
Blocking Migration of Polymorphonuclear Myeloid-Derived Suppressor Cells Inhibits Mouse Melanoma Progression

 , , , and
, , , and 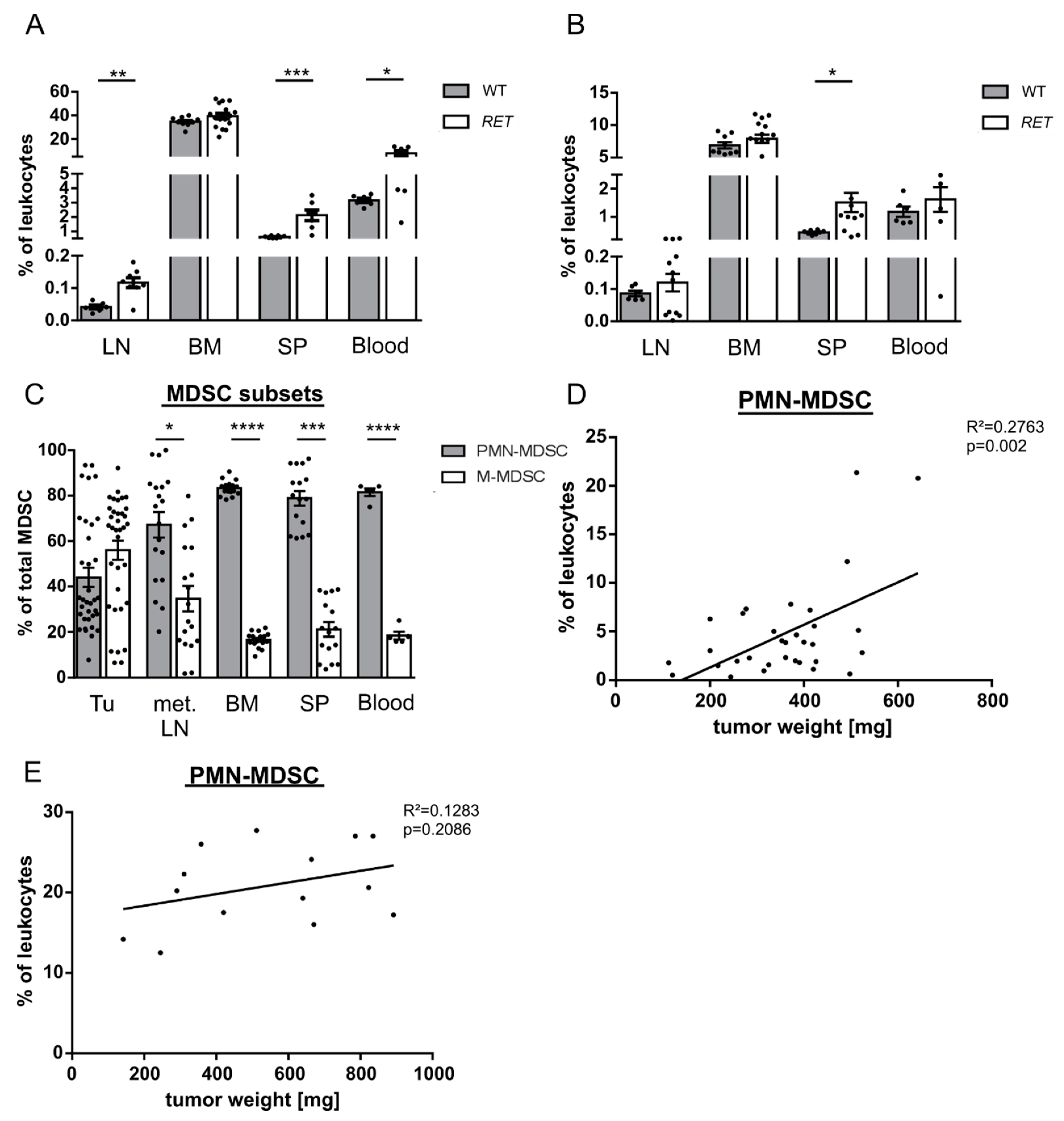{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. PMN-MDSC Subset Expands during Tumor Progression
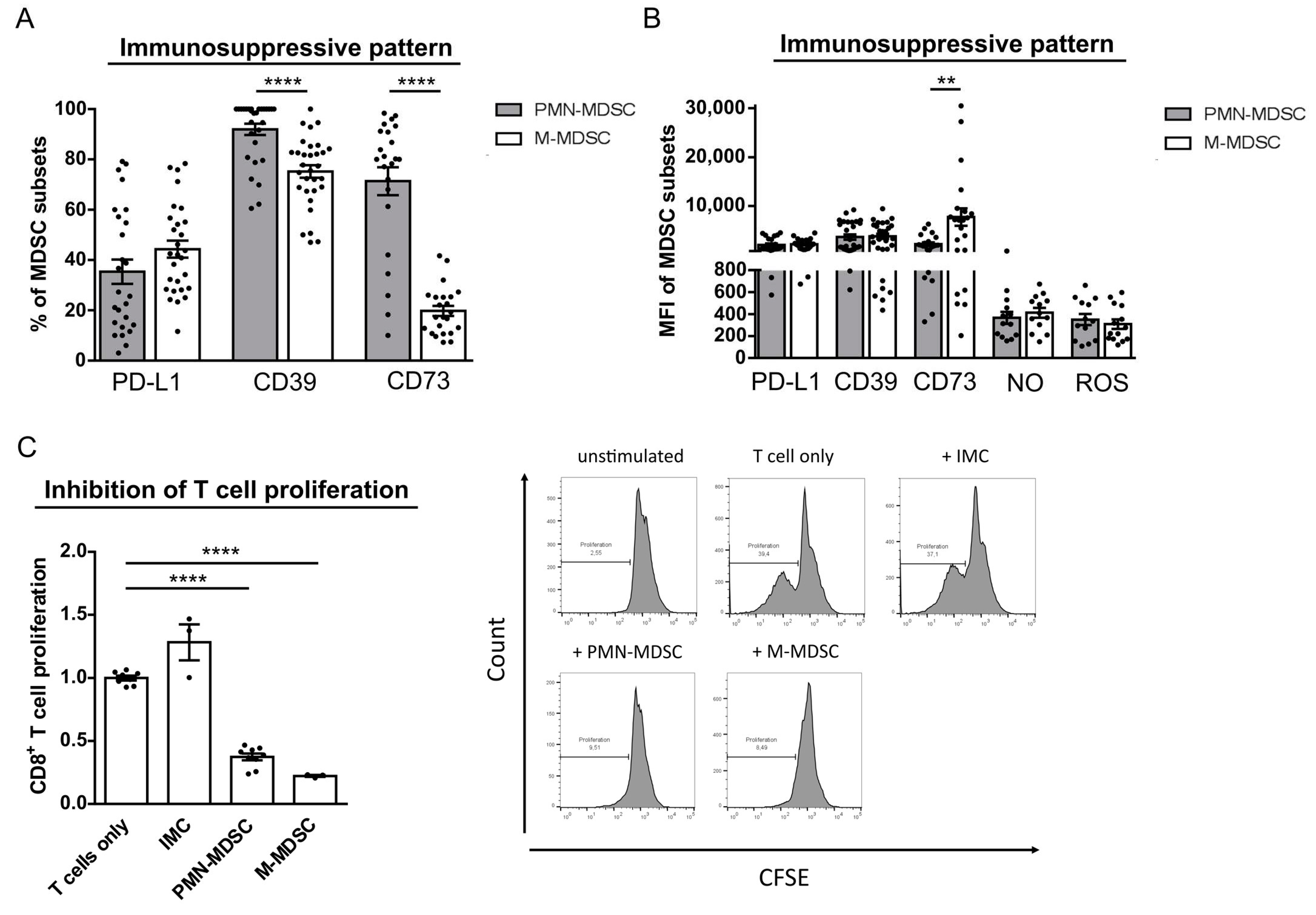
2.2. M-MDSC and PMN-MDSC Display a Comparable Immunosuppressive Phenotype in Primary Melanomas
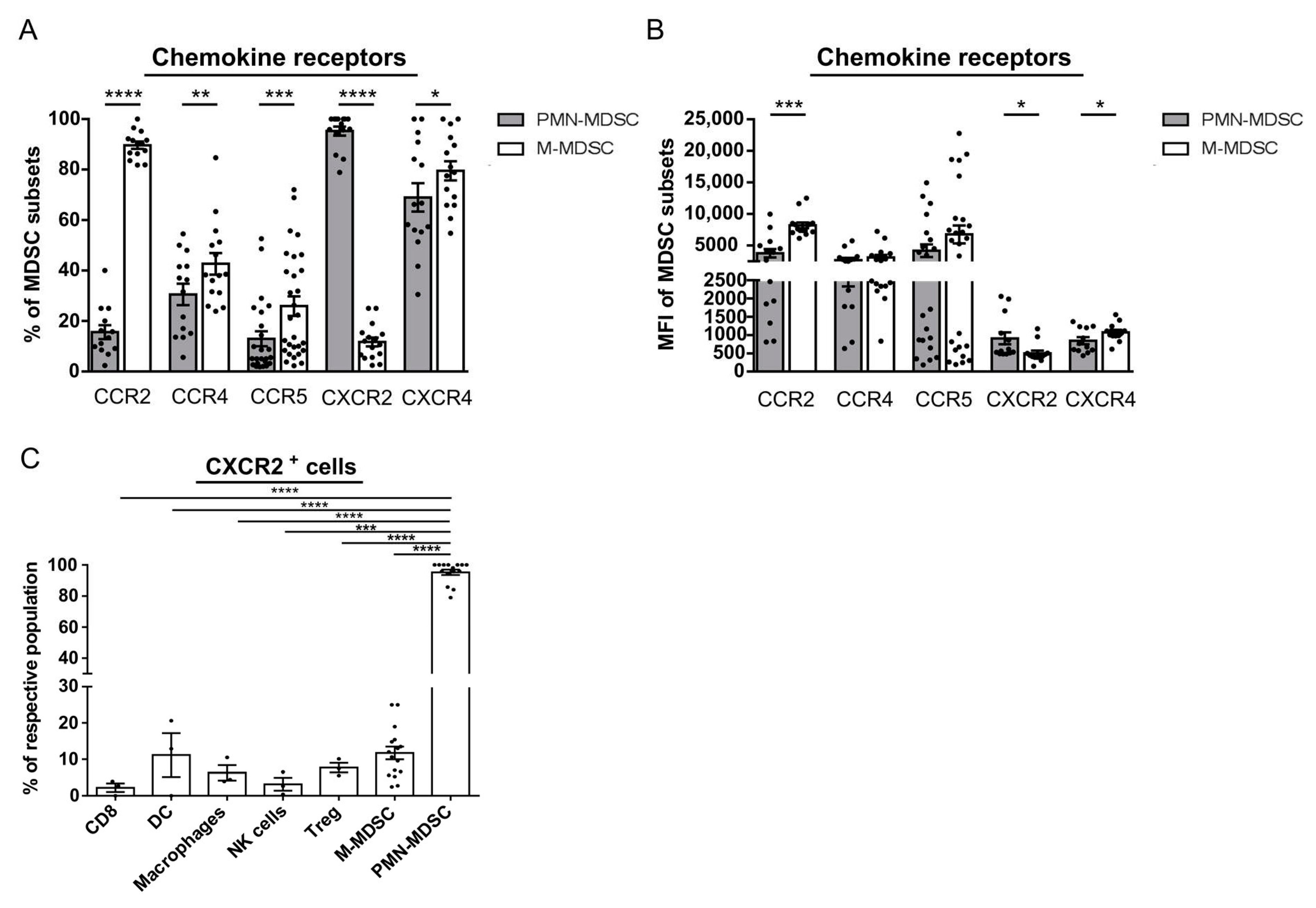
2.3. PMN-MDSC Selectively Express CXCR2
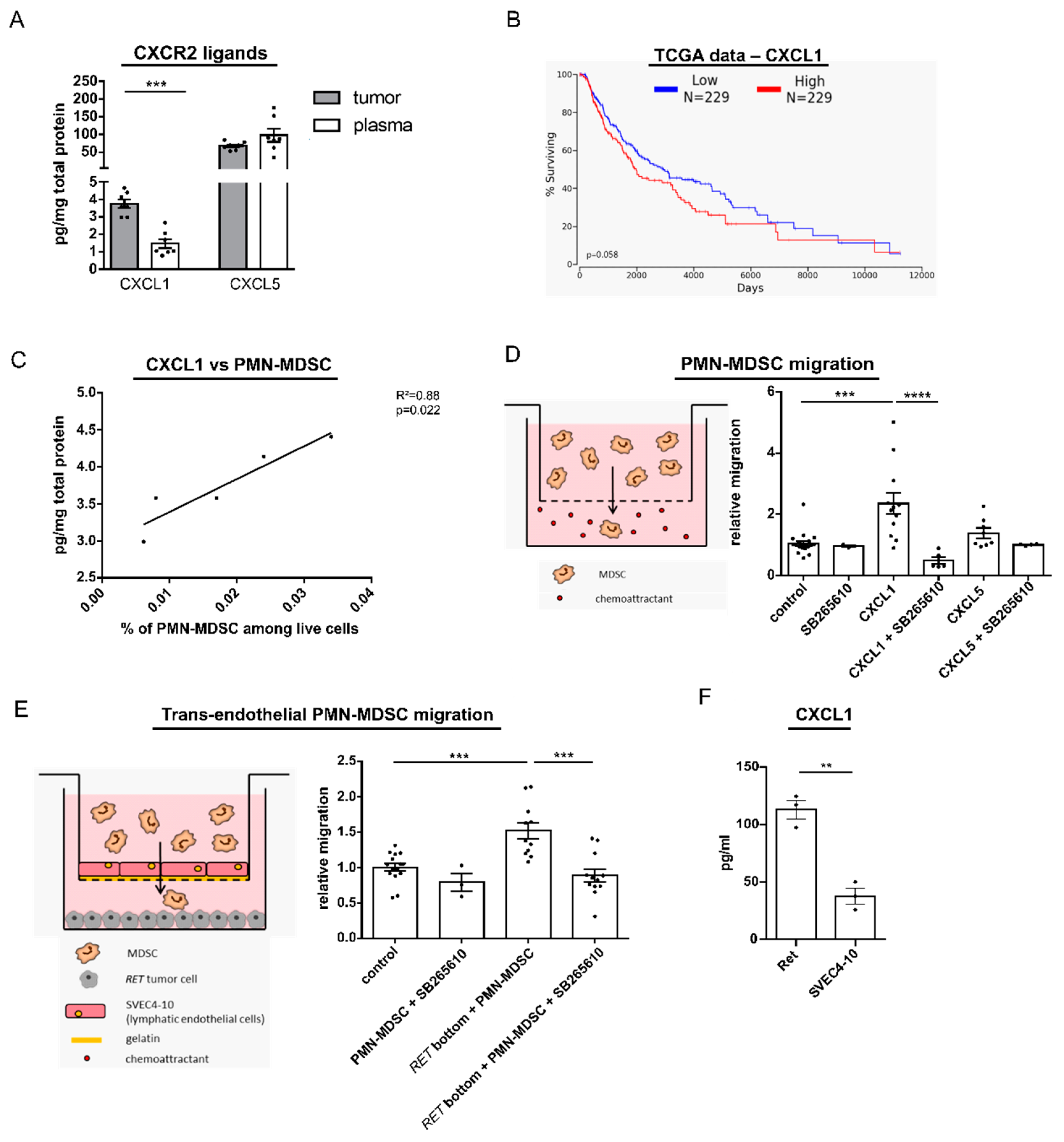
2.4. CXCL1 Mediates Migration of PMN-MDSC via CXCL1 In Vitro
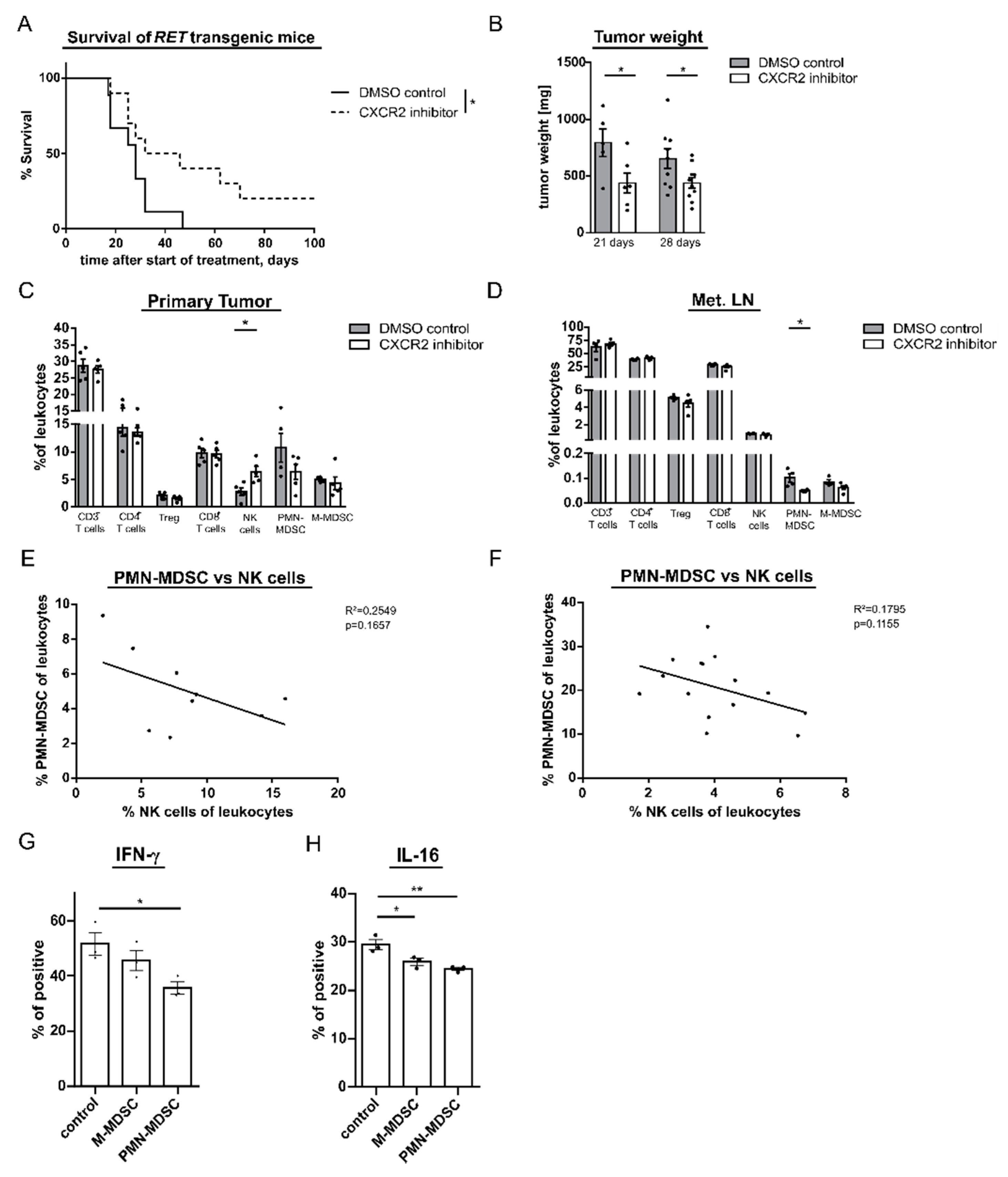
2.5. CXCR2 Inhibition Increases the Survival of Melanoma Bearing RET Transgenic Mice
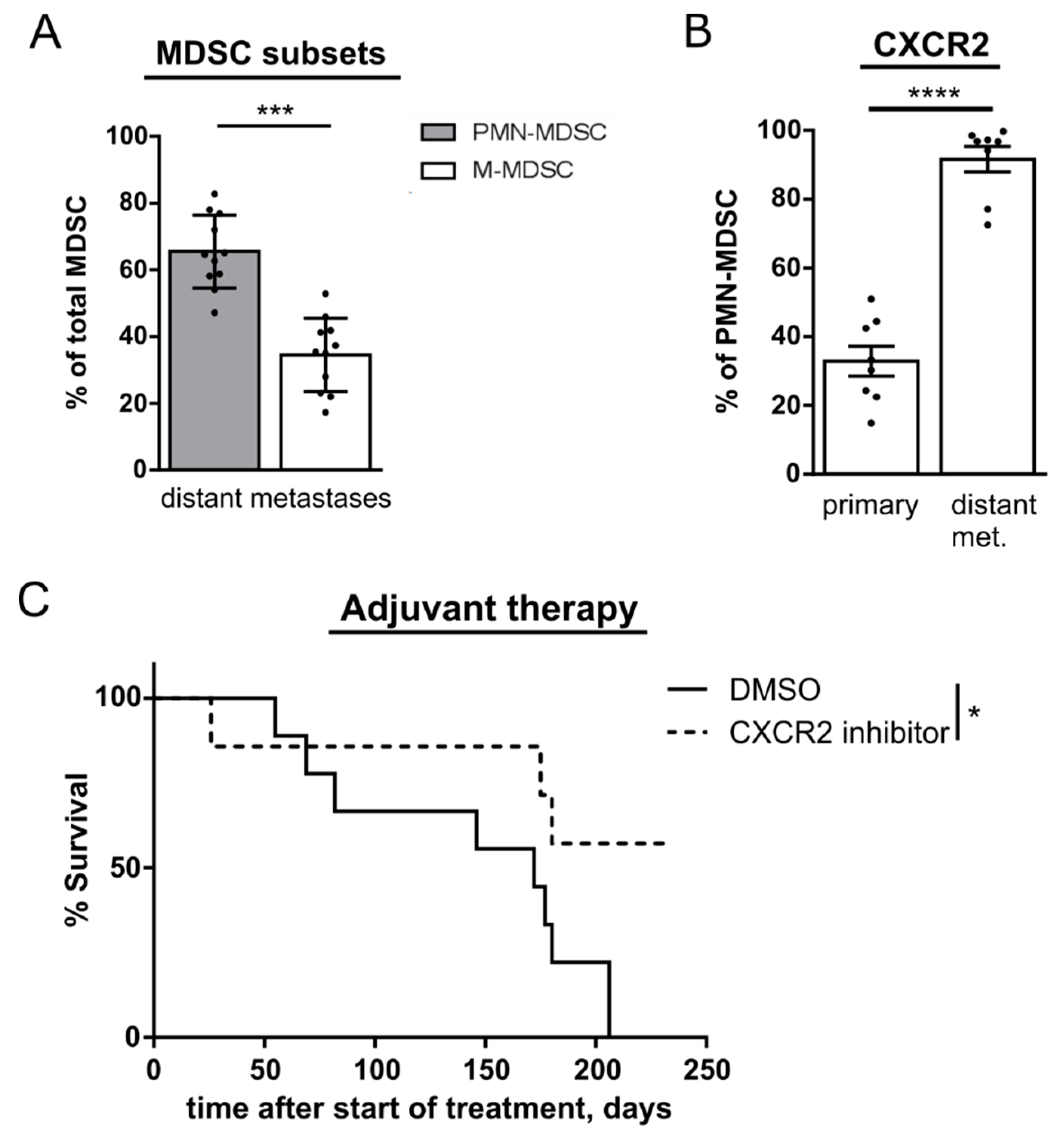
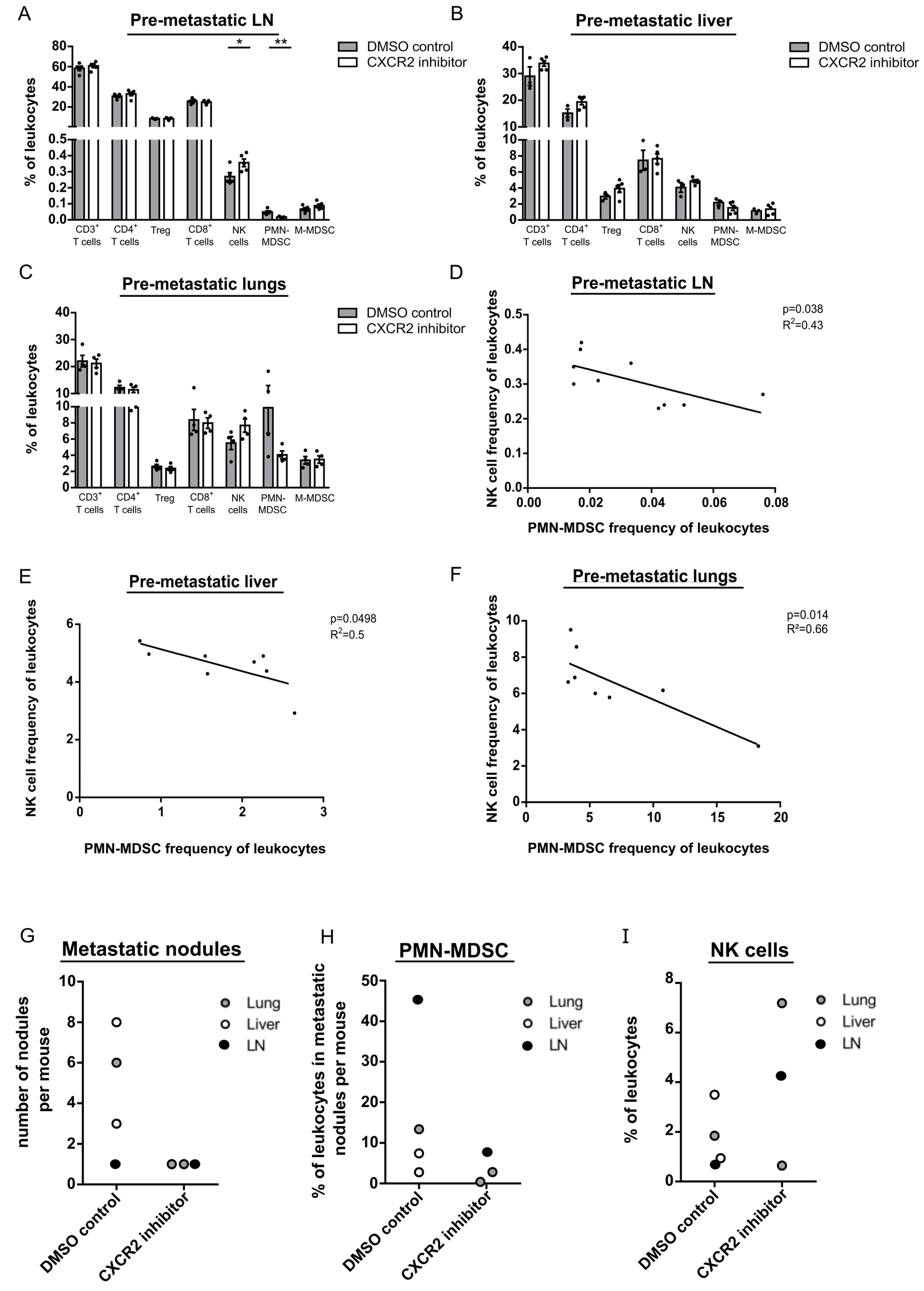
2.6. CXCR2 Inhibition Reduces Development of Metastases
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cell Culture
4.3. Isolation of Immature Myeloid Cells and MDSC Subsets
4.4. Flow Cytometry
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Bio-Plex Assay
4.7. Inhibition of T Cell Proliferation Assay
4.8. In Vitro Migration of MDSC Subsets from RET Transgenic Mice
4.9. Co-Culture of MDSC Subsets with NK Cells
4.10. Transplantation of AB12 Mesothelioma Cells
4.11. Therapy of Melanoma Bearing Mice
4.12. Therapy of Mesothelioma-Bearing Mice
4.13. Transplantation of Primary Skin Melanomas and Adjuvant Therapy
4.14. TCGA Data
