High-Dimensional Analysis of Single-Cell Flow Cytometry Data Predicts Relapse in Childhood Acute Lymphoblastic Leukaemia

 ,
,  ,
,  , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Flow Cytometer Machines and Antibodies
2.3. Genetic Data
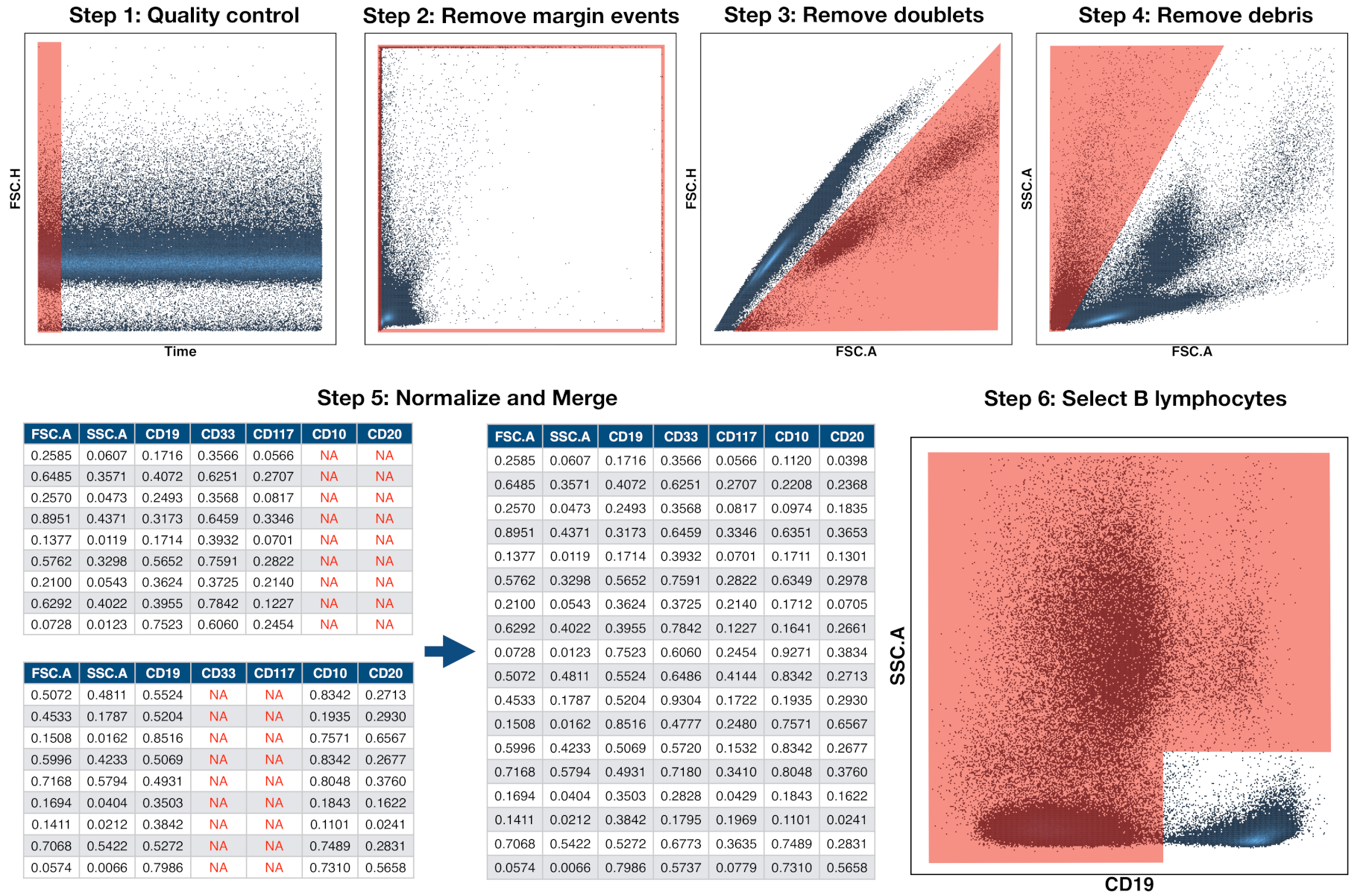
2.4. Preprocessing of Flow Cytometry Files
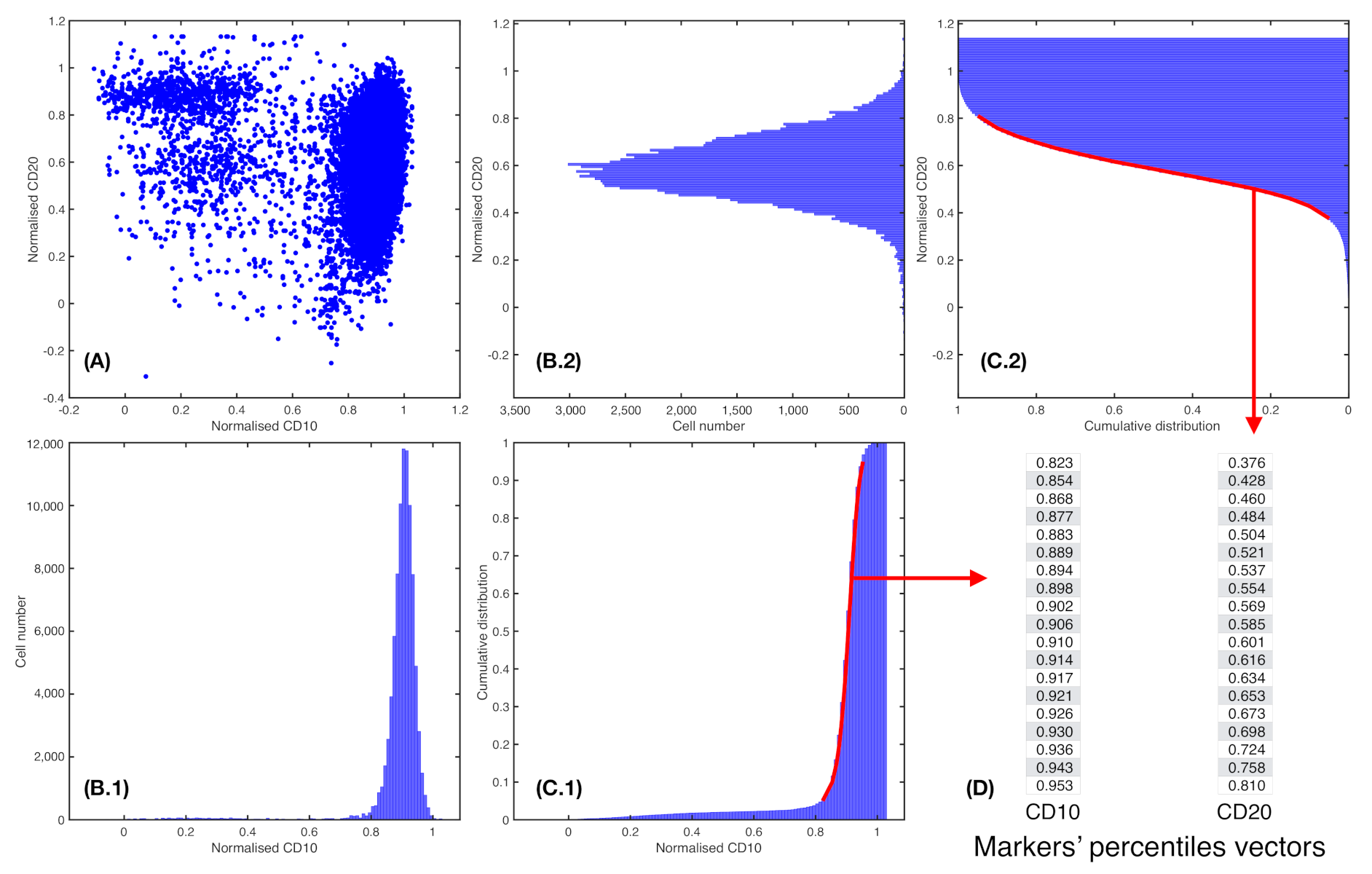
2.5. Marker Expression Characterisation
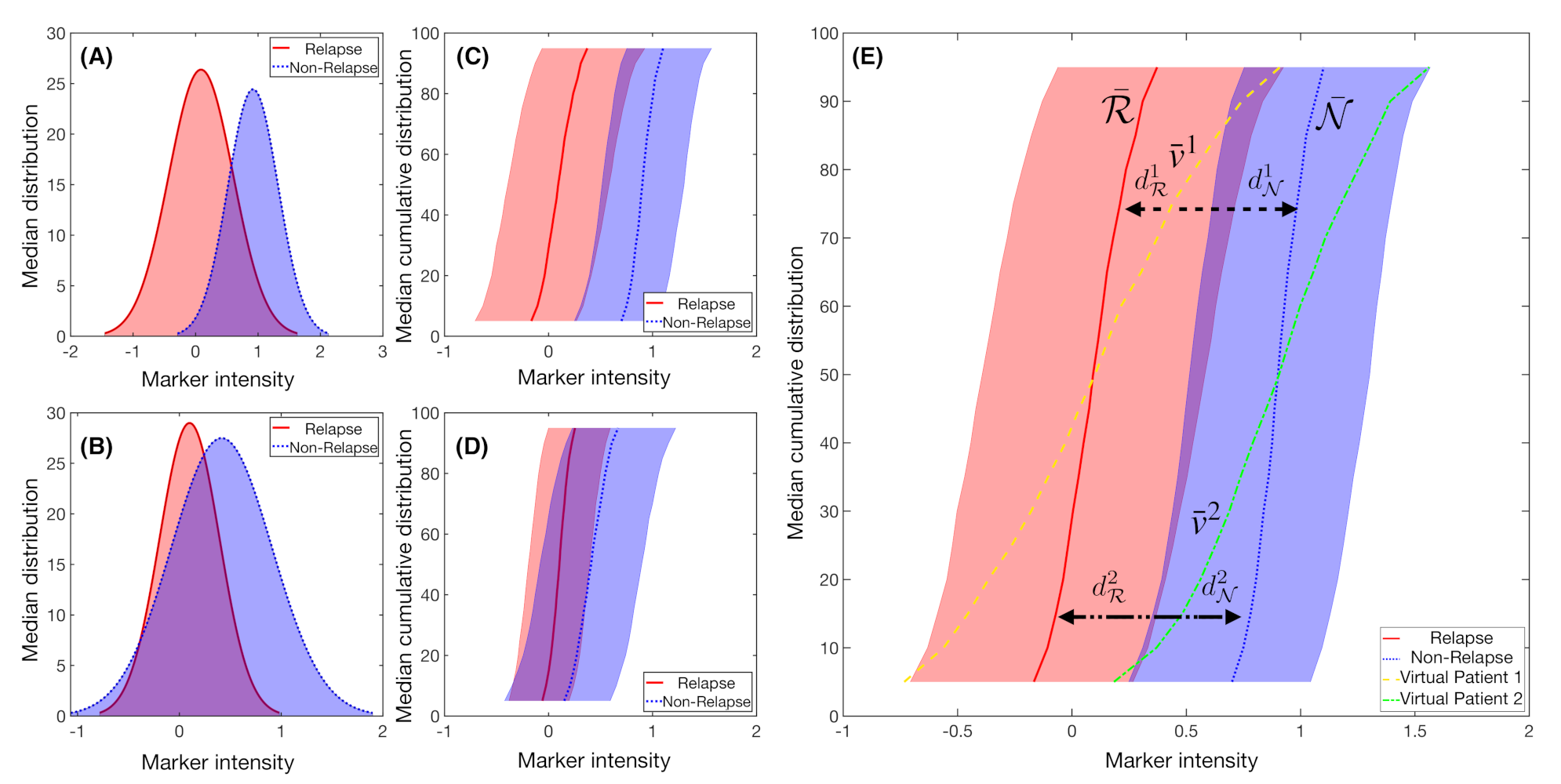
2.6. Fisher’s Linear Discriminant for Relapse Prediction
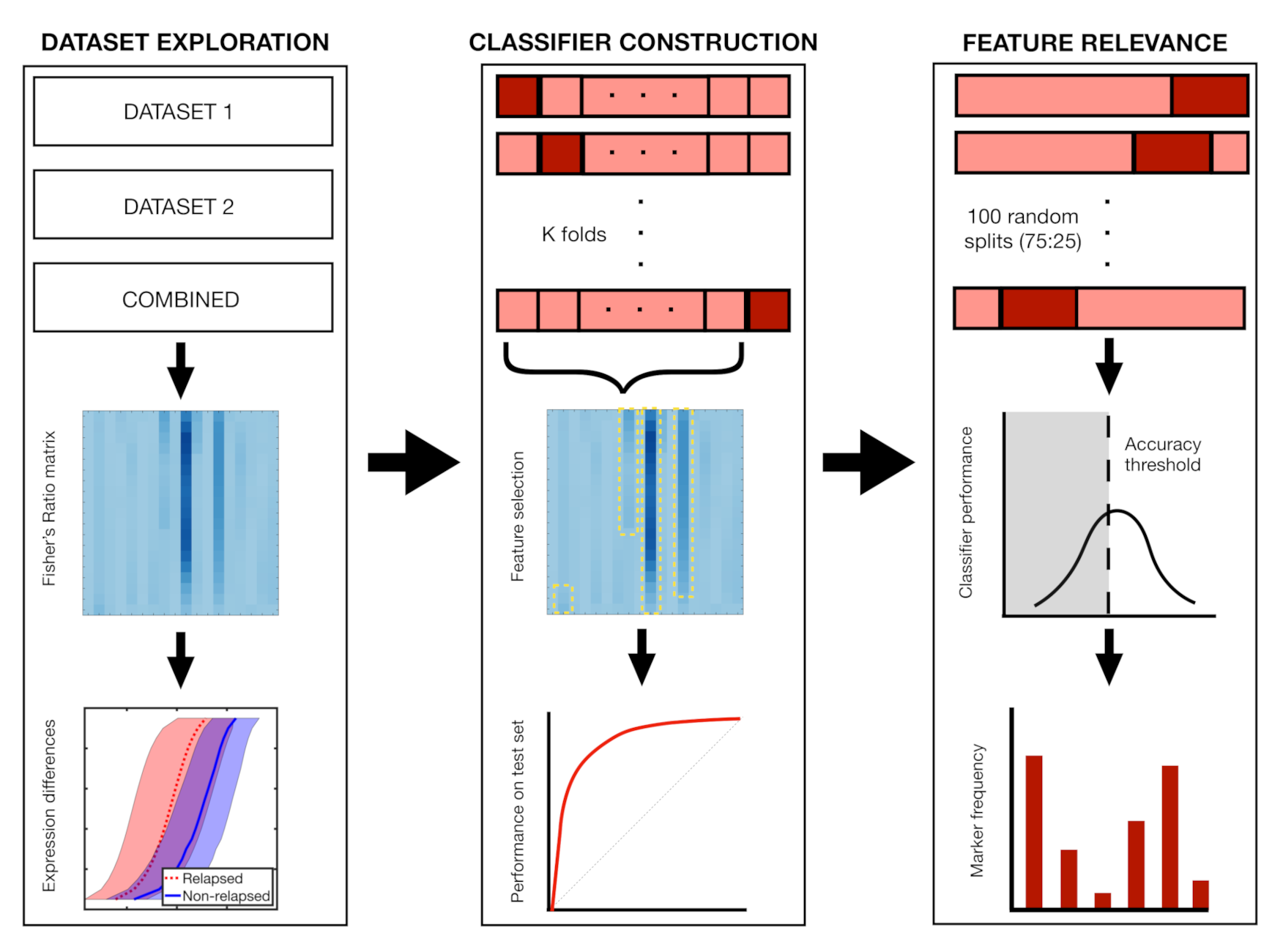
2.7. Classifier Construction and Feature Relevance
2.8. Statistical Analysis
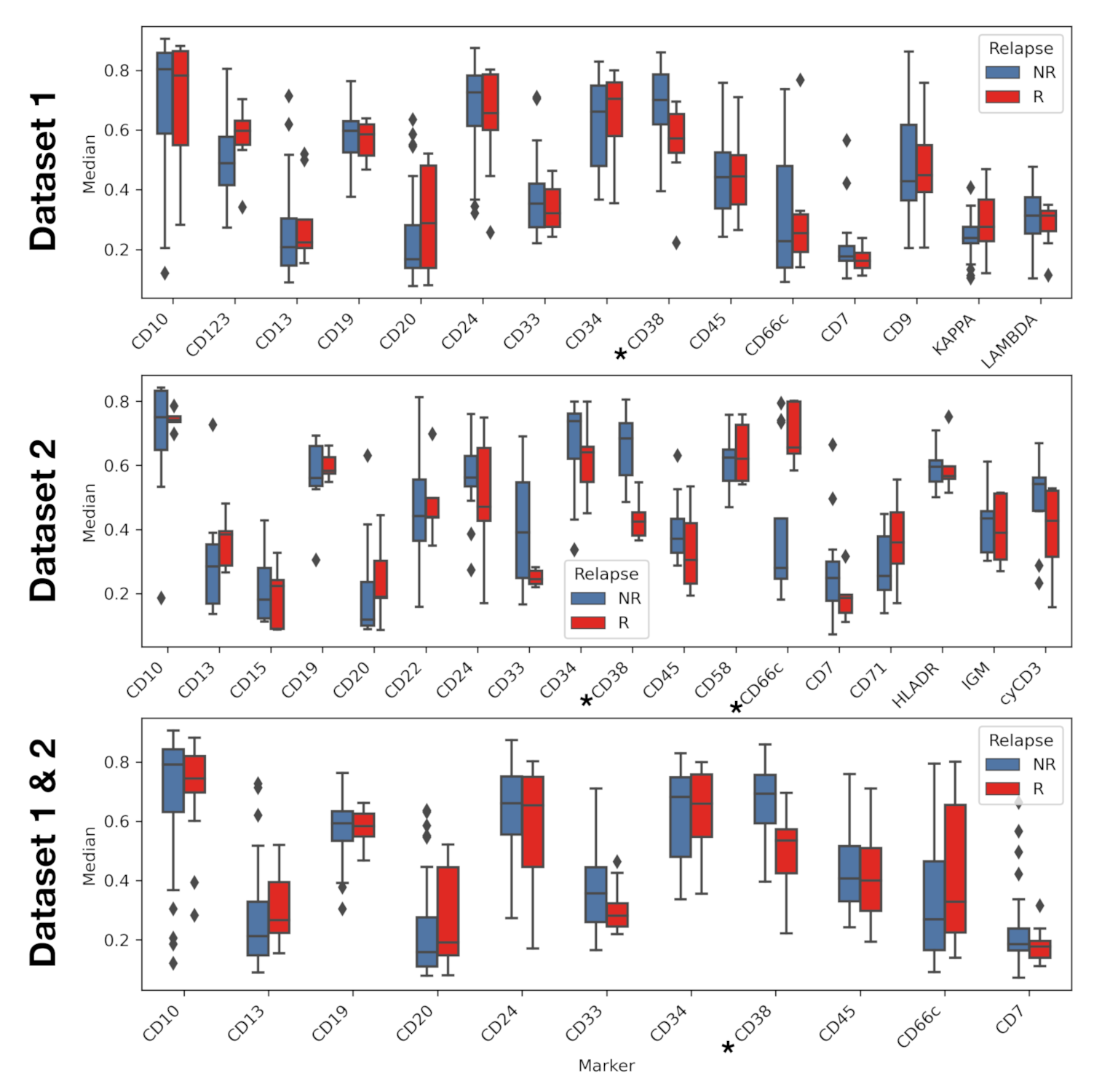
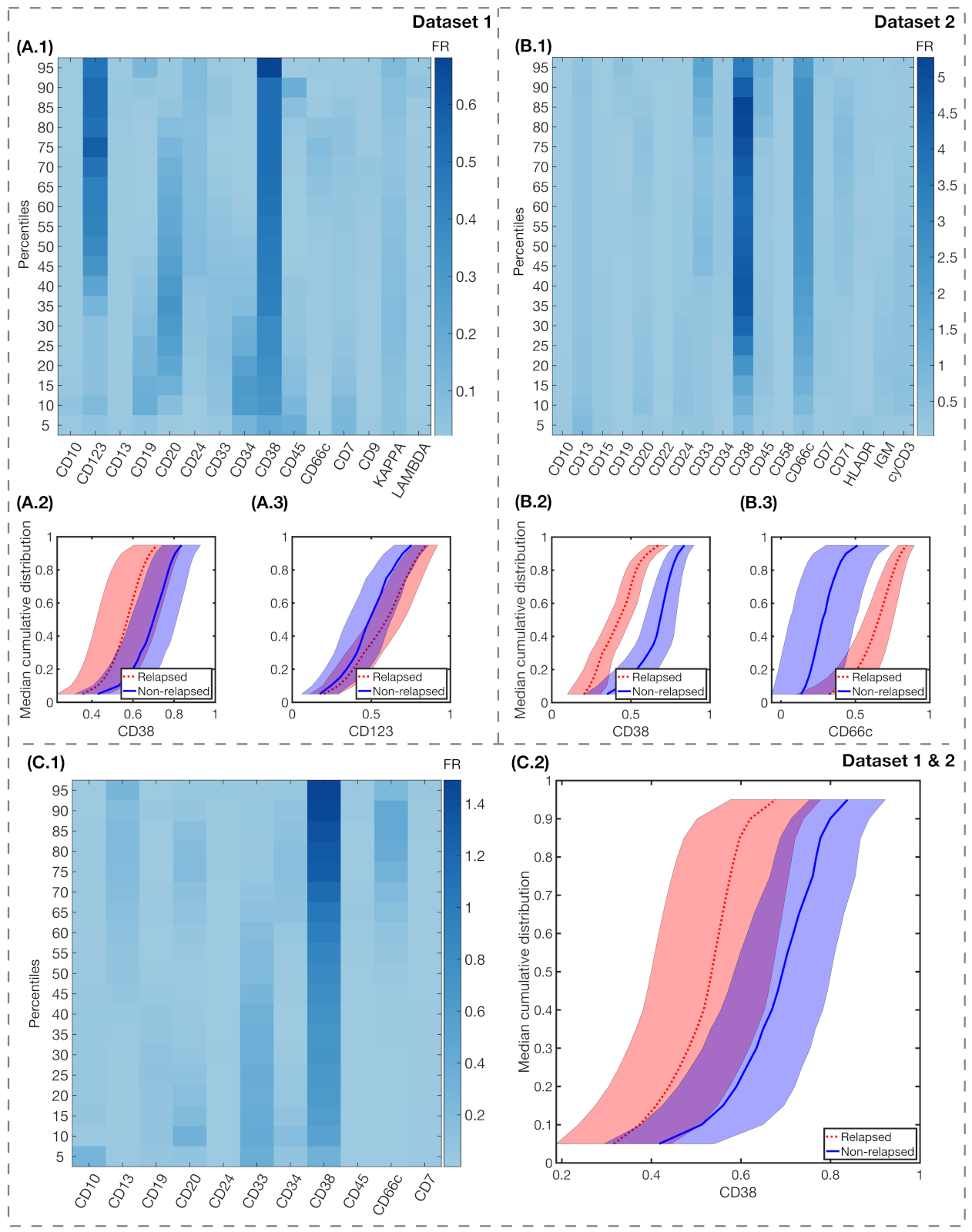
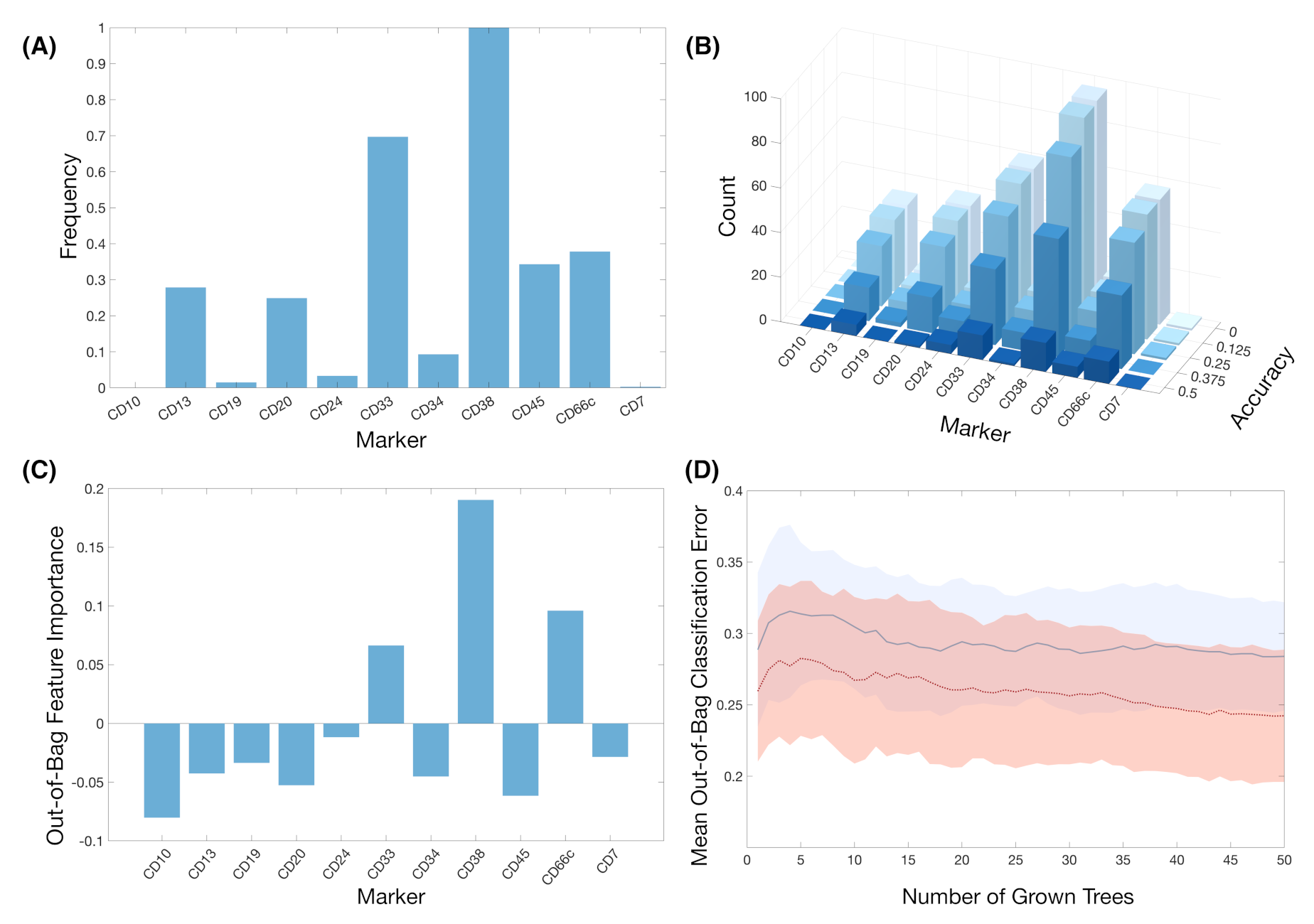
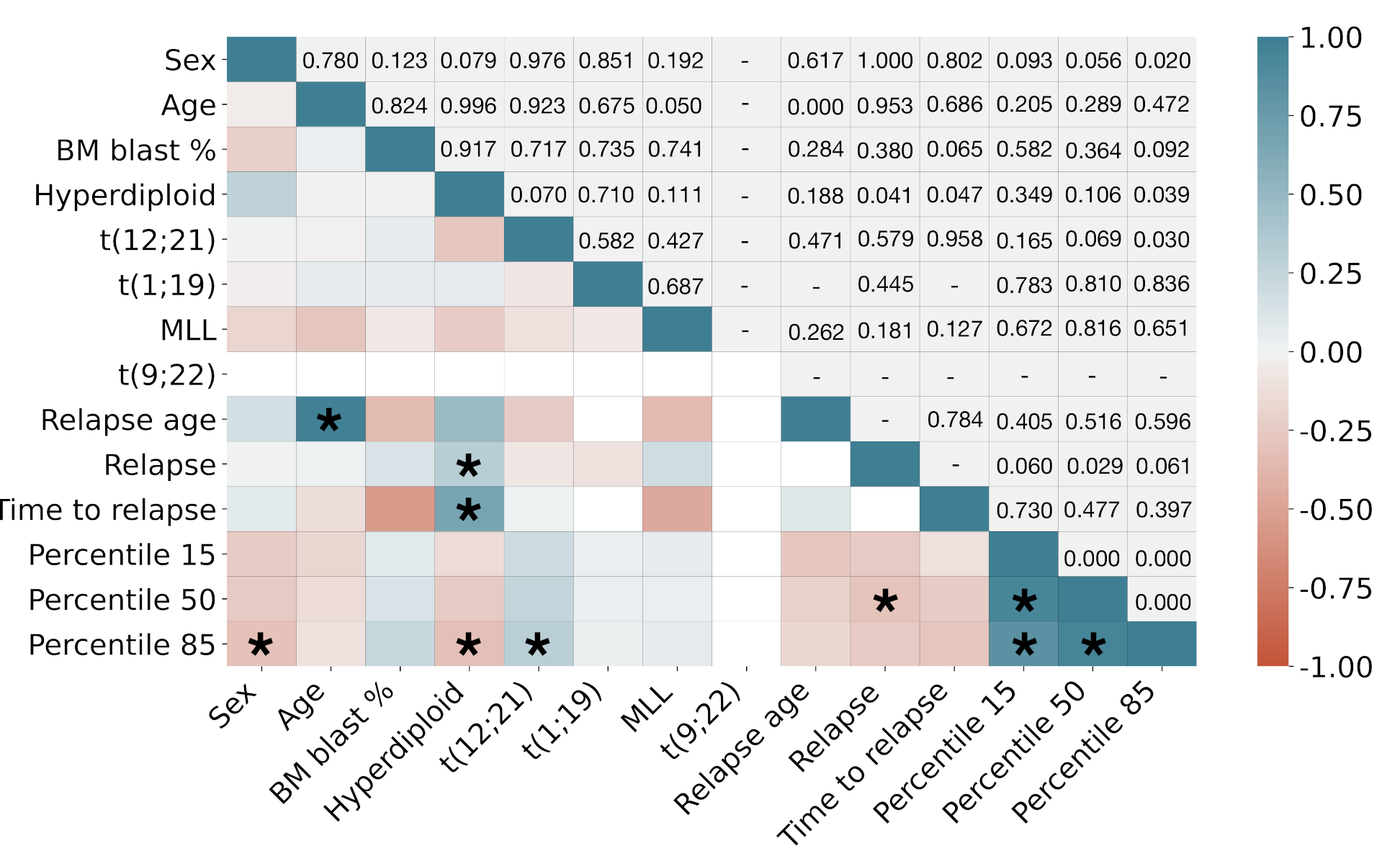
3. Results
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BM | Bone Marrow |
| MRD | Minimal Residual Disease |
| FCS | Flow Cytometry Standard |
| IRB | Institutional Review Board |
| CNS | Central Nervous System |
| IPT | Immunophenotypic |
| FR | Fisher’s Ratio |
| CD | Cluster of Differentiation |
| ALL | Acute Lymphoblastic Leukaemia |
| ROC | Receiver Operating Characteristic |
| AUC | Area Under Curve |
| LOOCV | Leave-One-Out cross-validation |
