Sonic Hedgehog Signature in Pediatric Primary Bone Tumors: Effects of the GLI Antagonist GANT61 on Ewing’s Sarcoma Tumor Growth

 ,
,  , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
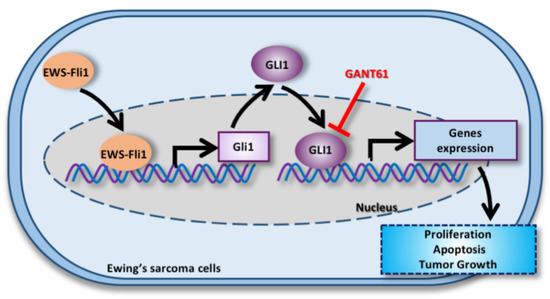
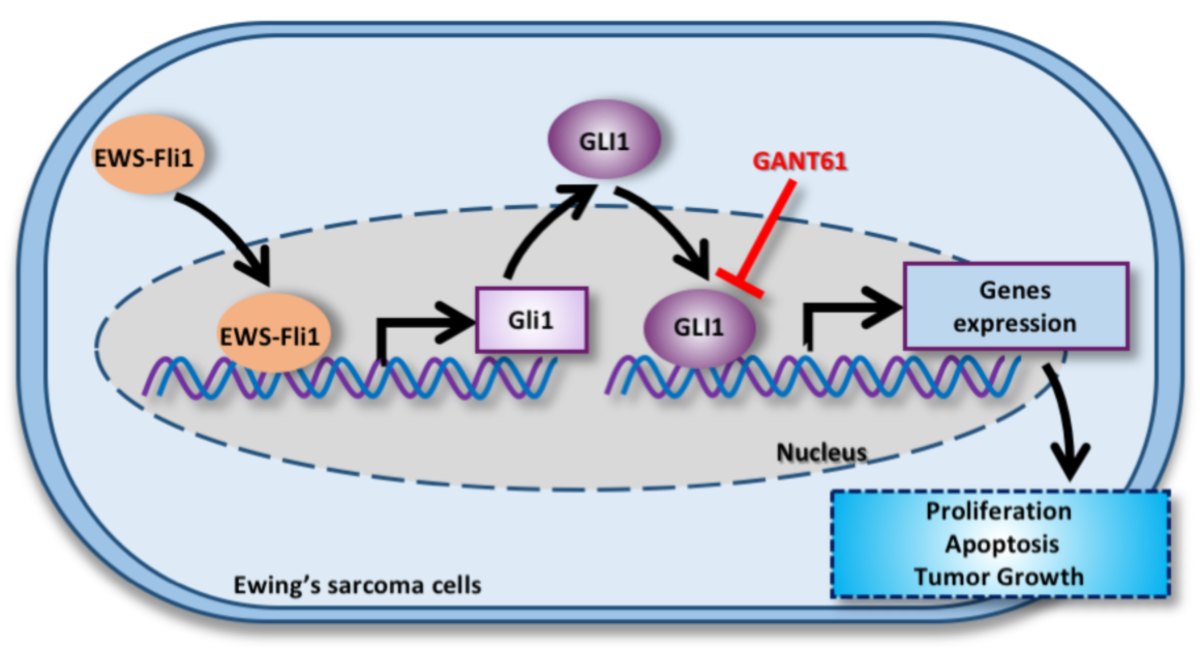
1. Introduction
2. Results
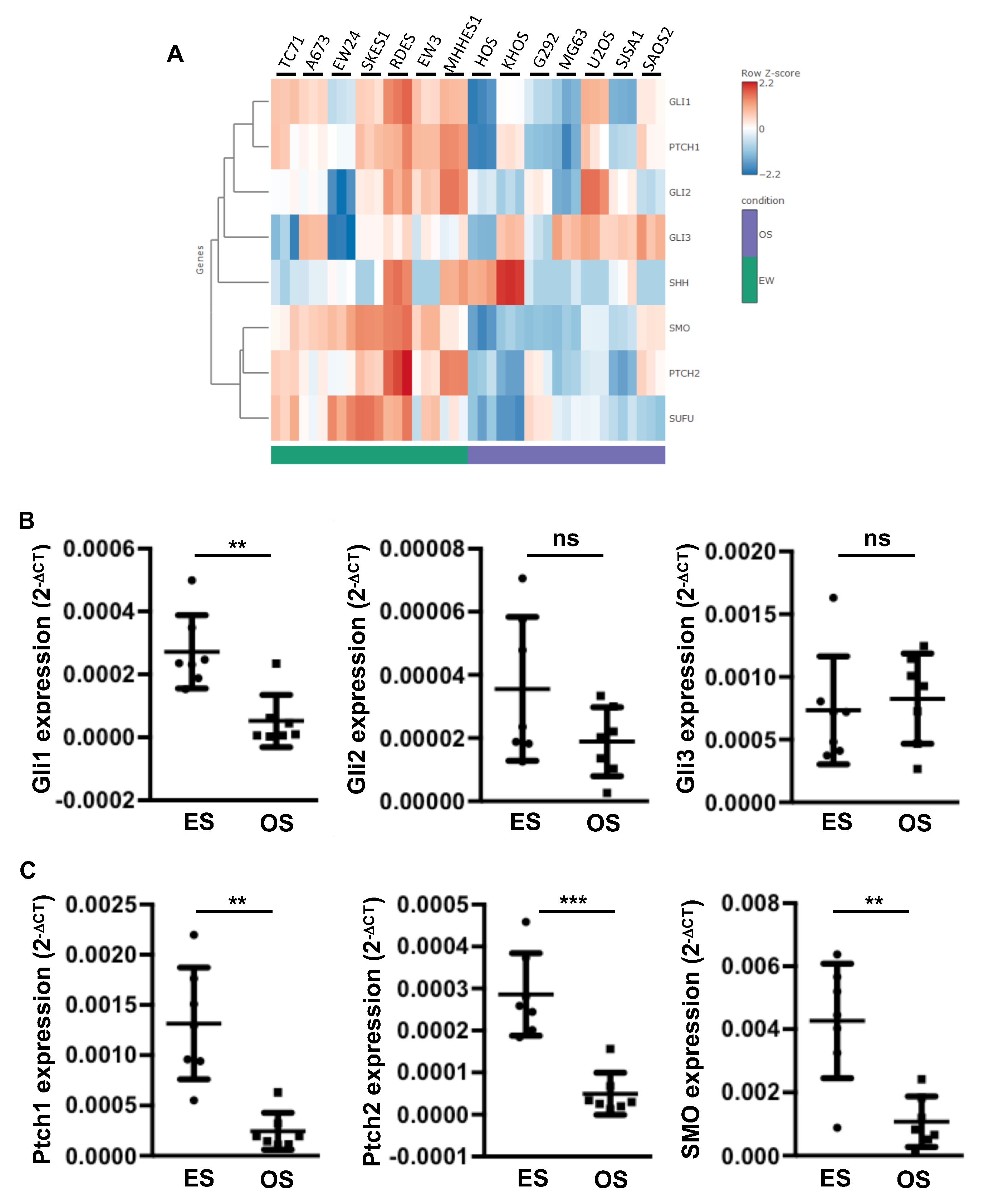
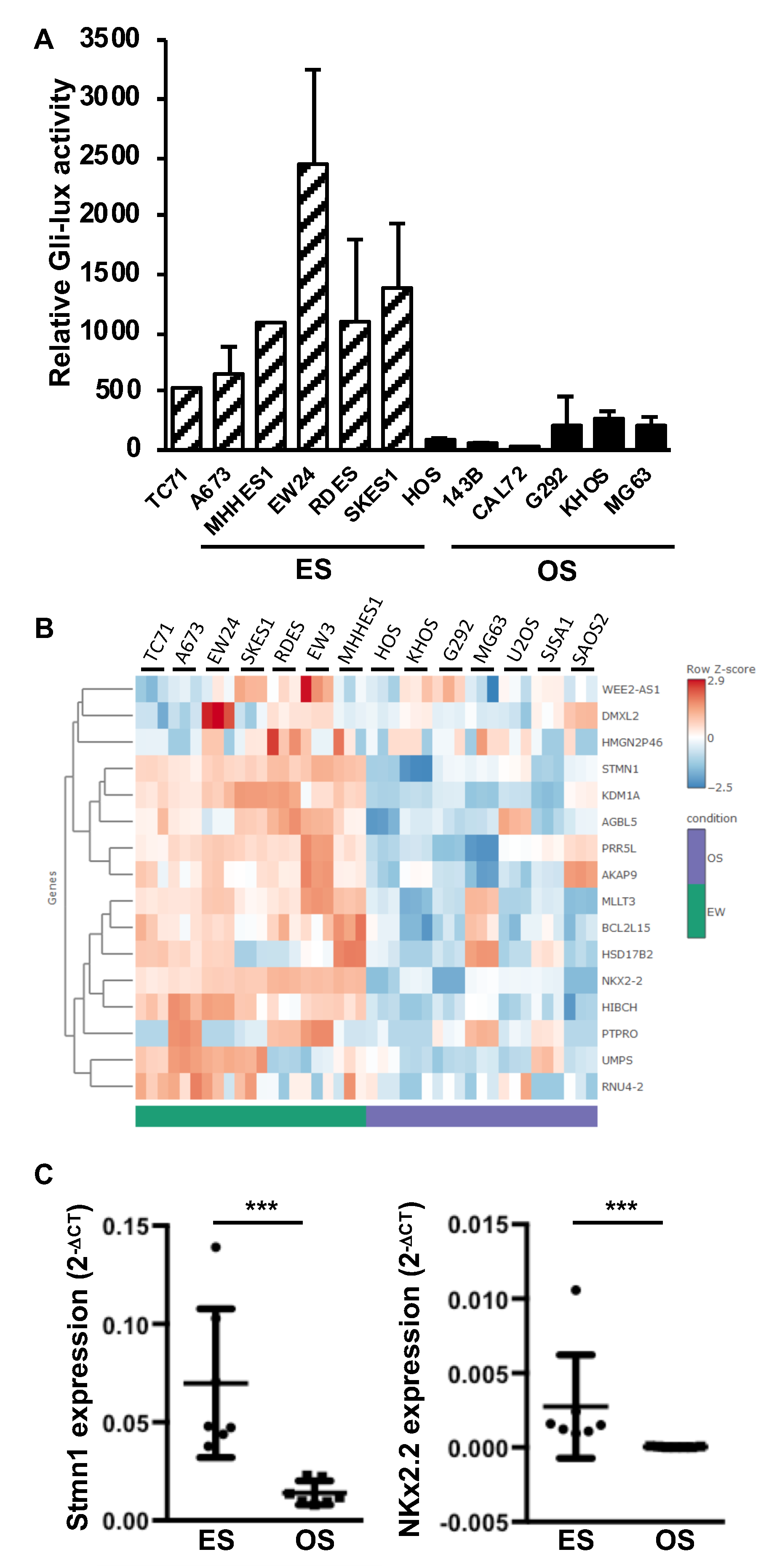
2.1. Elevation of Gli1 Target Gene Expression in ES Cell Lines Compared to Osteosarcoma (OS) Cell Lines
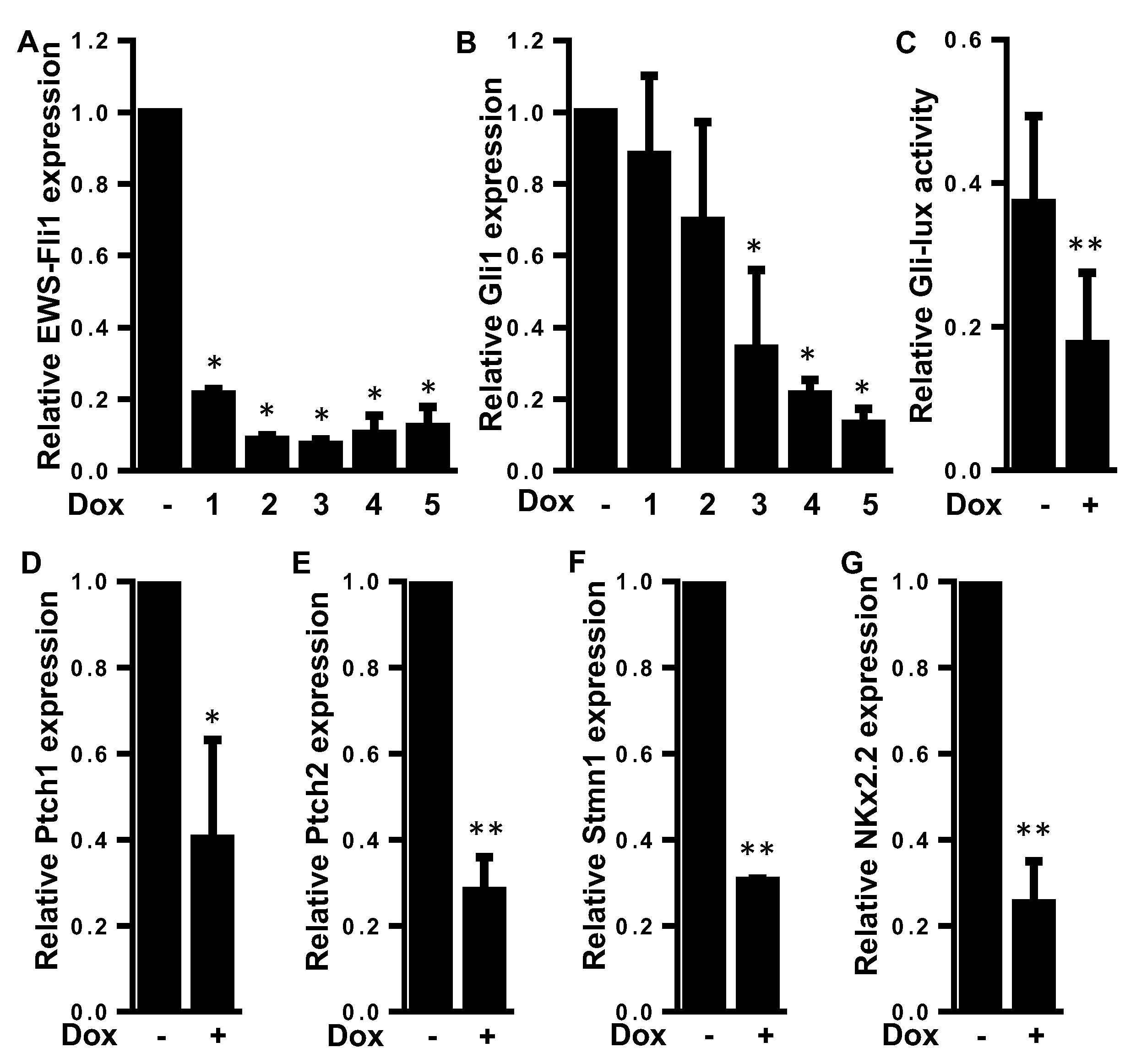
2.2. EWS-FLI1 Drives the Expression of Gli1 and the Gli Transcriptional Response in ES
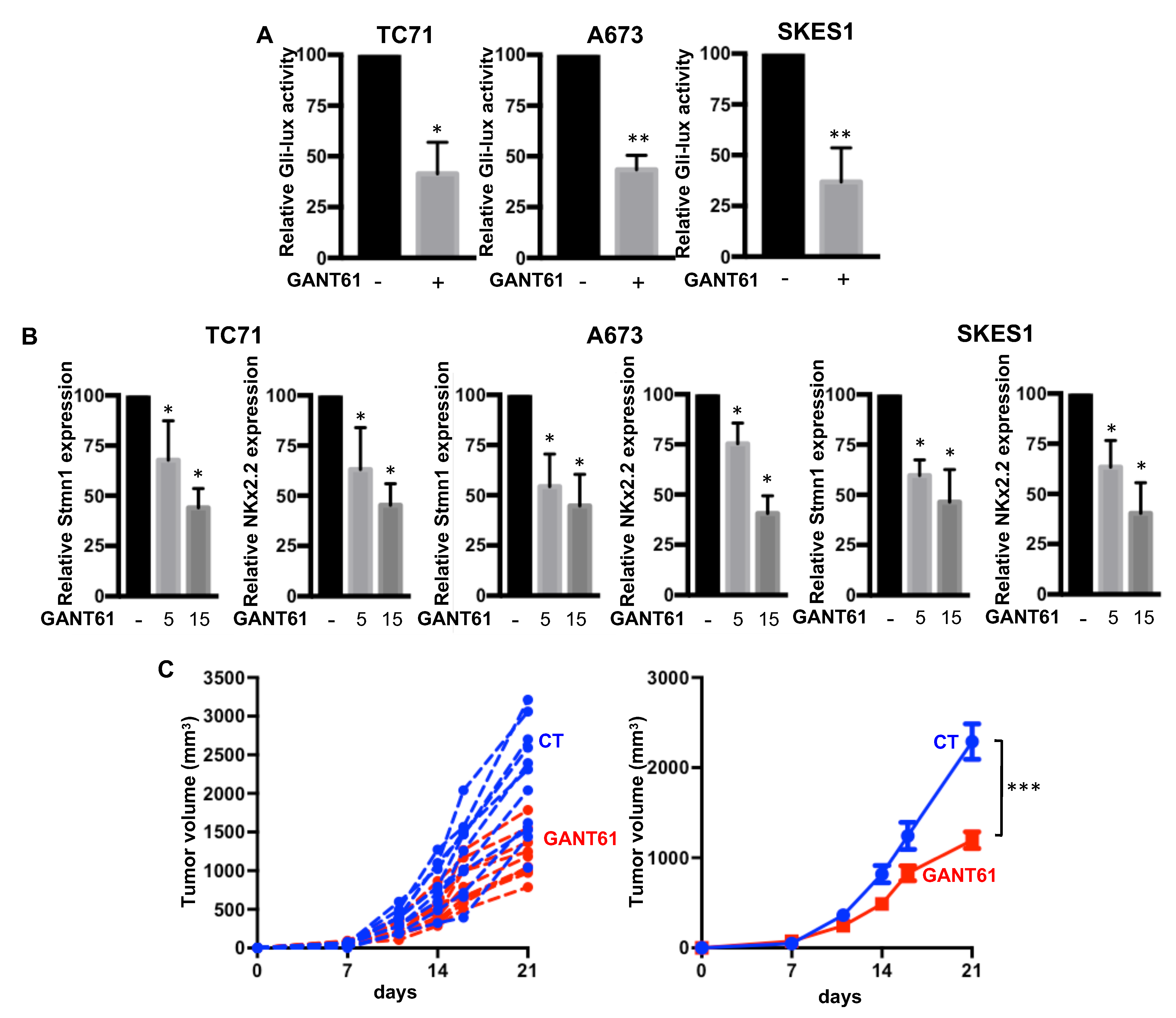
2.3. GANT61 Inhibits the Gli Signaling Pathway and Primary Tumor Growth in an Orthotopic Model of ES
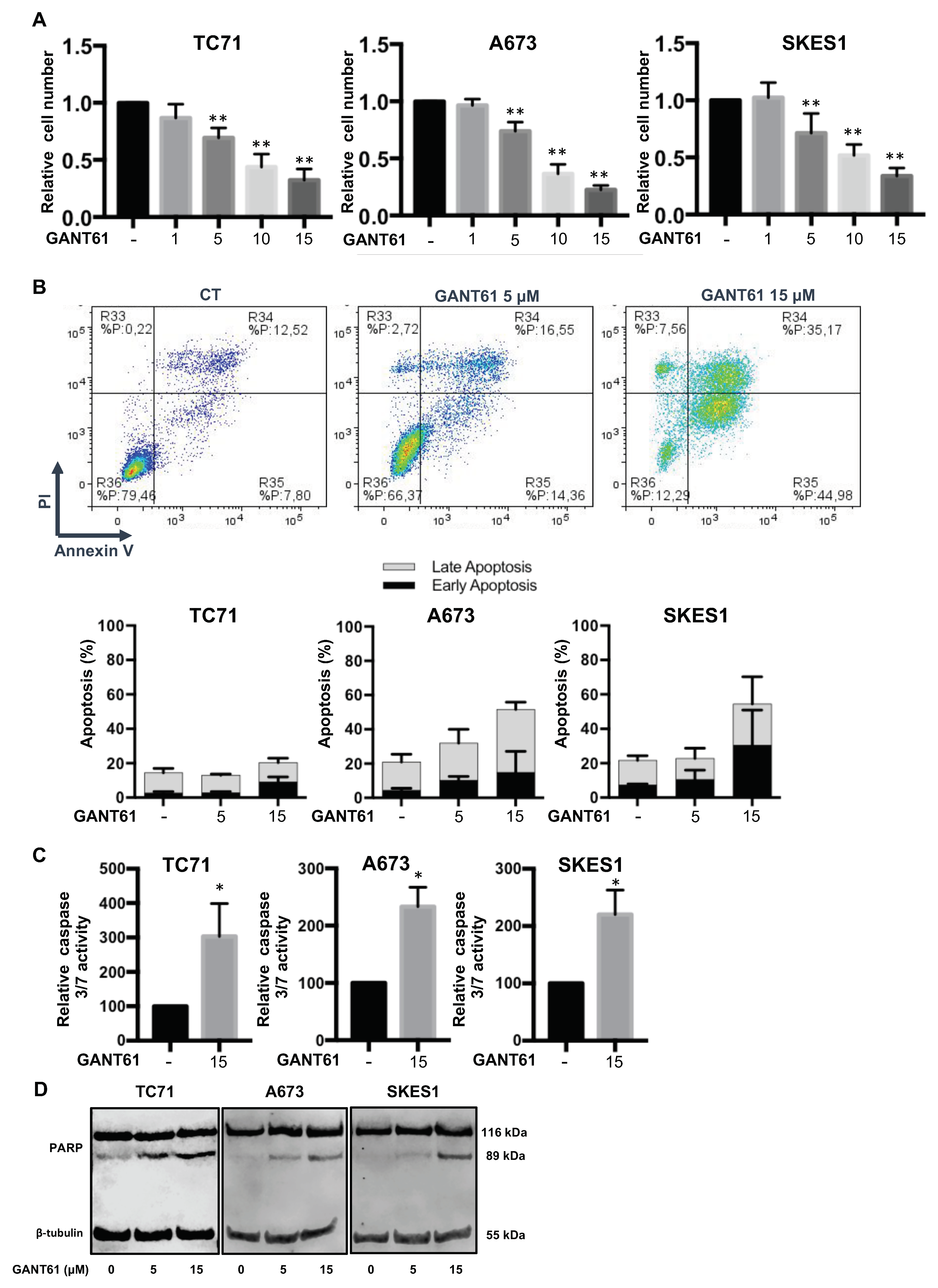
2.4. GANT61 Induces In Vitro Cell Death
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Real-Time Polymerase Chain Reaction
4.3. RNA Sequencing and Analysis
4.4. Transient Cell Transfections, Reporter Assays and Plasmid Constructs
4.5. Proliferation Assay
4.6. Annexin V Assay and Caspase Activity
4.7. Western Blot Analysis
4.8. Ewing’s Sarcoma (ES) Mouse Model
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heare, T.; Hensley, M.A.; Dell’Orfano, S. Bone tumors: Osteosarcoma and Ewing’s sarcoma. Curr. Opin. Pediatrics 2009, 21, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.D.; Luu, H.H. Osteosarcoma. Cancer Treat. Res. 2014, 162, 65–92. [Google Scholar] [CrossRef] [PubMed]
- Eccles, S.A.; Welch, D.R. Metastasis: Recent discoveries and novel treatment strategies. Lancet 2007, 369, 1742–1757. [Google Scholar] [CrossRef] [Green Version]