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Spatz, A.; Robert, C. Cutaneous melanoma. Lancet 2014, 383, 816–827. [Google Scholar] [CrossRef]
- Balch, C.M.; Buzaid, A.C.; Soong, S.J.; Atkins, M.B.; Cascinelli, N.; Coit, D.G.; Fleming, I.D.; Gershenwald, J.E.; Houghton, A.J.; Kirkwood, J.M.; et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J. Clin. Oncol. 2001, 19, 3635–3648. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Domingues, B.; Lopes, J.; Soares, P.; Populo, H. Melanoma treatment in review. ImmunoTargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Lanoy, E.; Besse, B. One or Two Immune Checkpoint Inhibitors? Cancer Cell 2019, 36, 579–581. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Solito, S.; Marigo, I.; Pinton, L.; Damuzzo, V.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N. Y. Acad. Sci. 2014, 1319, 47–65. [Google Scholar] [CrossRef]
- Parker, K.H.; Beury, D.W.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells: Critical Cells Driving Immune Suppression in the Tumor Microenvironment. Adv. Cancer Res. 2015, 128, 95–139. [Google Scholar] [CrossRef] [Green Version]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2018, 120, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Ballbach, M.; Dannert, A.; Singh, A.; Siegmund, D.M.; Handgretinger, R.; Piali, L.; Rieber, N.; Hartl, D. Expression of checkpoint molecules on myeloid-derived suppressor cells. Immunol. Lett. 2017, 192, 1–6. [Google Scholar] [CrossRef]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Cheng, X.; Zhang, L.; Lu, X.; Chaudhary, S.; Teng, R.; Frederickson, C.; Champion, M.M.; Zhao, R.; Cheng, L.; et al. