Targeting MDM2 for Neuroblastoma Therapy: In Vitro and In Vivo Anticancer Activity and Mechanism of Action

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
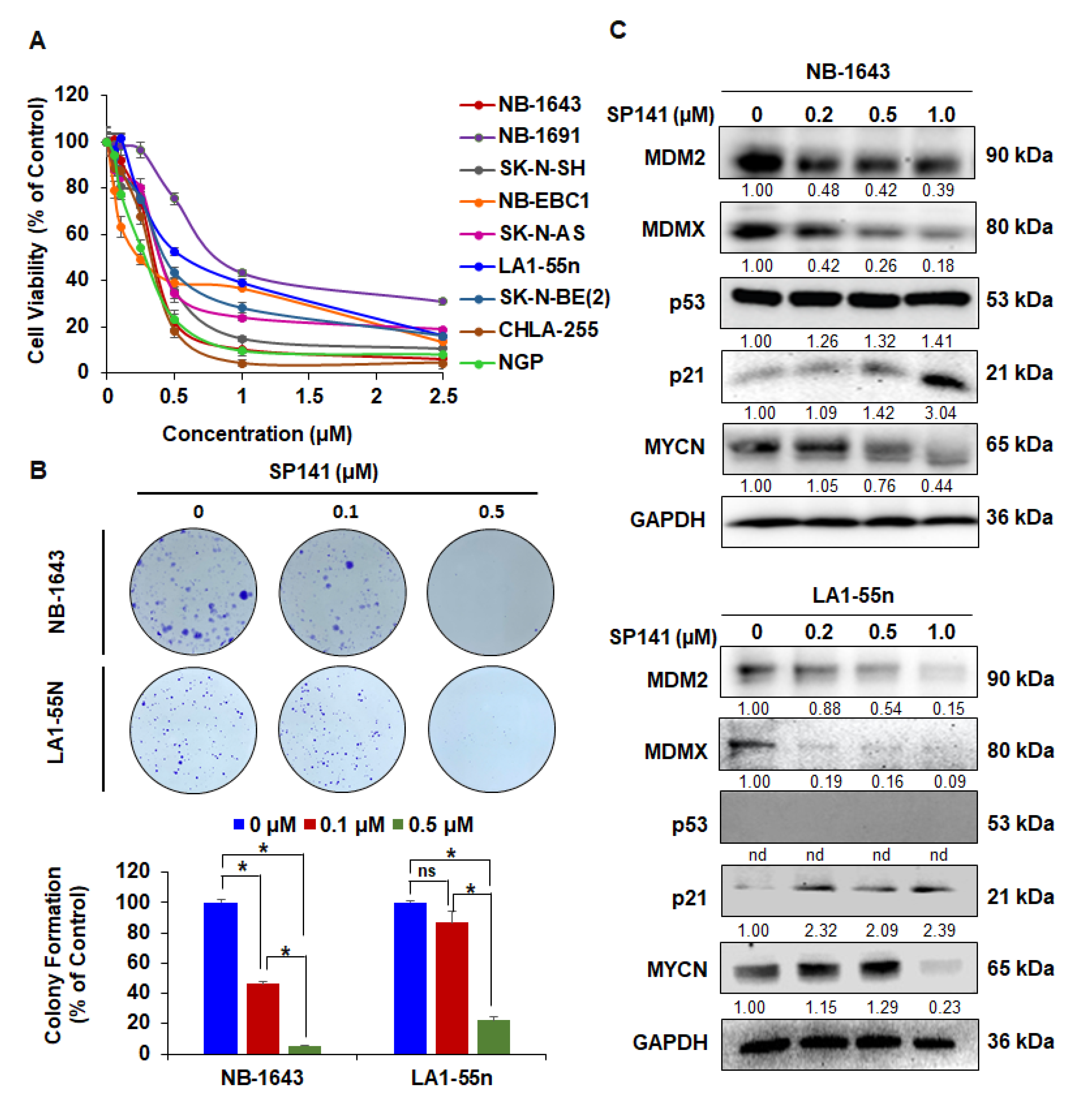
2.1. SP141 Inhibits MDM2 in Neuroblastoma Cells, Independent of p53
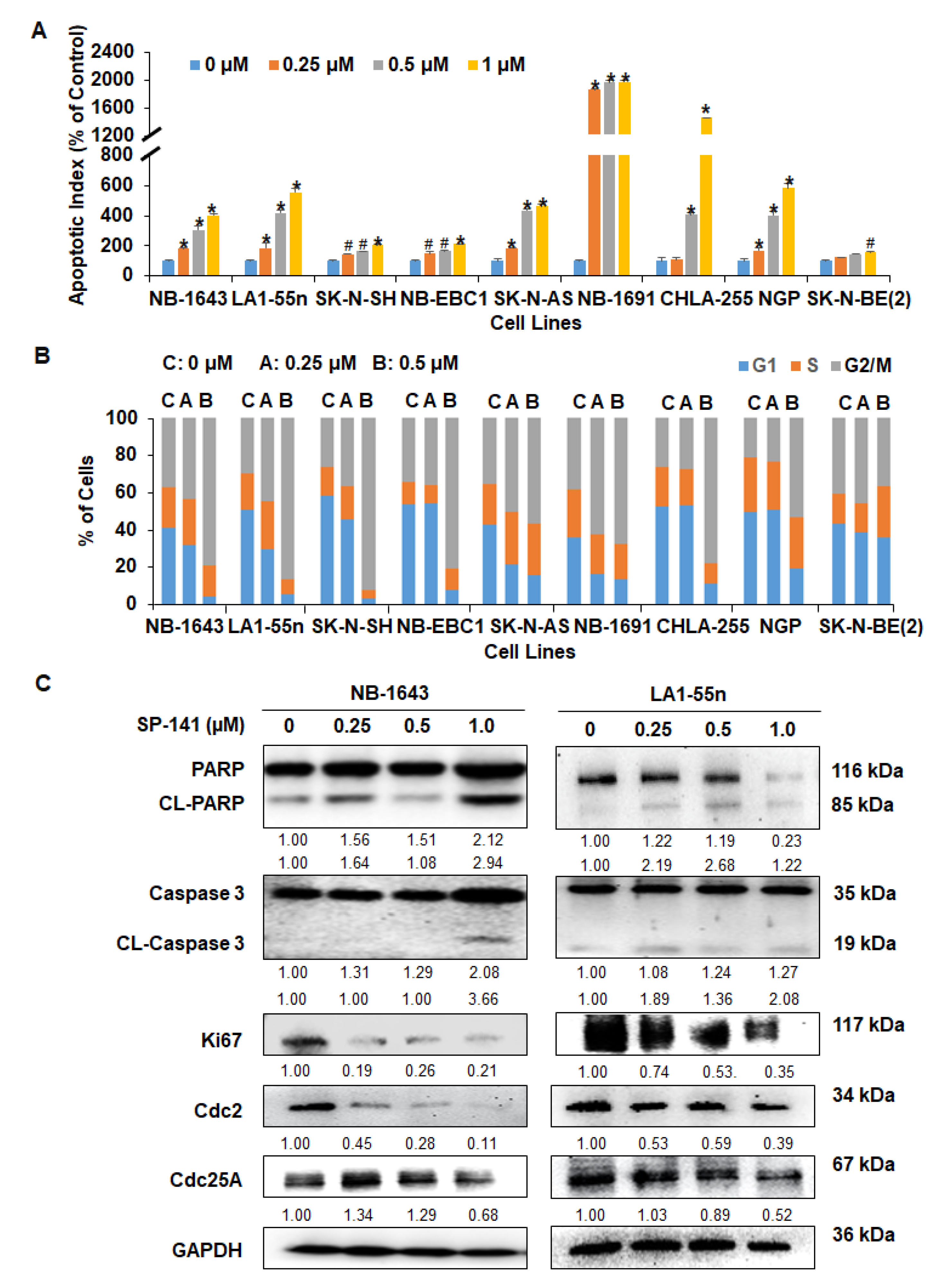
2.2. SP141 Induces Apoptosis and Cell Cycle Arrest in Neuroblastoma Cells
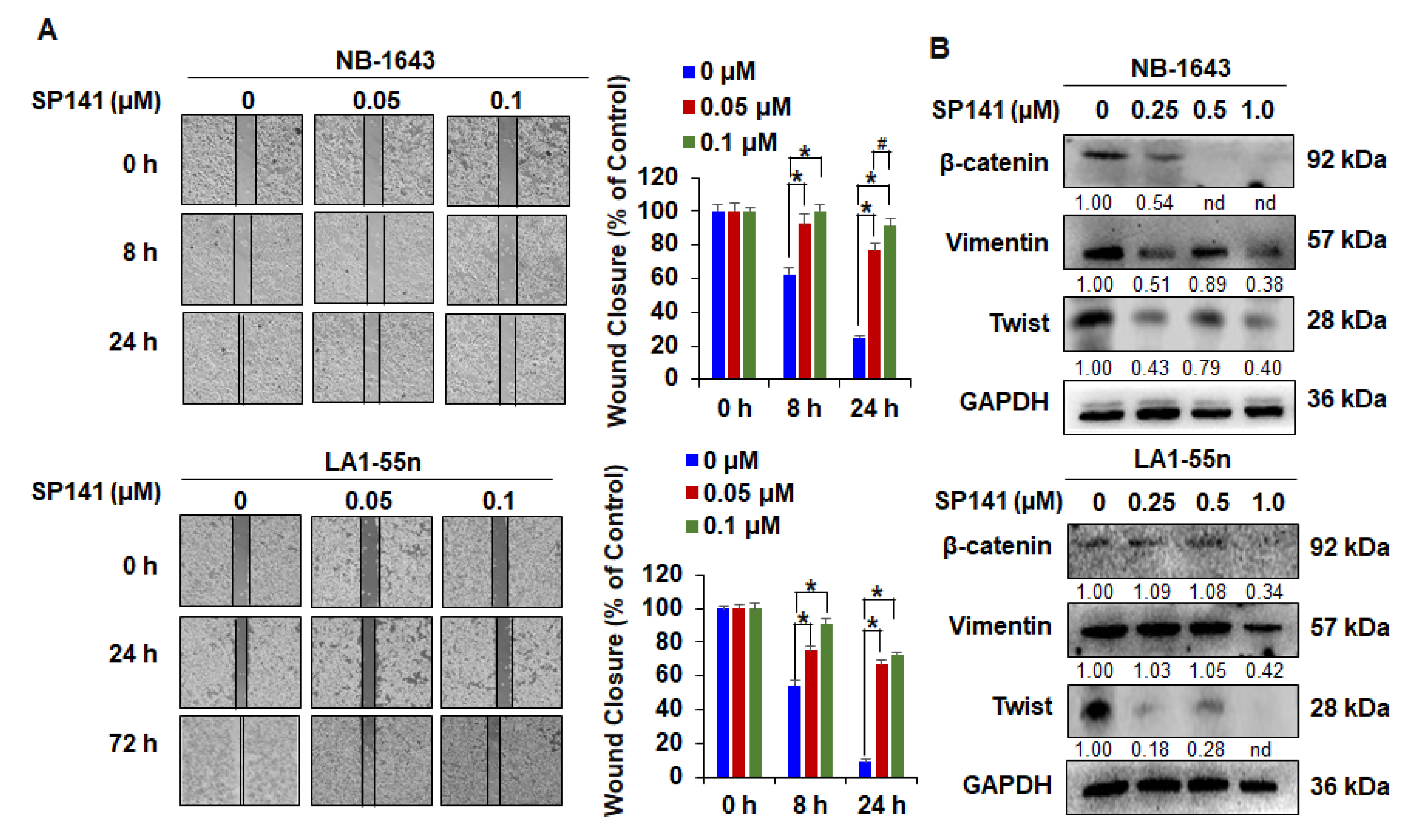
2.3. SP141 Inhibits the Migration of Neuroblastoma Cells
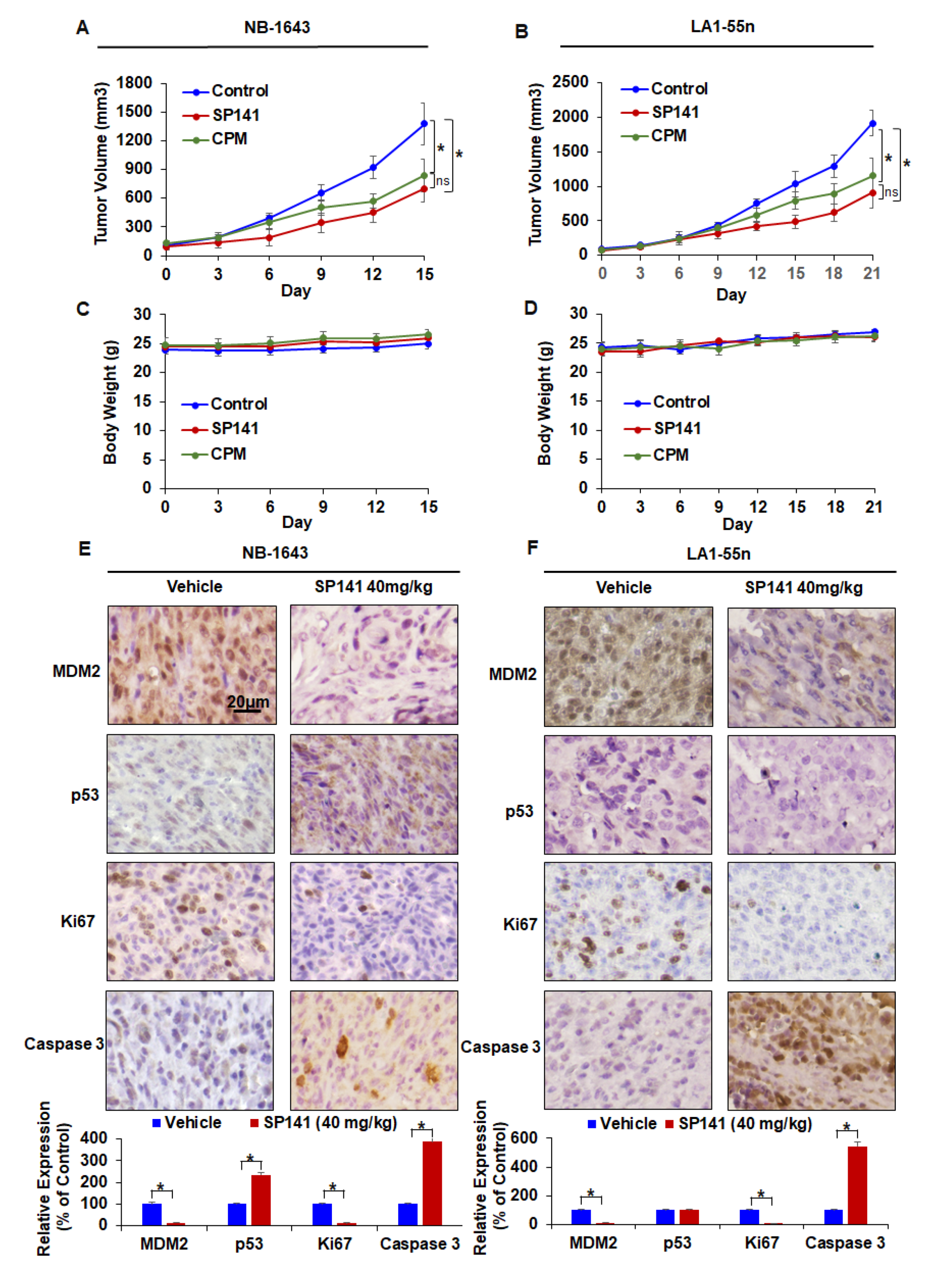
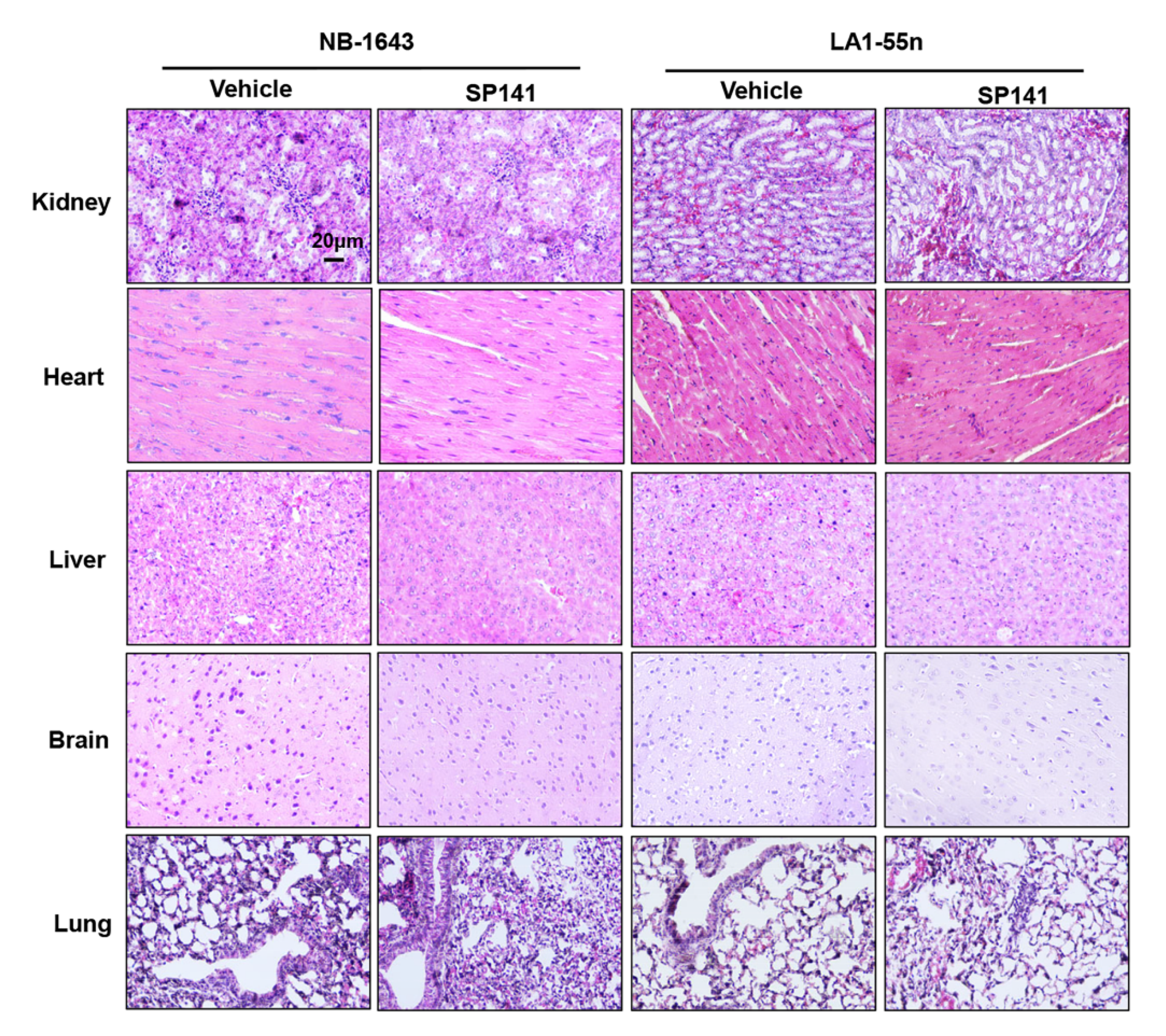
2.4. SP141 Inhibits Neuroblastoma Xenograft Tumor Growth
3. Discussion
4. Materials and Methods
4.1. Chemicals, Reagents, and Cell Lines
4.2. Assays for the In Vitro Effects of SP141
4.3. Western Blotting
4.4. In Vivo Xenograft Model for Human Cancer
4.5. H&E Staining and Immunohistochemistry
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Colon, N.C.; Chung, D.H. Neuroblastoma. Adv. Pediatr. 2011, 58, 297–311. [Google Scholar] [CrossRef] [Green Version]
- Louis, C.U.; Shohet, J.M. Neuroblastoma: Molecular pathogenesis and therapy. Annu. Rev. Med. 2015, 66, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Schwab, M.; Westermann, F.; Hero, B.; Berthold, F. Neuroblastoma: Biology and molecular and chromosomal pathology. Lancet Oncol. 2003, 4, 472–480. [Google Scholar] [CrossRef]
- Lakoma, A.; Barbieri, E.; Agarwal, S.; Jackson, J.; Chen, Z.; Kim, Y.; McVay, M.; Shohet, J.M.; Kim, E.S. The MDM2 small-molecule inhibitor RG7388 leads to potent tumor inhibition in p53 wild-type neuroblastoma. Cell Death Discov. 2015, 1, 15026. [Google Scholar] [CrossRef] [Green Version]
- Ngan, E.S.-W. Heterogeneity of neuroblastoma. Oncoscience 2015, 2, 837–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The international neuroblastoma risk group (INRG) classification system: An INRG task force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Sokol, E.; Desai, A.V. The Evolution of Risk Classification for Neuroblastoma. Children 2019, 6, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, V.; Foster, J. High-Risk Neuroblastoma Treatment Review. Children 2018, 5, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlakar, V.; Jurkovic-Mlakar, S.; Lopez, G.; Maris, J.M.; Ansari, M.; Gumy-Pause, F. 11q deletion in neuroblastoma: A review of biological and clinical implications. Mol. Cancer 2017, 16, 114. [Google Scholar] [CrossRef] [Green Version]