- Grier, H.E.; Krailo, M.D.; Tarbell, N.J.; Link, M.P.; Fryer, C.J.H.; Pritchard, D.J.; Gebhardt, M.C.; Dickman, P.S.; Perlman, E.J.; Meyers, P.A.; et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N. Engl. J. Med. 2003, 348, 694–701. [Google Scholar] [CrossRef] [Green Version]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.D.; Healey, J.H.; Bernstein, M.L.; Betcher, D.; Ferguson, W.S.; Gebhardt, M.C.; Goorin, A.M.; Harris, M.; et al. Osteosarcoma: The addition of muramyl tripeptide to chemotherapy improves overall survival—a report from the Children’s Oncology Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 633–638. [Google Scholar] [CrossRef]
- Bousquet, M.; Noirot, C.; Accadbled, F.; Sales de Gauzy, J.; Castex, M.P.; Brousset, P.; Gomez-Brouchet, A. Whole-exome sequencing in osteosarcoma reveals important heterogeneity of genetic alterations. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 738–744. [Google Scholar] [CrossRef]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Toomey, E.C.; Schiffman, J.D.; Lessnick, S.L. Recent advances in the molecular pathogenesis of Ewing’s sarcoma. Oncogene 2010, 29, 4504–4516. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; McManus, M.M.; Hughes, D.P.M. Understanding the Biology of Bone Sarcoma from Early Initiating Events through Late Events in Metastasis and Disease Progression. Front. Oncol. 2013, 3, 230. [Google Scholar] [CrossRef] [Green Version]
- Mortus, J.R.; Zhang, Y.; Hughes, D.P.M. Developmental pathways hijacked by osteosarcoma. Adv. Exp. Med. Biol. 2014, 804, 93–118. [Google Scholar] [CrossRef]
