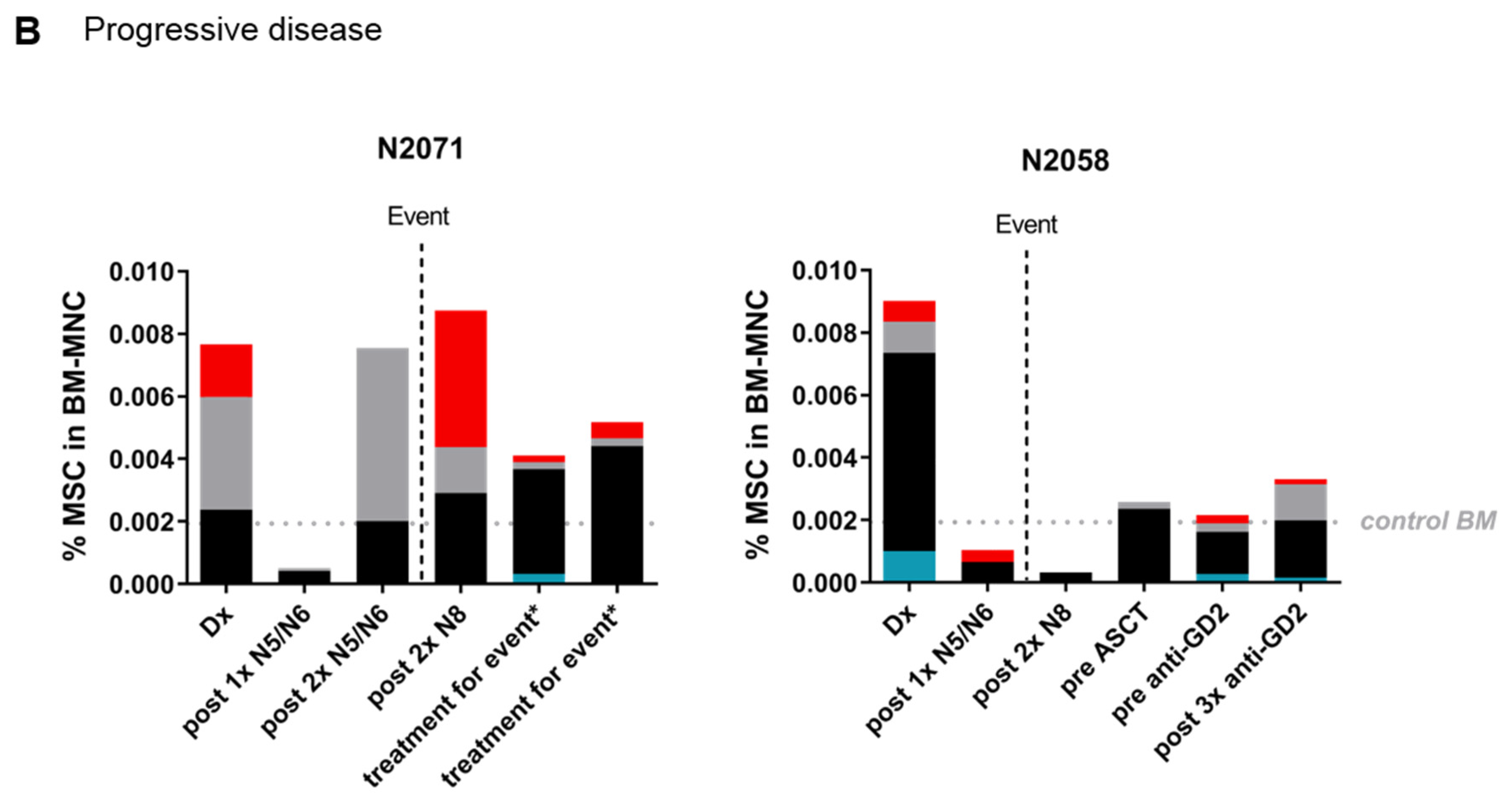
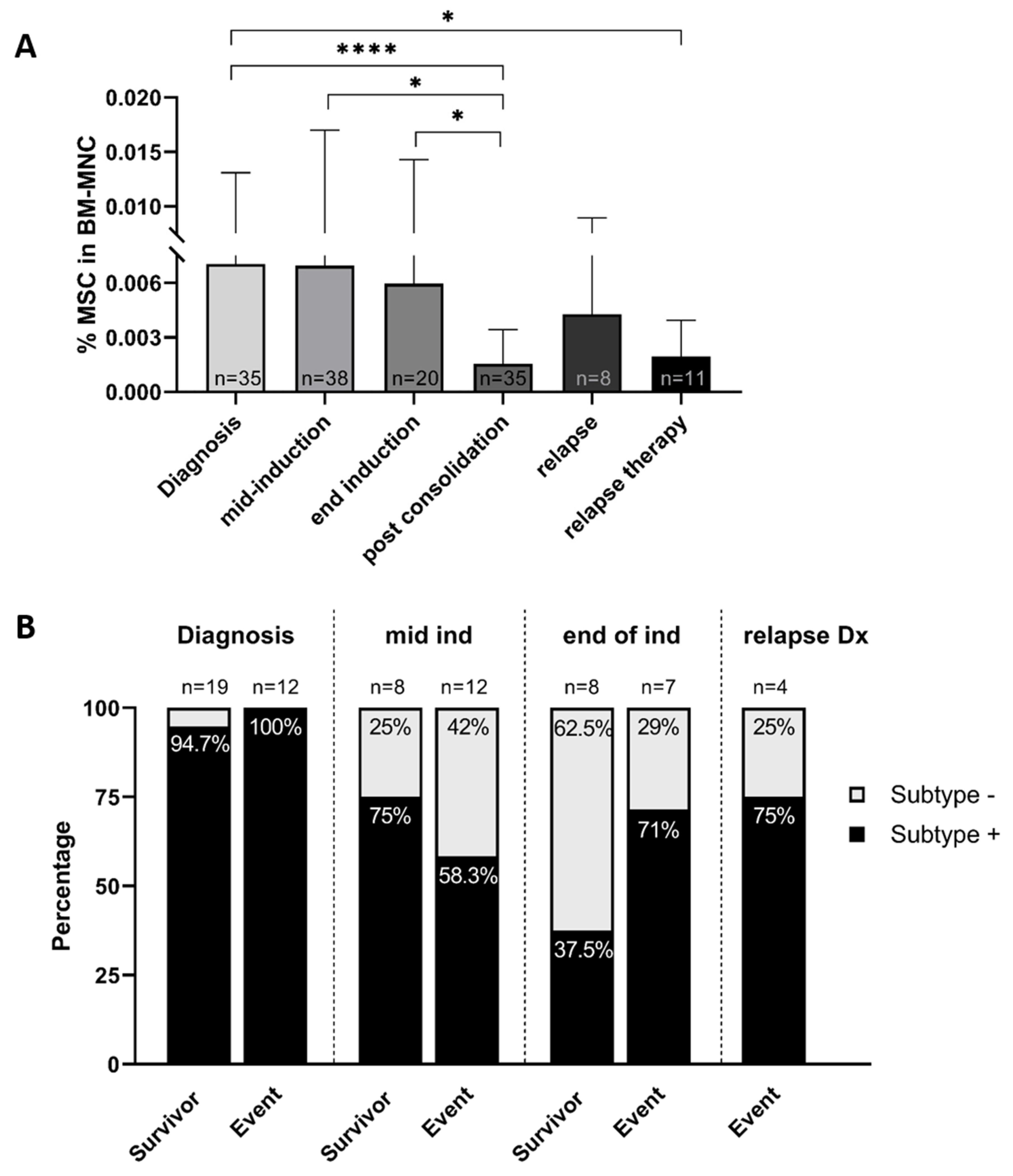
2.4. The MSC Compartment in the Course of Treatment
3. Discussion
4. Materials and Methods
4.1. Patient Samples and Treatment Regime
4.2. Adult and Fetal Bone Marrow MSCs
4.3. NB-BM Sample Preparation
4.4. Flow Cytometry
4.5. RNA, DNA and (RT) qPCR
4.6. DNA Extraction and qPCR
4.7. Cell Culture
4.8. Colony-Forming-Unit-Fibroblast (CFU-F) Assay
4.9. Differentiation Assays
4.10. Proliferation Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Park, J.R.; Eggert, A.; Caron, H. Neuroblastoma: Biology, prognosis, and treatment. Hematol. Oncol. Clin. N. Am. 2010, 24, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Mehes, G.; Luegmayr, A.; Ambros, I.M.; Ladenstein, R.; Ambros, P.F. Combined automatic immunological and molecular cytogenetic analysis allows exact identification and quantification of tumor cells in the bone marrow. Clin. Cancer Res. 2001, 7, 1969–1975. [Google Scholar] [PubMed]
- Van Zogchel, L.M.J.; van Wezel, E.M.; Stutterheim, J.; Zappeij-Kannegieter, L.; van Doornum, M.; Caron, H.; Fiocco, M.; van der Schoot, C.E.; Tytgat, G.A.M. Analysis of the prospective minimal residual disease monitoring study in gpoh-dcog high risk neuroblastoma. In Proceedings of the Advances in Neuroblastoma Research Association Meetings, San Francisco, CA, USA, 9–12 May 2018. [Google Scholar]
- Viprey, V.F.; Gregory, W.M.; Corrias, M.V.; Tchirkov, A.; Swerts, K.; Vicha, A.; Dallorso, S.; Brock, P.; Luksch, R.; Valteau-Couanet, D.; et al. Neuroblastoma mrnas predict outcome in children with stage 4 neuroblastoma: A european hr-nbl1/siopen study. J. Clin. Oncol. 2014, 32, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in risk classification and treatment strategies for neuroblastoma. J. Clin. Oncol 2015, 33, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Seeger, R.C.; Reynolds, C.P.; Gallego, R.; Stram, D.O.; Gerbing, R.B.; Matthay, K.K. Quantitative tumor cell content of bone marrow and blood as a predictor of outcome in stage iv neuroblastoma: A children’s cancer group study. J. Clin. Oncol. 2000, 18, 4067–4076. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Eber, M.R.; Berry, J.E.; Taichman, R.S. Bone marrow as a metastatic niche for disseminated tumor cells from solid tumors. Bonekey Rep. 2015, 4, 689. [Google Scholar] [CrossRef] [Green Version]
- Bielby, R.; Jones, E.; McGonagle, D. The role of mesenchymal stem cells in maintenance and repair of bone. Injury 2007, 38 (Suppl. 1), S26–S32. [Google Scholar] [CrossRef]