References
- Pizzo, P.A.; Poplack, D.G. Principles and practice of pediatric oncology. Lippincott Williams Wilkins 2015. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pui, C.-H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood acute lymphoblastic leukemia: Progress through collaboration. J. Clin. Oncol. 2015, 33, 2938. [Google Scholar] [CrossRef] [PubMed]
- Bhojwani, D.; Pui, C.-H. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013, 14, e205–e217. [Google Scholar] [CrossRef]
- Hunger, S.P.; Lu, X.; Devidas, M.; Camitta, B.M.; Gaynon, P.S.; Winick, N.J.; Reaman, G.H.; Carroll, W.L. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 1663. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef] [Green Version]
- Schultz, K.R.; Pullen, D.J.; Sather, H.N.; Shuster, J.J.; Devidas, M.; Borowitz, M.J.; Carroll, A.J.; Heerema, N.A.; Rubnitz, J.E.; Loh, M.L.; et al. Risk-and response-based classification of childhood B-precursor acute lymphoblastic leukemia: A combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children’s Cancer Group (CCG). Blood 2007, 109, 926–935. [Google Scholar] [CrossRef] [Green Version]
- van Dongen, J.J.M.; Lhermitte, L.; Böttcher, S.; Almeida, J.; der ver Velden, V.H.J.; Flores-Montero, J.; Rawstron, A.; Asnafi, V.; Lécrevisse, Q.; Lucio, P.; et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef] [Green Version]
- Weir, E.G.; Borowitz, M.J. Flow Cytometry in the Diagnosis of Acute Leukemia. In Seminars in Hematology; Elsevier: Amsterdam, The Netherlands, 2001; Volume 38, pp. 124–138. [Google Scholar] [CrossRef]
- Borowitz, M.J.; Devidas, M.; Hunger, S.P.; Bowman, W.P.; Carroll, A.J.; Carroll, W.L.; Linda, S.; Martin, P.L.; Pullen, D.J.; Viswanatha, D.; et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: A Children’s Oncology Group study. Blood 2008, 111, 5477–5485. [Google Scholar] [CrossRef] [Green Version]
- Conter, V.; Bartram, C.R.; Valsecchi, M.G.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Aricò, M.; Zimmermann, M.; Mann, G.; Rossi, G.D.; et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: Results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010, 115, 3206–3214. [Google Scholar] [CrossRef]
- Lugli, E.; Roederer, M.; Cossarizza, A. Data analysis in flow cytometry: The future just started. Cytom. Part A 2010, 77, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedreira, C.E.; Costa, E.S.; Lecrevisse, Q.; van Dongen, J.J.; Orfao, A. Overview of clinical flow cytometry data analysis: Recent advances and future challenges. Trends Biotechnol. 