- Lézot, F.; Corre, I.; Morice, S.; Rédini, F.; Verrecchia, F. SHH Signaling Pathway Drives Pediatric Bone Sarcoma Progression. Cells 2020, 9, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marigo, V.; Davey, R.A.; Zuo, Y.; Cunningham, J.M.; Tabin, C.J. Biochemical evidence that patched is the Hedgehog receptor. Nature 1996, 384, 176–179. [Google Scholar] [CrossRef]
- Incardona, J.P.; Lee, J.H.; Robertson, C.P.; Enga, K.; Kapur, R.P.; Roelink, H. Receptor-mediated endocytosis of soluble and membrane-tethered Sonic hedgehog by Patched-1. Proc. Natl. Acad. Sci. USA 2000, 97, 12044–12049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Shen, L.; Law, K.; Zhang, Z.; Liu, X.; Hua, H.; Li, S.; Huang, H.; Yue, S.; Hui, C.-C.; et al. Suppressor of Fused Chaperones Gli Proteins To Generate Transcriptional Responses to Sonic Hedgehog Signaling. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinzler, K.W.; Ruppert, J.M.; Bigner, S.H.; Vogelstein, B. The GLI gene is a member of the Kruppel family of zinc finger proteins. Nature 1988, 332, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Pavletich, N.P.; Pabo, C.O. Crystal structure of a five-finger GLI-DNA complex: New perspectives on zinc fingers. Science 1993, 261, 1701–1707. [Google Scholar] [CrossRef]
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.-J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [Green Version]