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc. Natl. Acad. Sci. USA 2018, 115, 10094–10099. [Google Scholar] [CrossRef] [Green Version]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.Y.; Ma, G.; Weber, K.J.; Ozao-Choy, J.; Wang, G.; Yin, B.; Divino, C.M.; Chen, S.H. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010, 70, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Ma, X.L.; Wei, Y.Q.; Wei, X.W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Obermajer, N.; Muthuswamy, R.; Odunsi, K.; Edwards, R.P.; Kalinski, P. PGE2-Induced CXCL12 Production and CXCR4 Expression Controls the Accumulation of Human MDSCs in Ovarian Cancer Environment. Cancer Res. 2011, 71, 7463–7470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trovato, R.; Canè, S.; Petrova, V.; Sartoris, S.; Ugel, S.; De Sanctis, F. The Engagement Between MDSCs and Metastases: Partners in Crime. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Toh, B.; Wang, X.; Keeble, J.; Sim, W.J.; Khoo, K.; Wong, W.C.; Kato, M.; Prevost-Blondel, A.; Thiery, J.P.; Abastado, J.P. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011, 9, e1001162. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.; Prekeris, R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front. Cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.H.; Pickup, M.; Pang, Y.; Gorska, A.E.; Li, Z.; Chytil, A.; Geng, Y.; Gray, J.W.; Moses, H.L.; Yang, L. