
The Adhesome Network: Key Components Shaping the Tumour Stroma



Biomedical Sciences Research Centre “Alexander Fleming”, Institute of Bioinnovation, 34 Fleming Str., 16672 Vari-Athens, Greece
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cancers 2021, 13(3), 525; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13030525
Submission received: 23 December 2020
/
Revised: 22 January 2021
/
Accepted: 25 January 2021
/
Published: 30 January 2021
(This article belongs to the Special Issue Matrix Effectors and Cancer)
Abstract
:Simple Summary
Tumours are not formed only by malignant cells but contain many other cell types, including endothelial and mural cells of blood vessels, immune cells and cancer-associated fibroblasts. These host cells, together with extracellular matrix, form the tumour stroma. Tumour growth and metastasis depends on interactions between cancer cells and tumour stroma. Cell adhesion to extracellular matrix is essential for tissue growth and homeostasis and is deregulated in many pathological conditions, including cancer. This review highlights the vital role of cell adhesion in malignancy and describes how adhesion components regulate tumour stroma responses and control cancer development.
Abstract
Beyond the conventional perception of solid tumours as mere masses of cancer cells, advanced cancer research focuses on the complex contributions of tumour-associated host cells that are known as “tumour microenvironment” (TME). It has been long appreciated that the tumour stroma, composed mainly of blood vessels, cancer-associated fibroblasts and immune cells, together with the extracellular matrix (ECM), define the tumour architecture and influence cancer cell properties. Besides soluble cues, that mediate the crosstalk between tumour and stroma cells, cell adhesion to ECM arises as a crucial determinant in cancer progression. In this review, we discuss how adhesome, the intracellular protein network formed at cell adhesions, regulate the TME and control malignancy. The role of adhesome extends beyond the physical attachment of cells to ECM and the regulation of cytoskeletal remodelling and acts as a signalling and mechanosensing hub, orchestrating cellular responses that shape the tumour milieu.
1. Introduction
Malignant cells are the main driving force of tumour formation and growth, yet they do not manifest the disease single-handed. It is now increasingly accepted that noncancerous cells located in the tumour niche are involved in the hallmark capabilities of cancer [1]. Key cellular players in the tumour stroma are vascular cells comprising blood and lymphatic vessels (endothelial cells and mural cells), cancer-associated fibroblasts (CAFs), and infiltrating immune cells. Additional stromal cell types of solid tumours include mesenchymal stem cells, adipocytes and neurons [2,3]. The complex contributions of these allegedly normal, constituent cells together with noncellular components, namely the extracellular matrix (ECM) and soluble factors, are collectively known as the tumour microenvironment (TME) (Figure 1).
As in normal tissue, all cells in the TME are attached and interact with ECM at specific sites, called cell–matrix adhesions. These structures are composed by the integrin family of ECM receptors and an intrigue protein network called adhesome that is directly linked to the cell cytoskeleton [4,5,6]. Adhesion sites physically attach the cell to its microenvironment and maintain cell homeostasis. Depending on the protein composition and arrangements within the adhesome, cell–matrix adhesions have distinct architecture, localisation and lifespan [7]. These dynamic characteristics enable adhesion sites to rapidly change cell morphology and modulate cell spreading to facilitate cell migration and invasion. Furthermore, cell–matrix adhesions co-ordinate cellular responses to extracellular stimuli through crosstalk with intracellular signal transduction pathways, hence influencing cell survival, proliferation and differentiation. Emerging data highlight the role of mechanical cues in regulating cell function. In this aspect, adhesome proteins constitute critical players in both sensing and transducing mechanical forces to cell cytoskeleton. Adhesome members include more than 232 adhesion-related proteins broadly divided into structural/adaptor proteins (such as talin, vinculin, ILK-PINCH-parvin, paxillin, zyxin, tensin, α-actinin, filamin and KANK) and signalling proteins (for example FAK, Pyk2, Src, RhoGTPases and myosin light chain kinase). These proteins could constitute intrinsic components of cell–matrix adhesions or transiently associate with the adhesion network, facilitating the coordination of cell behaviour [8,9,10,11,12]. Finally, adhesome components connect with different actin structures (stress fibres, cortical actin, lamellipodia, filopodia) and microtubules, to stabilise cell shape and facilitate cell migration and invasion.
Several experimental approaches, ranging from genomics, proteomics and bioinformatics, to classical biochemistry, cell biology and model organisms (fly, worm, mouse), have started to decipher both the composition and the role of adhesome members in orchestrating cell function in physiology and pathology. In this review, we elaborate on the functional impact of adhesome proteins in cancer, focusing on host cells located into the tumour niche: endothelial cells (ECs), mural cells, CAFs and immune cells (summarised in Table 1).
To delineate the involvement of critical adhesome components on tumour stroma, we refrain from reviewing the role of adhesion molecules in cancer cell properties. Instead, we present the current understanding of how adhesome components control tumour stroma attained from cell-specific gene deletion and inhibition studies in different tumour settings. When such knowledge is missing, we discuss findings from genetic animal studies that provide valuable insight into stroma cell-specific roles of adhesome members under physiological conditions (summarised in Table 2). Since there are excellent reviews on the role of integrins on TME [52,53,54,55,56], we focus on the function of intracellular adhesome components in the tumour stroma. For clarity, we classify the adhesome proteins into three distinct categories, namely the adhesome signalling, the adhesion organisation and the actin regulatory layer (Figure 2). This classification has limitations because it does not take into account the dynamic crosstalk and the combined multiple roles of many adhesome members. We believe, though, that this simplifying approach will provide a clearer framework to emphasise the essential role of cell–matrix adhesions on the TME.
2. Adhesome Function in Vasculature
Blood vessels are composed of ECs, forming the inner surface of vessels, and perivascular mural cells (pericytes and smooth muscle cells) surrounding the vascular network. Under physiological conditions, blood vessels supply tissues with nutrients and oxygen and discharge metabolic waste products [123]. Cancer cells co-opt blood vessels and favour angiogenesis—the development of new blood vessels from pre-existing ones—to promote tumour growth and obtain routes for metastasis [124,125]. Owing to deregulated angiogenic processes, the tumour-associated vasculature has abnormal structure and function [126,127]. Unlike normal vessels, tumour vessels lack hierarchical organisation and exhibit irregular branching, aberrant density, shunting, incoherent blood flow and leakiness [128], establishing a chaotic vascular network with impaired functionality.
2.1. Endothelial Cell Adhesome
ECs are strategically positioned to influence the TME because of their ability to proliferate and migrate to form new blood vessels, fuelling tumour growth. Moreover, ECs serve as gatekeepers, enabling circulating factors and immune cells to penetrate the tumour environment and tumour cells to enter the circulation in order to form metastasis. They contribute to cancer development through communication with other stroma components and cancer cells. ECs are also critical drivers of tumour ECM remodelling. Endothelial adhesome has emerged as a crucial player of tumour progression because it controls both cell–cell junctions and cell–ECM attachment, thus influencing cellular crosstalk, angiogenesis and leakage.
2.1.1. Adhesome Signalling
The most studied adhesome signalling member both in physiology and cancer is focal adhesion kinase (FAK). FAK is abundant in developing blood vessels suggesting that FAK may play a role in angiogenesis [129]. Indeed, overexpression of FAK in ECs enhanced angiogenesis during wound healing and recovery from hind limb muscle ischemia [130] and endothelial-specific deletion of FAK led to embryonic lethality between embryonic day (E) 10.5 and 11.5 due to defective angiogenesis [57,58]. The critical role of FAK in tumour growth and angiogenesis was established in later studies by inducible deletion of FAK in tumour endothelium [13,14]. Tarova et al. showed that the endothelial-specific ablation of FAK in adult mice reduced tumour growth by inhibiting tumour angiogenesis. Mechanistically, endothelial FAK deficiency reduced cell migration and proliferation and enhanced apoptosis, resulting in a less dense vascular network both in tumour and in the developing retina [13]. Similarly, Lee et al. showed that deleted FAK from adult ECs reduces glioma growth and decreases tumour vascular dilation, tortuosity, and permeability by stabilising brain EC junctions and astrocyte feet interactions [14]. FAK was shown to be dispensable for VEGF-induced angiogenesis in adult mice due to compensatory Pyk2 expression [60]. However, endothelial FAK ablation did not change Pyk2 levels in embryonic or cancer vascular development [13,14,59,131,132]. In addition to the importance of endothelial FAK expression in angiogenesis and tumour growth, endothelial-specific deletion of FAK enhanced tumour sensitivity to DNA-damaging drugs by increasing inflammatory cytokine production, indicating that endothelial FAK could be targeted in combinatorial anticancer treatment [133].
Several studies have shown that pharmacological inhibition of FAK results in decreased tumour growth [134,135,136,137]. A dose-dependent effect of FAK inhibition in tumour growth and angiogenesis, however, was demonstrated in a study using FAK-heterozygous mice that displayed increased tumour growth [15]. These findings raised the necessity for more careful characterisation of FAK function before clinical application. To delineate FAK function in vivo, different mutant mice were generated targeting the FAK kinase domain, the autophosphorylation tyrosine site Y397 (blocking Y397F or mimic Y397E phosphorylation), and the Src-phosphorylation site Y861 (blocking Y861F) [16,17,18,59,131,132,133,138]. Embryos that lack FAK kinase activity or FAK-Y397 phosphorylation died at E13.5–15.5, exhibiting haemorrhages, oedema and vascular remodelling defects [59,131,132]. In agreement with embryonic studies, FAK kinase activity in ECs was necessary to induce phosphorylation in tyrosine residue Y658 of VE-cadherin and thus destabilisation of adherens junctions (AJ) [16,17,59]. Inhibiting FAK kinase activity in tumour endothelium decreased vascular leakage, tumour growth and metastasis [16,17]. Interestingly, an endothelial-specific FAK Y397E mutant, that mimics the phosphorylation of FAK Y397, was sufficient to restore VE-cadherin Y658 phosphorylation in tumour ECs and induce tumour vascular permeability in FAK kinase-deficient mice [17]. Consistently, blocking FAK phosphorylation at tyrosine Y397 in ECs inhibited tumour growth and angiogenesis [18]. The endothelial-specific phosphorylation of FAK at tyrosine residue-861 impairs the VEGF-dependent angiogenesis at early stages of tumour development, but this effect is not sustained in late-stage tumours [18]. Taken together, these data demonstrate that an intricate balance of FAK regulation is required to obtain the desired anticancer outcome.
2.1.2. Adhesion Organisation
Despite the indispensable role of many adhesome members in cell adhesion organisation and their effect in cancer cell motility and survival, genetic evidence for their function in tumour-associated ECs is mainly unexplored. Indirect evidence exists for the involvement of kindlin2 and integrin linked kinase (ILK) in tumour angiogenesis [36,139]. Prostate tumours growing in kindlin2 heterozygous mice were smaller and developed significantly less tumour blood vessels compared to control littermates [36]. Consistently, the heterozygous expression of kindlin2 reduced VEGF responses in a Matrigel angiogenesis assay in vivo [36]. Moreover, kindlin2 partial reduction caused defective basement membrane and pericyte coverage, resulting in impaired vascular maturation and increased vessel leakage [36]. Later studies further showed that kindlin2 regulates endothelial barrier function [140]. These data raise the possibility that kindlin2 could affect the formation of metastasis.
ILK is an essential mediator of integrin signalling, regulating cytoskeletal organisation and dynamics [141] and cancer cell survival. In thyroid tumour xenografts, pharmacological inhibition of ILK triggered apoptosis in both tumour cells and ECs, thus inhibiting tumour volume and angiogenesis [40]. The same pharmacological approach was used to suppress ILK expression in glioblastoma cells resulting in smaller tumours and reduced vessel density [42,142]. In an orthotopic model of human prostate cancer, siRNA-mediated depletion of ILK reduced tumour growth and angiogenesis by inhibiting HIF1a and VEGF expression [139]. In a paracrine action, ILK knockdown in melanoma cells decreased angiogenesis by lowering the proinflammatory cytokine IL-6 [143]. Pharmacological inhibition of ILK also resulted in decreased VEGF-induced EC invasion into Matrigel plugs in vivo [139]. Collectively, these data indicate that ILK could affect tumour growth by regulating angiogenesis. In pancreatic cancer xenografts, however, pharmacological ILK inhibition reduced tumour growth without affecting tumour angiogenesis [41]. Direct evidence for the significance of ILK expression in tumour-associated ECs is still missing. Accumulating in vitro data indicates a significant function of ILK in ECs. ILK regulates EC migration and responses to angiogenic factors VEGF and EGF and is necessary for integrin α5β1-mediated adhesion and capillary tube formation in vitro [139,144,145]. Furthermore, ECs derived from embryonic stem cells lacking ILK expression could not form vessel-like structures due to perturbed organisation of microtubules and cortical actin filaments and defective caveolin-1 localisation [144]. In vivo studies further support the vital role of ILK in endothelial physiology. Endothelial-specific deletion of ILK in mice results in embryonic lethality due to defective placenta vascularisation [87]. Inducible EC deletion in postnatal mice decreased sprouting of the retinal vasculature, reduced EC proliferation and disrupted the blood–retina barrier causing increased vascular leakage [88]. Therefore, deciphering whether and how the endothelial expression of ILK affects tumour growth and angiogenesis could support ILK as a target for anticancer therapies.
Besides ILK, talin is a key adhesome organiser because it controls the cell–ECM attachment by activating integrins and directly binding to the actin cytoskeleton. In addition, talin’s unique mechano-sensitive structure serves as a scaffold for the recruitment of numerous adhesome molecules. Endothelial-deletion of talin1 results in embryonic lethality due to vascular defects, while inducible ablation of talin1 in adult endothelium causes severe haemorrhaging in the intestinal vasculature followed by death [77,78]. Recent studies also showed that integrin activation by talin1 plays an important role in tumour angiogenesis. Endothelial-specific expression of an integrin activation talin mutant (talin1 L325R) decreased subcutaneous melanoma tumour growth and angiogenesis [39]. In line with the role of talin in endothelial junctions, vinculin, a close interactor of talin, is required for the remodelling of AJs [146]. As a response to different barrier-disruptive or barrier-enhancing agonists, vinculin localisation at AJ affects the actomyosin dynamics and thus the intracellular forces that are essential for the integrity of EC barrier [147]. These findings make vinculin and talin key candidates for regulating tumour vascular permeability and metastasis, a notion that remains to be investigated.
2.1.3. Actin Regulatory Layer
Cytoskeletal dynamics are crucial for cell shape maintenance and cell membrane extensions during cell migration and invasion. Many adhesome components are involved in linking the adhesion protein network to actin and tubulin cytoskeleton. How these proteins influence tumour stroma is mostly unexplored. Rho-GTPases family are the widely accepted master controllers of actin dynamics. The best-characterised members are Cdc42, which affects filopodia formation, Rac which guides lamellipodia development, and RhoA, which promotes stress fibres [148,149]. Although extensively studied in cancer cell migration, invasion and metastasis, the stroma cell-specific roles of Rho-GTPases family are less understood [150]. D’Amico and colleagues demonstrated that deleting Rac1 specifically from tumour ECs did not alter tumour growth and angiogenesis in wild-type mice but significantly decreased tumour vascular formation and inhibited tumour development in mice lacking integrin β3 expression [48], indicating a context-dependent role of Rac1 in tumour ECs. Another study showed that silencing Rac1 expression in the whole tumour xenograft, reduced tumour growth and angiogenesis and inhibited VEGF-induced neovascularisation of Matrigel plugs in vivo [49]. During development, endothelial deletion of Rac1 resulted in embryonic lethality caused by defects in heart development, early vasculogenesis and lymphatic-blood vessel separation [43,151]. In developing retina vasculature, endothelial deletion of Rac1 decreased vessel branching and EC invasion necessary to form the deeper plexus [108]. Taken together, these findings indicate an intricate involvement of Rac1 in tumour development, driven by EC-specific expression.
Another member of the Cdc42 subfamily, RhoJ, is essential for pathological angiogenesis [152,153]. RhoJ has distinct vascular expression pattern which is regulated by the endothelial transcription factor ERG and is required for EC migration, proliferation, and tube formation [154,155]. Although mice with constitutive deletion of RhoJ are viable, comprehensive examination revealed reduced vessel branching during embryogenesis and a delay in retinal vascular development due to impaired EC migration [120]. The effect of RhoJ deficiency was stronger in tumour settings, where it drastically decreased tumour angiogenesis and growth [50]. These findings were confirmed by endothelial-specific targeting of RhoJ, which suppressed blood vessel formation and disrupted tumour vascular integrity and function [51]. These differences in developmental and pathological angiogenesis could be caused by compensatory mechanisms acting in different contexts. Similar compensation could exist between various members of the RhoA family since endothelial deletion of RhoA did not affect developmental angiogenesis [117].
Besides the Rho-GTPases family, other actin-binding proteins could have an EC-related impact in cancer progression. Endothelial-specific deletion of filamin A decreased tumour angiogenesis and fibrosarcoma development [47]. Similarly, depletion of filamin B in HUVECs prevented EC migration and VEGF-induced tube formation in vitro [156] while constitutive deletion of filamin B in vivo caused nonlethal defects in microvascular patterning [103]. Collectively, these findings imply an important role of filamins in regulating tumour growth and angiogenesis.
2.1.4. Emerging Adhesome Players in Tumour Endothelium
Evidence from genetic studies have demonstrated a vital role of other adhesome components in endothelial biology, making them intriguing targets to be further explored in tumour settings. For example, parvin family proteins, α- parvin and β-parvin, form a tri-partite complex called IPP, with ILK and particularly interesting new cysteine-histidine-rich protein (PINCH) proteins. Endothelial-specific deletion of α-parvin reduced angiogenic sprouting and impaired vessel stability during late embryonic developmental stages [91]. Similar defects were observed in postnatal retina vascular formation [91]. Moreover, mice with endothelial deletion of both α- and β-parvins die earlier than single α-parvin deficient mice, indicating a partial compensatory function between parvin proteins [92]. Detailed examination of the embryonic vasculature revealed an abnormal vascular network with reduced branching, dilated vessels, balloon-type endothelial clusters and discontinuous basement membrane leading to extensive haemorrhaging. Parvin deficient vessels had defects in endothelial cell–cell junctions, apical-basal polarity and pericyte–vessel coverage [92]. All these defects could manifest in tumour vasculature but further studies are required to establish the role of parvins in tumour ECs.
Moreover, the putative role of tensins, a family of intracellular adhesion proteins that links integrins to actin filaments in cancer, stems from in vivo studies. Specifically, tensin1 deficiency in mice, although viable, has been shown to reduce angiogenesis both in vivo Matrigel assay and in ex vivo aortic ring assay [96,157]. This effect is mediated by decreased EC proliferation, migration and tube formation caused by downregulation of RhoA activity [96]. Thus, one can postulate that tensins could affect tumour angiogenesis. Another adhesome member that is closely linked to actin is zyxin [158]. Zyxin could affect tumour haemorrhaging and growth because it has been shown to regulate the exocytosis of endothelial Weibel–Palade body (WPB) and the secretion of von Willebrand factor (vWF) [159]. Interestingly, zyxin is upregulated upon shear stress and translocates from cell–matrix adhesions to the nucleus where it affects gene expression [160], indicating alternative ways to influence tumour development. Besides zyxin, another structural component of adhesion sites that can translocate to the nucleus and affect transcription is the family of four and a half LIM domain protein (FHL). In vitro studies propose that FHL2 has an antiangiogenic role because it can bind and inhibit HIF1a transactivation and VEGF transcription [161,162] and impairs EC migration and survival [163,164]. However, angiogenesis in the ischemia hind limp model and during recovery from corneal injury is not elevated but decreased in FHL2 knockout mice [165,166]. More studies are needed to decipher the involvement of FHL family proteins in tumour vascular development and cancer progression.
Another adhesion component that can influence tumour growth by affecting both vessel formation and permeability, is paxillin. Paxillin interacts with many kinases, including FAK, Src, Erk, Akt, PKA, and tyrosine phosphatases [167,168] and depending on its phosphorylation status can induce the assembly or the disassembly of adhesion sites [169]. Moreover, paxillin can regulate cell migration and invasion by controlling the spatiotemporal dynamics of Rho-GTPase family proteins Rho, Rac and Cdc42 through the recruitment of several GEFs and GAPs and interactions with the GIT/PIX/PAK complex [170,171]. Consistent with this, paxillin knockdown using siRNA enhanced EC migration and invasion in vitro and increased retina vascular sprouting in vivo by reducing neuropilin2 expression [32]. Interestingly, tumour soluble factors decreased endothelial expression of both paxillin and neuropilin2 and increased angiogenesis in a Matrigel assay in vivo [32], indicating an antiangiogenic role for paxillin in tumour setting. Additional to modulating angiogenesis, paxillin could affect cancer development by its established role in regulating endothelial barrier function [172,173,174]. The phosphorylation of paxillin in tyrosine-31 and -118 sites leads to destabilisation of VE-cadherin and enhanced pulmonary vascular permeability in LPS-mediated lung injury [173]. Nevertheless, in the presence of barrier enhancing factors such as sphingosine-1-phosphate (S1P) and hepatocyte growth factor (HGF), paxillin reinforces endothelial barrier through the formation of lamellipodia [174]. Therefore, it would be informative to investigate the effect of endothelial deletion of paxillin in tumour vascular permeability and metastasis.