- Dennler, S.; André, J.; Verrecchia, F.; Mauviel, A. Cloning of the human GLI2 Promoter: Transcriptional activation by transforming growth factor-beta via SMAD3/beta-catenin cooperation. J. Biol. Chem. 2009, 284, 31523–31531. [Google Scholar] [CrossRef] [Green Version]
- Alexaki, V.-I.; Javelaud, D.; Van Kempen, L.C.L.; Mohammad, K.S.; Dennler, S.; Luciani, F.; Hoek, K.S.; Juàrez, P.; Goydos, J.S.; Fournier, P.J.; et al. GLI2-mediated melanoma invasion and metastasis. J. Natl. Cancer Inst. 2010, 102, 1148–1159. [Google Scholar] [CrossRef] [Green Version]
- Niyaz, M.; Khan, M.S.; Mudassar, S. Hedgehog Signaling: An Achilles’ Heel in Cancer. Transl. Oncol. 2019, 12, 1334–1344. [Google Scholar] [CrossRef]
- Giroux-Leprieur, E.; Costantini, A.; Ding, V.W.; He, B. Hedgehog Signaling in Lung Cancer: From Oncogenesis to Cancer Treatment Resistance. Int. J. Mol. Sci. 2018, 19, 2835. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Hui, C.-C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Pasca di Magliano, M.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Lo, H.-W. Hedgehog pathway and GLI1 isoforms in human cancer. Discov. Med. 2012, 13, 105–113. [Google Scholar]
- Villavicencio, E.H.; Walterhouse, D.O.; Iannaccone, P.M. The sonic hedgehog-patched-gli pathway in human development and disease. Am. J. Hum. Genet. 2000, 67, 1047–1054. [Google Scholar] [CrossRef]
- Ng, J.M.Y.; Curran, T. The Hedgehog’s tale: Developing strategies for targeting cancer. Nat. Rev. Cancer 2011, 11, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Üren, A. GLI1 Is a Direct Transcriptional Target of EWS-FLI1 Oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef] [Green Version]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.