2013, 31, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Aghaeepour, N.; Finak, G.; Hoos, H.; Mosmann, T.R.; Brinkman, R.; Gottardo, R.; Scheuermann, R.H. Critical assessment of automated flow cytometry data analysis techniques. Nat. Methods 2013, 10, 228–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeys, Y.; Gassen, S.V.; Lambrecht, B.N. Computational flow cytometry: Helping to make sense of high-dimensional immunology data. Nat. Rev. Immunol. 2016, 16, 449–462. [Google Scholar] [CrossRef]
- Radakovich, N.; Nagy, M.; Nazha, A. Machine learning in haematological malignancies. Lancet Haematol. 2020, 7, e541–e550. [Google Scholar] [CrossRef]
- Mahmood, N.; Shahid, S.; Bakhshi, T.; Riaz, S.; Ghufran, H.; Yaqoob, M. Identification of significant risks in pediatric acute lymphoblastic leukemia (all) through machine learning (ml) approach. Med Biol. Comput. 2020, 1–10. [Google Scholar] [CrossRef]
- Pan, L.; Liu, G.; Lin, F.; Zhong, S.; Xia, H.; Sun, X.; Liang, H. Machine learning applications for prediction of relapse in childhood acute lymphoblastic leukemia. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Good, Z.; Sarno, J.; Jager, A.; Samusik, N.; Aghaeepour, N.; Simonds, E.F.; White, L.; Lacayo, N.J.; Fantl, W.J.; Fazio, G.; et al. Single-cell developmental classification of b cell precursor acute lymphoblastic leukemia at diagnosis reveals predictors of relapse. Nat. Med. 2018, 24, 474. [Google Scholar] [CrossRef]
- Reiter, M.; Diem, M.; Schumich, A.; Maurer-Granofszky, M.; Karawajew, L.; Rossi, G.J.; Ratei, R.; Groeneveld-Krentz, S.; Sajaroff, O.E. Automated flow cytometric mrd assessment in childhood acute b- lymphoblastic leukemia using supervised machine learning. Cytom. Part A J. Int. Soc. Anal. 2019, 95, 966–975. [Google Scholar] [CrossRef]
- Parks, D.R.; Roederer, M.; Moore, W.A. A new “logicle” display method avoids deceptive effects of logarithmic scaling for low signals and compensated data. Cytom. Part A J. Int. Soc. Anal. Cytol. 2006, 69, 541–551. [Google Scholar] [CrossRef]
- Hahne, F.; LeMeur, N.; Brinkman, R.R.; Ellis, B.; Haaland, P.; Sarkar, D.; Spidlen, J.; Strain, E.; Gentleman, R. Flowcore: A bioconductor package for high throughput flow cytometry. BMC Bioinform. 