Indications for a role of Cdc42 subfamily in tumour stroma stems from developmental studies showing that endothelial deletion of Cdc42 results in embryonic lethality with impaired vascular function [104,105]. Specifically, endothelial loss of cdc42 reduced EC migration and survival by decreasing the surface availability of VEGFR2, and thus angiogenic responses [105]. A more detailed examination revealed that endothelial deletion of cdc42 disrupted lumen formation during embryonic angiogenesis and reduced vascular sprouting and filopodia formation in the developing retina. These defects were caused by a disorganised actin cytoskeleton, loss of cell adhesion and endothelial polarity [104]. Besides the Rho family GTPases, other actin regulators could influence tumour vascular function. Specifically, VASP is upregulated during capillary morphogenesis in vitro [175] and it regulates the tethering of actin filaments during the formation of endothelial cell–matrix and cell–cell contacts [176]. Several studies, also, have demonstrated an essential role of VASP in maintaining endothelial barrier function in response to several stimuli that also affect tumour vasculature, including nitric oxide (NO) and hypoxia [177,178,179]. Due to functional compensation between the Ena, VASP and Ena/VASP-like proteins, single knockout mice have no major defect. The triple knockout mouse, though, is lethal and displays impaired endothelial cell junctions, leading to oedema and vascular leakage [97], indicating an important and yet undiscovered function in tumour vascular permeability and metastasis.
2.2. Mural Adhesome
Mural cells, comprising vascular smooth muscle cells (vSMCs) and mesenchymal-like cells called pericytes, are critical constituents of the TME [180]. In the TME, both vSMCs and pericytes are loosely associated, enabling vessel leakage [127,181]. Emerging data suggest a critical role of pericyte adhesion in regulating tumour growth and metastasis. In particular, deletion of integrin αvβ3 specifically from mural cells increased tumour development and metastasis by paracrine survival and tumour promoting signals without causing major defects in the tumour vasculature [182]. Another study showed that metastatic cells exploit integrins, ILK and YAP to dislodge pericytes, spread on capillaries, and form colonies in distant organs [183]. Despite these findings, very little is known about how adhesome components intrinsically influence mural function in cancer.
2.2.1. Adhesome Signalling
Recent studies using genetic deletion approaches have addressed the role of mural FAK in tumour angiogenesis and cancer development. In particular, mural FAK deficiency enhanced tumour growth and angiogenesis in syngeneic subcutaneous mouse models and spontaneously arising RIP-Tag2 pancreatic tumours [19]. In both models, FAK deficiency weakened the association of pericytes with tumour blood vessels and increased tumour angiogenesis. Mechanistically, the loss of pericyte FAK enhanced the expression of the proangiogenic and tumorigenic cytokine Cyr61, via a Gas6/Axl axis, driving tumour progression. These results were corroborated by human melanoma studies, where a high correlation was shown between tumour size and loss of pericyte-FAK [19]. As in ECs, pericyte FAK phosphorylation appears to have distinct roles in tumour growth and angiogenesis. Whereas phosphorylation of mural FAK at Y397 does not seem to affect angiogenesis and tumour growth, the phosphorylation at Y861 is important for blocking vessel regression and enhancing tumour survival [20].
2.2.2. Adhesion Organisation
The impact of adhesome structural components in mural function during cancer development has not been studied. Nevertheless, evidence from mural specific genetic ablation of key adhesome members advocates a critical role in tumour vascular function. A direct paradigm constitutes the mural ablation of ILK, which results in the defective formation of the vessel wall and embryonic lethality. Mechanistically, ILK deficiency enhances phosphorylation of myosin light chain (MLC) through the activation of Rho/ROCK signalling pathway and enhances vSMC contraction both in vitro and in vivo [89]. The same Rho/ROCK signalling pathway is also activated in the absence of α-parvin. Deletion of α-parvin in mice is embryonic lethal with defects in vascular remodelling and reduced vSMC/pericyte spreading on blood vessels due to hypercontractility [93]. Elevated contractility was also observed in pericytes overexpressing talin in vitro. This contractile phenotype was shown to be dependent on calpain-mediated signalling, as calpain pharmacological inhibition and expression of a talin mutant that is resistant in calpain cleavage reversed this effect [184]. In contrast, zyxin-null vSMCs display reduced responses to contractile agonists and exhibit enhanced proliferation, migration and invasion and are resistant to stress-induced apoptosis [185]. More investigation is needed to address the functional role of these key adhesome members in tumour pericyte function and cancer development.
3. Adhesome Function in CAFs
Fibroblasts are spindle-shaped cells of mesenchymal origin and have a pivotal role in tissue repair through a reversible transition to activated fibroblasts (also termed as myofibroblasts) [186]. CAFs represent one of the most dominant components of the tumour stroma in several malignancies including breast [187], liver [188], gastrointestinal [189], and pancreatic [190] cancer. In contrast to normal fibroblasts, the majority of CAFs remain in an “activated” state characterised by enhanced contractility, proliferation and ECM deposition [191,192]. Advances in single-cell scale approaches revealed that CAFs are a heterogeneous population with functional diversity deriving from multiple origins [193,194]. Currently, unique markers for CAFs have not been defined, and therefore CAFs are mainly recognised by the expression of myofibroblast proteins, such as a-smooth muscle actin (aSMA), fibroblast activation protein (FAP), fibroblast-specific protein 1 (FSP1), and vimentin [195]. Several studies have revealed that CAFs have either a pro- or an antitumorigenic potential within the TME, implying a dual role of CAFs in cancer progression [196].
3.1. Adhesome Signalling
Several studies show that CAFs display altered adhesion-mediated signalling that favours their activation and their protumorigenic capacity. For example, microarray gene- profiling in cultured CAFs and matched normal fibroblasts from nonsmall cell lung carcinoma (NSCLC) patients, together with data of differentially expressed stromal/CAF genes from various solid tumours revealed significant enrichment in focal adhesion pathways [197], with integrins and FAK posing central nodes [197,198]. Moreover, primary CAFs from NSCLC patients displayed elevated expression of integrin β1 and FAK-Y397 phosphorylation, driving proliferative responses to matrix rigidity and differential accumulation in squamous cell carcinoma vs. adenocarcinoma, in vivo [199]. Similarly, patient-derived oral squamous cell carcinoma (OSCC) CAFs overexpress FAK and siRNA mediated FAK depletion in CAFs reduced monocyte chemoattractant protein 1 (MCP1) production and decreased cancer cell invasion in vitro [200]. Recently, it was reported that FAK kinase activity regulates the TGFβR2 recycling and hepatic stellate cell (HSC) activation to tumour-promoting CAFs in vitro and in mice [23]. In other studies, chemical inhibition of FAK resulted in tumour stasis by reducing stroma proliferation and limiting the presence of FAP+ and aSMA+ CAFs [21,22]. Moreover, FAK activity in CAFs was recently found to drive tumour metastasis and augment an immunosuppressive TME [25,26]. Taken together, these studies provide evidence for a role of FAK in mediating the tumour-promoting actions of CAFs in vivo. Strikingly, a recent study has further addressed the direct involvement of FAK in CAF properties and discovered a tumour suppressive role for CAF-FAK by regulating cancer cell metabolism [24]. Specifically, conditional deletion of FAK from FSP1-expressing CAFs increased tumour growth in both orthotopic and spontaneous breast cancer mouse models. The enhanced tumour development in FSP-Cre+;FAKfl/fl knockout mice was caused by altered CAF chemokine production and paracrine signalling, which increased malignant cell glycolysis. These findings were corroborated by clinical observations that low FAK stromal expression is positively correlated with poor overall survival in human breast and pancreatic patients [24].
3.2. Adhesion Organisation
Besides FAK, recent studies revealed that kindlin2 is necessary for bladder CAF activation and pancreatic stellate cell (PSC) proliferation, migration and cytokine production [37,201]. Kindlin2 siRNA-mediated ablation in isolated human PSCs decreased pancreatic cancer cell proliferation and migration in vitro and growth of tumour/CAFs co-implantation in athymic mice [37]. Similarly, kindlin2 silencing suppressed CAF-mediated cancer cell survival, migration and epithelial to mesenchymal transition [201]. Altered adhesion properties are crucial not only for the differentiation of fibroblasts into CAFs and their migratory capacity but also for the production and organisation of ECM, that governs tumour rigidity and response to treatment [202]. Besides the role of integrins, emerging data indicate an involvement of other adhesome components in CAF-mediated ECM remodelling. For example, CAFs exhibit an increased number of adhesion sites and higher turnover rates of large vinculin-containing adhesions that enhance contractility and traction force generation necessary for aligning fibronectin (FN) fibres and creating migratory tracks for cancer cells [203]. A separate study implicated talin1 and kindlin2 for the activation of β1 integrins and the cooperation with the DDR2 collagen receptor to enable CAF-mediated collagen matrix assembly and linear organisation [204].
3.3. Actin Regulatory Layer
Given the essential role of actomyosin cytoskeleton in eliciting CAF contraction, actin-related proteins have emerged as critical effectors of CAF function. Microarray analysis in CAFs isolated from patients with distinct breast cancer molecular types revealed that CAFs from Her2-positive tumours had significant upregulation of integrin and actin-related pathways including Rho GTPases and kinases [205]. A separate study showed that blocking the mechanical activity of CAFs by inhibiting ROCK kinase decreases vascularisation in a 3D assay, indicating an additional way by which adhesome components regulate CAF-tumour promoting function [206]. Furthermore, a key role for VASP in HSC activation into myofibroblasts and CAF-related tumour progression has been shown in vitro and in vivo experiments. Specifically, siRNA knockdown of VASP in mouse embryonic fibroblasts failed to induce tumour progression and an αSMA-enriched microenvironment in a subcutaneous mouse tumour/CAF coengraftment model [45]. Another study, showed that VASP expression is increased in HSCs associated with liver metastasis both in experimental mice and cancer patient samples and this is required for TGFβ signalling, differentiation of normal HSCs to myofibroblasts and CAF paracrine function [46].
In addition to VASP, several studies have established that increased expression of a long palladin isoform is mostly restricted in the tumour stroma, and elevated palladin levels in CAFs are associated with poor prognosis and unfavourable chemotherapeutic efficacy of pancreatic and renal cancer [207,208,209,210]. It has been shown that palladin has a prominent role in mechanosensing, and adhesion dynamics of pancreatic CAFs [211,212]. Furthermore, palladin can control the activity of Cdc42 and increase the formation of adhesive membrane protrusions, called invadopodia, that can remodel ECM and promote tumour development in vivo [44,213]. Taken together, these data suggest that palladin is a key regulator of CAF tumour-supportive properties.
4. Adhesome Function in Immune Cells
Tumour-infiltrating immune cells (TICs) contribute to many if not most hallmarks of cancer [1]. The “proinflammatory” and cytotoxic activities of immune cells lead to cancer cell destruction, while distinct leukocyte subsets acquire inhibitory functions that benefit tumour growth and immune evasion. The abundance and constitution of TICs vary considerably depending on the tumour type [3]. The spatial organisation of immune infiltrates within the TME is now increasingly recognised as a dynamic nexus of chemokine-driven cell motility rather than simple segregation into peritumoral, stromal and intratumoral location [214]. Here, we classify TICs into two main categories according to their recruitment in innate (macrophages, natural killer cells, monocytes, myeloid-derived suppressor cells, neutrophils, dendritic cells or DCs), or adaptive (T and B lymphocytes) immune responses to review the role of adhesome components in immune TME composition and function.
4.1. Innate Immune Cell Adhesome
Inside the TME, tumour-associated macrophages (TAMs) are the most prevalent immune constituent. They serve as a mix of tissue-resident and monocyte-derived cells that are involved in every step of malignant progression, from the primary tumour formation to metastatic colonisation [215,216]. In a general term, two fundamental phenotypes of TAMs have been documented, termed as the M1 (“classically” activated, proinflammatory, tumouricidal) and the M2 (“alternatively” activated, anti-inflammatory, tumour-promotive) type, but accumulating evidence has also described plasticity between TAM states and multiple distinct phenotypes combining features of both M1 and M2 polarisation [217]. Besides TAM, other leukocyte populations including recruited monocytes, tumour-associated neutrophils (TANs), myeloid-derived suppressor cells (MDSCs), dendritic cells and natural killer (NK) cells are critical components of innate tumour responses. Current research has now begun to scratch the surface of adhesome-mediated regulation within the immune TME.
4.1.1. Adhesome Signalling
Early in vitro studies showed that FAK and the related Pyk2 signalling are vital for macrophage responses [218,219], including differentiation [220], cytokine production [221,222], migration [66,223], and phagocytosis [224,225]. Besides FAK and Pyk2, paxillin phosphorylation has been implicated in cytokine-induced neutrophil activation and macrophage phagocytosis, in vitro [226,227,228]. However, the impact of paxillin signalling on individual innate immune subsets in vivo and the regulation of TME within different tumour types remains to be explored.
Genetic ablation of FAK and Pyk2 in myeloid lineages demonstrated their essential role in macrophage recruitment during inflammation [61,229]. Likewise, genetic studies in neutrophils revealed that FAK and Pyk2 regulate neutrophil chemotaxis, degranulation and host-killing responses [62,67]. Pyk2 kinase activity has also been involved in regulating chemokine-driven DC motility in vitro [230,231] and a recent study attempted to delineate how Pyk2 activity defines the ratio of monocyte subsets at steady state [68]. Taken together, these studies suggest that FAK/Pyk2 signalling could play an important role in tumour innate responses.
Indeed, inhibiting FAK kinase activity in murine models of breast and pancreatic cancer reduced tumour growth and limited CD45+ immune cells [27] and TAM infiltration [21,22,28], indicating a tumour-promoting FAK function. Accordingly, high levels of FAK phosphorylation within tumour tissue have been correlated with elevated granulocyte recruitment in human pancreatic cancer and FAK inhibition decreased tumour penetration of both monocytic MDSCs and polymorphonuclear/granulocytic MDSCs [22]. However, these studies were mainly focused on cancer cell-mediated effects of FAK inhibition, and a more detailed examination is needed to dissect the effects of FAK inhibition on individual innate immune subsets within different tumour types. A recent study showed that mechanical stretch preconditioning of macrophages activates FAK and induces M1 polarisation. Intratumoural injection of mechanical stretched macrophages decreased tumour growth in vivo [232], suggesting an antitumour role for FAK in macrophages. In agreement with a multifaceted action of FAK in the tumour stroma, myeloid cell-specific deletion of FAK in LysM-Cre mice with spontaneously developed mammary carcinomas was associated with both pro- and antitumorigenic functions depending on the stage of malignancy [29]. In particular, FAK depletion in myeloid cells retards early tumour progression, but it accelerates outgrowth after primary tumour formation by reducing tumour-infiltrating NK cell abundance [29].
NK cells are innate lymphocytes with antitumour cytolytic activity [233]. Previous in vitro studies showed that Pyk2 kinase activity along with paxillin activation contributes to NK cytotoxic efficiency [234,235,236,237] and inhibition of Pyk2 activity decreased NK trans-endothelial migration [69], indicating that Pyk2 and paxillin signalling could potentially mediate NK cell extravasation and tumour infiltration limiting tumour progression. Although the increased density of intratumoral NK cells has been associated with a beneficial prognosis [238,239,240], immune cell recruitment in the tumour niche does not necessarily coincide with functionality. Tumours discover ways to evade the cytotoxic function of NK cells and impel them to enter an anergic state [241]. Therefore, further in vivo studies are required to determine the functional significance of adhesome signalling in NK-mediated tumour responses.
4.1.2. Adhesion Organisation
Additionally to the role of adhesion scaffolding proteins, including talin, vinculin and α-actinin on phagosome structure [226,242], and function [243,244], adhesion organisation is critical for podosome assembly and innate immune cell migration [245,246,247]. Apart from the facilitation of cell migration, podosomes in DCs have been proposed to participate in antigen representation [248]. In agreement with this concept, genetic deletion of talin1 in DCs revealed that talin1 plays a vital role in DC antigen sampling, DC activation and priming of adaptive immunity [80] but not in DC migration and arrival to lymph nodes in vivo [249]. However, genetic deletion of talin1 in skin resident DCs, decreased T cell-mediated DC activation and chemotaxis to lymph nodes upon inflammatory stimuli, hindering skin antimicrobial and immune responses [81]. Although these studies highlight an important function of talin in DCs, the role of DC talin expression in cancer remains elusive. Talin could also affect the innate TME by regulating the function of NK cells and TANs. Talin-depleted NK cells had impaired αLβ2 integrin-mediated adhesion and cytotoxicity [82], whereas talin deficiency in myeloid lineages impaired integrin activation, neutrophil tethering and rolling along the vessel wall and extravasation into inflammatory sites [50,120,156]. Methylation of talin by Ezh2, also decreased adhesion of innate leukocytes, extravasation and organ infiltration in response to inflammatory stimuli [250,251]. Interestingly, a recent study showed that Ezh2 inhibition in mice increased the number of immunosuppressive CD11b+Gr1+ cells within subcutaneous tumours [252]. In-depth research is yet needed to unravel whether changes in leukocyte differentiation and infiltration within tumours are associated with post-translational modifications in adhesome members.
Additional adhesome components have been implicated to innate immune cell function. FHL2-deficient mice displayed increased DC migration in response to chemokine signalling, hinting at a suppressive role of FHL2 in immune responses [95]. Similarly, conditional depletion of filamin A from myeloid lineages increased neutrophil adhesion and spreading under basal conditions without though affecting immune cell distribution in different organs, adhesion underflow or the generation of reactive oxygen species [101]. However, in an in vivo peritonitis model, filamin A deletion impaired neutrophil chemotaxis, revealing a role of filamin A in inflammatory responses and potentially in cancer [102]. Further studies are required to reveal the function of these proteins in cancer immunity.
Moreover, overexpression of ILK attenuated leukocyte adhesion on ECs [253] and in vitro deletion of ILK in immortalised macrophages decreased their survival [254]. Other proteins of the adhesion network including vinculin and γ-parvin, although they disrupt leukocyte adhesion in vitro [226,243,255], are dispensable for leukocyte function in vivo [86,94]. Interestingly, mutations of the kindlin3 gene have been linked to leukocyte adhesion deficiency syndrome [256], and kindlin3-null mice display impaired integrin activation as well as neutrophil adhesion and arrest on the vascular wall [73,74]. Minimal kindlin3 expression (5% of normal levels) is sufficient for physiological neutrophil function but inadequate for integrin activation and neutrophil responses in injury or inflammation [75]. Collectively, these studies indicate an intricate function of adhesion components and highlight the need for a thorough examination in a cancer setting. Indeed, inducible deletion of kindlin3 from myeloid lineages in a murine melanoma tumour model impaired the ability of nonclassical monocytes to patrol and scavenge tumour cells. Furthermore, it decreased NK cell and DC recruitment and proliferation, thus promoting the development of metastatic lesions in vivo [38].
4.1.3. Actin Regulatory Layer
Several studies have addressed the role of Rho GTPase family in innate cell adhesion and migration. Specifically, the Rho-GTPases members, RhoA, Rac and Cdc42 have been implicated in podosome assembly with diverse effects in migration [69,257,258]. Although ablation of Rac1 or Rac2 from myeloid lineages does not affect macrophage migration [109,110], the Rho/Pyk2/cofilin axis regulates chemokine-induced DC motility [231]. Rac activation by Pyk2 has also been reported to affect NK cell transendothelial migration [69] and the production of reactive oxygen species in neutrophils [259]. Other cytoskeletal regulators, including Wiskott–Aldrich syndrome protein (WASP) and VASP, were found to be required for actin reorganisation during macrophage phagocytosis [260]. Recent data also have interrelated VASP with NK cell-mediated killing [261] and the regulation of polymorphonuclear neutrophil adhesion in response to cytokine stimulation [99].
Collectively, these findings reveal that albeit compensatory mechanisms, actin regulators influence innate immune functions in a cell type-specific manner and could have an impact on cancer. However, which actin-related proteins are essential and how they shape innate immunity during cancer remains to be investigated.
4.2. Adaptive Immune Cell Adhesome
Antitumour adaptive immunity relies mainly on the functional diversity of T cell subsets and the crosstalk between T cells, antigen presenting cells (APCs) and other stromal/immune populations [262]. Upon antigen recognition by T cell receptors (TCR) through major histocompatibility complex (MHC) molecules, costimulation and cytokine signals, naïve T cells are conventionally activated and become effector T cells (TEFF cells). Among TEFF cells, CD8+ cytotoxic T lymphocytes (CTLs) play a crucial role in tumour surveillance and kill cancer cells through granule exocytosis (granzymes and perforin secretion) and ligand/death receptor-mediated apoptosis [263]. Concomitantly, CD4+ T cells shape tumour immune responses at the tumour site and in lymphoid organs either by supporting cancer cell elimination (T helper cells or Th, follicular helper T cells and CD4+ CTLs) or by promoting an immunosuppressing TME and tumour growth (mainly CD4+ regulatory T cells or Treg cells) [264]. Although the abundance of intratumoural T cells, especially CD8+ cells, provides a positive prognostic and predictive value for cancer patients [265], T-cell-mediated tumour clearance is often restricted. Intrinsic (transcriptional, epigenetic, metabolic factors) and extrinsic (interaction with immunosuppressive cells and soluble factors within the TME) parameters renders T cells dysfunctional (anergic, exhausted or senescent), leading to cancer immune evasion [266,267].