-W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.-C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin Invest. 2011, 121, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.-K.; Kim, H.-J.; Lee, Y.-S.; Han, M.-E.; Yoon, S.; Baek, S.-Y.; Kim, B.-S.; Kim, J.-B.; Oh, S.-O. Hedgehog signaling regulates proliferation of prostate cancer cells via stathmin1. Clin. Exp. Med. 2010, 10, 51–57. [Google Scholar] [CrossRef]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog signaling pathway in cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.M.R.; Fuchs, B. Hedgehog signaling inhibitors as anti-cancer agents in osteosarcoma. Cancers 2015, 7, 784–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohseny, A.B.; Cai, Y.; Kuijjer, M.; Xiao, W.; van den Akker, B.; de Andrea, C.E.; Jacobs, R.; ten Dijke, P.; Hogendoorn, P.C.W.; Cleton-Jansen, A.-M. The activities of Smad and Gli mediated signalling pathways in high-grade conventional osteosarcoma. Eur. J. Cancer 2012, 48, 3429–3438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirotsu, M.; Setoguchi, T.; Sasaki, H.; Matsunoshita, Y.; Gao, H.; Nagao, H.; Kunigou, O.; Komiya, S. Smoothened as a new therapeutic target for human osteosarcoma. Mol. Cancer 2010, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.-Q.; Chu, X.-D. GANT61 plays antitumor effects by inducing oxidative stress through the miRNA-1286/RAB31 axis in osteosarcoma. Cell Biol. Int. 2020. [Google Scholar] [CrossRef]