- Gatta, G.; Botta, L.; Rossi, S.; Aareleid, T.; Bielska-Lasota, M.; Clavel, J.; Dimitrova, N.; Jakab, Z.; Kaatsch, P.; Lacour, B.; et al. Childhood cancer survival in Europe 1999–2007: Results of EUROCARE-5—a population-based study. Lancet Oncol. 2014, 15, 35–47. [Google Scholar] [CrossRef]
- Tonini, G.P.; Capasso, M. Genetic predisposition and chromosome instability in neuroblastoma. Cancer Metastasis Rev. 2020, 39, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; Kreissman, S.G.; London, W.B.; Naranjo, A.; Cohn, S.L.; Hogarty, M.D.; Tenney, S.C.; Haas-Kogan, D.; Shaw, P.J.; Kraveka, J.M.; et al. Effect of Tandem Autologous Stem Cell Transplant vs Single Transplant on Event-Free Survival in Patients With High-Risk Neuroblastoma: A Randomized Clinical Trial. Jama 2019, 322, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Zage, P.E. Novel Therapies for Relapsed and Refractory Neuroblastoma. Children 2018, 5, 148. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Fernandez, R.; Watters, K.; Piskareva, O.; Stallings, R.L.; Bray, I. The role of genetic and epigenetic alterations in neuroblastoma disease pathogenesis. Pediatr. Surg. Int. 2013, 29, 101–119. [Google Scholar] [CrossRef] [Green Version]
- Nicolai, S.; Pieraccioli, M.; Peschiaroli, A.; Melino, G.; Raschellà, G. Neuroblastoma: Oncogenic mechanisms and therapeutic exploitation of necroptosis. Cell Death Dis. 2015, 6, e2010. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Y.H.P.; Muller, W.J. Oncogenes and Tumor Suppressor Genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a003236. [Google Scholar] [CrossRef] [Green Version]
- Corvi, R.; Savelyeva, L.; Breit, S.; Wenzel, A.; Handgretinger, R.; Barak, J.; Oren, M.; Amler, L.; Schwab, M. Non-syntenic amplification of MDM2 and MYCN in human neuroblastoma. Oncogene 1995, 10, 1081–1086. [Google Scholar]