Additionally to T cells, B cells are crucial players of cancer adaptive immunity. Both tumour-promoting and antitumorigenic actions have been attributed to B cells [268]. B cells produce tumour reactive antibodies that facilitate tumour killing by NK and CD4+ CD8+ T cells and phagocytosis by macrophages. On the other hand, B regulating cells and the production of autoantibodies can suppress Th1 and CD8+ cytotoxic effects, leading to tumour development. Future studies will define the context-dependent B cell function and explore the use of B cell-based therapies for cancer treatment [269].
4.2.1. Adhesome Signalling
Numerous studies have implicated FAK and Pyk2 in downstream TCR signalling [270], suggesting a central role in T-cell function. Pharmacological inhibition of Pyk2 and RNAi-mediated knockdown in T cells decreased CTL migration [271], cytokine production and CD4+ effector responses in vitro [272,273]. Deletion of Pyk2 in vivo did not affect T cell development but impaired T cell activation and generation of short term CD8+ TEFF [274]. Moreover, downregulation of Pyk2 and paxillin disrupted microadhesion structures around TCR microclusters at immunological synapses and impaired T-cell activation [275]. Similarly, overexpression of FAK in the Jurkat T cell line increased integrin β1 mediated migration, whereas mutation in FAK Y397 phosphorylation site decreased T cell migration [276]. Pharmacological inhibition of FAK in T cells impaired their adhesion, proliferation and activation [64]. In contrast to the in vitro studies, genetic deletion of FAK from CD4+ T cells did not affect T cell development, proliferation or activation, possibly due to residual remaining FAK levels [64]. Consistent with this, FAK heterozygous mice also do not develop any T cell defects [63].
Regarding B cell immunity, FAK and Pyk2 phosphorylation affects progenitor B cell adhesion and spreading [277,278,279] and specific deletion of FAK was shown to regulate B cell homing and retention in bone marrow as well as B cell survival and proliferation [65]. Likewise, Pyk2 deficiency impaired B cell migration into the marginal zone of the spleen and humoral responses to thymic antigen responses [70]. However, the involvement of FAK/Pyk2 in tumour adaptive responses is not well-established. An immunomodulatory function of FAK in tumour stroma has been suggested using FAK inhibitors in vivo. Administration of FAK inhibitors or FAK-specific depletion in cancer cells caused tumour regression with increased CD4+ and CD8+ T cell infiltration and decreased Tregs in vivo. Specifically, CD8+ T cell function was correlated with the efficacy of FAK inhibition [30,31]. The inhibitory role of FAK appeared to be cancer-cell specific and was attributed to its kinase activity within the nucleus and the transcriptional regulation of cytokines production [31,280]. Similar changes in intratumoural CTLs were observed in pancreatic tumour-bearing mice treated with FAK inhibitor [22]. This inverse correlation between FAK activity and CD8+ infiltration was also reflected in lesions of pancreatic cancer patients [22]. Of note, FAK inhibition combined with chemotherapy or immunotherapy (adoptive T cell transfer or immune checkpoint inhibitors) brought beneficial results in tumour response and survival of treatment-resistant animals [22,30,281]. Although these studies highlight the role of FAK in T cell-mediated cancer immunity and can pave the way for more effective combination therapies against cancer, more comprehensive examination is required to distinguish the importance of FAK function in immune vs. cancer cells.
In addition to FAK and Pyk2, inhibition of paxillin phosphorylation decreased the tumour-targeting function of CD8+ T cells, in vitro, and paxillin knockdown reduced the adhesion and spreading of tissue-resident memory T cells (TRM) isolated from human lung tumours [282]. TRM cells emerge as a prominent, non-recirculating subset of CD8+ T lymphocytes that reside into tissues to rapidly re-encounter with pathogens and cancer [283]. The exact role of TRM cells in antitumoural responses is now beginning to be appreciated [284] and further investigation is needed to understand how adhesome molecules are involved in immunological memory.
Increasing evidence suggests that the GIT/PAK/PIX signalling pathway influences tumour-resident lymphocytes. Mice deficient in αPIX present defective immune responses, with reduced T and B cell proliferation, increased motility and disrupted immune synapse formation and positive selection [71,72]. In a tumour setting, mouse Tregs lacking GIT2 were unable to induce tumour development when compared with wild-type Tregs in an adoptive T cell transfer mouse model [35]. Further in vitro experiments showed that GIT2 knockdown in human Tregs inhibits TEFF proliferation and decreases the surface levels of CD86 costimulatory molecule on B cells. PAK1/2 is also activated in Tregs in a CTLA4/PKCη-dependent manner [35]. Transcriptomics analysis of various CD4+ T cell subsets from patient tumours, normal surrounding tissue and peripheral blood revealed that tumour-localising Tregs have unique signatures of upregulated genes including pak2 [285]. Additionally, mice lacking PAK1 had increased number of spleen T and B cells in contrast to wild type mice developing cancer in the small intestine, distal colon and rectum, indicating that PAK1 might contribute to the tumour eradication by immune cells [34]. Accordingly, PAK1 depletion decreased tumour incidence rates and size in mice developing spontaneous intestinal-colorectal tumorigenesis [34]. It also enhanced CD4+ and CD8+ T cell recruitment in a mouse model of pancreatic adenocarcinoma, prolonging mouse survival [33].
4.2.2. Adhesion Organisation
Besides adhesome signalling, emerging data demonstrate that adhesion scaffolding proteins have an essential function in adaptive immunity and therefore, could impact cancer development. T cell specific deletion of talin1 displayed defective immunological synapses and transient T cell-APC interactions leading to reduced T cell activation and proliferation [83]. Another in vivo study showed that talin1 is necessary for Treg survival and function [84]. Furthermore, B cell specific ablation of talin1 revealed that, although talin does not affect the maturation of B cells, it is required for humoral responses to T cell dependent antigen and emigration to lymph nodes [85]. The partner of talin, vinculin, also localises to immunological synapses at B cells and is required for B cell stable adhesion and migration [286]. Recent data also implicated filamin A in T cell trafficking to lymph nodes and inflamed skin in vivo [100]. Additionally, T cell specific ILK deletion decreased chemokine-mediated T cell trafficking and survival [90]. A context-dependent role of kindlin3 was revealed through in vivo studies. T cells lacking kindlin3 failed to arrest and transmigrate through blood vessels under normal shear stress conditions. However, an inflamed vasculature, as observed in cancer, provided sufficient integrin ligands to overcome kindlin3 deficiency and rescued the defective extravasation [76]. Taken together, these findings indicate a putative role of integrin-interacting proteins in adaptive antitumour responses.
4.2.3. Actin Regulatory Layer
Genetic ablation studies in mice have revealed that Rho GTPase family members regulate essential T cell functions, including activation, cell division, migration, and adhesion [114,287,288,289]. Mice lacking both Rac1 and Rac2 present defects in T cell development, resulting in decreased CD4+ and CD8+ thymocytes [111,112]. Other studies also showed that combined Rac1 and Rac2 function regulate T-cell migration and homing to lymph nodes, thymus and spleen [115]. A similar phenotype is observed in the absence of Vav1, Vav2 and Vav3 guanine nucleotide exchange factors of Rac [112]. Likewise, gene targeting of cdc42 in mice impairs the initial stages of T cell development, proliferation and migration but augments effector and memory T cell differentiation [106,107], indicating a stage-specific role with putative implications in cancer. While other Rho GTPases, including RhoC and RhoG, do not affect T cell function [121,122], specific deletion of RhoA in thymocytes leads to reduced proliferation, survival and differentiation of T cells. Moreover, it abolishes Th1 inflammation responses, resulting in amelioration of autoimmune diseases [118]. These findings imply a putative important, yet unexplored, function in tumour suppressive immunity. Additionally, RhoA affects B cell development but not BCR-dependent proliferation [119]. Similar to RhoA, Cdc42 deficiency and double Rac1/Rac2 deletion have reduced numbers of transitional, marginal zone and follicular B cells and decreased BCR-mediated survival and proliferation [116,289,290]. However, the importance of B cell expression of Rho GTPases in shaping cancer immunity is not explored yet.
Besides Rho-GTPases, evidence exists for other actin regulators, including the ERM family of ezrin-radixin-moesin and the Ena/VASP proteins in T cell function. Specifically, ERM inactivation and removal from the TCR microstructures via the Rac1/Vav signalling axis is important for efficient T cell-APC association [291,292] suggesting that ERM proteins could facilitate immune evasion in cancer. Cytoskeletal remodelling is important for T cell function as is indicated by mutations in WASP that disrupt T cell cytoskeleton in Wiskott–Aldrich syndrome patients [293] and cause defective TCR signalling and activation in Jurkat T cells [294]. Moreover, Ena/VASP combined deficiency in mice is dispensable for T cell development, adhesion and crawling on the vascular endothelium. Nevertheless, recruitment of CD4+ activated T cells to inflammatory sites is severely compromised in double Ena/VASP knockout mice due to defective transendothelial migration [98], suggesting a suppressing mechanism of cancer immunity. A more detailed examination is needed to decipher the role of actin regulators in shaping cancer adaptive immunity.
5. Conclusions
It is becoming increasingly evident that the tumour stroma is not a bystander but rather a key driver in tumour development. Malignancy is controlled by the cumulative action of the different TME constituents. In the dynamic crosstalk between cancer cells and TME components, cell adhesion to the ECM has a dominant function. Despite the entrenched role of adhesome members in cancer cells, which adhesome proteins are important to regulate TME cellular players, ECs, mural cells, CAFs and immune cells- is poorly understood. While comprehensive approaches dissecting cancer-intrinsic vs. tumour-associated host effects of adhesome components are still lacking, evidence derived from developmental studies could shed light into the functional requirement of adhesome in controlling TME and tumour progression. It is envisaged that the impact of individual adhesome members in cancer could be cell-type-dependent, with both tumour promoting and suppressive effects elicited by the same protein in different TME cellular components and cancer cells. Therefore, gene-targeting approaches are needed to define the requirement of adhesome proteins in specific cell types of TME. Deciphering how cell adhesion organisation and signalling determines tumour stroma architecture and impact on malignancy will set the stage for effective interventions in cancer prognosis and treatment. Specifically, evidence exists about the role of ECM rigidity in defining cancer outcome and studies have revealed that radiation treatment could lead to fibrosis. One avenue for exploration could be to dissect how adhesome members could impact on tumour fibrosis, affect ECM remodelling and define cancer responses to treatment.
Author Contributions
Visualisation—planning and overview, V.K.; literature search, M.A.K., P.A.N.; writing—original draft preparation, M.A.K., P.A.N.; writing—final draft, review and editing, V.K.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by World Wide Cancer Research (WWCR), grant number 14-1260 and “A Greek Research Infrastructure for Visualizing and Monitoring Fundamental Biological Processes (BIO- IMAGING-GR)” (MIS 5002755), implemented under the Action “Reinforcement of the Research and Innovation Infrastructure,” funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and cofinanced by Greece and the European Union (European Regional Development Fund) and “The Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “First Call for H.F.R.I. Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment grant” (Project Number: HFRI-FM17-2644)”.
Acknowledgments
Figures were prepared using BioRender (https://biorender.com/). The authors thank Kostourou Lab members, Myrto Denaxa and Christos Zervas for helpful comments and suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance. J. Cell. Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, H.; Remark, R.; Gnjatic, S.; Merad, M. Host Tissue Determinants of Tumour Immunity. Nat. Rev. Cancer 2019, 19, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Yamada, K.M. Molecular Architecture and Function of Matrix Adhesions. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, C.A.; Bergeron, K.-F.; Whittle, J.; Brandhorst, B.P.; Burke, R.D.; Hynes, R.O. The Echinoderm Adhesome. Dev. Biol. 2006, 300, 252–266. [Google Scholar] [CrossRef] [Green Version]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional Atlas of the Integrin Adhesome. Nat. Cell Biol. 2007, 9, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell Adhesion: Integrating Cytoskeletal Dynamics and Cellular Tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef]
- Byron, A.; Frame, M.C. Adhesion Protein Networks Reveal Functions Proximal and Distal to Cell-Matrix Contacts. Curr. Opin. Cell Biol. 2016, 39, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Byron, A.; Morgan, M.R.; Humphries, M.J. Adhesion Signalling Complexes. Curr. Biol. 2010, 20, R1063–R1067. [Google Scholar] [CrossRef] [Green Version]
- Horton, E.R.; Byron, A.; Askari, J.A.; Ng, D.H.J.; Millon-Frémillon, A.; Robertson, J.; Koper, E.J.; Paul, N.R.; Warwood, S.; Knight, D.; et al. Definition of a Consensus Integrin Adhesome and Its Dynamics during Adhesion Complex Assembly and Disassembly. Nat. Cell Biol. 2015, 17, 1577–1587. [Google Scholar] [CrossRef] [Green Version]
- Horton, E.R.; Astudillo, P.; Humphries, M.J.; Humphries, J.D. Mechanosensitivity of Integrin Adhesion Complexes: Role of the Consensus Adhesome. Exp. Cell Res. 2016, 343, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, E.R.; Humphries, J.D.; James, J.; Jones, M.C.; Askari, J.A.; Humphries, M.J. The Integrin Adhesome Network at a Glance. J. Cell Sci. 2016, 129, 4159–4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavora, B.; Batista, S.; Reynolds, L.E.; Jadeja, S.; Robinson, S.; Kostourou, V.; Hart, I.; Fruttiger, M.; Parsons, M.; Hodivala-Dilke, K.M. Endothelial FAK Is Required for Tumour Angiogenesis. EMBO Mol. Med. 2010, 2, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Borboa, A.K.; Chun, H.B.; Baird, A.; Eliceiri, B.P. Conditional Deletion of the Focal Adhesion Kinase FAK Alters Remodeling of the Blood-Brain Barrier in Glioma. Cancer Res. 2010, 70, 10131–10140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostourou, V.; Lechertier, T.; Reynolds, L.E.; Lees, D.M.; Baker, M.; Jones, D.T.; Tavora, B.; Ramjaun, A.R.; Birdsey, G.M.; Robinson, S.D.; et al. FAK-Heterozygous Mice Display Enhanced Tumour Angiogenesis. Nat. Commun. 2013, 4, 2020. [Google Scholar] [CrossRef] [Green Version]
- Jean, C.; Chen, X.L.; Nam, J.-O.; Tancioni, I.; Uryu, S.; Lawson, C.; Ward, K.K.; Walsh, C.T.; Miller, N.L.G.; Ghassemian, M.; et al. Inhibition of Endothelial FAK Activity Prevents Tumor Metastasis by Enhancing Barrier Function. J. Cell Biol. 2014, 204, 247–263. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulou, A.N.; Lees, D.M.; Bodrug, N.; Lechertier, T.; Fernandez, I.; D’Amico, G.; Dukinfield, M.; Batista, S.; Tavora, B.; Serrels, B.; et al. Focal Adhesion Kinase (FAK) Tyrosine 397E Mutation Restores the Vascular Leakage Defect in Endothelium-Specific FAK-Kinase Dead Mice: Endothelial FAKY397E Mutation and Tumour Vascular Leakage. J. Pathol. 2017, 242, 358–370. [Google Scholar] [CrossRef] [Green Version]
- Pedrosa, A.-R.; Bodrug, N.; Gomez-Escudero, J.; Carter, E.P.; Reynolds, L.E.; Georgiou, P.N.; Fernandez, I.; Lees, D.M.; Kostourou, V.; Alexopoulou, A.N.; et al. Tumor Angiogenesis Is Differentially Regulated by Phosphorylation of Endothelial Cell Focal Adhesion Kinase Tyrosines-397 and -861. Cancer Res. 2019, 79, 4371–4386. [Google Scholar] [CrossRef] [Green Version]
- Lechertier, T.; Reynolds, L.E.; Kim, H.; Pedrosa, A.R.; Gómez-Escudero, J.; Muñoz-Félix, J.M.; Batista, S.; Dukinfield, M.; Demircioglu, F.; Wong, P.P.; et al. Pericyte FAK Negatively Regulates Gas6/Axl Signalling to Suppress Tumour Angiogenesis and Tumour Growth. Nat. Commun. 2020, 11, 2810. [Google Scholar] [CrossRef] [PubMed]
- Lees, D.M.; Reynolds, L.E.; Pedrosa, A.R.; Roy-Luzarraga, M.; Hodivala-Dilke, K.M. Phosphorylation of Pericyte FAK-Y861 Affects Tumour Cell Apoptosis and Tumour Blood Vessel Regression. bioRxiv 2020. [Google Scholar] [CrossRef]