- Vokes, S.A.; Ji, H.; Wong, W.H.; McMahon, A.P. A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev. 2008, 22, 2651–2663. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, J.P.; Du, F.; Zhang, S.; Powell, M.B.; Falkenstein, K.N.; Ji, H.; Vokes, S.A. Spatiotemporal regulation of GLI target genes in the mammalian limb bud. Dev. Biol. 2015, 406, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Kasper, M.; Schnidar, H.; Neill, G.W.; Hanneder, M.; Klingler, S.; Blaas, L.; Schmid, C.; Hauser-Kronberger, C.; Regl, G.; Philpott, M.P.; et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol. Cell Biol. 2006, 26, 6283–6298. [Google Scholar] [CrossRef] [Green Version]
- Seto, M.; Ohta, M.; Asaoka, Y.; Ikenoue, T.; Tada, M.; Miyabayashi, K.; Mohri, D.; Tanaka, Y.; Ijichi, H.; Tateishi, K.; et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol. Carcinog. 2009, 48, 703–712. [Google Scholar] [CrossRef]
- Tanaka, K.; Iwakuma, T.; Harimaya, K.; Sato, H.; Iwamoto, Y. EWS-Fli1 antisense oligodeoxynucleotide inhibits proliferation of human Ewing’s sarcoma and primitive neuroectodermal tumor cells. J. Clin Invest. 1997, 99, 239–247. [Google Scholar] [CrossRef]
- Pishas, K.I.; Lessnick, S.L. Recent advances in targeted therapy for Ewing sarcoma. F1000Res 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, E.M.; Uren, A. A new era for an ancient drug: Arsenic trioxide and Hedgehog signaling. Vitam. Horm. 2012, 88, 333–354. [Google Scholar] [CrossRef]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [Green Version]