- Majumdar, M.K.; Thiede, M.A.; Haynesworth, S.E.; Bruder, S.P.; Gerson, S.L. Human marrow-derived mesenchymal stem cells (mscs) express hematopoietic cytokines and support long-term hematopoiesis when differentiated toward stromal and osteogenic lineages. J. Hematother. Stem. Cell. Res. 2000, 9, 841–848. [Google Scholar] [CrossRef]
- Korn, C.; Mendez-Ferrer, S. Myeloid malignancies and the microenvironment. Blood 2017, 129, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Kim Lyerly, H.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340–373. [Google Scholar] [CrossRef]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal stem cell-derived exosomes stimulate cycling quiescence and early breast cancer dormancy in bone marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef] [Green Version]
- Schelker, R.C.; Iberl, S.; Muller, G.; Hart, C.; Herr, W.; Grassinger, J. Tgf-beta1 and cxcl12 modulate proliferation and chemotherapy sensitivity of acute myeloid leukemia cells co-cultured with multipotent mesenchymal stromal cells. Hematology 2018, 23, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baglio, S.R.; Lagerweij, T.; Perez-Lanzon, M.; Ho, X.D.; Leveille, N.; Melo, S.A.; Cleton-Jansen, A.M.; Jordanova, E.S.; Roncuzzi, L.; Greco, M.; et al. Blocking tumor-educated msc paracrine activity halts osteosarcoma progression. Clin. Cancer Res. 2017, 23, 3721–3733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Takam Kamga, P.; Bassi, G.; Cassaro, A.; Midolo, M.; Di Trapani, M.; Gatti, A.; Carusone, R.; Resci, F.; Perbellini, O.; Gottardi, M.; et al. Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget 2016, 7, 21713–21727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reagan, M.R.; Rosen, C.J. Navigating the bone marrow niche: Translational insights and cancer-driven dysfunction. Nat. Rev. Rheumatol. 2016, 12, 154–168. [Google Scholar] [CrossRef]
- Borriello, L.; Nakata, R.; Sheard, M.A.; Fernandez, G.E.; Sposto, R.; Malvar, J.; Blavier, L.; Shimada, H.; Asgharzadeh, S.; Seeger, R.C.; et al. Cancer-associated fibroblasts share characteristics and protumorigenic activity with mesenchymal stromal cells. Cancer Res. 2017, 77, 5142–5157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakata, R.; Shimada, H.; Fernandez, G.E.; Fanter, R.; Fabbri, M.; Malvar, J.; Zimmermann, P.; DeClerck, Y.A. Contribution of neuroblastoma-derived exosomes to the production of pro-tumorigenic signals by bone marrow mesenchymal stromal cells. J. Extracell Vesicles 2017, 6, 1332941. [Google Scholar] [CrossRef]
- Silverman, A.M.; Nakata, R.; Shimada, H.; Sposto, R.; DeClerck, Y.A. A galectin-3-dependent pathway upregulates interleukin-6 in the microenvironment of human neuroblastoma. Cancer Res. 2012, 72, 2228–2238. [Google Scholar] [CrossRef] [Green Version]