- Zafar, A.; Wang, W.; Liu, G.; Xian, W.; McKeon, F.; Zhou, J.; Zhang, R. Targeting the p53-MDM2 pathway for neuroblastoma therapy: Rays of hope. Cancer Lett. 2021, 496, 16–29. [Google Scholar] [CrossRef]
- Cattelani, S.; Defferrari, R.; Marsilio, S.; Bussolari, R.; Candini, O.; Corradini, F.; Ferrari-Amorotti, G.; Guerzoni, C.; Pecorari, L.; Menin, C.; et al. Impact of a Single Nucleotide Polymorphism in the MDM2 Gene on Neuroblastoma Development and Aggressiveness: Results of a Pilot Study on 239 Patients. Clin. Cancer Res. 2008, 14, 3248–3253. [Google Scholar] [CrossRef] [Green Version]
- Cattelani, S.; Ferrari-Amorotti, G.; Soliera, A.R.; Manzotti, G.; Raschellà, G.; Calabretta, B. Neuroblastoma: Role of MDM2 and SNP309 as Markers; Springer: Dordrecht, The Netherlands, 2013. [Google Scholar]
- Slack, A.; Chen, Z.; Tonelli, R.; Pule, M.; Hunt, L.; Pession, A.; Shohet, J.M. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc. Natl. Acad. Sci. USA 2005, 102, 731–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaika, A.; Marchenko, N.; Moll, U.M. Cytoplasmically “Sequestered” Wild Type p53 Protein Is Resistant to Mdm2-mediated Degradation. J. Biol. Chem. 1999, 274, 27474–27480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Gu, L.; He, J.; Zhang, H.; Zhou, M. MDM2 Regulates Vascular Endothelial Growth Factor mRNA Stabilization in Hypoxia. Mol. Cell. Biol. 2011, 31, 4928–4937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Zhang, H.; He, J.; Li, J.; Huang, M.; Zhou, M. MDM2 regulates MYCN mRNA stabilization and translation in human neuroblastoma cells. Oncogene 2012, 31, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Keshelava, N.; Zuo, J.J.; Chen, P.; Waidyaratne, S.N.; Luna, M.C.; Gomer, C.J.; Triche, T.J.; Reynolds, C.P. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001, 61, 6185–6193. [Google Scholar]
- Van Maerken, T.; Ferdinande, L.; Taildeman, J.; Lambertz, I.; Yigit, N.; Vercruysse, L.; Rihani, A.; Michaelis, M.; Cinatl, J.; Cuvelier, C.A.; et al. Antitumor Activity of the Selective MDM2 Antagonist Nutlin-3 against Chemoresistant Neuroblastoma with Wild-Type p53. J. Natl. Cancer Inst. 2009, 101, 1562–1574. [Google Scholar] [CrossRef]