- Agyeman, A.; Jha, B.K.; Mazumdar, T.; Houghton, J.A. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget 2014, 5, 4492–4503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickström, M.; Dyberg, C.; Shimokawa, T.; Milosevic, J.; Baryawno, N.; Fuskevåg, O.M.; Larsson, R.; Kogner, P.; Zaphiropoulos, P.G.; Johnsen, J.I. Targeting the hedgehog signal transduction pathway at the level of GLI inhibits neuroblastoma cell growth in vitro and in vivo. Int. J. Cancer 2013, 132, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Walter, V.; Hayes, D.N.; Onaitis, M. Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin. Cancer Res. 2014, 20, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Peng, X.; Yuan, X.; Huang, D.; Chen, J.; Lu, Q.; Lv, N.; Luo, S. Suppression of growth and migration by blocking the Hedgehog signaling pathway in gastric cancer cells. Cell Oncol. 2013, 36, 421–435. [Google Scholar] [CrossRef]
- Yoon, J.W.; Lamm, M.; Chandler, C.; Iannaccone, P.; Walterhouse, D. Up-regulation of GLI1 in vincristine-resistant rhabdomyosarcoma and Ewing sarcoma. BMC Cancer 2020, 20, 511. [Google Scholar] [CrossRef]
- Tirode, F.; Laud-Duval, K.; Prieur, A.; Delorme, B.; Charbord, P.; Delattre, O. Mesenchymal Stem Cell Features of Ewing Tumors. Cancer Cell 2007, 11, 421–429. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward | Reverse |
|---|---|---|
| GAPDH | TGG GTG TGA ACC ATG AGA AGT ATG | GGT GCA GGA GGC ATT GCT |
| B2M | TTC TGG CCT GGA GGC TAT C | TCA GGA AAT TTG ACT TTC CAT TC |
| GLI1 | CCA ACT CCA CAG GCA TAC AGG AT | CAC AGA TTC AGG CTC ACG CTT C |
| GLI2 | AAG TCA CTC AAG GAT TCC TGC TCA | GTT TTC CAG GAT GGA GCC ACT T |
| GLI3 | CGC GAC TGA ACC CCA TTC TAC | GTG TTG TTG GAC TGT GTG CCA TT |