- Shankar, V.; Hori, H.; Kihira, K.; Lei, Q.; Toyoda, H.; Iwamoto, S.; Komada, Y. Mesenchymal stromal cell secretome up-regulates 47 kda cxcr4 expression, and induce invasiveness in neuroblastoma cell lines. PLoS ONE 2015, 10, e0120069. [Google Scholar] [CrossRef]
- Lifshitz, V.; Priceman, S.J.; Li, W.; Cherryholmes, G.; Lee, H.; Makovski-Silverstein, A.; Borriello, L.; DeClerck, Y.A.; Yu, H. Sphingosine-1-phosphate receptor-1 promotes environment-mediated and acquired chemoresistance. Mol. Cancer 2017, 16, 2516–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.W.; Sheard, M.A.; Malvar, J.; Fernandez, G.E.; DeClerck, Y.A.; Blavier, L.; Shimada, H.; Theuer, C.P.; Sposto, R.; Seeger, R.C. Anti-cd105 antibody eliminates tumor microenvironment cells and enhances anti-gd2 antibody immunotherapy of neuroblastoma with activated natural killer cells. Clin. Cancer Res. 2019, 25, 4761–4774. [Google Scholar] [CrossRef] [Green Version]
- Ara, T.; Nakata, R.; Sheard, M.A.; Shimada, H.; Buettner, R.; Groshen, S.G.; Ji, L.; Yu, H.; Jove, R.; Seeger, R.C.; et al. Critical role of stat3 in il-6-mediated drug resistance in human neuroblastoma. Cancer Res. 2013, 73, 3852–3864. [Google Scholar] [CrossRef] [Green Version]
- Debruyne, D.N.; Bhatnagar, N.; Sharma, B.; Luther, W.; Moore, N.F.; Cheung, N.K.; Gray, N.S.; George, R.E. Alk inhibitor resistance in alk(f1174l)-driven neuroblastoma is associated with axl activation and induction of emt. Oncogene 2016, 35, 3681–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef]
- Stutterheim, J.; Gerritsen, A.; Zappeij-Kannegieter, L.; Kleijn, I.; Dee, R.; Hooft, L.; van Noesel, M.M.; Bierings, M.; Berthold, F.; Versteeg, R.; et al. Phox2b is a novel and specific marker for minimal residual disease testing in neuroblastoma. J. Clin. Oncol. 2008, 26, 5443–5449. [Google Scholar] [CrossRef] [PubMed]
- Stutterheim, J.; Ichou, F.A.; den Ouden, E.; Versteeg, R.; Caron, H.N.; Tytgat, G.A.; van der Schoot, C.E. Methylated rassf1a is the first specific DNA marker for minimal residual disease testing in neuroblastoma. Clin. Cancer Res. 2012, 18, 808–814. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Bozzi, F.; Collini, P.; Aiello, A.; Barzano, E.; Gambirasio, F.; Podda, M.; Meazza, C.; Ferrari, A.; Luksch, R. Flow cytometric phenotype of rhabdomyosarcoma bone marrow metastatic cells and its implication in differential diagnosis with neuroblastoma. Anticancer Res. 2008, 28, 1565–1569. [Google Scholar]
- Seeger, R.C.; Danon, Y.L.; Rayner, S.A.; Hoover, F. Definition of a thy-1 determinant on human neuroblastoma, glioma, sarcoma, and teratoma cells with a monoclonal antibody. J. Immunol. 1982, 128, 983–989. [Google Scholar] [PubMed]