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of Focal Adhesion Kinase by PF-562,271 Inhibits the Growth and Metastasis of Pancreatic Cancer Concomitant with Altering the Tumor Microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting Focal Adhesion Kinase Renders Pancreatic Cancers Responsive to Checkpoint Immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Q.; Tu, K.; Wang, Y.; Wang, X.; Liu, D.; Chen, C.; Liu, D.; Yang, R.; Qiu, W.; et al. Focal Adhesion Kinase Promotes Hepatic Stellate Cell Activation by Regulating Plasma Membrane Localization of TGFβ Receptor 2. Hepatol. Commun. 2020, 4, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Demircioglu, F.; Wang, J.; Candido, J.; Costa, A.S.H.; Casado, P.; de Luxan Delgado, B.; Reynolds, L.E.; Gomez-Escudero, J.; Newport, E.; Rajeeve, V.; et al. Cancer Associated Fibroblast FAK Regulates Malignant Cell Metabolism. Nat. Commun. 2020, 11, 1290. [Google Scholar] [CrossRef] [Green Version]
- Zaghdoudi, S.; Decaup, E.; Belhabib, I.; Samain, R.; Cassant-Sourdy, S.; Rochotte, J.; Brunel, A.; Schlaepfer, D.; Cros, J.; Neuzillet, C.; et al. FAK Activity in Cancer-Associated Fibroblasts Is a Prognostic Marker and a Druggable Key Metastatic Player in Pancreatic Cancer. EMBO Mol. Med. 2020, 12, e12010. [Google Scholar] [CrossRef]
- Wu, H.-J.; Hao, M.; Yeo, S.K.; Guan, J.-L. FAK Signaling in Cancer-Associated Fibroblasts Promotes Breast Cancer Cell Migration and Metastasis by Exosomal MiRNAs-Mediated Intercellular Communication. Oncogene 2020, 39, 2539–2549. [Google Scholar] [CrossRef]
- Walsh, C.; Tanjoni, I.; Uryu, S.; Tomar, A.; Nam, J.-O.; Luo, H.; Phillips, A.; Patel, N.; Kwok, C.; McMahon, G.; et al. Oral Delivery of PND-1186 FAK Inhibitor Decreases Tumor Growth and Spontaneous Breast to Lung Metastasis in Pre-Clinical Models. Cancer Biol. Ther. 2010, 9, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Wendt, M.K.; Schiemann, W.P. Therapeutic Targeting of the Focal Adhesion Complex Prevents Oncogenic TGF-Beta Signaling and Metastasis. Breast Cancer Res. 2009, 11, R68. [Google Scholar] [CrossRef] [Green Version]
- Llewellyn, R.A.; Gutknecht, M.F.; Thomas, K.S.; Conaway, M.R.; Bouton, A.H. Focal Adhesion Kinase (FAK) Deficiency in Mononuclear Phagocytes Alters Murine Breast Tumor Progression. Am. J. Cancer Res. 2018, 8, 675–687. [Google Scholar]
- Canel, M.; Taggart, D.; Sims, A.H.; Lonergan, D.W.; Waizenegger, I.C.; Serrels, A. T-Cell Co-Stimulation in Combination with Targeting FAK Drives Enhanced Anti-Tumor Immunity. eLife 2020, 9, e48092. [Google Scholar] [CrossRef]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-Tumor Immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- German, A.E.; Mammoto, T.; Jiang, E.; Ingber, D.E.; Mammoto, A. Paxillin Controls Endothelial Cell Migration and Tumor Angiogenesis by Altering Neuropilin 2 Expression. J. Cell Sci. 2014, 127, 1672–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Zhan, Y.; Huynh, N.; Dumesny, C.; Wang, X.; Asadi, K.; Herrmann, D.; Timpson, P.; Yang, Y.; Walsh, K.; et al. Inhibition of PAK1 Suppresses Pancreatic Cancer by Stimulation of Anti-Tumour Immunity through down-Regulation of PD-L1. Cancer Lett. 2020, 472, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Huynh, N.; Wang, K.; Yim, M.; Dumesny, C.J.; Sandrin, M.S.; Baldwin, G.S.; Nikfarjam, M.; He, H. Depletion of P21-Activated Kinase 1 up-Regulates the Immune System of APC∆14/+ Mice and Inhibits Intestinal Tumorigenesis. BMC Cancer 2017, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pedros, C.; Canonigo-Balancio, A.J.; Kong, K.-F.; Altman, A. Requirement of Treg-Intrinsic CTLA4/PKCη Signaling Pathway for Suppressing Tumor Immunity. JCI Insight 2017, 2, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluskota, E.; Dowling, J.J.; Gordon, N.; Golden, J.A.; Szpak, D.; West, X.Z.; Nestor, C.; Ma, Y.-Q.; Bialkowska, K.; Byzova, T.; et al. The Integrin Coactivator Kindlin-2 Plays a Critical Role in Angiogenesis in Mice and Zebrafish. Blood 2011, 117, 4978–4987. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, N.; Masamune, A.; Hamada, S.; Kikuta, K.; Takikawa, T.; Motoi, F.; Unno, M.; Shimosegawa, T. Kindlin-2 in Pancreatic Stellate Cells Promotes the Progression of Pancreatic Cancer. Cancer Lett. 2017, 390, 103–114. [Google Scholar] [CrossRef]
- Marcovecchio, P.M.; Zhu, Y.P.; Hanna, R.N.; Dinh, H.Q.; Tacke, R.; Wu, R.; McArdle, S.; Reynolds, S.; Araujo, D.J.; Ley, K.; et al. Frontline Science: Kindlin-3 Is Essential for Patrolling and Phagocytosis Functions of Nonclassical Monocytes during Metastatic Cancer Surveillance. J. Leukoc. Biol. 2020, 107, 883–892. [Google Scholar] [CrossRef]
- Pulous, F.E.; Carnevale, J.C.; Al-Yafeai, Z.; Pearson, B.H.; Hamilton, J.A.G.; Henry, C.J.; Wayne Orr, A.; Petrich, B.G. Talin-Dependent Integrin Activation Is Required for Endothelial Proliferation and Postnatal Angiogenesis. Angiogenesis 2020, 1–14. [Google Scholar] [CrossRef]
- Younes, M.N. Integrin-Linked Kinase Is a Potential Therapeutic Target for Anaplastic Thyroid Cancer. Mol. Cancer Ther. 2005, 4, 1146–1156. [Google Scholar] [CrossRef] [Green Version]
- Yau, C.Y.F.; Wheeler, J.J.; Sutton, K.L.; Hedley, D.W. Inhibition of Integrin-Linked Kinase by a Selective Small Molecule Inhibitor, QLT0254, Inhibits the PI3K/PKB/MTOR, Stat3, and FKHR Pathways and Tumor Growth, and Enhances Gemcitabine-Induced Apoptosis in Human Orthotopic Primary Pancreatic Cancer Xenografts. Cancer Res. 2005, 65, 1497–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, L.A.; Woo, J.; Huxham, L.A.; Verreault, M.; Dragowska, W.H.; Chiu, G.; Rajput, A.; Kyle, A.H.; Kalra, J.; Yapp, D.; et al. Suppression of VEGF Secretion and Changes in Glioblastoma Multiforme Microenvironment by Inhibition of Integrin-Linked Kinase (ILK). Mol. Cancer Ther. 2008, 7, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Palmby, T.R.; Gavard, J.; Amornphimoltham, P.; Zheng, Y.; Gui, J.S. An Essential Role for Rac1 in Endothelial Cell Function and Vascular Development. FASEB J. 2008, 22, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Goicoechea, S.; García-Mata, R.; Staub, J.; Valdivia, A.; Sharek, L.; McCulloch, C.; Hwang, R.; Urrutia, R.; Yeh, J.; Kim, H.; et al. Palladin Promotes Invasion of Pancreatic Cancer Cells by Enhancing Invadopodia Formation in Cancer-Associated Fibroblasts. Oncogene 2014, 33, 1265–1273. [Google Scholar] [CrossRef] [Green Version]
- Kang, N.; Yaqoob, U.; Geng, Z.; Bloch, K.; Liu, C.; Gomez, T.; Billadeau, D.; Shah, V. Focal Adhesion Assembly in Myofibroblasts Fosters a Microenvironment That Promotes Tumor Growth. Am. J. Pathol. 2010, 177, 1888–1900. [Google Scholar] [CrossRef]
- Tu, K.; Li, J.; Verma, V.K.; Liu, C.; Billadeau, D.D.; Lamprecht, G.; Xiang, X.; Guo, L.; Dhanasekaran, R.; Roberts, L.R.; et al. VASP Promotes TGF-β Activation of Hepatic Stellate Cells by Regulating Rab11 Dependent Plasma Membrane Targeting of TGF-β Receptors. Hepatology 2015, 61, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Nallapalli, R.K.; Ibrahim, M.X.; Zhou, A.X.; Bandaru, S.; Sunkara, S.N.; Redfors, B.; Pazooki, D.; Zhang, Y.; Borén, J.; Cao, Y.; et al. Targeting Filamin A Reduces K-RAS–Induced Lung Adenocarcinomas and Endothelial Response to Tumor Growth in Mice. Mol. Cancer 2012, 11, 50. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, G.; Robinson, S.D.; Germain, M.; Reynolds, L.E.; Thomas, G.J.; Elia, G.; Saunders, G.; Fruttiger, M.; Tybulewicz, V.; Mavria, G.; et al. Endothelial-Rac1 Is Not Required for Tumor Angiogenesis Unless Avβ3-Integrin Is Absent. PLoS ONE 2010, 5, e9766. [Google Scholar] [CrossRef] [Green Version]
- Vader, P.; van der Meel, R.; Symons, M.H.; Fens, M.H.A.M.; Pieters, E.; Wilschut, K.J.; Storm, G.; Jarzabek, M.; Gallagher, W.M.; Schiffelers, R.M.; et al. Examining the Role of Rac1 in Tumor Angiogenesis and Growth: A Clinically Relevant RNAi-Mediated Approach. Angiogenesis 2011, 14, 457–466. [Google Scholar] [CrossRef]
- Wilson, E.; Leszczynska, K.; Poulter, N.S.; Edelmann, F.; Salisbury, V.A.; Noy, P.J.; Bacon, A.; Rappoport, J.Z.; Heath, J.K.; Bicknell, R.; et al. RhoJ Interacts with the GIT–PIX Complex and Regulates Focal Adhesion Disassembly. J. Cell Sci. 2014, 127, 3039–3051. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Yang, H.; Fukushima, Y.; Saw, P.E.; Lee, J.; Park, J.-S.; Park, I.; Jung, J.; Kataoka, H.; Lee, D.; et al. Vascular RhoJ Is an Effective and Selective Target for Tumor Angiogenesis and Vascular Disruption. Cancer Cell 2014, 25, 102–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brassart-Pasco, S.; Brézillon, S.; Brassart, B.; Ramont, L.; Oudart, J.-B.; Monboisse, J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.-Z.; Jin, W.-L. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal. Transduct. Target. Ther. 2020, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in Angiogenesis and Lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidi, H.; Ivaska, J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Braren, R.; Hu, H.; Kim, Y.H.; Beggs, H.E.; Reichardt, L.F.; Wang, R. Endothelial FAK Is Essential for Vascular Network Stability, Cell Survival, and Lamellipodial Formation. J. Cell Biol. 2006, 172, 151–162. [Google Scholar] [CrossRef]
- Shen, T.-L.; Park, A.Y.-J.; Alcaraz, A.; Peng, X.; Jang, I.; Koni, P.; Flavell, R.A.; Gu, H.; Guan, J.-L. Conditional Knockout of Focal Adhesion Kinase in Endothelial Cells Reveals Its Role in Angiogenesis and Vascular Development in Late Embryogenesis. J. Cell Biol. 2005, 169, 941–952. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Peng, X.; Sun, S.; Park, A.Y.J.; Guan, J.-L. Role of Kinase-Independent and -Dependent Functions of FAK in Endothelial Cell Survival and Barrier Function during Embryonic Development. J. Cell Biol. 2010, 189, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Weis, S.M.; Lim, S.-T.; Lutu-Fuga, K.M.; Barnes, L.A.; Chen, X.L.; Göthert, J.R.; Shen, T.-L.; Guan, J.-L.; Schlaepfer, D.D.; Cheresh, D.A. Compensatory Role for Pyk2 during Angiogenesis in Adult Mice Lacking Endothelial Cell FAK. J. Cell Biol. 2008, 181, 43–50. [Google Scholar] [CrossRef]
- Owen, K.A.; Pixley, F.J.; Thomas, K.S.; Vicente-Manzanares, M.; Ray, B.J.; Horwitz, A.F.; Parsons, J.T.; Beggs, H.E.; Stanley, E.R.; Bouton, A.H. Regulation of Lamellipodial Persistence, Adhesion Turnover, and Motility in Macrophages by Focal Adhesion Kinase. J. Cell Biol. 2007, 179, 1275–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasorn, A.; Alcaide, P.; Jia, Y.; Subramanian, K.K.; Sarraj, B.; Li, Y.; Loison, F.; Hattori, H.; Silberstein, L.E.; Luscinskas, W.F.; et al. Focal Adhesion Kinase Regulates Pathogen-Killing Capability and Life Span of Neutrophils via Mediating Both Adhesion-Dependent and -Independent Cellular Signals. J. Immunol. 2009, 183, 1032–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanazawa, S.; Ilic, D.; Hashiyama, M.; Okada, M.; Noumura, T.; Aizawa, S.; Suda, T. Impaired Development of CD4+ CD8+ Thymocytes by Csk-’knock-in’ into Fyn Locus. Oncogene 1996, 13, 199–204. [Google Scholar] [PubMed]
- Wiemer, A.J.; Wernimont, S.A.; Cung, T.-D.; Bennin, D.A.; Beggs, H.E.; Huttenlocher, A. The Focal Adhesion Kinase Inhibitor PF-562,271 Impairs Primary CD4+ T Cell Activation. Biochem. Pharmacol. 2013, 86, 770–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-Y.; Wolfram, P.; Canty, K.; Harley, B.; Nombela-Arrieta, C.; Pivarnik, G.; Manis, J.; Beggs, H.E.; Silberstein, L.E. Focal Adhesion Kinase Regulates the Localization and Retention of Pro-B Cells in Bone Marrow Microenvironments. J. Immunol. 2013, 190, 1094–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okigaki, M.; Davis, C.; Falasca, M.; Harroch, S.; Felsenfeld, D.P.; Sheetz, M.P.; Schlessinger, J. Pyk2 Regulates Multiple Signaling Events Crucial for Macrophage Morphology and Migration. PNAS 2003, 100, 10740–10745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamen, L.A.; Schlessinger, J.; Lowell, C.A. Pyk2 Is Required for Neutrophil Degranulation and Host Defense Responses to Bacterial Infection. J. Immunol. 2011, 186, 1656–1665. [Google Scholar] [CrossRef] [Green Version]
- Llewellyn, R.A.; Thomas, K.S.; Gutknecht, M.F.; Bouton, A.H. The Nonreceptor Protein Tyrosine Kinase Pyk2 Promotes the Turnover of Monocytes at Steady State. J. Leukoc. Biol. 2017, 102, 1069–1080. [Google Scholar] [CrossRef]
- Gismondi, A.; Jacobelli, J.; Strippoli, R.; Mainiero, F.; Soriani, A.; Cifaldi, L.; Piccoli, M.; Frati, L.; Santoni, A. Proline-Rich Tyrosine Kinase 2 and Rac Activation by Chemokine and Integrin Receptors Controls NK Cell Transendothelial Migration. J. Immunol. 2003, 170, 3065–3073. [Google Scholar] [CrossRef] [Green Version]
- Guinamard, R.; Okigaki, M.; Schlessinger, J.; Ravetch, J.V. Absence of Marginal Zone B Cells in Pyk-2-Deficient Mice Defines Their Role in the Humoral Response. Nat. Immunol. 2000, 1, 31–36. [Google Scholar] [CrossRef]
- Missy, K.; Hu, B.; Schilling, K.; Harenberg, A.; Sakk, V.; Kuchenbecker, K.; Kutsche, K.; Fischer, K.-D. APIX Rho GTPase Guanine Nucleotide Exchange Factor Regulates Lymphocyte Functions and Antigen Receptor Signaling. Mol. Cell. Biol. 2008, 28, 3776–3789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korthals, M.; Schilling, K.; Reichardt, P.; Mamula, D.; Schlüter, T.; Steiner, M.; Langnäse, K.; Thomas, U.; Gundelfinger, E.; Premont, R.T.; et al. APIX RhoGEF Supports Positive Selection by Restraining Migration and Promoting Arrest of Thymocytes. J. Immunol. 2014, 192, 3228–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, C.T.; Rossaint, J.; Moser, M.; Petrich, B.G.; Zarbock, A.; Monkley, S.J.; Critchley, D.R.; Ginsberg, M.H.; Fässler, R.; Ley, K. Distinct Roles for Talin-1 and Kindlin-3 in LFA-1 Extension and Affinity Regulation. Blood 2012, 119, 4275–4282. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Bauer, M.; Schmid, S.; Ruppert, R.; Schmidt, S.; Sixt, M.; Wang, H.-V.; Sperandio, M.; Fässler, R. Kindlin-3 Is Required for Beta2 Integrin-Mediated Leukocyte Adhesion to Endothelial Cells. Nat. Med. 2009, 15, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Klapproth, S.; Moretti, F.A.; Zeiler, M.; Ruppert, R.; Breithaupt, U.; Mueller, S.; Haas, R.; Mann, M.; Sperandio, M.; Fässler, R.; et al. Minimal Amounts of Kindlin-3 Suffice for Basal Platelet and Leukocyte Functions in Mice. Blood 2015, 126, 2592–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, F.A.; Moser, M.; Lyck, R.; Abadier, M.; Ruppert, R.; Engelhardt, B.; Fässler, R. Kindlin-3 Regulates Integrin Activation and Adhesion Reinforcement of Effector T Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 17005–17010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkley, S.J.; Kostourou, V.; Spence, L.; Petrich, B.; Coleman, S.; Ginsberg, M.H.; Pritchard, C.A.; Critchley, D.R. Endothelial Cell Talin1 Is Essential for Embryonic Angiogenesis. Dev. Biol. 2011, 349, 494–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulous, F.E.; Grimsley-Myers, C.M.; Kansal, S.; Kowalczyk, A.P.; Petrich, B.G. Talin-Dependent Integrin Activation Regulates VE-Cadherin Localization and Endothelial Cell Barrier Function. Circ. Res. 2019, 124, 891–903. [Google Scholar] [CrossRef]
- Bromberger, T.; Klapproth, S.; Rohwedder, I.; Zhu, L.; Mittmann, L.; Reichel, C.A.; Sperandio, M.; Qin, J.; Moser, M. Direct Rap1/Talin1 Interaction Regulates Platelet and Neutrophil Integrin Activity in Mice. Blood 2018, 132, 2754–2762. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wu, H.; An, J.; Ballantyne, C.M.; Cyster, J.G. Critical Role of Integrin CD11c in Splenic Dendritic Cell Capture of Missing-Self CD47 Cells to Induce Adaptive Immunity. PNAS 2018, 115, 6786–6791. [Google Scholar] [CrossRef] [Green Version]
- Lim, T.J.F.; Bunjamin, M.; Ruedl, C.; Su, I.-H. Talin1 Controls Dendritic Cell Activation by Regulating TLR Complex Assembly and Signaling. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Mace, E.M.; Monkley, S.J.; Critchley, D.R.; Takei, F. A Dual Role for Talin in NK Cell Cytotoxicity: Activation of LFA-1-Mediated Cell Adhesion and Polarization of NK Cells. J. Immunol. 2009, 182, 948–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernimont, S.A.; Wiemer, A.J.; Bennin, D.A.; Monkley, S.J.; Ludwig, T.; Critchley, D.R.; Huttenlocher, A. Contact-Dependent T Cell Activation and T Cell Stopping Require Talin1. J. Immunol. 2011, 187, 6256–6267. [Google Scholar] [CrossRef] [PubMed]
- Klann, J.E.; Remedios, K.A.; Kim, S.H.; Metz, P.J.; Lopez, J.; Mack, L.A.; Zheng, Y.; Ginsberg, M.H.; Petrich, B.G.; Chang, J.T. Talin Plays a Critical Role in the Maintenance of the Regulatory T Cell Pool. J. Immunol. 2017, 198, 4639–4651. [Google Scholar] [CrossRef] [Green Version]
- Manevich-Mendelson, E.; Grabovsky, V.; Feigelson, S.W.; Cinamon, G.; Gore, Y.; Goverse, G.; Monkley, S.J.; Margalit, R.; Melamed, D.; Mebius, R.E.; et al. Talin1 Is Required for Integrin-Dependent B Lymphocyte Homing to Lymph Nodes and the Bone Marrow but Not for Follicular B-Cell Maturation in the Spleen. Blood 2010, 116, 5907–5918. [Google Scholar] [CrossRef] [Green Version]