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010, 70, 6139–6149. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Takahashi, M.; Akhand, A.A.; Liu, W.; Dai, Y.; Shimizu, S.; Iwamoto, T.; Suzuki, H.; Nakashima, I. Transgenic mouse model for skin malignant melanoma. Oncogene 1998, 17, 1885–1888. [Google Scholar] [CrossRef]
- Abschuetz, O.; Osen, W.; Frank, K.; Kato, M.; Schadendorf, D.; Umansky, V. T-cell mediated immune responses induced in ret transgenic mouse model of malignant melanoma. Cancers 2012, 4, 490–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, C.; Sevko, A.; Ramacher, M.; Bazhin, A.V.; Falk, C.S.; Osen, W.; Borrello, I.; Kato, M.; Schadendorf, D.; Baniyash, M.; et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc. Natl. Acad. Sci. USA 2011, 108, 17111–17116. [Google Scholar] [CrossRef] [Green Version]
- Blattner, C.; Fleming, V.; Weber, R.; Himmelhan, B.; Altevogt, P.; Gebhardt, C.; Schulze, T.J.; Razon, H.; Hawila, E.; Wildbaum, G.; et al. CCR5+ myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res. 2018, 78, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Liu, L.; Chiu, M.S.; Cheung, K.-W.; Yan, C.W.; Yu, Z.; Lee, B.K.; Liu, W.; Man, K.; Chen, Z. Virotherapy-recruited PMN-MDSC infiltration of mesothelioma blocks antitumor CTL by IL-10-mediated dendritic cell suppression. Oncoimmunology 2019, 8, e1518672. [Google Scholar] [CrossRef] [Green Version]