| Ptch1 | CCC CTG TAC GAA GTG GAC ACT CTC | AAG GAA GAT CAC CAC TAC CTT GGC T |
| Ptch2 | GAT GGG GCC ATC TCC ACA TT | CGC CGC AAA GAA GTA CCT TAC A |
| SMO | GCT ACT TCC TCA TCC GAG GAG TCA | GGC GCA GCA TGG TCT CGT T |
| STMN1 | TGG TGC TCA GAG TGT GGT CA | TCA CCT GGA TAT CAG AAG AAG CCA |
| NKx2-2 | GCA CCC CTC CTG GAG TTA GAA AC | CCA ACC CAG TGC CTC TCT CTG |
| EWS-FLI1 | GCC AAG CTC CAA GTC AAT ATA GC | GAG GCC AGA ATT CAT GTT ATT GC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mullard, M.; Cadé, M.; Morice, S.; Dupuy, M.; Danieau, G.; Amiaud, J.; Renault, S.; Lézot, F.; Brion, R.; Thepault, R.A.; et al. Sonic Hedgehog Signature in Pediatric Primary Bone Tumors: Effects of the GLI Antagonist GANT61 on Ewing’s Sarcoma Tumor Growth. Cancers 2020, 12, 3438. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113438
Mullard M, Cadé M, Morice S, Dupuy M, Danieau G, Amiaud J, Renault S, Lézot F, Brion R, Thepault RA, et al. Sonic Hedgehog Signature in Pediatric Primary Bone Tumors: Effects of the GLI Antagonist GANT61 on Ewing’s Sarcoma Tumor Growth. Cancers. 2020; 12(11):3438. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113438
Chicago/Turabian StyleMullard, Mathilde, Marie Cadé, Sarah Morice, Maryne Dupuy, Geoffroy Danieau, Jérome Amiaud, Sarah Renault, Frédéric Lézot, Régis Brion, Rose Anne Thepault, and et al. 2020. "Sonic Hedgehog Signature in Pediatric Primary Bone Tumors: Effects of the GLI Antagonist GANT61 on Ewing’s Sarcoma Tumor Growth" Cancers 12, no. 11: 3438. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113438