- Wilson, Z.S.; Witt, H.; Hazlett, L.; Harman, M.; Neumann, B.M.; Whitman, A.; Patel, M.; Ross, R.S.; Franck, C.; Reichner, J.S.; et al. Context-Dependent Role of Vinculin in Neutrophil Adhesion, Motility and Trafficking. Sci. Rep. 2020, 10, 2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, E.B.; Liu, E.; Sinha, S.; Cook, S.; Milstone, D.S.; MacRae, C.A.; Mariotti, M.; Kuhlencordt, P.J.; Force, T.; Rosenzweig, A.; et al. Integrin-Linked Kinase Regulates Endothelial Cell Survival and Vascular Development. Mol. Cell. Biol. 2004, 24, 8134–8144. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Yamamoto, H.; Mohn, L.; Ambühl, L.; Kanai, K.; Schmidt, I.; Kim, K.-P.; Fraccaroli, A.; Feil, S.; Junge, H.J.; et al. Integrin-Linked Kinase Controls Retinal Angiogenesis and Is Linked to Wnt Signaling and Exudative Vitreoretinopathy. Nat. Commun. 2019, 10, 5243. [Google Scholar] [CrossRef]
- Kogata, N.; Tribe, R.M.; Fässler, R.; Way, M.; Adams, R.H. Integrin-Linked Kinase Controls Vascular Wall Formation by Negatively Regulating Rho/ROCK-Mediated Vascular Smooth Muscle Cell Contraction. Genes Dev. 2009, 23, 2278–2283. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Sinha, S.; Williams, C.; Cyrille, M.; Heller, E.; Snapper, S.B.; Georgopoulos, K.; St-Arnaud, R.; Force, T.; Dedhar, S.; et al. Targeted Deletion of Integrin-Linked Kinase Reveals a Role in T-Cell Chemotaxis and Survival. Mol. Cell Biol. 2005, 25, 11145–11155. [Google Scholar] [CrossRef] [Green Version]
- Fraccaroli, A.; Pitter, B.; Taha, A.A.; Seebach, J.; Huveneers, S.; Kirsch, J.; Casaroli-Marano, R.P.; Zahler, S.; Pohl, U.; Gerhardt, H.; et al. Endothelial Alpha-Parvin Controls Integrity of Developing Vasculature and Is Required for Maintenance of Cell-Cell Junctions. Circ. Res. 2015, 117, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Pitter, B.; Werner, A.-C.; Montanez, E. Parvins Are Required for Endothelial Cell-Cell Junctions and Cell Polarity During Embryonic Blood Vessel Formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1147–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montanez, E.; Wickström, S.A.; Altstätter, J.; Chu, H.; Fässler, R. α-Parvin Controls Vascular Mural Cell Recruitment to Vessel Wall by Regulating RhoA/ROCK Signalling. EMBO J. 2009, 28, 3132–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, H.; Thievessen, I.; Sixt, M.; Lämmermann, T.; Waisman, A.; Braun, A.; Noegel, A.A.; Fässler, R. γ-Parvin Is Dispensable for Hematopoiesis, Leukocyte Trafficking, and T-Cell-Dependent Antibody Response. Mol. Cell Biol. 2006, 26, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- König, K.; Diehl, L.; Rommerscheidt-Fuss, U.; Golletz, C.; Quast, T.; Kahl, P.; Kolanus, W.; Knolle, P.; Buettner, R.; Heukamp, L.C. Four-and-a-Half LIM Domain Protein 2 Is a Novel Regulator of Sphingosine 1-Phosphate Receptor 1 in CCL19-Induced Dendritic Cell Migration. J. Immunol. 2010, 185, 1466–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, Y.-P.; Sun, P.; Wang, A.; Lo, S.H. Tensin1 Positively Regulates RhoA Activity through Its Interaction with DLC1. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 3258–3265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furman, C.; Sieminski, A.L.; Kwiatkowski, A.V.; Rubinson, D.A.; Vasile, E.; Bronson, R.T.; Fässler, R.; Gertler, F.B. Ena/VASP Is Required for Endothelial Barrier Function in Vivo. J. Cell Biol. 2007, 179, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Estin, M.L.; Thompson, S.B.; Traxinger, B.; Fisher, M.H.; Friedman, R.S.; Jacobelli, J. Ena/VASP Proteins Regulate Activated T-Cell Trafficking by Promoting Diapedesis during Transendothelial Migration. PNAS 2017, 114, E2901–E2910. [Google Scholar] [CrossRef] [Green Version]
- Deevi, R.K.; Koney-Dash, M.; Kissenpfennig, A.; Johnston, J.A.; Schuh, K.; Walter, U.; Dib, K. Vasodilator-Stimulated Phosphoprotein Regulates Inside-Out Signaling of Β2 Integrins in Neutrophils. J. Immunol. 2010, 184, 6575–6584. [Google Scholar] [CrossRef] [Green Version]
- Savinko, T.; Guenther, C.; Uotila, L.M.; Llort Asens, M.; Yao, S.; Tojkander, S.; Fagerholm, S.C. Filamin A Is Required for Optimal T Cell Integrin-Mediated Force Transmission, Flow Adhesion, and T Cell Trafficking. J. Immunol. 2018, 200, 3109–3116. [Google Scholar] [CrossRef] [Green Version]
- Uotila, L.M.; Guenther, C.; Savinko, T.; Lehti, T.A.; Fagerholm, S.C. Filamin A Regulates Neutrophil Adhesion, Production of Reactive Oxygen Species, and Neutrophil Extracellular Trap Release. J. Immunol. 2017, 199, 3644–3653. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Forster, C.; Nakamura, F.; Glogauer, M. Filamin-A Regulates Neutrophil Uropod Retraction through RhoA during Chemotaxis. PLoS ONE 2013, 8, e79009. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Tian, F.; Sandzen, J.; Cao, R.; Flaberg, E.; Szekely, L.; Cao, Y.; Ohlsson, C.; Bergo, M.O.; Boren, J.; et al. Filamin B Deficiency in Mice Results in Skeletal Malformations and Impaired Microvascular Development. Proc. Natl. Acad. Sci. USA 2007, 104, 3919–3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, D.M.; Xu, K.; Meadows, S.M.; Zheng, Y.; Norden, P.R.; Davis, G.E.; Cleaver, O. Cdc42 Is Required for Cytoskeletal Support of Endothelial Cell Adhesion during Blood Vessel Formation in Mice. Development 2015, 142, 3058–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Liu, Y.; Lin, Q.; Li, J.; Druso, J.E.; Antonyak, M.A.; Meininger, C.J.; Zhang, S.L.; Dostal, D.E.; Guan, J.-L.; et al. Deletion of Cdc42 Enhances ADAM17-Mediated Vascular Endothelial Growth Factor Receptor 2 Shedding and Impairs Vascular Endothelial Cell Survival and Vasculogenesis. Mol. Cell. Biol. 2013, 33, 4181–4197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Zhang, S.; Tripathi, P.; Mattner, J.; Phelan, J.; Sproles, A.; Mo, J.; Wills-Karp, M.; Grimes, H.L.; Hildeman, D.; et al. Distinct Roles of Cdc42 in Thymopoiesis and Effector and Memory T Cell Differentiation. PLoS ONE 2011, 6, e18002. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Hildeman, D.; Tripathi, P.; Velu, C.S.; Grimes, H.L.; Zheng, Y. Coordination of IL-7 Receptor and T-Cell Receptor Signaling by Cell-Division Cycle 42 in T-Cell Homeostasis. Proc. Natl. Acad. Sci. USA 2010, 107, 18505–18510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nohata, N.; Uchida, Y.; Stratman, A.; Adams, R.; Zheng, Y.; Weinstein, B.; Mukouyama, Y.; Gutkind, J.S. Temporal-Specific Roles of Rac1 during Vascular Development and Retinal Angiogenesis. Dev. Biol. 2016, 411, 183–194. [Google Scholar] [CrossRef]
- Wells, C.M.; Walmsley, M.; Ooi, S.; Tybulewicz, V.; Ridley, A.J. Rac1-Deficient Macrophages Exhibit Defects in Cell Spreading and Membrane Ruffling but Not Migration. J. Cell Sci. 2004, 117, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, A.P.; Wells, C.M.; Smith, S.D.; Vega, F.M.; Henderson, R.B.; Tybulewicz, V.L.; Ridley, A.J. Rac1 and Rac2 Regulate Macrophage Morphology but Are Not Essential for Migration. J. Cell Sci. 2006, 119, 2749–2757. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Cancelas, J.A.; Hildeman, D.; Williams, D.A.; Zheng, Y. Rac GTPase Isoforms Rac1 and Rac2 Play a Redundant and Crucial Role in T-Cell Development. Blood 2008, 112, 1767–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, C.; Corsoni-Tadrzak, A.; Ruf, S.; de Boer, J.; Williams, A.; Turner, M.; Kioussis, D.; Tybulewicz, V.L.J. Rac GTPases Play Critical Roles in Early T-Cell Development. Blood 2009, 113, 3990–3998. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, M.J.; Ooi, S.K.T.; Reynolds, L.F.; Smith, S.H.; Ruf, S.; Mathiot, A.; Vanes, L.; Williams, D.A.; Cancro, M.P.; Tybulewicz, V.L.J. Critical Roles for Rac1 and Rac2 GTPases in B Cell Development and Signaling. Science 2003, 302, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Croker, B.A.; Tarlinton, D.M.; Cluse, L.A.; Tuxen, A.J.; Light, A.; Yang, F.-C.; Williams, D.A.; Roberts, A.W. The Rac2 Guanosine Triphosphatase Regulates B Lymphocyte Antigen Receptor Responses and Chemotaxis and Is Required for Establishment of B-1a and Marginal Zone B Lymphocytes. J. Immunol. 2002, 168, 3376–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faroudi, M.; Hons, M.; Zachacz, A.; Dumont, C.; Lyck, R.; Stein, J.V.; Tybulewicz, V.L.J. Critical Roles for Rac GTPases in T-Cell Migration to and within Lymph Nodes. Blood 2010, 116, 5536–5547. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.B.; Grys, K.; Vehlow, A.; de Bettignies, C.; Zachacz, A.; Henley, T.; Turner, M.; Batista, F.; Tybulewicz, V.L.J. A Novel Rac-Dependent Checkpoint in B Cell Development Controls Entry into the Splenic White Pulp and Cell Survival. J. Exp. Med. 2010, 207, 837–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahra, F.T.; Sajib, M.S.; Ichiyama, Y.; Akwii, R.G.; Tullar, P.E.; Cobos, C.; Minchew, S.A.; Doçi, C.L.; Zheng, Y.; Kubota, Y.; et al. Endothelial RhoA GTPase Is Essential for in Vitro Endothelial Functions but Dispensable for Physiological in Vivo Angiogenesis. Sci. Rep. 2019, 9, 11666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manresa-Arraut, A.; Johansen, F.F.; Brakebusch, C.; Issazadeh-Navikas, S.; Hasseldam, H. RhoA Drives T-Cell Activation and Encephalitogenic Potential in an Animal Model of Multiple Sclerosis. Front. Immunol. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhou, X.; Lang, R.A.; Guo, F. RhoA of the Rho Family Small GTPases Is Essential for B Lymphocyte Development. PLoS ONE 2012, 7, e33773. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, Y.; Nishiyama, K.; Kataoka, H.; Fruttiger, M.; Fukuhara, S.; Nishida, K.; Mochizuki, N.; Kurihara, H.; Nishikawa, S.-I.; Uemura, A. RhoJ Integrates Attractive and Repulsive Cues in Directional Migration of Endothelial Cells. EMBO J. 2020, 39, 1–18. [Google Scholar] [CrossRef]
- Hakem, A. RhoC Is Dispensable for Embryogenesis and Tumor Initiation but Essential for Metastasis. Genes Dev. 2005, 19, 1974–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigorito, E.; Bell, S.; Hebeis, B.J.; Reynolds, H.; McAdam, S.; Emson, P.C.; McKenzie, A.; Turner, M. Immunological Function in Mice Lacking the Rac-Related GTPase RhoG. Mol. Cell Biol. 2004, 24, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the Tumour Vasculature: Insights from Physiological Angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular Mechanisms and Clinical Applications of Angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the Angiogenic Switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Nagy, J.A.; Chang, S.-H.; Dvorak, A.M.; Dvorak, H.F. Why Are Tumour Blood Vessels Abnormal and Why Is It Important to Know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef]
- Polte, T.R.; Hanks, S.K.; Naftilan, A.J. Focal Adhesion Kinase Is Abundant in Developing Blood Vessels and Elevation of Its Phosphotyrosine Content in Vascular Smooth Muscle Cells Is a Rapid Response to Angiotensin II. J. Cell. Biochem. 1994, 55, 106–119. [Google Scholar] [CrossRef]
- Peng, X.; Ueda, H.; Zhou, H.; Stokol, T.; Shen, T.; Alcaraz, A.; Nagy, T.; Vassalli, J.; Guan, J. Overexpression of Focal Adhesion Kinase in Vascular Endothelial Cells Promotes Angiogenesis in Transgenic Mice. Cardiovasc. Res. 2004, 64, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Corsi, J.-M.; Houbron, C.; Billuart, P.; Brunet, I.; Bouvrée, K.; Eichmann, A.; Girault, J.-A.; Enslen, H. Autophosphorylation-Independent and -Dependent Functions of Focal Adhesion Kinase during Development. J. Biol. Chem. 2009, 284, 34769–34776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.-T.; Chen, X.L.; Tomar, A.; Miller, N.L.G.; Yoo, J.; Schlaepfer, D.D. Knock-in Mutation Reveals an Essential Role for Focal Adhesion Kinase Activity in Blood Vessel Morphogenesis and Cell Motility-Polarity but Not Cell Proliferation. J. Biol. Chem. 2010, 285, 21526–21536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavora, B.; Reynolds, L.E.; Batista, S.; Demircioglu, F.; Fernandez, I.; Lechertier, T.; Lees, D.M.; Wong, P.; Alexopoulou, A.; Elia, G.; et al. Endothelial–Cell FAK Targeting Sensitizes Tumours to DNA–Damaging Therapy. Nature 2014, 514, 112–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK Signaling in Human Cancer as a Target for Therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Schwock, J.; Dhani, N.; Hedley, D.W. Targeting Focal Adhesion Kinase Signaling in Tumor Growth and Metastasis. Expert Opin. Ther. Targets 2010, 14, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in Cancer: Mechanistic Findings and Clinical Applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yi, Q.; Tang, L. The Roles of Nuclear Focal Adhesion Kinase (FAK) on Cancer: A Focused Review. J. Exp. Clin. Cancer Res. 2019, 38, 250. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.L.; Nam, J.-O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.-T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-Induced Vascular Permeability Is Mediated by FAK. Dev. Cell 2012, 22, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Cruet-Hennequart, S.; Troussard, A.; Fazli, L.; Costello, P.; Sutton, K.; Wheeler, J.; Gleave, M.; Sanghera, J.; Dedhar, S. Regulation of Tumor Angiogenesis by Integrin-Linked Kinase (ILK). Cancer Cell 2004, 5, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Pluskota, E.; Bledzka, K.M.; Bialkowska, K.; Szpak, D.; Soloviev, D.A.; Jones, S.V.; Verbovetskiy, D.; Plow, E.F. Kindlin-2 Interacts with Endothelial Adherens Junctions to Support Vascular Barrier Integrity: Kindlin-2 Regulates Endothelial Barier Function. J. Physiol. 2017, 595, 6443–6462. [Google Scholar] [CrossRef] [Green Version]
- Legate, K.R.; Montañez, E.; Kudlacek, O.; Fässler, R. ILK, PINCH and Parvin: The TIPP of Integrin Signalling. Nat. Rev. Mol. Cell Biol. 2006, 7, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Koul, D.; Shen, R.; Bergh, S.; Lu, Y.; de Groot, J.F.; Liu, T.J.; Mills, G.B.; Yung, W.K.A. Targeting Integrin-Linked Kinase Inhibits Akt Signaling Pathways and Decreases Tumor Progression of Human Glioblastoma. Mol. Cancer Ther. 2005, 4, 1681–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, A.A.; Jafarnejad, S.M.; Zhou, J.; Li, G. Integrin-Linked Kinase Regulates Melanoma Angiogenesis by Activating NF-ΚB/Interleukin-6 Signaling Pathway. Oncogene 2011, 30, 2778–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malan, D.; Elischer, A.; Hesse, M.; Wickström, S.A.; Fleischmann, B.K.; Bloch, W. Deletion of Integrin Linked Kinase in Endothelial Cells Results in Defective RTK Signaling Caused by Caveolin 1 Mislocalization. Development 2013, 140, 987–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vouret-Craviari, V.; Boulter, E.; Grall, D.; Matthews, C.; Obberghen-Schilling, E.V. ILK Is Required for the Assembly of Matrix-Forming Adhesions and Capillary Morphogenesis in Endothelial Cells. J. Cell Sci. 2004, 117, 4559–4569. [Google Scholar] [CrossRef] [Green Version]
- Huveneers, S.; Oldenburg, J.; Spanjaard, E.; van der Krogt, G.; Grigoriev, I.; Akhmanova, A.; Rehmann, H.; de Rooij, J. Vinculin Associates with Endothelial VE-Cadherin Junctions to Control Force-Dependent Remodeling. J. Cell Biol. 2012, 196, 641–652. [Google Scholar] [CrossRef] [Green Version]
- Birukova, A.A.; Shah, A.S.; Tian, Y.; Moldobaeva, N.; Birukov, K.G. Dual Role of Vinculin in Barrier-Disruptive and Barrier-Enhancing Endothelial Cell Responses. Cell. Signal. 2016, 28, 541–551. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, Rac, and Cdc42 GTPases Regulate the Assembly of Multimolecular Focal Complexes Associated with Actin Stress Fibers, Lamellipodia, and Filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Wojciak-Stothard, B.; Ridley, A. Rho GTPases and the Regulation of Endothelial Permeability. Vasc. Pharmacol. 2002, 39, 187–199. [Google Scholar] [CrossRef]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and Roles in Cancer Cell Biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, G.; Jones, D.T.; Nye, E.; Sapienza, K.; Ramjuan, A.R.; Reynolds, L.E.; Robinson, S.D.; Kostourou, V.; Martinez, D.; Aubyn, D.; et al. Regulation of Lymphatic-Blood Vessel Separation by Endothelial Rac1. Development 2009, 136, 4043–4053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leszczynska, K.; Kaur, S.; Wilson, E.; Bicknell, R.; Heath, V. The Role of RhoJ in Endothelial Cell Biology and Angiogenesis. Biochem. Soc. Trans. 2011, 39, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.-T.; Li, G.; Xiao, H.-T. The Role of RhoJ in Endothelial Cell Biology and Tumor Pathology. Biomed. Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.; Leszczynska, K.; Abraham, S.; Scarcia, M.; Hiltbrunner, S.; Marshall, C.; Mavria, G.; Bicknell, R.; Heath, V. RhoJ/TCL Regulates Endothelial Motility and Tube Formation and Modulates Actomyosin Contractility and Focal Adhesion Numbers. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 657–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Sacharidou, A.; Stratman, A.N.; Le Bras, A.; Zwiers, P.J.; Spokes, K.; Bhasin, M.; Shih, S.-C.; Nagy, J.A.; Molema, G.; et al. RhoJ Is an Endothelial Cell-Restricted Rho GTPase That Mediates Vascular Morphogenesis and Is Regulated by the Transcription Factor ERG. Blood 2011, 118, 1145–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Valle-Pérez, B.; Martínez, V.G.; Lacasa-Salavert, C.; Figueras, A.; Shapiro, S.S.; Takafuta, T.; Casanovas, O.; Capellà, G.; Ventura, F.; Viñals, F. Filamin B Plays a Key Role in Vascular Endothelial Growth Factor-Induced Endothelial Cell Motility through Its Interaction with Rac-1 and Vav-2. J. Biol. Chem. 2010, 285, 10748–10760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, S.H.; Yu, Q.-C.; Degenstein, L.; Chen, L.B.; Fuchs, E. Progressive Kidney Degeneration in Mice Lacking Tensin. J. Cell Biol. 1997, 136, 1349–1361. [Google Scholar] [CrossRef] [Green Version]