2009, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Pedreira, C.E.; Costa, E.S.; Barrena, S.; Lecrevisse, Q.; Almeida, J.; van Dongen, J.J.; Orfao, A. Generation of flow cytometry data files with a potentially infinite number of dimensions. Cytom. Part A J. Int. Soc. Anal. Cytol. 2008, 73, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Finn, W.; Scott, C. Statistical file matching of flow cytometry data. J. Biomed. Inform. 2011, 44, 663–676. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, K.; Aghaeepour, N.; Parker, J.; Hogge, D.; Karsan, A.; Dalal, B.; Brinkman, R.R. Deep profiling of multitube flow cytometry data. Bioinformatics 2015, 31, 1623–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, A.L.; Lambotte, O.; Grand, R.L.; Cosma, A.; Tchitchek, N. Cytobackbone: An algorithm for merging of phenotypic information from different cytometric profiles. Bioinformatics 2019, 35, 4187–4189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, M.; Taghiyar, M.J.; Chong, L.; Finak, G.; Gottardo, R.; Brinkman, R.R. flowDensity: Reproducing manual gating of flow cytometry data by automated density-based cell population identification. Bioinformatics 2015, 31, 606–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkay, L. Available online: https://www.mathworks.com/matlabcentral/fileexchange/9608-fca_readfcs (accessed on 30 June 2020).