- Van Wezel, E.M.; van Zogchel, L.M.J.; van Wijk, J.; Timmerman, I.; Vo, N.K.; Zappeij-Kannegieter, L.; de Carolis, B.; Simon, T.; van Noesel, M.M.; Molenaar, J.J.; et al. Mesenchymal neuroblastoma cells are undetected by current mrna marker panels: The development of a specific neuroblastoma mesenchymal mrd panel. JCO Precis. Oncol. 2019, 3. [Google Scholar]
- Buhring, H.J.; Treml, S.; Cerabona, F.; de Zwart, P.; Kanz, L.; Sobiesiak, M. Phenotypic characterization of distinct human bone marrow-derived msc subsets. Ann. N. Y. Acad. Sci. 2009, 1176, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Tormin, A.; Li, O.; Brune, J.C.; Walsh, S.; Schutz, B.; Ehinger, M.; Ditzel, N.; Kassem, M.; Scheding, S. Cd146 expression on primary nonhematopoietic bone marrow stem cells is correlated with in situ localization. Blood 2011, 117, 5067–5077. [Google Scholar] [CrossRef] [Green Version]
- Maijenburg, M.W.; Kleijer, M.; Vermeul, K.; Mul, E.P.; Van Alphen, F.P.; van der Schoot, C.E.; Voermans, C. The composition of the mesenchymal stromal cell compartment in human bone marrow changes during development and aging. Haematologica 2012, 97, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Berthold, F.; Faldum, A.; Ernst, A.; Boos, J.; Dilloo, D.; Eggert, A.; Fischer, M.; Fruhwald, M.; Henze, G.; Klingebiel, T.; et al. Extended induction chemotherapy does not improve the outcome for high-risk neuroblastoma patients: Results of the randomized open-label gpoh trial nb2004-hr. Ann. Oncol. 2020, 31, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Activity Study of Bevacizumab with Temozolomide ± Irinotecan for Neuroblastoma in Children. Available online: https://ClinicalTrials.gov/show/NCT02308527 (accessed on 21 September 2020).
- Forest, A.E.; Shiozawa, Y.; Pienta, K.J.; Taichman, R.S. The hematopoietic stem cell niche and bone metastasis. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Kraal, K.; Timmerman, I.; Kansen, H.M.; van den Bos, C.; Zsiros, J.; van den Berg, H.; Somers, S.; Braakman, E.; Peek, A.M.L.; van Noesel, M.M.; et al. Peripheral stem cell apheresis is feasible post (131)iodine-metaiodobenzylguanidine-therapy in high-risk neuroblastoma, but results in delayed platelet reconstitution. Clin. Cancer Res. 2019, 25, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamran, N.; Li, Y.; Sierra, M.; Alghamri, M.S.; Kadiyala, P.; Appelman, H.D.; Edwards, M.; Lowenstein, P.R.; Castro, M.G. Melanoma induced immunosuppression is mediated by hematopoietic dysregulation. Oncoimmunology 2018, 7, e1408750. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Zhao, Y.; Fei, C.; Zhao, S.; Zheng, Q.; Su, J.; Wu, D.; Li, X.; Chang, C. Dicer1 downregulation by multiple myeloma cells promotes the senescence and tumor-supporting capacity and decreases the differentiation potential of mesenchymal stem cells. Cell. Death Dis. 2018, 9, 512. [Google Scholar] [CrossRef]