- Salazar, N.; Zabel, B.A. Support of tumor endothelial cells by chemokine receptors. Front. Immunol. 2019, 10, 147. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Metzger, P.; Kirchleitner, S.V.; Kluge, M.; Koenig, L.M.; Hörth, C.; Rambuscheck, C.A.; Böhmer, D.; Ahlfeld, J.; Kobold, S.; Friedel, C.C.; et al. Immunostimulatory RNA leads to functional reprogramming of myeloid-derived suppressor cells in pancreatic cancer. J. Immunother. Cancer 2019, 7, 288. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.-I.; Kumar, V.; Collazo, M.; Nefedova, Y.; Condamine, T.; Cheng, P.; Villagra, A.; Antonia, S.; McCaffrey, J.C.; Fishman, M.; et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat. Immunol. 2013, 14, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Mastio, J.; Condamine, T.; Dominguez, G.; Kossenkov, A.V.; Donthireddy, L.; Veglia, F.; Lin, C.; Wang, F.; Fu, S.; Zhou, J.; et al. Identification of monocyte-like precursors of granulocytes in cancer as a mechanism for accumulation of PMN-MDSCs. J. Exp. Med. 2019, 216, 2150–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitkovsky, M.V.; Hatfield, S.; Abbott, R.; Belikoff, B.; Lukashev, D.; Ohta, A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol. Res. 2014, 2, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Xu, X.; Qiao, M.; Li, X.; Zhao, C.; Zhou, F.; Gao, G.; Wu, F.; Chen, X.; Su, C.; et al. Comprehensive evaluation of NT5E/CD73 expression and its prognostic significance in distinct types of cancers. BMC Cancer 2018, 18, 267. [Google Scholar] [CrossRef] [Green