- Wang, S.; Li, D.; Song, X.; Wei, Y.; Li, H. A feature selection method based on improved fisher’s discriminant ratio for text sentiment classification. Expert Syst. Appl. 2011, 38, 8696–8702. [Google Scholar] [CrossRef]
- Rodríguez, J.D.; Pérez, A.; Lozano, J.A. Sensitivity Analysis of k-Fold Cross Validation in Prediction Error Estimation. IEEE Trans. Pattern Anal. Mach. Intell. 2010, 32, 569–575. [Google Scholar] [CrossRef]
- Vallat, R. Pingouin: Statistics in Python. J. Open Source Softw. 2018, 3, 1026. [Google Scholar] [CrossRef]
- Pérez-García, V.M.; Fitzpatrick, S.; Pérez-Romasanta, L.A.; Pesic, M.; Schucht, P.; Arana, E.; Sánchez-Gómez, P. Applied mathematics and nonlinear sciences in the war on cancer. Appl. Math. Nonlinear Sci. 2016, 1, 423–436. [Google Scholar] [CrossRef] [Green Version]
- Radakovich, N.; Nagy, M.; Nazha, A. Artificial intelligence in hematology: Current challenges and opportunities. Networks 2020, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the adp ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deaglio, S.; Mehta, K.; Malavasi, F. Human CD38: A (r) evolutionary story of enzymes and receptors. Leuk. Res. 2001, 25, 1–12. [Google Scholar] [CrossRef]
- Ibrahim, S.; Keating, M.; Do, K.-A.; O’Brien, S.; Huh, Y.O.; Jilani, I.; Lerner, S.; Kantarjian, H.M.; Albitar, M. CD38 expression as an important prognostic factor in b-cell chronic lymphocytic leukemia. Blood J. Am. Soc. Hematol. 2001, 98, 181–186. [Google Scholar] [CrossRef]
- Keyhani, A.; Huh, Y.O.; Jendiroba, D.; Pagliaro, L.; Cortez, J.; Pierce, S.; Pearlman, M.; Estey, E.; Kantarjian, H.; Freireich, E.J. Increased CD38 expression is associated with favorable prognosis in adult acute leukemia. Leuk. Res. 2000, 24, 153–159. [Google Scholar] [CrossRef]
- Naik, J.; Themeli, M.; de Jong-Korlaar, R.; Ruiter, R.W.; Poddighe, P.J.; Yuan, H.; de Bruijn, J.D.; Ossenkoppele, G.J.; Zweegman, S.; Smit, L.; et al. CD38 as a therapeutic target for adult acute myeloid leukemia and t-cell acute lymphoblastic leukemia. Haematologica 2019, 104, e100. [Google Scholar] [CrossRef]
- George, A.A.; Franklin, J.; Kerkof, K.; Shah, A.J.; Price, M.; Tsark, E.; Bockstoce, D.; Yao, D.; Hart, N.; Carcich, S.; et al. Detection of leukemic cells in the CD34+ CD38- bone marrow progenitor population in children with acute lymphoblastic leukemia. Blood J. Am. Soc. Hematol. 2001, 97, 3925–3930. [Google Scholar] [CrossRef]
- Kong, Y.; Yoshida, S.; Saito, Y.; Doi, T.; Nagatoshi, Y.; Fukata, M.; Saito, N.; Yang, S.; Iwamoto, C.; Okamura, J.; et al. CD34+ CD38+ CD19+ as well as CD34+ CD38- CD19+ cells are leukemia-initiating cells with self-renewal capacity in human b-precursor all. Leukemia 2008, 22, 1207–1213. [Google Scholar] [CrossRef] [Green Version]
- Taussig, D.C.; Miraki-Moud, F.; Anjos-Afonso, F.; Pearce, D.J.; Allen, K.; Ridler, C.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G.; et al. Anti-CD38 antibody–mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood 2008, 112, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Wojcik, B.; Bothur, S.; Knecht, C.; Falkenburg, J.F.; Schroeder, T.; Serve, H.; Ottmann, O.G.; Rieger, M.A. Plastic CD34 and CD38 expression in adult b–cell precursor acute lymphoblastic leukemia explains ambiguity of leukemia-initiating stem cell populations. Leukemia 2017, 31, 731–734. [Google Scholar] [CrossRef]
- Ebinger, M.; Witte, K.-E.; Ahlers, J.; Schäfer, I.; André, M.; Kerst, G.; Scheel-Walter, H.-G.; Lang, P.; Handgretinger, R. High frequency of immature cells at diagnosis predicts high minimal residual disease level in childhood acute lymphoblastic leukemia. Leuk. Res. 2010, 34, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Shman, T.V.; Movchan, L.V.; Aleinikova, O.V. Frequencies of immature CD34+ CD38- and CD34+ CD38- CD19+ blasts correlate with minimal residual disease level in pediatric b-cell precursor acute lymphoblastic leukemia. Leuk. Lymphoma 2013, 54, 2560–2562. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Liu, S.; Li, K.; Zhou, X.; Zhang, P.; Zou, L. High proportion of CD34+/CD38- cells is positively correlated with poor prognosis in newly diagnosed childhood acute lymphoblastic leukemia. Leuk. Lymphoma 2014, 54, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wu, D.; Lin, S.; Li, P. CD34 and CD38 are prognostic biomarkers for acute b lymphoblastic leukemia. Biomark. Res. 2016, 4, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owaidah, T.M.; Rawas, F.I.; Elkum, N.B. Expression of CD66c and CD25 in acute lymphoblastic leukemia as a predictor of the presence of BCR/ABL rearrangement. Hematol. Stem Cell Ther. 2008, 1, 34–37. [Google Scholar] [CrossRef] [Green Version]
- Guillaume, N.; Penther, D.; Vaida, I.; Gruson, B.; Harrivel, V.; Claisse, J.F.; Capiod, J.C.; Lefrere, J.J.; Damaj, G. CD66c expression in B-cell acute lymphoblastic leukemia: Strength and weakness. Int. J. Lab. Hematol. 2011, 33, 92–96. [Google Scholar] [CrossRef]
- Kiyokawa, N.; Iijima, K.; Tomita, O.; Miharu, M.; Hasegawa, D.; Kobayashi, K.; Okita, H.; Kajiwara, M.; Shimada, H.; Inukai, T.; et al. Significance of CD66c expression in childhood acute lymphoblastic leukemia. Leuk. Res. 2014, 38, 42–48. [Google Scholar] [CrossRef]