- Desbourdes, L.; Javary, J.; Charbonnier, T.; Ishac, N.; Bourgeais, J.; Iltis, A.; Chomel, J.C.; Turhan, A.; Guilloton, F.; Tarte, K.; et al. Alteration analysis of bone marrow mesenchymal stromal cells from de novo acute myeloid leukemia patients at diagnosis. Stem. Cells Dev. 2017, 26, 709–722. [Google Scholar] [CrossRef]
- Cordeiro-Spinetti, E.; de Mello, W.; Trindade, L.S.; Taub, D.D.; Taichman, R.S.; Balduino, A. Human bone marrow mesenchymal progenitors: Perspectives on an optimized in vitro manipulation. Front. Cell. Dev. Biol. 2014, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Roodman, G.D. Mechanisms of bone metastasis. Discov. Med. 2004, 4, 144–148. [Google Scholar] [CrossRef]
- Conforti, A.; Biagini, S.; Del Bufalo, F.; Sirleto, P.; Angioni, A.; Starc, N.; Li Pira, G.; Moretta, F.; Proia, A.; Contoli, B.; et al. Biological, functional and genetic characterization of bone marrow-derived mesenchymal stromal cells from pediatric patients affected by acute lymphoblastic leukemia. PLoS ONE 2013, 8, e76989. [Google Scholar] [CrossRef]
- Battula, V.L.; Le, P.M.; Sun, J.C.; Nguyen, K.; Yuan, B.; Zhou, X.; Sonnylal, S.; McQueen, T.; Ruvolo, V.; Michel, K.A.; et al. Aml-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight 2017, 2, 13. [Google Scholar] [CrossRef]
- Hall, C.L.; Bafico, A.; Dai, J.; Aaronson, S.A.; Keller, E.T. Prostate cancer cells promote osteoblastic bone metastases through wnts. Cancer Res. 2005, 65, 7554–7560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wessely, A.; Waltera, A.; Reichert, T.E.; Stockl, S.; Grassel, S.; Bauer, R.J. Induction of alp and mmp9 activity facilitates invasive behavior in heterogeneous human bmsc and hnscc 3d spheroids. Faseb. J. 2019, 33, 11884–11893. [Google Scholar] [CrossRef] [Green Version]
- HaDuong, J.H.; Blavier, L.; Baniwal, S.K.; Frenkel, B.; Malvar, J.; Punj, V.; Sposto, R.; DeClerck, Y.A. Interaction between bone marrow stromal cells and neuroblastoma cells leads to a vegfa-mediated osteoblastogenesis. Int. J. Cancer 2015, 137, 797–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Zhang, J.; Dai, J.; Dehne, L.A.; Mizokami, A.; Yao, Z.; Keller, E.T. Osteoblasts induce prostate cancer proliferation and psa expression through interleukin-6-mediated activation of the androgen receptor. Clin. Exp. Metastasis 2004, 21, 399–408. [Google Scholar] [CrossRef]