- Lu, J.; Guan, S.; Zhao, Y.; Yu, Y.; Wang, Y.; Shi, Y.; Mao, X.; Yang, K.L.; Sun, W.; Xu, X.; et al. Novel MDM2 inhibitor SAR405838 (MI-773) induces p53-mediated apoptosis in neuroblastoma. Oncotarget 2016, 7, 82757–82769. [Google Scholar] [CrossRef] [Green Version]
- Gamble, L.D.; Kees, U.R.; Tweddle, D.A.; Lunec, J. MYCN sensitizes neuroblastoma to the MDM2-p53 antagonists Nutlin-3 and MI-63. Oncogene 2012, 31, 752–763. [Google Scholar] [CrossRef] [Green Version]
- Burmakin, M.; Shi, Y.; Hedström, E.; Kogner, P.; Selivanova, G. Dual Targeting of Wild-Type and Mutant p53 by Small Molecule RITA Results in the Inhibition of N-Myc and Key Survival Oncogenes and Kills Neuroblastoma Cells In Vivo and In Vitro. Clin. Cancer Res. 2013, 19, 5092–5103. [Google Scholar] [CrossRef] [Green Version]
- Al-Ghabkari, A.; Narendran, A. In Vitro Characterization of a Potent p53-MDM2 Inhibitor, RG7112 in Neuroblastoma Cancer Cell Lines. Cancer Biother. Radiopharm. 2019, 34, 252–257. [Google Scholar] [CrossRef]
- Giustiniano, M.; Daniele, S.; Pelliccia, S.; La Pietra, V.; Pietrobono, D.; Brancaccio, D.; Cosconati, S.; Messere, A.; Giuntini, S.; Cerofolini, L.; et al. Computer-Aided Identification and Lead Optimization of Dual Murine Double Minute 2 and 4 Binders: Structure–Activity Relationship Studies and Pharmacological Activity. J. Med. Chem. 2017, 60, 8115–8130. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Qin, J.-J.; Voruganti, S.; Srivenugopal, K.S.; Nag, S.; Patil, S.; Sharma, H.; Wang, M.-H.; Wang, H.; Buolamwini, J.K.; et al. The pyrido[b]indole MDM2 inhibitor SP-141 exerts potent therapeutic effects in breast cancer models. Nat. Commun. 2014, 5, 5086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Qin, J.-J.; Voruganti, S.; Wang, M.-H.; Sharma, H.; Patil, S.; Zhou, J.; Wang, H.; Mukhopadhyay, D.; Buolamwini, J.K.; et al. Identification of a New Class of MDM2 Inhibitor That Inhibits Growth of Orthotopic Pancreatic Tumors in Mice. Gastroenterology 2014, 147, 893–902.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Hu, B.; Qin, J.-J.; Cheng, J.-W.; Li, X.; Rajaei, M.; Fan, J.; Yang, X.; Zhang, R. A novel inhibitor of MDM2 oncogene blocks metastasis of hepatocellular carcinoma and overcomes chemoresistance. Genes Dis. 2019, 6, 419–430. [Google Scholar] [CrossRef]