Version]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Limagne, E.; Richard, C.; Thibaudin, M.; Fumet, J.D.; Truntzer, C.; Lagrange, A.; Favier, L.; Coudert, B.; Ghiringhelli, F. Tim-3/galectin-9 pathway and mMDSC control primary and secondary resistances to PD-1 blockade in lung cancer patients. Oncoimmunology 2019, 8, e1564505. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2+ myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef] [Green Version]
- Hawila, E.; Razon, H.; Wildbaum, G.; Blattner, C.; Sapir, Y.; Shaked, Y.; Umansky, V.; Karin, N. CCR5 Directs the Mobilization of CD11b +Gr1 +Ly6Clow Polymorphonuclear Myeloid Cells from the Bone Marrow to the Blood to Support Tumor Development. Cell Rep. 2017, 21, 2212–2222. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Clavijo, P.E.; Robbins, Y.; Patel, P.; Friedman, J.; Greene, S.; Das, R.; Silvin, C.; Van Waes, C.; Horn, L.A.; et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 2019, 4, e126853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Cheng, J.; Fu, B.; Liu, W.; Chen, G.; Zhang, Q.; Yang, Y. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene 2017, 36, 1090–1101. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front. Immunol. 2019, 10, 771. [Google Scholar] [CrossRef]
- Pedersen, L.; Idorn, M.; Olofsson, G.H.; Lauenborg, B.; Nookaew, I.; Hansen, R.H.; Johannesen, H.H.; Becker, J.C.; Pedersen, K.S.; Dethlefsen, C.; et al. Voluntary Running Suppresses Tumor Growth through Epinephrine- and IL-6-Dependent NK Cell Mobilization and Redistribution. Cell Metab. 