- Larsen, K.H.; Frederiksen, C.M.; Burns, J.S.; Abdallah, B.M.; Kassem, M. Identifying a molecular phenotype for bone marrow stromal cells with in vivo bone-forming capacity. J. Bone Min. Res. 2010, 25, 796–808. [Google Scholar]
- Correa, D.; Somoza, R.A.; Lin, P.; Schiemann, W.P.; Caplan, A.I. Mesenchymal stem cells regulate melanoma cancer cells extravasation to bone and liver at their perivascular niche. Int. J. Cancer 2016, 138, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Kucerova, L.; Zmajkovic, J.; Toro, L.; Skolekova, S.; Demkova, L.; Matuskova, M. Tumor-driven molecular changes in human mesenchymal stromal cells. Cancer Microenviron. 2015, 8, 1–14. [Google Scholar] [CrossRef]
- Schiano, C.; Grimaldi, V.; Casamassimi, A.; Infante, T.; Esposito, A.; Giovane, A.; Napoli, C. Different expression of cd146 in human normal and osteosarcoma cell lines. Med. Oncol. 2012, 29, 2998–3002. [Google Scholar] [CrossRef]
- Piskareva, O.; Harvey, H.; Nolan, J.; Conlon, R.; Alcock, L.; Buckley, P.; Dowling, P.; Henry, M.; O’Sullivan, F.; Bray, I.; et al. The development of cisplatin resistance in neuroblastoma is accompanied by epithelial to mesenchymal transition in vitro. Cancer Lett. 2015, 364, 142–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burchill, S.A.; Beiske, K.; Shimada, H.; Ambros, P.F.; Seeger, R.; Tytgat, G.A.; Brock, P.R.; Haber, M.; Park, J.R.; Berthold, F. Recommendations for the standardization of bone marrow disease assessment and reporting in children with neuroblastoma on behalf of the international neuroblastoma response criteria bone marrow working group. Cancer 2017, 123, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.K.; Ostrovnaya, I.; Kuk, D.; Cheung, I.Y. Bone marrow minimal residual disease was an early response marker and a consistent independent predictor of survival after anti-gd2 immunotherapy. J. Clin. Oncol. 2015, 33, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fried, W.; Chamberlin, W.; Kedo, A.; Barone, J. Effects of radiation on hematopoietic stroma. Exp. Hematol. 1976, 4, 310–314. [Google Scholar] [PubMed]
- Domenech, J.; Roingeard, F.; Herault, O.; Truglio, D.; Desbois, I.; Colombat, P.; Binet, C. Changes in the functional capacity of marrow stromal cells after autologous bone marrow transplantation. Leuk. Lymphoma 1998, 29, 533–546. [Google Scholar] [CrossRef]
- Somaiah, C.; Kumar, A.; Sharma, R.; Sharma, A.; Anand, T.; Bhattacharyya, J.; Das, D.; Deka Talukdar, S.; Jaganathan, B.G. Mesenchymal stem cells show functional defect and decreased anti-cancer effect after exposure to chemotherapeutic drugs. J. Biomed. Sci. 2018, 25, 5. [Google Scholar] [CrossRef] [Green Version]