- Yoshigi, M.; Hoffman, L.M.; Jensen, C.C.; Yost, H.J.; Beckerle, M.C. Mechanical Force Mobilizes Zyxin from Focal Adhesions to Actin Filaments and Regulates Cytoskeletal Reinforcement. J. Cell Biol. 2005, 171, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Li, P.; Yang, Z.; Huang, X.; Wei, G.; Sun, Y.; Kang, X.; Hu, X.; Deng, Q.; Chen, L.; et al. Zyxin Regulates Endothelial von Willebrand Factor Secretion by Reorganizing Actin Filaments around Exocytic Granules. Nat. Commun. 2017, 8, 14639. [Google Scholar] [CrossRef]
- Wójtowicz, A.; Babu, S.S.; Li, L.; Gretz, N.; Hecker, M.; Cattaruzza, M. Zyxin Mediation of Stretch-Induced Gene Expression in Human Endothelial Cells. Circ. Res. 2010, 107, 898–902. [Google Scholar] [CrossRef] [Green Version]
- Hubbi, M.E.; Gilkes, D.M.; Baek, J.H.; Semenza, G.L. Four-and-a-Half LIM Domain Proteins Inhibit Transactivation by Hypoxia-Inducible Factor 1. J. Biol. Chem. 2012, 287, 6139–6149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Qin, X.; Zhu, Z.; Mu, J.; Zhu, L.; Wu, K.; Jiao, H.; Xu, X.; Ye, Q. FHL Family Members Suppress Vascular Endothelial Growth Factor Expression through Blockade of Dimerization of HIF1α and HIF1β. IUBMB Life 2012, 64, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimian, T.; Arfa, O.; Simeone, S.; Lemarié, C.A.; Lehoux, S.; Wassmann, S. Inhibition of Four-and-a-Half LIM Domain Protein-2 Increases Survival, Migratory Capacity, and Paracrine Function of Human Early Outgrowth Cells Through Activation of the Sphingosine Kinase-1 Pathway: Implications for Endothelial Regeneration. Circ. Res. 2014, 114, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, H.; Nakagami, H.; Takami, Y.; Koriyama, H.; Mori, M.; Tamai, K.; Sun, J.; Nagao, K.; Morishita, R.; Kaneda, Y. FHL-2 Suppresses VEGF-Induced Phosphatidylinositol 3-Kinase/Akt Activation via Interaction With Sphingosine Kinase-1. ATVB 2009, 29, 909–914. [Google Scholar] [CrossRef] [Green Version]
- Chu, P.-H.; Yeh, L.-K.; Lin, H.-C.; Jung, S.-M.; Ma, D.H.-K.; Wang, I.-J.; Wu, H.-H.; Shiu, T.-F.; Chen, J. Deletion of the FHL2 Gene Attenuating Neovascularization after Corneal Injury. Invest. Ophthalmol. Vis. Sci. 2008, 49, 5314. [Google Scholar] [CrossRef]
- Huang, P.-H.; Chen, C.-Y.; Lin, C.-P.; Wang, C.-H.; Tsai, H.-Y.; Lo, W.-Y.; Leu, H.-B.; Chen, J.-W.; Lin, S.-J.; Chu, P.-H. Deletion of FHL2 Gene Impaired Ischemia-Induced Blood Flow Recovery by Modulating Circulating Proangiogenic Cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Deakin, N.O.; Turner, C.E. Paxillin Comes of Age. J. Cell Sci. 2008, 121, 2435–2444. [Google Scholar] [CrossRef] [Green Version]
- López-Colomé, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; López, E. Paxillin: A Crossroad in Pathological Cell Migration. J. Hematol. Oncol. 2017, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.E. Paxillin and Focal Adhesion Signalling. Nat. Cell Biol. 2000, 2, E231–E236. [Google Scholar] [CrossRef]
- Nayal, A.; Webb, D.J.; Brown, C.M.; Schaefer, E.M.; Vicente-Manzanares, M.; Horwitz, A.R. Paxillin Phosphorylation at Ser273 Localizes a GIT1-PIX-PAK Complex and Regulates Adhesion and Protrusion Dynamics. J. Cell Biol. 2006, 173, 587–589. [Google Scholar] [CrossRef]
- Nishiya, N.; Kiosses, W.B.; Han, J.; Ginsberg, M.H. An A4 Integrin–Paxillin–Arf-GAP Complex Restricts Rac Activation to the Leading Edge of Migrating Cells. Nat. Cell Biol. 2005, 7, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.A.; Sharma, R.; Roccamatisi, D.L.; Zhang, H.; Petri, B.; Kubes, P.; Colarusso, P.; Patel, K.D. Endothelial Paxillin and Focal Adhesion Kinase (FAK) Play a Critical Role in Neutrophil Transmigration: Innate Immunity. Eur. J. Immunol. 2012, 42, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Usatyuk, P.V.; Lele, A.; Harijith, A.; Gregorio, C.C.; Garcia, J.G.N.; Salgia, R.; Natarajan, V. C-Abl Mediated Tyrosine Phosphorylation of Paxillin Regulates LPS-Induced Endothelial Dysfunction and Lung Injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1025–L1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, P.; Usatyuk, P.V.; Jacobson, J.; Cress, A.E.; Garcia, J.G.N.; Salgia, R.; Natarajan, V. Role Played by Paxillin and Paxillin Tyrosine Phosphorylation in Hepatocyte Growth Factor/Sphingosine-1-Phosphate-Mediated Reactive Oxygen Species Generation, Lamellipodia Formation, and Endothelial Barrier Function. Pulm. Circ. 2015, 5, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, R.; Bell, S.E.; Davis, G.E. Coordinate Induction of the Actin Cytoskeletal Regulatory Proteins Gelsolin, Vasodilator-Stimulated Phosphoprotein, and Profilin during Capillary Morphogenesisin Vitro. Exp. Cell Res. 1999, 249, 22–32. [Google Scholar] [CrossRef]
- Price, C.J.; Brindle, N.P.J. Vasodilator-Stimulated Phosphoprotein Is Involved in Stress-Fiber and Membrane Ruffle Formation in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2051–2056. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, N.; Burger, S.; Golenhofen, N.; Walter, U.; Drenckhahn, D.; Waschke, J. The Role of VASP in Regulation of CAMP- and Rac 1-Mediated Endothelial Barrier Stabilization. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, C178–C188. [Google Scholar] [CrossRef]
- Schmit, M.A.; Mirakaj, V.; Stangassinger, M.; König, K.; Köhler, D.; Rosenberger, P. Vasodilator Phosphostimulated Protein (VASP) Protects Endothelial Barrier Function During Hypoxia. Inflammation 2012, 35, 566–573. [Google Scholar] [CrossRef] [Green Version]
- Sporbert, A.; Mertsch, K.; Smolenski, A.; Haseloff, R.F.; Schönfelder, G.; Paul, M.; Ruth, P.; Walter, U.; Blasig, I.E. Phosphorylation of Vasodilator-Stimulated Phosphoprotein: A Consequence of Nitric Oxide- and CGMP-Mediated Signal Transduction in Brain Capillary Endothelial Cells and Astrocytes. Mol. Brain Res. 1999, 67, 258–266. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in Pericytes on Blood Vessels and Endothelial Sprouts in Tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.-P.; Muñoz-Félix, J.M.; Hijazi, M.; Kim, H.; Robinson, S.D.; De Luxán-Delgado, B.; Rodríguez-Hernández, I.; Maiques, O.; Meng, Y.-M.; Meng, Q.; et al. Cancer Burden Is Controlled by Mural Cell-Β3-Integrin Regulated Crosstalk with Tumor Cells. Cell 2020, 181, 1346–1363.e21. [Google Scholar] [CrossRef]
- Er, E.E.; Valiente, M.; Ganesh, K.; Zou, Y.; Agrawal, S.; Hu, J.; Griscom, B.; Rosenblum, M.; Boire, A.; Brogi, E.; et al. Pericyte-like Spreading by Disseminated Cancer Cells Activates YAP and MRTF for Metastatic Colonization. Nat. Cell Biol. 2018, 20, 966–978. [Google Scholar] [CrossRef]
- Kotecki, M.; Zeiger, A.; Van Vliet, K.; Herman, I. Calpain- and Talin-Dependent Control of Microvascular Pericyte Contractility and Cellular Stiffness. Microvasc. Res. 2010, 80, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Kollar, B.; Nahar, T.; Suresh Babu, S.; Wojtowicz, A.; Sticht, C.; Gretz, N.; Wagner, A.H.; Korff, T.; Hecker, M. Loss of the Mechanotransducer Zyxin Promotes a Synthetic Phenotype of Vascular Smooth Muscle Cells. J. Am. Heart Assoc. 2015, 4, e001712. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Houthuijzen, J.M.; Jonkers, J. Cancer-Associated Fibroblasts as Key Regulators of the Breast Cancer Tumor Microenvironment. Cancer Metastasis Rev. 2018, 37, 577–597. [Google Scholar] [CrossRef]
- Yin, Z.; Dong, C.; Jiang, K.; Xu, Z.; Li, R.; Guo, K.; Shao, S.; Wang, L. Heterogeneity of Cancer-Associated Fibroblasts and Roles in the Progression, Prognosis, and Therapy of Hepatocellular Carcinoma. J. Hematol. Oncol. 2019, 12, 1–9. [Google Scholar] [CrossRef]
- Kobayashi, H.; Enomoto, A.; Woods, S.L.; Burt, A.D.; Takahashi, M.; Worthley, D.L. Cancer-Associated Fibroblasts in Gastrointestinal Cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 282–295. [Google Scholar] [CrossRef]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in Cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning Foes to Friends: Targeting Cancer-Associated Fibroblasts. Nature Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Gieniec, K.A.; Butler, L.M.; Worthley, D.L.; Woods, S.L. Cancer-Associated Fibroblasts—Heroes or Villains? Br. J. Cancer 2019, 121, 293–302. [Google Scholar] [CrossRef]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers Basel 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A Peek into Cancer-Associated Fibroblasts: Origins, Functions and Translational Impact. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.-Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic Gene-Expression Signature of Carcinoma-Associated Fibroblasts in Non-Small Cell Lung Cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef] [Green Version]
- Navab, R.; Strumpf, D.; To, C.; Pasko, E.; Kim, K.S.; Park, C.J.; Hai, J.; Liu, J.; Jonkman, J.; Barczyk, M.; et al. Integrin A11β1 Regulates Cancer Stromal Stiffness and Promotes Tumorigenicity and Metastasis in Non-Small Cell Lung Cancer. Oncogene 2016, 35, 1899–1908. [Google Scholar] [CrossRef] [Green Version]
- Puig, M.; Lugo, R.; Gabasa, M.; Giménez, A.; Velásquez, A.; Galgoczy, R.; Ramírez, J.; Gómez-Caro, A.; Busnadiego, Ó.; Rodríguez-Pascual, F.; et al. Matrix Stiffening and Β1 Integrin Drive Subtype-Specific Fibroblast Accumulation in Lung Cancer. Mol. Cancer Res. 2015, 13, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Min, A.; Zhu, C.; Wang, J.; Peng, S.; Shuai, C.; Gao, S.; Tang, Z.; Su, T. Focal Adhesion Kinase Knockdown in Carcinoma-Associated Fibroblasts Inhibits Oral Squamous Cell Carcinoma Metastasis via Downregulating MCP-1/CCL2 Expression. J. Biochem. Mol. Toxicol. 2015, 29, 70–76. [Google Scholar] [CrossRef]
- Wu, J.; Yu, C.; Cai, L.; Lu, Y.; Jiang, L.; Liu, C.; Li, Y.; Feng, F.; Gao, Z.; Zhu, Z.; et al. Effects of Increased Kindlin-2 Expression in Bladder Cancer Stromal Fibroblasts. Oncotarget 2017, 8, 50692–50703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-Associated Fibroblasts Drive the Progression of Metastasis through Both Paracrine and Mechanical Pressure on Cancer Tissue. Mol. Cancer Res. 2012, 10, 1403–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-Associated Fibroblasts Promote Directional Cancer Cell Migration by Aligning Fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, S.V.; Grither, W.R.; Brenot, A.; Hwang, P.Y.; Barcus, C.E.; Ernst, M.; Pence, P.; Walter, C.; Pathak, A.; Longmore, G.D. DDR2 Controls Breast Tumor Stiffness and Metastasis by Regulating Integrin Mediated Mechanotransduction in CAFs. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Tchou, J.; Kossenkov, A.V.; Chang, L.; Satija, C.; Herlyn, M.; Showe, L.C.; Puré, E. Human Breast Cancer Associated Fibroblasts Exhibit Subtype Specific Gene Expression Profiles. BMC Med. Genom. 2012, 5, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sewell-Loftin, M.K.; Bayer, S.V.H.; Crist, E.; Hughes, T.; Joison, S.M.; Longmore, G.D.; George, S.C. Cancer-Associated Fibroblasts Support Vascular Growth through Mechanical Force. Sci. Rep. 2017, 7, 12574. [Google Scholar] [CrossRef]
- Goicoechea, S.M.; Bednarski, B.; Stack, C.; Cowan, D.W.; Volmar, K.; Thorne, L.; Cukierman, E.; Rustgi, A.K.; Brentnall, T.; Hwang, R.F.; et al. Isoform-Specific Upregulation of Palladin in Human and Murine Pancreas Tumors. PLoS ONE 2010, 5, e10347. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Bassi, D.E.; Simons, J.D.; Devarajan, K.; Al-Saleem, T.; Uzzo, R.G.; Cukierman, E. Elevated Expression of Stromal Palladin Predicts Poor Clinical Outcome in Renal Cell Carcinoma. PLoS ONE 2011, 6, e21494. [Google Scholar] [CrossRef]
- Salaria, S.N.; Illei, P.; Sharma, R.; Walter, K.M.; Klein, A.P.; Eshleman, J.R.; Maitra, A.; Schulick, R.; Winter, J.; Ouellette, M.M.; et al. Palladin Is Overexpressed in the Non-Neoplastic Stroma of Infiltrating Ductal Adenocarcinomas of the Pancreas, but Is Only Rarely Overexpressed in Neoplastic Cells. Cancer Biol. Ther. 2007, 6, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Sato, D.; Tsuchikawa, T.; Mitsuhashi, T.; Hatanaka, Y.; Marukawa, K.; Morooka, A.; Nakamura, T.; Shichinohe, T.; Matsuno, Y.; Hirano, S. Stromal Palladin Expression Is an Independent Prognostic Factor in Pancreatic Ductal Adenocarcinoma. PLoS ONE 2016, 11, e0152523. [Google Scholar] [CrossRef] [Green Version]
- Azatov, M.; Goicoechea, S.M.; Otey, C.A.; Upadhyaya, A. The Actin Crosslinking Protein Palladin Modulates Force Generation and Mechanosensitivity of Tumor Associated Fibroblasts. Sci. Rep. 2016, 6, 28805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLane, J.S.; Ligon, L.A. Palladin Mediates Stiffness-Induced Fibroblast Activation in the Tumor Microenvironment. Biophys. J. 2015, 109, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentnall, T.A.; Lai, L.A.; Coleman, J.; Bronner, M.P.; Pan, S.; Chen, R. Arousal of Cancer-Associated Stroma: Overexpression of Palladin Activates Fibroblasts to Promote Tumor Invasion. PLoS ONE 2012, 7, e30219. [Google Scholar] [CrossRef] [Green Version]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The Prognostic Value of Infiltrating Immune Cells in Cancer. Br. J. Cancer 2017, 117, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kielbassa, K.; Vegna, S.; Ramirez, C.; Akkari, L. Understanding the Origin and Diversity of Macrophages to Tailor Their Targeting in Solid Cancers. Front. Immunol. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; McKay, D.; Pollard, J.W.; Lewis, C.E. Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer Res. 2018, 78, 5492–5503. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.M.; Ridley, A.J. Lipopolysaccharide Induces Actin Reorganization and Tyrosine Phosphorylation of Pyk2 and Paxillin in Monocytes and Macrophages. J. Immunol. 2000, 164, 2028–2036. [Google Scholar] [CrossRef]
- Xi, C.-X.; Xiong, F.; Zhou, Z.; Mei, L.; Xiong, W.-C. PYK2 Interacts with MyD88 and Regulates MyD88-Mediated NF-ΚB Activation in Macrophages. J. Leukoc. Biol. 2010, 87, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Kume, A.; Nishiura, H.; Suda, J.; Suda, T. Focal Adhesion Kinase Upregulated by Granulocyte-Macrophage Colony-Stimulating Factor but Not by Interleukin-3 in Differentiating Myeloid Cells. Blood 1997, 89, 3434–3442. [Google Scholar] [CrossRef]
- He, X.; Chen, X.; Li, B.; Ji, J.; Chen, S. FAK Inhibitors Induce Cell Multinucleation and Dramatically Increase Pro-Tumoral Cytokine Expression in RAW 264.7 Macrophages. FEBS Lett. 2017, 591, 3861–3871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisel, M.B.; Druet, V.A.; Sibilia, J.; Klein, J.-P.; Quesniaux, V.; Wachsmann, D. Cross Talk between MyD88 and Focal Adhesion Kinase Pathways. J. Immunol. 2005, 174, 7393–7397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, L.T.; Rodan, G.A. PYK2 Is an Adhesion Kinase in Macrophages, Localized in Podosomes and Activated by Beta(2)-Integrin Ligation. Cell Motil. Cytoskel. 2000, 47, 174–188. [Google Scholar] [CrossRef]
- Kedzierska, K.; Vardaxis, N.J.; Jaworowski, A.; Crowe, S.M. FcγR-Mediated Phagocytosis by Human Macrophages Involves Hck, Syk, and Pyk2 and Is Augmented by GM-CSF. J. Leukoc. Biol. 2001, 70, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.A.; Thomas, K.S.; Bouton, A.H. The Differential Expression of Yersinia Pseudotuberculosis Adhesins Determines the Requirement for FAK and/or Pyk2 during Bacterial Phagocytosis by Macrophages. Cell. Microbiol. 2007, 9, 596–609. [Google Scholar] [CrossRef]
- Allen, L.A.; Aderem, A. Molecular Definition of Distinct Cytoskeletal Structures Involved in Complement- and Fc Receptor-Mediated Phagocytosis in Macrophages. J. Exp. Med. 1996, 184, 627–637. [Google Scholar] [CrossRef]
- Fuortes, M.; Jin, W.W.; Nathan, C. Beta 2 Integrin-Dependent Tyrosine Phosphorylation of Paxillin in Human Neutrophils Treated with Tumor Necrosis Factor. J. Cell Biol. 1994, 127, 1477–1483. [Google Scholar] [CrossRef]
- Greenberg, S.; Chang, P.; Silverstein, S.C. Tyrosine Phosphorylation of the Gamma Subunit of Fc Gamma Receptors, P72syk, and Paxillin during Fc Receptor-Mediated Phagocytosis in Macrophages. J. Biol. Chem. 1994, 269, 3897–3902. [Google Scholar] [CrossRef]
- Abshire, M.Y.; Thomas, K.S.; Owen, K.A.; Bouton, A.H. Macrophage Motility Requires Distinct A5β1/FAK and A4β1/Paxillin Signaling Events. J. Leukoc. Biol. 2011, 89, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Rhee, I.; Zhong, M.-C.; Reizis, B.; Cheong, C.; Veillette, A. Control of Dendritic Cell Migration, T Cell-Dependent Immunity, and Autoimmunity by Protein Tyrosine Phosphatase PTPN12 Expressed in Dendritic Cells. Mol. Cell Biol. 2014, 34, 888–899. [Google Scholar] [CrossRef] [Green Version]