- Punganuru, S.R.; Arutla, V.; Zhao, W.; Rajaei, M.; Deokar, H.; Zhang, R.; Buolamwini, J.K.; Srivenugopal, K.S.; Wang, W. Targeted Brain Tumor Therapy by Inhibiting the MDM2 Oncogene: In Vitro and In Vivo Antitumor Activity and Mechanism of Action. Cells 2020, 9, 1592. [Google Scholar] [CrossRef]
- McKenzie, P.P.; Guichard, S.M.; Middlemas, D.S.; A Ashmun, R.; Danks, M.K.; Harris, L.C. Wild-type p53 can induce p21 and apoptosis in neuroblastoma cells but the DNA damage-induced G1 checkpoint function is attenuated. Clin. Cancer Res. 1999, 5, 4199–4207. [Google Scholar]
- Li, H.; Yu, Y.; Zhao, Y.; Wu, D.; Yu, X.; Lu, J.; Chen, Z.; Zhang, H.; Hu, Y.; Zhai, Y.; et al. Small molecule inhibitor agerafenib effectively suppresses neuroblastoma tumor growth in mouse models via inhibiting ERK MAPK signaling. Cancer Lett. 2019, 457, 129–141. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Rayburn, E.; Zhang, R.; He, J.; Wang, H. MDM2 and Human Malignancies: Expression, Clinical Pathology, Prognostic Markers, and Implications for Chemotherapy. Curr. Cancer Drug Targets 2005, 5, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254. [Google Scholar] [CrossRef]
- Qin, J.-J.; Li, X.; Hunt, C.; Wang, W.; Wang, H.; Zhang, R. Natural products targeting the p53-MDM2 pathway and mutant p53: Recent advances and implications in cancer medicine. Genes Dis. 2018, 5, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Z.; Hill, D.L.; Chen, X.; Wang, H.; Zhang, R. Genistein, a Dietary Isoflavone, Down-Regulates the MDM2 Oncogene at Both Transcriptional and Posttranslational Levels. Cancer Res. 2005, 65, 8200–8208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zhang, Z.; Hill, D.L.; Wang, H.; Zhang, R. Curcumin, a Dietary Component, Has Anticancer, Chemosensitization, and Radiosensitization Effects by Down-regulating the MDM2 Oncogene through the PI3K/mTOR/ETS2 Pathway. Cancer Res. 2007, 67, 1988–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nag, S.A.; Qin, J.-J.; Wang, W.; Wang, M.-H.; Wang, H.; Zhang, R. Ginsenosides as anticancer agents: In vitro and in vivo activities, structure–activity relationships, and molecular mechanisms of action. Front. Pharmacol. 