- Prata Kde, L.; Orellana, M.D.; De Santis, G.C.; Kashima, S.; Fontes, A.M.; Carrara Rde, C.; Palma, P.V.; Neder, L.; Covas, D.T. Effects of high-dose chemotherapy on bone marrow multipotent mesenchymal stromal cells isolated from lymphoma patients. Exp. Hematol. 2010, 38, 292–300. [Google Scholar] [CrossRef]
- Maijenburg, M.W.; Noort, W.A.; Kleijer, M.; Kompier, C.J.; Weijer, K.; van Buul, J.D.; van der Schoot, C.E.; Voermans, C. Cell cycle and tissue of origin contribute to the migratory behaviour of human fetal and adult mesenchymal stromal cells. Br. J. Haematol. 2010, 148, 428–440. [Google Scholar] [CrossRef]
- Stutterheim, J.; Gerritsen, A.; Zappeij-Kannegieter, L.; Yalcin, B.; Dee, R.; van Noesel, M.M.; Berthold, F.; Versteeg, R.; Caron, H.N.; van der Schoot, C.E.; et al. Detecting minimal residual disease in neuroblastoma: The superiority of a panel of real-time quantitative pcr markers. Clin. Chem. 2009, 55, 1316–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bate-Eya, L.T.; Ebus, M.E.; Koster, J.; den Hartog, I.J.; Zwijnenburg, D.A.; Schild, L.; van der Ploeg, I.; Dolman, M.E.; Caron, H.N.; Versteeg, R.; et al. Newly-derived neuroblastoma cell lines propagated in serum-free media recapitulate the genotype and phenotype of primary neuroblastoma tumours. Eur. J. Cancer 2014, 50, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Van Nes, J.; Chan, A.; van Groningen, T.; van Sluis, P.; Koster, J.; Versteeg, R. A notch3 transcriptional module induces cell motility in neuroblastoma. Clin. Cancer Res. 2013, 19, 3485–3494. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hochheuser, C.; van Zogchel, L.M.J.; Kleijer, M.; Kuijk, C.; Tol, S.; van der Schoot, C.E.; Voermans, C.; Tytgat, G.A.M.; Timmerman, I. The Metastatic Bone Marrow Niche in Neuroblastoma: Altered Phenotype and Function of Mesenchymal Stromal Cells. Cancers 2020, 12, 3231. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113231
Hochheuser C, van Zogchel LMJ, Kleijer M, Kuijk C, Tol S, van der Schoot CE, Voermans C, Tytgat GAM, Timmerman I. The Metastatic Bone Marrow Niche in Neuroblastoma: Altered Phenotype and Function of Mesenchymal Stromal Cells. Cancers. 2020; 12(11):3231. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113231
Chicago/Turabian StyleHochheuser, Caroline, Lieke M. J. van Zogchel, Marion Kleijer, Carlijn Kuijk, Simon Tol, C. Ellen van der Schoot, Carlijn Voermans, Godelieve A. M. Tytgat, and Ilse Timmerman. 2020. "The Metastatic Bone Marrow Niche in Neuroblastoma: Altered Phenotype and Function of Mesenchymal Stromal Cells" Cancers 12, no. 11: 3231. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113231