- Riol-Blanco, L.; Sánchez-Sánchez, N.; Torres, A.; Tejedor, A.; Narumiya, S.; Corbí, A.L.; Sánchez-Mateos, P.; Rodríguez-Fernández, J.L. The Chemokine Receptor CCR7 Activates in Dendritic Cells Two Signaling Modules That Independently Regulate Chemotaxis and Migratory Speed. J. Immunol. 2005, 174, 4070–4080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, S.; Fang, B.; Zhang, Y.; Wang, C.; Zhou, J.; Niu, C.; Gao, Y.; Zhao, D.; He, J.; Wang, J.; et al. Mechanical Stretch Promotes Tumoricidal M1 Polarization via the FAK/NF-ΚB Signaling Pathway. FASEB J. 2019, 33, 13254–13266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural Killer Cells and Other Innate Lymphoid Cells in Cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Gismondi, A.; Jacobelli, J.; Mainiero, F.; Paolini, R.; Piccoli, M.; Frati, L.; Santoni, A. Cutting Edge: Functional Role for Proline-Rich Tyrosine Kinase 2 in NK Cell-Mediated Natural Cytotoxicity. J. Immunol. 2000, 164, 2272–2276. [Google Scholar] [CrossRef] [PubMed]
- March, M.E.; Long, E.O. Β2 Integrin Induces TCR ζ–Syk–PLC-γ Phosphorylation and Paxillin-Dependent Granule Polarization in Human NK Cells. J. Immunol. 2011, 186. [Google Scholar] [CrossRef] [Green Version]
- Sancho, D.; Nieto, M.; Llano, M.; Rodríguez-Fernández, J.L.; Tejedor, R.; Avraham, S.; Cabañas, C.; López-Botet, M.; Sánchez-Madrid, F. The Tyrosine Kinase PYK-2/RAFTK Regulates Natural Killer (NK) Cell Cytotoxic Response, and Is Translocated and Activated upon Specific Target Cell Recognition and Killing. J. Cell Biol. 2000, 149, 1249–1262. [Google Scholar] [CrossRef]
- Steblyanko, M.; Anikeeva, N.; Campbell, K.S.; Keen, J.H.; Sykulev, Y. Integrins Influence the Size and Dynamics of Signaling Microclusters in a Pyk2-Dependent Manner. J. Biol. Chem. 2015, 290, 11833–11842. [Google Scholar] [CrossRef] [Green Version]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The Prognostic Significance of Intratumoral Natural Killer Cells in Patients with Colorectal Carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef]
- Ishigami, S.; Natsugoe, S.; Tokuda, K.; Nakajo, A.; Che, X.; Iwashige, H.; Aridome, K.; Hokita, S.; Aikou, T. Prognostic Value of Intratumoral Natural Killer Cells in Gastric Carcinoma. Cancer 2000, 88, 577–583. [Google Scholar] [CrossRef]
- Villegas, F.R.; Coca, S.; Villarrubia, V.G.; Jiménez, R.; Chillón, M.J.; Jareño, J.; Zuil, M.; Callol, L. Prognostic Significance of Tumor Infiltrating Natural Killer Cells Subset CD57 in Patients with Squamous Cell Lung Cancer. Lung Cancer 2002, 35, 23–28. [Google Scholar] [CrossRef]
- Bassani, B.; Baci, D.; Gallazzi, M.; Poggi, A.; Bruno, A.; Mortara, L. Natural Killer Cells as Key Players of Tumor Progression and Angiogenesis: Old and Novel Tools to Divert Their Pro-Tumor Activities into Potent Anti-Tumor Effects. Cancers Basel 2019, 11, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, S.; Burridge, K.; Silverstein, S.C. Colocalization of F-Actin and Talin during Fc Receptor-Mediated Phagocytosis in Mouse Macrophages. J. Exp. Med. 1990, 172, 1853–1856. [Google Scholar] [CrossRef] [PubMed]
- Jaumouillé, V.; Cartagena-Rivera, A.X.; Waterman, C.M. Coupling of β 2 Integrins to Actin by a Mechanosensitive Molecular Clutch Drives Complement Receptor-Mediated Phagocytosis. Nat. Cell Biol. 2019, 21, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Wiedemann, A.; Tzircotis, G.; Monkley, S.J.; Critchley, D.R.; Caron, E. An Essential Role for Talin during AMβ2-Mediated Phagocytosis. Mol. Biol. Cell 2007, 18, 976–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaidano, G.; Bergui, L.; Schena, M.; Gaboli, M.; Cremona, O.; Marchisio, P.C.; Caligaris-Cappio, F. Integrin Distribution and Cytoskeleton Organization in Normal and Malignant Monocytes. Leukemia 1990, 4, 682–687. [Google Scholar] [PubMed]
- Joosten, B.; Willemse, M.; Fransen, J.; Cambi, A.; van den Dries, K. Super-Resolution Correlative Light and Electron Microscopy (SR-CLEM) Reveals Novel Ultrastructural Insights Into Dendritic Cell Podosomes. Front. Immunol. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Kopp, P. Podosomes at a Glance. J. Cell Sci. 2005, 118, 2079–2082. [Google Scholar] [CrossRef] [Green Version]
- Baranov, M.; Ter Beest, M.; Reinieren-Beeren, I.; Cambi, A.; Figdor, C.G.; van den Bogaart, G. Podosomes of Dendritic Cells Facilitate Antigen Sampling. J. Cell. Sci. 2014, 127, 1052–1064. [Google Scholar] [CrossRef] [Green Version]
- Lämmermann, T.; Bader, B.L.; Monkley, S.J.; Worbs, T.; Wedlich-Söldner, R.; Hirsch, K.; Keller, M.; Förster, R.; Critchley, D.R.; Fässler, R.; et al. Rapid Leukocyte Migration by Integrin-Independent Flowing and Squeezing. Nature 2008, 453, 51–55. [Google Scholar] [CrossRef]
- Gunawan, M.; Venkatesan, N.; Loh, J.T.; Wong, J.F.; Berger, H.; Neo, W.H.; Li, L.Y.J.; La Win, M.K.; Yau, Y.H.; Guo, T.; et al. The Methyltransferase Ezh2 Controls Cell Adhesion and Migration through Direct Methylation of the Extranuclear Regulatory Protein Talin. Nat. Immunol. 2015, 16, 505–516. [Google Scholar] [CrossRef]
- Lim, T.J.F.; Su, I.-H. Talin1 Methylation Is Required for Neutrophil Infiltration and Lipopolysaccharide-Induced Lethality. J. Immunol. 2018, 201, 3651–3661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Wang, Z.; Zhou, J.; Huang, J.; Zhou, L.; Luo, J.; Wan, Y.Y.; Long, H.; Zhu, B. EZH2 Inhibitor GSK126 Suppresses Antitumor Immunity by Driving Production of Myeloid-Derived Suppressor Cells. Cancer Res. 2019, 79, 2009–2020. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, E.B.; Sinha, S.; Li, L.; Dedhar, S.; Force, T.; Rosenzweig, A.; Gerszten, R.E. Role of Integrin-Linked Kinase in Leukocyte Recruitment. J. Biol. Chem. 2002, 277, 16371–16375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troussard, A.A.; Mawji, N.M.; Ong, C.; Mui, A.; St -Arnaud, R.; Dedhar, S. Conditional Knock-out of Integrin-Linked Kinase Demonstrates an Essential Role in Protein Kinase B/Akt Activation. J. Biol. Chem. 2003, 278, 22374–22378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimi, R.; Yamaji, S.; Suzuki, A.; Mishima, W.; Okamura, M.; Obana, T.; Matsuda, C.; Miwa, Y.; Ohno, S.; Ishigatsubo, Y. The γ-Parvin-Integrin-Linked Kinase Complex Is Critically Involved in Leukocyte-Substrate Interaction. J. Immunol. 2006, 176, 3611–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, L.; Howarth, K.; McDowall, A.; Patzak, I.; Evans, R.; Ussar, S.; Moser, M.; Metin, A.; Fried, M.; Tomlinson, I.; et al. Leukocyte Adhesion Deficiency-III Is Caused by Mutations in KINDLIN3 Affecting Integrin Activation. Nat. Med. 2009, 15, 306–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, W.E.; Zicha, D.; Ridley, A.J.; Jones, G.E. A Role for Cdc42 in Macrophage Chemotaxis. J. Cell Biol. 1998, 141, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Burns, S.; Thrasher, A.J.; Blundell, M.P.; Machesky, L.; Jones, G.E. Configuration of Human Dendritic Cell Cytoskeleton by Rho GTPases, the WAS Protein, and Differentiation. Blood 2001, 98, 1142–1149. [Google Scholar] [CrossRef]
- Zhao, T.; Bokoch, G.M. Critical Role of Proline-Rich Tyrosine Kinase 2 in Reversion of the Adhesion-Mediated Suppression of Reactive Oxygen Species Generation by Human Neutrophils. J. Immunol. 2005, 174, 8049–8055. [Google Scholar] [CrossRef] [Green Version]
- Coppolino, M.G.; Krause, M.; Hagendorff, P.; Monner, D.A.; Trimble, W.; Grinstein, S.; Wehland, J.; Sechi, A.S. Evidence for a Molecular Complex Consisting of Fyb/SLAP, SLP-76, Nck, VASP and WASP That Links the Actin Cytoskeleton to Fcgamma Receptor Signalling during Phagocytosis. J. Cell. Sci. 2001, 114, 4307–4318. [Google Scholar]
- Wilton, K.M.; Billadeau, D.D. VASP Regulates NK Cell Lytic Granule Convergence. J. Immunol. 2018, 201, 2899–2909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, S.G.; Rabinovich, P.M. T Cell Reprogramming Against Cancer. Methods Mol. Biol. 2020, 2097, 3–44. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-J.; Cantor, H. CD4 T-Cell Subsets and Tumor Immunity: The Helpful and the Not-so-Helpful. Cancer Immunol. Res. 2014, 2, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ Cytotoxic T Lymphocytes in Cancer Immunotherapy: A Review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.-J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yuen, G.J.; Demissie, E.; Pillai, S. B Lymphocytes and Cancer: A Love-Hate Relationship. Trends Cancer 2016, 2, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Tsou, P.; Katayama, H.; Ostrin, E.J.; Hanash, S.M. The Emerging Role of B Cells in Tumor Immunity. Cancer Res. 2016, 76, 5597–5601. [Google Scholar] [CrossRef] [Green Version]
- Chapman, N.M.; Houtman, J.C.D. Functions of the FAK Family Kinases in T Cells: Beyond Actin Cytoskeletal Rearrangement. Immunol. Res. 2014, 59, 23–34. [Google Scholar] [CrossRef]
- Cheung, S.M.S.; Ostergaard, H.L. Pyk2 Controls Integrin-Dependent CTL Migration through Regulation of De-Adhesion. J. Immunol. 2016, 197, 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Conolly, S.F.; Reinl, E.L.; Houtman, J.C. Focal Adhesion Kinase Negatively Regulates Lck Function Downstream of the T Cell Antigen Receptor. J. Immunol. 2013, 191, 6208–6221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, N.M.; Yoder, A.N.; Barbón, K.M.; Bilal, M.Y.; Connolly, S.F.; Houtman, J.C.D. Proline-Rich Tyrosine Kinase 2 Controls PI3-Kinase Activation Downstream of the T Cell Antigen Receptor in Human T Cells. J. Leukoc. Biol. 2015, 97, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beinke, S.; Phee, H.; Clingan, J.M.; Schlessinger, J.; Matloubian, M.; Weiss, A. Proline-Rich Tyrosine Kinase-2 Is Critical for CD8 T-Cell Short-Lived Effector Fate. PNAS 2010, 107, 16234–16239. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto-Tane, A.; Sakuma, M.; Ike, H.; Yokosuka, T.; Kimura, Y.; Ohara, O.; Saito, T. Micro–Adhesion Rings Surrounding TCR Microclusters Are Essential for T Cell Activation. J. Exp. Med. 2016, 213, 1609–1625. [Google Scholar] [CrossRef]
- Van Seventer, G.A.; Salmen, H.J.; Law, S.F.; O’Neill, G.M.; Mullen, M.M.; Franz, A.M.; Kanner, S.B.; Golemis, E.A.; van Seventer, J.M. Focal Adhesion Kinase Regulates Beta1 Integrin-Dependent T Cell Migration through an HEF1 Effector Pathway. Eur. J. Immunol. 2001, 31, 1417–1427. [Google Scholar] [CrossRef]
- Astier, A.; Avraham, H.; Manie, S.N.; Groopman, J.; Canty, T.; Avraham, S.; Freedman, A.S. The Related Adhesion Focal Tyrosine Kinase Is Tyrosine-Phosphorylated after Beta1-Integrin Stimulation in B Cells and Binds to P130cas. J. Biol. Chem. 1997, 272, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Le, Y.; Zhu, B.-M.; Harley, B.; Park, S.-Y.; Kobayashi, T.; Manis, J.P.; Luo, H.R.; Yoshimura, A.; Hennighausen, L.; Silberstein, L.E. SOCS3 Protein Developmentally Regulates the Chemokine Receptor CXCR4-FAK Signaling Pathway during B Lymphopoiesis. Immunity 2007, 27, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Tse, K.W.K.; Dang-Lawson, M.; Lee, R.L.; Vong, D.; Bulic, A.; Buckbinder, L.; Gold, M.R. B Cell Receptor-Induced Phosphorylation of Pyk2 and Focal Adhesion Kinase Involves Integrins and the Rap GTPases and Is Required for B Cell Spreading. J. Biol. Chem. 2009, 284, 22865–22877. [Google Scholar] [CrossRef] [Green Version]
- Serrels, B.; McGivern, N.; Canel, M.; Byron, A.; Johnson, S.C.; McSorley, H.J.; Quinn, N.; Taggart, D.; Von Kreigsheim, A.; Anderton, S.M.; et al. IL-33 and ST2 Mediate FAK-Dependent Antitumor Immune Evasion through Transcriptional Networks. Sci. Signal. 2017, 10, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Symeonides, S.N.; Anderton, S.M.; Serrels, A. FAK-Inhibition Opens the Door to Checkpoint Immunotherapy in Pancreatic Cancer. J. Immunother. Cancer 2017, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.; Corgnac, S.; Boutet, M.; Gros, G.; Validire, P.; Bismuth, G.; Mami-Chouaib, F. Paxillin Binding to the Cytoplasmic Domain of CD103 Promotes Cell Adhesion and Effector Functions for CD8+ Resident Memory T Cells in Tumors. Cancer Res. 2017, 77, 7072–7082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, M.; Ito, M.; Srirat, T.; Kondo, T.; Yoshimura, A. Memory T Cell, Exhaustion, and Tumor Immunity. Immunol. Med. 2020, 43, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corgnac, S.; Boutet, M.; Kfoury, M.; Naltet, C.; Mami-Chouaib, F. The Emerging Role of CD8+ Tissue Resident Memory T (TRM) Cells in Antitumor Immunity: A Unique Functional Contribution of the CD103 Integrin. Front. Immunol. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Simone, M.; Arrigoni, A.; Rossetti, G.; Gruarin, P.; Ranzani, V.; Politano, C.; Bonnal, R.J.P.; Provasi, E.; Sarnicola, M.L.; Panzeri, I.; et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity 2016, 45, 1135–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Guinoa, J.S.; Barrio, L.; Carrasco, Y.R. Vinculin Arrests Motile B Cells by Stabilizing Integrin Clustering at the Immune Synapse. J. Immunol. 2013, 191, 2742–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New Insights into Their Functions from in Vivo Studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Saoudi, A.; Kassem, S.; Dejean, A.; Gaud, G. Rho-GTPases as Key Regulators of T Lymphocyte Biology. Small GTPases 2014, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Tybulewicz, V.L.J.; Henderson, R.B. Rho Family GTPases and Their Regulators in Lymphocytes. Nat. Rev. Immunol. 2009, 9, 630–644. [Google Scholar] [CrossRef]
- Guo, F.; Velu, C.S.; Grimes, H.L.; Zheng, Y. Rho GTPase Cdc42 Is Essential for B-Lymphocyte Development and Activation. Blood 2009, 114, 2909–2916. [Google Scholar] [CrossRef] [Green Version]
- Faure, S.; Salazar-Fontana, L.I.; Semichon, M.; Tybulewicz, V.L.J.; Bismuth, G.; Trautmann, A.; Germain, R.N.; Delon, J. ERM Proteins Regulate Cytoskeleton Relaxation Promoting T Cell-APC Conjugation. Nat. Immunol. 2004, 5, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Roumier, A.; Olivo-Marin, J.C.; Arpin, M.; Michel, F.; Martin, M.; Mangeat, P.; Acuto, O.; Dautry-Varsat, A.; Alcover, A. The Membrane-Microfilament Linker Ezrin Is Involved in the Formation of the Immunological Synapse and in T Cell Activation. Immunity 2001, 15, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Derry, J.M.; Ochs, H.D.; Francke, U. Isolation of a Novel Gene Mutated in Wiskott-Aldrich Syndrome. Cell 1994, 78, 635–644. [Google Scholar] [CrossRef]
- Krause, M.; Sechi, A.S.; Konradt, M.; Monner, D.; Gertler, F.B.; Wehland, J. Fyn-Binding Protein (Fyb)/SLP-76-Associated Protein (SLAP), Ena/Vasodilator-Stimulated Phosphoprotein (VASP) Proteins and the Arp2/3 Complex Link T Cell Receptor (TCR) Signaling to the Actin Cytoskeleton. J. Cell Biol. 2000, 149, 181–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Tumour microenvironment (TME). Besides cancer cells (depicted in grey), distinct cell types and extracellular matrix (ECM) constitute the TME that controls cancer development and progression. Abbreviations: CAF, cancer-associated fibroblast; TAM, tumour-associated macrophages; MDSCs, myeloid-derived suppressor cells; NK cell, natural killer cell; EC, endothelial cell; DC, dendritic cell; TAN, tumour-associated neutrophils; ECM, extracellular matrix.
Figure 1.
Tumour microenvironment (TME). Besides cancer cells (depicted in grey), distinct cell types and extracellular matrix (ECM) constitute the TME that controls cancer development and progression. Abbreviations: CAF, cancer-associated fibroblast; TAM, tumour-associated macrophages; MDSCs, myeloid-derived suppressor cells; NK cell, natural killer cell; EC, endothelial cell; DC, dendritic cell; TAN, tumour-associated neutrophils; ECM, extracellular matrix.

Figure 2.
Adhesome in the tumour microenvironment. (A) Adhesome molecular architecture. Members of the adhesome, the complex protein network formed at integrin-mediated cell–matrix adhesions (red cylinders at the bottom of the cell), are divided in three levels: the adhesome signalling, the adhesion organisation and the actin regulatory layer (pink boxes). (B) Adhesome components and the tumour stroma. The distinct cell types of the TME, with in vivo studied roles of adhesome members are depicted on the right side of each box (layer). Abbreviations: CAF, cancer-associated fibroblast; TAM, tumour-associated macrophage; NK, natural killer; EC, endothelial cell; DC, dendritic cell; TAN, tumour-associated neutrophils; ECM, extracellular matrix.
Figure 2.
Adhesome in the tumour microenvironment. (A) Adhesome molecular architecture. Members of the adhesome, the complex protein network formed at integrin-mediated cell–matrix adhesions (red cylinders at the bottom of the cell), are divided in three levels: the adhesome signalling, the adhesion organisation and the actin regulatory layer (pink boxes). (B) Adhesome components and the tumour stroma. The distinct cell types of the TME, with in vivo studied roles of adhesome members are depicted on the right side of each box (layer). Abbreviations: CAF, cancer-associated fibroblast; TAM, tumour-associated macrophage; NK, natural killer; EC, endothelial cell; DC, dendritic cell; TAN, tumour-associated neutrophils; ECM, extracellular matrix.


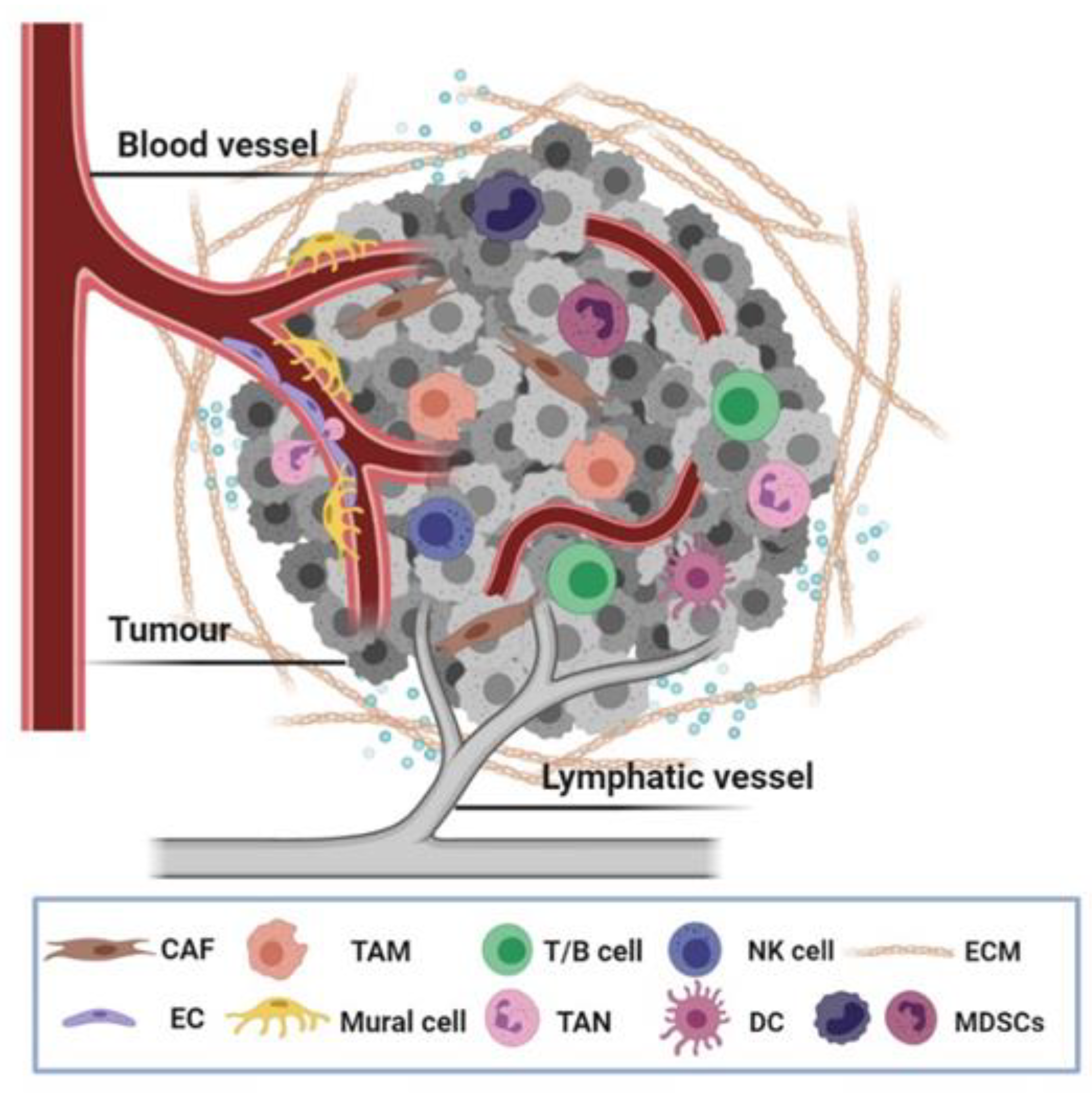{kind=link}
{kind=link}
Table 1.
Adhesome and cancer: in vivo studies investigating the effect of genetic deletion or pharmacological inhibition of adhesome members in specific tumour stroma cell types.
Table 1.
Adhesome and cancer: in vivo studies investigating the effect of genetic deletion or pharmacological inhibition of adhesome members in specific tumour stroma cell types.