2012, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Qin, J.-J.; Li, X.; Tao, G.; Wang, Q.; Wu, X.; Zhou, J.; Zi, X.; Zhang, R. Prevention of prostate cancer by natural product MDM2 inhibitor GS25: In vitro and in vivo activities and molecular mechanisms. Carcinogenesis 2018, 39, 1026–1036. [Google Scholar] [CrossRef]
- Wang, W.; Rayburn, E.R.; Hao, M.; Zhao, Y.; Hill, D.L.; Zhang, R.; Wang, H. Experimental therapy of prostate cancer with novel natural product anti-cancer ginsenosides. Prostate 2008, 68, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Rayburn, E.R.; Hang, J.; Zhao, Y.; Wang, H.; Zhang, R. Anti-lung cancer effects of novel ginsenoside 25-OCH3-PPD. Lung Cancer 2009, 65, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Rayburn, E.R.; Zhao, Y.; Wang, H.; Zhang, R. Novel ginsenosides 25-OH-PPD and 25-OCH 3 -PPD as experimental therapy for pancreatic cancer: Anticancer activity and mechanisms of action. Cancer Lett. 2009, 278, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, H.; Rayburn, E.R.; Zhao, Y.; Hill, D.L.; Zhang, R. 20(S)-25-methoxyl-dammarane-3β, 12β, 20-triol, a novel natural product for prostate cancer therapy: Activity in vitro and in vivo and mechanisms of action. Br. J. Cancer 2008, 98, 792–802. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, X.; Qin, J.-J.; Voruganti, S.; Nag, S.A.; Wang, M.-H.; Wang, H.; Zhang, R. Natural Product Ginsenoside 25-OCH3-PPD Inhibits Breast Cancer Growth and Metastasis through Down-Regulating MDM2. PLoS ONE 2012, 7, e41586. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, Y.; Rayburn, E.R.; Hill, D.L.; Wang, H.; Zhang, R. In vitro anti-cancer activity and structure–activity relationships of natural products isolated from fruits of Panax ginseng. Cancer Chemother. Pharmacol. 2007, 59, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Deokar, H.; Deokar, M.; Wang, W.; Zhang, R.; Buolamwini, J.K. QSAR studies of new pyrido[3,4-b]indole derivatives as inhibitors of colon and pancreatic cancer cell proliferation. Med. Chem. Res. 2018, 27, 2466–2481. [Google Scholar] [CrossRef] [PubMed]