2016, 23, 554–562. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.I.; Park, S.M.; Park, S.; Kim, G.; Lee, H.J.; Son, J.; Hong, M.H.; Ghaderpour, A.; Baik, B.; Islam, J.; et al. Peripheral natural killer cells and myeloid-derived suppressor cells correlate with anti-PD-1 responses in non-small cell lung cancer. Sci. Rep. 2020, 10, 9050. [Google Scholar] [CrossRef]
- Chen, J.Y.; Lai, Y.S.; Chu, P.Y.; Chan, S.H.; Wang, L.H.; Hung, W.C. Cancer-Derived VEGF-C Increases Chemokine Production in Lymphatic Endothelial Cells to Promote CXCR2-Dependent Cancer Invasion and MDSC Recruitment. Cancers 2019, 11, 1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Falk, C.; Osen, W.; Kato, M.; Schadendorf, D.; Umansky, V. Activation of p38 mitogen-activated protein kinase drives dendritic cells to become tolerogenic in ret transgenic mice spontaneously developing melanoma. Clin. Cancer Res. 2009, 15, 4382–4390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, K.A.; Edidin, M. A mouse lymphoid endothelial cell line immortalized by simian virus 40 binds lymphocytes and retains functional characteristics of normal endothelial cells. J. Immunol. 1990, 144, 521–525. [Google Scholar]
- Marigo, I.; Bosio, E.; Solito, S.; Mesa, C.; Fernandez, A.; Dolcetti, L.; Ugel, S.; Sonda, N.; Bicciato, S.; Falisi, E.; et al. Tumor-Induced Tolerance and Immune Suppression Depend on the C/EBPβ Transcription Factor. Immunity 2010, 32, 790–802. [Google Scholar] [CrossRef]