| Molecule | Cell Type | Murine Tumour Model | Intervention | Effect | References |
|---|---|---|---|---|---|
| Adhesome signalling | |||||
| FAK/Pyk2 | ECs | Subcutaneous tumours (B16F0 or CMT19T) | Pdgfb iCreERT2; FAKfl/fl | anti-tumourigenic: vessel density (↓), no Pyk2 compensation | [13] |
| Orthotopic patient-derived human glioma (DBTRG) | Tie2 CreERT2; FAKfl/fl | anti-tumourigenic: vascular normalisation, no Pyk2 compensation | [14] | ||
| Subcutaneous tumours (B16F0 or CMT19T) | FAKhet | pro-tumourigenic: vessel density (↑) | [15] | ||
| Subcutaneous tumours (B16F10), Metastasis models: I.V or spontaneous metastasis after B16F10 injection | SCL CreERT; FAKfl/KD | metastasis (↓) tumour growth (−) | [16] | ||
| Subcutaneous tumours (B16F0) | PDGFb iCreERT; FAKfl/fl; R26FAKKD/KD (FAK kinase-dead mice) | anti-tumourigenic: vessel density (↓), vascular permeability (↓), VE−CAD pY658 levels (↓) | [17] | ||
| Subcutaneous tumours (B16F0) | PDGFb iCreERT; FAKfl/fl; R26FAKDM/DM (FAK DM mice: KD with a putatively phosphomimetic Y397E mutation) | anti-tumourigenic: vessel density (↓), vascular permeability (−), VE−CAD pY658 levels (−) | [17] | ||
| Subcutaneous tumours (B16F0 or CMT19T) | PDGFb iCreERT; FAKfl/fl; R26FAKY397F/Y397F | anti-tumourigenic: vessel density (↓) | [18] | ||
| Subcutaneous tumours (B16F0 or CMT19T) | PDGFb iCreERT; FAKfl/fl; R26FAKY861F/Y8617F | no effect: tumour growth (−), vessel density (−) | [18] | ||
| Pericytes | Subcutaneous tumours (B16F0 or LLC) | PDGFRβ Cre; FAKfl/fl | pro-tumourigenic: vessel density (↑) | [19] | |
| Subcutaneous tumours (LLC) | PDGFRβ Cre; FAKY397F/Y397F | no effect: tumour growth (−), vessel density (−) | [20] | ||
| Subcutaneous tumours (B16F0 or LLC) | PDGFRβ Cre; FAKY861F/Y861F | anti-tumourigenic: vessel density (↓) | [20] | ||
| CAFs | Orthotopic xenografts of pancreatic cancer (MPanc-96 or MAD08-608) | Inhibition (FAK/Pyk2 inhibitor: PF-562,271) | anti-tumourigenic: CAFs migration and infiltration (↓) | [21] | |
| Genetically engineered mice of pancreatic cancer (KPC and KPPC) | Inhibition (FAK inhibitor: VS-4718) | tumour stasis: intratumoural aSMA+ and FAP+ cells (↓), FAP+ cell proliferation (↓) | [22] | ||
| Subcutaneous tumours (HT29) | Inhibition (FAK inhibitor: PF-573,228) or HT29 expressing FAKY397F construct | anti-tumourigenic | [23] | ||
| Syngeneic orthotopic breast (E0771) and pancreatic (TB32048) tumours/MMTV-PyMT mouse model of breast cancer | FSP Cre+; FAKfl/fl mice/MMTV+; FSP Cre+; FAKfl/fl | pro-tumourigenic: CAF−mediated changes in malignant metabolism (↑) | [24] | ||
| Pancreatic cancer cell and fibroblast syngeneic and orthotopic cografting | FAKKD(kinase−dead) fibroblasts, inhibition (FAK inhibitor: PF562,271) | anti-tumourigenic: tumour growth (−), lung metastasis (↓), fibrosis (↓), M2 macrophage polarisation and migration (↓), | [25] | ||
| MMTV-PyMT mouse model of spontaneous breast cancer | MMTV-PyMT; Col1a2 CreER; FAKfl/fl | tumour development and growth (−), breast cancer metastasis (↓), exosome amount and functions (↓) | [26] | ||
| TAMs/monocytes | Orthotopic pancreatic tumours (MPanc-96 or MAD08-608) | Inhibition (FAK/PYK2 inhibitor: PF-562,271) | anti-tumourigenic: TAMs (F4/80+) migration and infiltration (↓) | [21] | |
| Genetically engineered mice of pancreatic cancer(KPC and KPPC) | Inhibition (FAK inhibitor: VS-4718) | tumour stasis: intratumoural F4/80+ and CD206+ macrophages (↓) | [22] | ||
| Syngeneic orthotopic breast tumours (4T1 or MDA-MB-231) | Inhibition (FAK inhibitor: PND-1186) | anti-tumourigenic: metastasis (↓), leukocyte infiltration (↓) | [27] | ||
| Orthotopic breast tumours (4T1) | Inhibition (FAK inhibitors: PF-562271 and PF-573,228) | anti-tumourigenic: intratumoural F4/80+ macrophages (↓) | [28] | ||
| MMTV-PyMT mouse model of spontaneous breast cancer | PyVmT+/−;LysMwt/cre; FAKΔmyeloid | pro−and anti-tumorigenic: depending on the stage of malignancy, carcinoma stage: NK cells (↓) | [29] | ||
| Subcutaneous tumours (SCC) | Inhibition (FAK inhibitor: BI 853,520) | anti−tumorigenic: PD−L2 surface expression on TAMs (↓) | [30] | ||
| MDSCs/TANs | Genetically engineered mice of pancreatic cancer (KPC and KPPC) | Inhibition (FAK inhibitor: VS-4718) | tumour stasis: recruitment of Gr1+ granulocytes (↓) | [22] | |
| Subcutaneous tumours (SCC cells) | Inhibition (FAK inhibitor: BI 853,520) | anti-tumorigenic: PD−L2 surface expression on M−MDSCs (↓) | [30] | ||
| CD3+/CD4+/CD8+ T cells | Subcutaneous tumours (SCC cells) | SSC FAK−/− cells, inhibition (FAK inhibitor: VS-4718) | anti-tumourigenic: CD4+ and CD8+ cell recruitment (↑) | [31] | |
| Genetically engineered mice of pancreatic cancer (KPC and KPPC) | Inhibition (FAK inhibitor: VS-4718) | tumour stasis: CD4+ and CD8+ cell recruitment (↑) | [22] | ||
| Subcutaneous tumours (SCC cells) | Inhibition (FAK inhibitor: BI 853,520) | anti-tumorigenic: ICOS surface expression in CD8+ cells (↑) | [30] | ||
| Tregs | Genetically engineered mice of pancreatic cancer (KPC and KPPC) | SSC FAK−/− cells, inhibition (FAK inhibitor: VS-4718) | anti-tumourigenic: infiltration of CD4+FoxP3+CD25+ Tregs in the tumour niche (↓) | [31] | |
| Genetically engineered mice of pancreatic cancer (KPC and KPPC) | Inhibition (FAK inhibitor: VS-4718) | anti-tumorigenic: infiltration of CD4+FOXP3+ TREGS in the tumour niche (↓) | [22] | ||
| Subcutaneous tumours (SCC) | Inhibition (FAK inhibitor: BI 853,520) | anti-tumorigenic: Tregs (↓) | [30] | ||
| Paxillin | ECs | Matrigel plugs implanted subcutaneously (LLC) | siRNA | matrigel plugs: angiogenesis (↑) | [32] |
| GIT/PIX/PAK | CAFs | Genetically engineered mice of pancreatic cancer (KPC) | PAK1−/− | anti-tumourigenic: intratumoural aSMA+ and desmin+ cells (↓) | [33] |
| CD3+/CD4+/CD8+ T cells | Genetically engineered mice of pancreatic cancer (KPC) | PAK1−/− | anti-tumourigenic: CD3+, CD4+ and CD8+ cell recruitment (↑) | [33] | |
| Genetically engineered mice of intestinal-colorectal cancer APCΔ14/+ | PAK1−/−, inhibition (PAK inhibitor PF-3758,309) | anti-tumourigenic: CD3+, CD4+ and CD8+ cell recruitment (↑) | [34] | ||
| Tregs | Subcutaneous tumours (B16F10 or TRAMP-C1) | GIT2−/− | anti-tumourigenic | [35] | |
| Adhesion organisation | |||||
| Kindlin2 | ECs | Subcutaneous tumours (RM1) | Kindlin2+/− | anti-tumourigenic: vessel density (↓) | [36] |
| CAFs | Subcutaneous coengraftments (SUIT-2 and PSCs) | siRNA | anti-tumourigenic | [37] | |
| Kindlin3 | TAMs/monocytes | Intravenous injections of B16F10 cells | Mx1 Cre+/− (Poly(I:C)); CX3CR1gfp/+; kindlin3fl/fl | pro-tumorigenic: monocyte patrolling (↓), NK cells (↓), | [38] |
| Talin | ECs | Subcutaneous tumours (B16F0) | Cdh5 CreERT2; talin1f/L325R | anti-tumourigenic: vessel density (↓) | [39] |
| ILK | ECs | Thyroid (DRO) tumour xenografts | Inhibition (ILK inhibitor: QLT0267) | anti-tumourigenic: vessel density (↓) | [40] |
| Orthotopic pancreatic tumour xenografts | Inhibition (ILK inhibitor: QLT0254) | anti-tumourigenic: vessel density (−) | [41] | ||
| Glioblastoma (U87MG) tumour xenografts | Inhibition (ILK inhibitor: QLT0267) | anti-tumourigenic: vessel density (↓) | [42] | ||
| Subcutaneous tumours (PC3) | Inhibition (ILK inhibitor: KP-307-2) | anti-tumourigenic: vessel density (↓) | [43] | ||
| Actin regulatory layer | |||||
| Palladin | CAFs | Orthotopic xenografts of pancreatic cancer (AsPC-1 coimplantation with palladin KD CAFs) | siRNA | anti-tumourigenic | [44] |
| VASP | CAFs | Subcutaneous tumours (LLC coimplantation with VASP KD MEFs) | siRNA and EVH2 VASP mutant | anti-tumourigenic: intratumoural aSMA+ and desmin+ cells (↓) | [45] |
| Subcutaneous tumours (HT-29 coimplantation with VASP KD HSCs) | siRNA | anti-tumourigenic: intratumoural aSMA+ cells (↓) | [46] | ||
| Filamin | ECs | Subcutaneous tumours (T241 or B16F0) | VE-Cad Cre; flnAfl/fl | anti-tumourigenic: vessel density (↓) | [47] |
| Rac1 | ECs | Subcutaneous tumours (B16F0) | PDGFb iCreERT; Rac1fl/fl | no effect: tumour growth (−), vessel density (−) | [48] |
| Subcutaneous tumours (Neuro2a cells) | siRNA | anti-tumourigenic: vessel density (↓) | [49] | ||
| RhoJ | ECs | Subcutaneous tumours (LLC) | RhoJKO | anti-tumourigenic: vessel density (↓) | [50] |
| Subcutaneous tumours (B16F0 or LLC) | Cad5 CreERT2; RhoJfl/fl | anti-tumourigenic: vessel density (↓) | [51] | ||
↓ denotes reduction; ↑ denotes increase; - denotes no difference; Bold, the adhesome layers, as discussed in the text; Orange, adhesome signalling; Blue, Adhesion organization; Green, actin regulatory layer; italic, mouse models that have been used in the cited studies.
Table 2.
Adhesome and physiology: in vivo studies describing the effect of deletion of adhesome members in specific cell types that participate in tumour microenvironment. Abbreviations: HSCs, hematopoietic stem cells.
Table 2.
Adhesome and physiology: in vivo studies describing the effect of deletion of adhesome members in specific cell types that participate in tumour microenvironment. Abbreviations: HSCs, hematopoietic stem cells.
| Molecule | Cell Type | Mouse Model | Effect | References |
|---|---|---|---|---|
| Adhesome signalling | ||||
| Paxillin | ECs | siRNA | Retinal angiogenesis (↑), vessel sprouting (↑) | [32] |
| FAK | ECs | Tie2 Cre; FAKfl/fl | EC migration (↓), EC proliferation (↓), EC retraction and death (↑), haemorrhage, vessel growth (↓), vessel regression (↑), embryonic lethality (E10.5–E11.5) | [57,58] |
| Tie2 Cre; FAKflox/KD | Vascular permeability (↑), VE-cadherin Y658 phosphorylation (↓), embryonic lethality (E13.5) | [59] | ||
| End-SCL CreERT; FAKfl/fl | No effect-Pyk2 compensation (adult mice) | [60] | ||
| Myeloid lineages → macrophages | LysM cre; FAKfl/fl | Macrophages: adhesion (↓), migration and invasion (↓), recruitment (↓) | [61] | |
| Myeloid lineages→ neutrophils | LysM cre; FAKfl/fl | Adhesion to fibronectin or ICAM-1 (↓), life span (↓), pathogen-killing capability (↓) | [62] | |
| T cells | FAK+/− | Normal T cell development | [63] | |
| CD4 Cre; FAKfl/fl | Normal T cell development | [64] | ||
| B cells | CD19 Cre; Fak fl/fl | Progenitor B and immature B cells (↓), homing to the BM (↓), retention in BM (↓) | [65] | |
| Pyk2 | Macrophages | Pyk2–/– | Macrophages: cell polarisation (↓), cell contractility (↓), size of lamellipodia (↑), migration (↓), | [66] |
| Neutrophils | Pyk2–/– | Neutrophil migration (↓), degranulation (↓) | [67] | |
| Monocytes | Pyk2–/– | Number of BM monocyte-lineage cells (↓), Pyk2 promotes the turnover of monocytes at steady state | [68] | |
| T cells | Pyk2–/– | Normal T cell development | [66] | |
| Pyk2–/– | CD8 T cell activation (↓), LFA-1-dependent CD8 T cell adhesion and migration (↓), CD8+ TEFF (↓) | [69] | ||
| B cells | Pyk2–/– | B cell migration into the marginal zone of spleen (↓), humoral responses to T dependent antigen (↓) | [70] | |
| a-PIX | Immune cells→ T and B cells | a-PIX−/− | Mature lymphocytes (↓), T cell proliferation (↓), B cell proliferation (↓), B or T motility (↑), disrupted immune synapse | [71] |
| T cells | a-PIX−/− | Thymocytes motility (↑), T cell positive selection (↓) | [72] | |
| Adhesion organisation | ||||
| Kindlin3 | Neutrophils | kindlin3−/− | Neutrophil: firm adhesion (↓), vascular arrest (↓), recruitment (↓), bleeding, death | [73,74] |
| HSCs→ neutrophils | Mx1 Cre (Poly(I:C)); kindlin3fl/fl | Leukocyte adhesion (↓), bleeding, death, 5% protein expression viable | [75] | |
| T cells | kindlin3−/−, Mx1 Cre (Poly(I:C)); kindlin3fl/fl, CD4 Cre; kindlin3fl/fl | Thumocyte proliferation (↓), T cell homing to thymus (↓),T cell adhesion at low vascular shear stress (↓) | [76] | |
| Talin1 | ECs | Tie2 Cre; talin1fl/fl | Haemorrhaging, vessel density (↓), small and round ECs, embryonic lethality (E10.5) | [77] |
| Cdh5 CreERT2; talin1 | Haemorrhaging of intestine vasculature, death (adult mice) | [78] | ||
| Cdh5 CreERT2; talin1 | Retinal angiogenesis (↓), haemorrhaging, EC proliferation (↓), embryonic lethality | [39] | ||
| Cdh5 CreERT2; talin1fl/L325R | Retinal angiogenesis (↓) | [39] | ||
| Neutrophils, platelets | Rap1 binding deficient talin1 knock-in | Neutrophil adhesion (↓), leukocyte adhesion and extravasation (↓) | [79] | |
| HSCs → neutrophils | Mx1 Cre (Poly(I:C)); talin1fl/fl | Neutrophil slow rolling and vascular arrest (↓), neutrophil recruitment (↓) | [73] | |
| Dendritic cells | CD11c Cre; talin1fl/fl | DC activation (↓), T cell and B cell priming responses (↓) | [80] | |
| CD11c Cre; talin1fl/fl | DC migration and activation (↓) | [81] | ||
| NK | Talin1−/− ES -> differentiate into NK | NK LFA-1-mediated-adhesion (↓), NK cytotoxicity (↓): only retained for selective target cells lacking ICAM-1 | [82] | |
| T cells | CD4 Cre; talin1fl/fl | T cell activation and proliferation (↓), T cell–APC interactions (transient) | [83] | |
| CD4 Cre; talin1fl/fl | T regs (↓), T cell–DCs interactions (↓) | [84] | ||
| B cells | CD19 Cre; talin1−/fl | Normal B cell development and maturation, humoral responses to T-dependent antigens (↓), B cells homing to lymph nodes and BM (bone marrow) (↓) | [85] | |
| Vinculin | HSCs → neutrophils | Mx1 Cre (Poly(I:C)); Vclfl/fl | Normal neutrophil recruitment, neutrophil spreading (↓) | [86] |
| ILK | ECs | Tie2 Cre; ILKfl/fl | Labyrinthine vascularisation (↓), EC apoptosis (↑), embryonic lethality (E9.5–E10) | [87] |
| PDGFb iCreERT; ILKfl/fl | Vessel sprouting (↓), EC proliferation (↓), disruption of the blood–retina barrier | [88] | ||
| vSMCs | Pdgfrb Cre; ILK fl/fl | vSMCs: contraction (↑), migration (↓), focal adhesion assembly (↓) | [89] | |
| T cells | Lck Cre+/ILKfl/fl | T cell chemotaxis (↓), T cell survival (↓) | [90] | |
| Parvin | ECs | Tie2 Cre; α-pvfl/fl | Haemorrhaging, vessel density (↓), embryonic lethality (E13.5) | [91] |
| Cdh5 CreERT2; α-pvfl/fl | Vessel density (↓), vessel sprouting (↓), EC proliferation (↓), vessel regression (↑) | [91] | ||
| Tie2 Cre; α-pvfl/fl; β-pv−/− | Haemorrhaging, vessel branching (↓), vessel diameter (↑), vessel density (↓), embryonic lethality (E10.5–E12.5) | [92] | ||
| vSMCs | a-pv−/− | vSMCs: contraction (↑), directed migration (↓) | [93] | |
| Hematopoietic cells | γ-pv−/− | Normal haematopoiesis | [94] | |
| FHL2 | Dendritic cells | FHL2−/− | DC migration (↑) | [95] |
| Tensin | ECs | TNS1−/− | EC migration (↓), EC proliferation (↓) | [96] |
| Actin regulatory layer | ||||
| Ena/VASP family | ECs | Ena/VASP triple null | Oedema, haemorrhaging, stress fibre formation (↓), vascular integrity (↓), embryonic lethality (E18.5) | [97] |
| EVL/VASP | T cells | EVL−/−; VASP−/− | Normal T cell development, activated T cell trafficking (↓), T cell trans endothelial migration (↓) | [98] |
| VASP | Polymorphonuclear leukocytes or neutrophils (PMNs) | VASP−/− | Neutrophil/PMNs adhesion (↓) | [99] |
| Filamin A | T cells | CD4 Cre; flnAfl/fl | Normal T cell development, Teff flow adhesion (↓), T cell trafficking (↓) | [100] |
| Myeloid lineages → neutrophils | LysM Cre; flnAfl/fl | Neutrophil adhesion (↑), normal adhesion underflow | [101] | |
| Myeloid lineages → neutrophils | LysM Cre; flnAfl/fl | Neutrophil recruitment due to inflammatory response (↓) | [102] | |
| Filamin B | ECs | flnb−/− | Microvascular development (↓) | [103] |
| Cdc42 | ECs | Tie2 Cre; Cdc42fl/fl | Haemorrhaging, vascular integrity (↓), EC migration (↓), EC proliferation (↓), embryonic lethality (E9–10) | [104,105] |
| Cad5 CreERT2; Cdc42fl/fl | Retinal angiogenesis (↓), vessel lumen formation (↓), EC adhesion (↓), embryonic lethality | [104] | ||
| T cells | Lck Cre; Cdc42fl/fl | T cell development (↓), CD4+CD8+ double-positive (DP) (↑), CD4+ and CD8+ single-positive (↓), T cell migration (↓), T cell survival (↓), CD8+ Teff (↑), CD8+ T memory (↑) | [106] | |
| Lck Cre; Cdc42fl/fl | T cell survival (↓), mature CD4+ and CD8+ T cells in thymus, spleen and lymph nodes (↓), T cell effector/memory (↑) | [107] | ||
| Rac1 | ECs | Tie2 Cre; Rac1fl/fl | Vessel density (↓), EC adhesion (↓), EC migration (↓), embryonic lethality (E9.5) | [43] |
| Cad5 CreERT2; Rac1fl/fl | Haemorrhaging, vessel density (↓), retinal angiogenesis (↓), embryonic lethality | [108] | ||
| HSCs→ macrophages | Mx1 Cre (Poly(I:C)) ;Rac1fl/fl | Macrophages: elongated morphology, normal migration speed and chemotaxis | [109] | |
| Mx1 Cre (Poly(I:C)) ;Rac1fl/fl | Macrophages: trans endothelial migration (↓) | [110] | ||
| HSCs → T cells | Mx1 Cre (Poly(I:C)) ;Rac1fl/fl | T cells (↓), B cells (↑), bone marrow lymphopoiesis (↓), CD4+CD8+ T cells (↓) | [111] | |
| T cells | Lck Cre;Rac1fl/fl | Normal T cell development | [111] | |
| CD2 Cre;Rac1fl/fl | Normal T cell development | [112] | ||
| B cells | CD19 Cre;Rac1 fl/fl | Normal B cell development | [113] | |
| Rac2 | Macrophages | Rac2−/− | Macrophages: normal trans endothelial migration, elongated morphology, adhesion (↓) | [110] |
| T cells | Rac2−/− | Normal T cell development, T lymphocyte migration (↓), chemotaxis (↓), CD4+ and CD8+ T lymphocytes in spleen, CD8+ T lymphocytes in lymph nodes (↑) | [114] | |
| Rac2−/− | Normal T cell development | [112] | ||
| Rac1 and Rac1 | HSCs→ macrophages | Mx1 Cre (Poly(I:C)); Rac1fl/fl; Rac2−/− | Trans endothelial migration (↓), normal migration speed and chemotaxis, stellate or elongated morphology | [110] |
| HSCs→ T cells | Mx1 Cre (Poly(I:C)); Rac1fl/fl; Rac2−/− | Common lymphoid progenitor (CLP, Lin/IL-7Ra) (↓) | [111] | |
| T cells | Lck Cre; Rac1fl/fl; Rac2−/− | T cell proliferation (↓), T cell survival (↓), adhesion (↓), migration (↓), immature CD4+CD8+ and mature CD4+ in thymus (↓), CD4+ and CD8+ in spleen (↓) | [111] | |
| CD2 Cre; Rac1fl/fl; Rac2−/− | T cell development (↓), CD4+CD8+ double-positive (DP) (↓), CD4+CD8− single-positive (4SP) (↓), CD4−CD8+ single-positive (8SP) (↓) | [112] | ||
| dLck iCre; Rac1fl/fl; Rac2−/− | T cells (↓), T cell chemotaxis (↓), T cell adhesion (↓), homing to secondary lymphoid organs (↓) | [115] | ||
| B cells | CD19 Cre; Rac1 fl/fl; Rac2−/− | Normal B cell development in bone marrow, B cell development in spleen (↓), B cell proliferation (↓), B cell survival (↓) | [113] | |
| CD19 Cre; Rac1 fl/fl; Rac2−/− | B cell development in bone marrow (↓), B cell development in spleen (↓), B cell chemotaxis (↓) | [116] | ||
| RhoA | ECs | Tie2 Cre; RhoAfl/fl | Normal retinal angiogenesis | [117] |
| Cad5 CreERT2; RhoAfl/fl | Normal retinal angiogenesis | [117] | ||
| T cells | Lck Cre; RhoAfl/fl | T cell proliferation (↓), T cell survival n (↓), T cell differentiation n (↓), mature CD4 CD8 T cells in thymus and spleen (↓), trans endothelial migration (↓), Th1 inflammation responses (↓) | [118] | |
| B cells | CD19 Cre; RhoA fl/fl | B cell development (↓) | [119] | |
| HSCs→ B cells | Mx1 Cre (Poly(I:C)); RhoAfl/fl | B cell development (↓) | [119] | |
| Rhoj | ECs | Pdgfb iCreERT2:Rhojfl/fl | Delayed retinal angiogenesis, EC motility (↓) | [120] |
| RhoG, RhoC | B, T cells | RhoG−/−, RhoC−/− | Normal T and B cell development | [121,122] |
↓ reduction; ↑ increase; Bold, the adhesome layers, as discussed in the text; Orange, Adhesome signalling; Blue, Adhesion organization; Green, Actin regulatory layer; italic, mouse models that have been used in the cited studies.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nikolopoulou, P.A.; Koufaki, M.A.; Kostourou, V. The Adhesome Network: Key Components Shaping the Tumour Stroma. Cancers 2021, 13, 525. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13030525
AMA Style
Nikolopoulou PA, Koufaki MA, Kostourou V. The Adhesome Network: Key Components Shaping the Tumour Stroma. Cancers. 2021; 13(3):525. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13030525
Chicago/Turabian StyleNikolopoulou, Pinelopi A., Maria A. Koufaki, and Vassiliki Kostourou. 2021. "The Adhesome Network: Key Components Shaping the Tumour Stroma" Cancers 13, no. 3: 525. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13030525
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.