- APatil, S.A.; Addo, J.K.; Deokar, H.; Sun, S.; Wang, J.; Li, W.; Suttle, D.P.; Wang, W.; Zhang, R.; Buolamwini, J.K. Synthesis, Biological Evaluation and Modeling Studies of New Pyrido[3,4-b] indole Derivatives as Broad-Spectrum Potent Anticancer Agents. Drug Des. 2017, 6, 143. [Google Scholar] [CrossRef]
- Qin, J.-J.; Wang, W.; Li, X.; Deokar, H.; Buolamwini, J.K.; Zhang, R. Inhibiting β-Catenin by β-Carboline-Type MDM2 Inhibitor for Pancreatic Cancer Therapy. Front. Pharmacol. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.-J.; Wang, W.; Sarkar, S.; Zhang, R. Oral delivery of anti-MDM2 inhibitor SP141-loaded FcRn-targeted nanoparticles to treat breast cancer and metastasis. J. Control. Release 2016, 237, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadkarni, D.H.; Wang, F.; Wang, W.; Rayburn, E.R.; Ezell, S.J.; Murugesan, S.; Velu, S.E.; Zhang, R. Synthesis and In Vitro Anti-Lung Cancer Activity of Novel 1, 3, 4, 8- Tetrahydropyrrolo [4, 3, 2-de]quinolin-8(1H)-o ne Alkaloid Analogs. Med. Chem. 2009, 5, 227–236. [Google Scholar] [CrossRef]
- Wang, F.; Ezell, S.J.; Zhang, Y.; Wang, W.; Rayburn, E.R.; Nadkarni, D.H.; Murugesan, S.; Velu, S.E.; Zhang, R. FBA-TPQ, a novel marine-derived compound as experimental therapy for prostate cancer. Investig. N. Drugs 2010, 28, 234–241. [Google Scholar] [CrossRef]
- Wang, W.; Cheng, J.-W.; Qin, J.-J.; Hu, B.; Li, X.; Nijampatnam, B.; Velu, S.E.; Fan, J.; Yang, X.-R.; Zhang, R. MDM2-NFAT1 dual inhibitor, MA242: Effective against hepatocellular carcinoma, independent of p53. Cancer Lett. 2019, 459, 156–167. [Google Scholar] [CrossRef]
- Wang, W.; Qin, J.-J.; Voruganti, S.; Nijampatnam, B.; Velu, S.E.; Ruan, K.; Hu, M.; Zhou, J.; Zhang, R. Discovery and Characterization of Dual Inhibitors of MDM2 and NFAT1 for Pancreatic Cancer Therapy. Cancer Res. 2018, 78, 5656–5667. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Rayburn, E.R.; Velu, S.E.; Chen, D.; Nadkarni, D.H.; Murugesan, S.; Chen, D.; Zhang, R. A novel synthetic iminoquinone, BA-TPQ, as an anti-breast cancer agent: In vitro and in vivo activity and mechanisms of action. Breast Cancer Res. Treat. 