- Gengenbacher, N.; Singhal, M.; Mogler, C.; Hai, L.; Milde, L.; Abdul Pari, A.A.; Besemfelder, E.; Fricke, C.; Baumann, D.; Gehrs, S.; et al. Timed Ang2-targeted therapy identifies the Angiopoietin-Tie pathway as key regulator of fatal lymphogenous metastasis. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Anaya, J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci. 2016, 2, e67. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groth, C.; Arpinati, L.; Shaul, M.E.; Winkler, N.; Diester, K.; Gengenbacher, N.; Weber, R.; Arkhypov, I.; Lasser, S.; Petrova, V.; et al. Blocking Migration of Polymorphonuclear Myeloid-Derived Suppressor Cells Inhibits Mouse Melanoma Progression. Cancers 2021, 13, 726. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13040726
Groth C, Arpinati L, Shaul ME, Winkler N, Diester K, Gengenbacher N, Weber R, Arkhypov I, Lasser S, Petrova V, et al. Blocking Migration of Polymorphonuclear Myeloid-Derived Suppressor Cells Inhibits Mouse Melanoma Progression. Cancers. 2021; 13(4):726. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13040726
Chicago/Turabian StyleGroth, Christopher, Ludovica Arpinati, Merav E. Shaul, Nina Winkler, Klara Diester, Nicolas Gengenbacher, Rebekka Weber, Ihor Arkhypov, Samantha Lasser, Vera Petrova, and et al. 2021. "Blocking Migration of Polymorphonuclear Myeloid-Derived Suppressor Cells Inhibits Mouse Melanoma Progression" Cancers 13, no. 4: 726. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13040726