- Mejstříková, E.; Kalina, T.; Trka, J.; Starỳ, J.; Hrušák, O. Correlation of CD33 with poorer prognosis in childhood all implicates a potential of anti-CD33 frontline therapy. Leukemia 2005, 19, 1092–1094. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Frankel, A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark. Res. 2014, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Zhu, M.; Huang, Y.; Wei, S.; Xie, J.; Xiao, Y. CD123 and its potential clinical application in leukemias. Life Sci. 2015, 59–64. [Google Scholar] [CrossRef]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, E.; Audette, C.; Kovtun, Y.; Daver, N.; Wang, S.A.; Pierce, S.; Konoplev, S.N.; Khogeer, H.; Jorgensen, J.L.; Konopleva, M.; et al. CD123 expression patterns and selective targeting with a CD123-targeted antibody-drug conjugate (imgn632) in acute lymphoblastic leukemia. Haematologica 2019, 104, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Djokic, M.; Björklund, E.; Blennow, E.; Mazur, J.; Söderhäll, S.; Porwit, A. Overexpression of CD123 correlates with the hyperdiploid genotype in acute lymphoblastic leukemia. Haematologica 2009, 94, 1016–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, K.; Case, M.; Minto, L.; Bailey, S.; Bown, N.; Jesson, J.; Lawson, S.; Vormoor, J.; Irving, J. Flow minimal residual disease monitoring of candidate leukemic stem cells defined by the immunophenotype, CD34+ CD38lowCD19+ in B-lineage childhood acute lymphoblastic leukemia. Haematologica 2010, 95, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bras, A.E.; Beishuizen, A.; Langerak, A.W.; Jongen-Lavrencic, M.; Marvelde, J.G.T.; van den Heuvel-Eibrink, M.M.; Zwaan, C.M.; van Dongen, J.J.; van der Velden, V.H. CD38 expression in paediatric leukaemia and lymphoma: Implications for antibody targeted therapy. Br. J. Haematol. 2018, 180, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset 1 (HVR) * (N = 38) | Dataset 2 (HNJ) ** (N = 18) | Total (N = 56) | |
|---|---|---|---|
| Sex—no. (%) | |||
| Male | 23 (60) | 9 (50) | 32 (57) |
| Female | 15 (40) | 9 (50) | 24 (43) |
| Age at diagnosis—yr/mo | |||
| Median | 3/8 | 3/7 | 3/9 |
| Range | 0/2–12/11 | 1/6–8/8 | 0/2–12/11 |
| Long term status—no. (%) | |||
| Relapse | 8 (21) | 5 (27) | 13 (23) |
| No relapse | 30 (79) | 13 (73) | 43 (77) |
| Immunophenotype—no. (%) | |||
| Common | 24 (63) | 11 (61) | 35 (62) |
| Pre-B | 3 (8) | 2 (11) | 5 (9) |
| Pro-B | 10 (26) | 1 (5) | 11 (20) |
| Mixed | 1 (3) | 2 (11) | 3 (5) |
| BM blasts at diagnosis—% | |||
| Median | 81 | 90 | 84 |
| Range | 11–96 | 33–95 | 11–96 |
| Karyotype—no. (%) | |||
| Hyperdiploid (>50) | 12 (32) | 2 (11) | 14 (25) |
| Normal (40–50) | 15 (39) | 13 (72) | 28 (50) |
| Hypodiploid (<40) | 1 (3) | 0 (0) | 1 (2) |
| Chromosomic alterations—no. (%) | |||
| ETV6/RUNX1 t(12;21) | 5 (13) | 3 (23) | 8 (14) |
| TCF3/PBX1 t(1;19) | 1 (3) | 1 (6) | 2 (4) |
| MLL/AF4 t(4;11) | 1 (3) | 0 (0) | 1 (2) |
| MLL rearrangement | 3 (8) | 0 (6) | 3 (5) |
| BCR/ABL1 t(9;22) | 0 (0) | 0 (0) | 0 (0) |
| Method | Accuracy | Sensitivity | Specificity | PPV | NPV | AUC | |
|---|---|---|---|---|---|---|---|
| Dataset 1 | LOOCV | 0.75 ± 0.04 | 0.74 ± 0.05 | 0.76 ± 0.05 | 0.76 ± 0.04 | 0.75 ± 0.04 | 0.76 ± 0.02 |
| 2-Fold | 0.59 ± 0.1 | 0.63 ± 0.14 | 0.43 ± 0.2 | 0.81 ± 0.04 | 0.24 ± 0.12 | 0.56 ± 0.1 | |
| 4-Fold | 0.62 ± 0.07 | 0.63 ± 0.1 | 0.58 ± 0.12 | 0.85 ± 0.03 | 0.3 ± 0.06 | 0.65 ± 0.06 | |
| 6-Fold | 0.64 ± 0.05 | 0.66 ± 0.05 | 0.58 ± 0.13 | 0.85 ± 0.04 | 0.31 ± 0.06 | 0.67 ± 0.06 | |
| 8-Fold | 0.7 ± 0.04 | 0.7 ± 0.04 | 0.71 ± 0.06 | 0.9 ± 0.02 | 0.39 ± 0.04 | 0.72 ± 0.03 | |
| Dataset 2 | LOOCV | 0.66 ± 0.06 | 0.95 ± 0.05 | 0.37 ± 0.1 | 0.6 ± 0.04 | 0.88 ± 0.1 | 0.89 ± 0.05 |
| 2-Fold | 0.72 ± 0.07 | 0.95 ± 0.06 | 0.13 ± 0.22 | 0.74 ± 0.05 | 0.42 ± 0.41 | 0.68 ± 0.16 | |
| 4-Fold | 0.78 ± 0.04 | 0.95 ± 0.05 | 0.34 ± 0.15 | 0.79 ± 0.03 | 0.81 ± 0.2 | 0.86 ± 0.06 | |
| Datasets 1 & 2 | LOOCV | 0.69 ± 0.05 | 0.62 ± 0.09 | 0.75 ± 0.09 | 0.72 ± 0.07 | 0.67 ± 0.05 | 0.78 ± 0.04 |
| 2-Fold | 0.64 ± 0.13 | 0.6 ± 0.17 | 0.75 ± 0.12 | 0.87 ± 0.09 | 0.38 ± 0.08 | 0.73 ± 0.11 | |
| 4-Fold | 0.69 ± 0.01 | 0.67 ± 0.02 | 0.77 ± 0.01 | 0.91 ± 0.01 | 0.41 ± 0.01 | 0.77 ± 0.04 | |
| 6-Fold | 0.7 ± 0.02 | 0.68 ± 0.02 | 0.77 ± 0.01 | 0.91 ± 0.01 | 0.42 ± 0.02 | 0.79 ± 0.02 | |
| 8-Fold | 0.7 ± 0.01 | 0.68 ± 0.02 | 0.77 ± 0.01 | 0.91 ± 0.01 | 0.42 ± 0.02 | 0.79 ± 0.02 | |
| 10-Fold | 0.7 ± 0.01 | 0.68 ± 0.02 | 0.77 ± 0.01 | 0.91 ± 0.01 | 0.42 ± 0.01 | 0.8 ± 0.02 | |
| 12-Fold | 0.69 ± 0.01 | 0.67 ± 0.02 | 0.77 ± 0.01 | 0.91 ± 0.01 | 0.41 ± 0.01 | 0.79 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chulián, S.; Martínez-Rubio, Á.; Pérez-García, V.M.; Rosa, M.; Blázquez Goñi, C.; Rodríguez Gutiérrez, J.F.; Hermosín-Ramos, L.; Molinos Quintana, Á.; Caballero-Velázquez, T.; Ramírez-Orellana, M.; et al. High-Dimensional Analysis of Single-Cell Flow Cytometry Data Predicts Relapse in Childhood Acute Lymphoblastic Leukaemia. Cancers 2021, 13, 17. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13010017
Chulián S, Martínez-Rubio Á, Pérez-García VM, Rosa M, Blázquez Goñi C, Rodríguez Gutiérrez JF, Hermosín-Ramos L, Molinos Quintana Á, Caballero-Velázquez T, Ramírez-Orellana M, et al. High-Dimensional Analysis of Single-Cell Flow Cytometry Data Predicts Relapse in Childhood Acute Lymphoblastic Leukaemia. Cancers. 2021; 13(1):17. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13010017
Chicago/Turabian StyleChulián, Salvador, Álvaro Martínez-Rubio, Víctor M. Pérez-García, María Rosa, Cristina Blázquez Goñi, Juan Francisco Rodríguez Gutiérrez, Lourdes Hermosín-Ramos, Águeda Molinos Quintana, Teresa Caballero-Velázquez, Manuel Ramírez-Orellana, and et al. 2021. "High-Dimensional Analysis of Single-Cell Flow Cytometry Data Predicts Relapse in Childhood Acute Lymphoblastic Leukaemia" Cancers 13, no. 1: 17. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13010017