2010, 123, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Rayburn, E.R.; Velu, S.E.; Nadkarni, D.H.; Murugesan, S.; Zhang, R. In vitro and in vivo anticancer activity of novel synthetic makaluvamine analogues. Clin. Cancer Res. 2009, 15, 3511–3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wang, H.; Li, M.; Agrawal, S.; Chen, X.; Zhang, R. MDM2 Is a Negative Regulator of p21WAF1/CIP1, Independent of p53. J. Biol. Chem. 2004, 279, 16000–16006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Lee, H.; Zeng, S.X.; Dai, M.; Lu, H. MDM2 promotes p21waf1/cip1 proteasomal turnover independently of ubiquitylation. EMBO J. 2003, 22, 6365–6377. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neuroblastoma Cell Lines | |||
| Cell Lines | p53 Status | Multidrug-Resistant | IC50 (μM) |
| NB-1643 | WT | − | 0.36 |
| SK-N-SH | WT | − | 0.32 |
| NB-EBC1 | WT | − | 0.26 |
| CHLA-255 | WT | − | 0.42 |
| NGP | WT | − | 0.30 |
| NB-1691 | WT | Yes [37] | 0.89 |
| LA1-55n | Null | − | 0.62 |
| SK-N-AS | MT | − | 0.41 |
| SK-N-BE (2) | MT | Yes [38] | 0.29 |
| Normal Fibroblast Cell Line | |||
| Cell Line | p53 Status | Drug-Resistant | IC50 (μM) |
| IMR90 | WT | − | 13.22 [34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Wang, X.; Rajaei, M.; Youn, J.Y.; Zafar, A.; Deokar, H.; Buolamwini, J.K.; Yang, J.; Foster, J.H.; Zhou, J.; et al. Targeting MDM2 for Neuroblastoma Therapy: In Vitro and In Vivo Anticancer Activity and Mechanism of Action. Cancers 2020, 12, 3651. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123651
Wang W, Wang X, Rajaei M, Youn JY, Zafar A, Deokar H, Buolamwini JK, Yang J, Foster JH, Zhou J, et al. Targeting MDM2 for Neuroblastoma Therapy: In Vitro and In Vivo Anticancer Activity and Mechanism of Action. Cancers. 2020; 12(12):3651. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123651
Chicago/Turabian StyleWang, Wei, Xinjie Wang, Mehrdad Rajaei, Ji Youn Youn, Atif Zafar, Hemantkumar Deokar, John K. Buolamwini, Jianhua Yang, Jennifer H. Foster, Jia Zhou, and et al. 2020. "Targeting MDM2 for Neuroblastoma Therapy: In Vitro and In Vivo Anticancer Activity and Mechanism of Action" Cancers 12, no. 12: 3651. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123651