
The Extracellular Small Leucine-Rich Proteoglycan Biglycan Is a Key Player in Gastric Cancer Aggressiveness

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Gastric Cancer Tissue Expression Analysis: Functional Annotation and Correlation Profiles
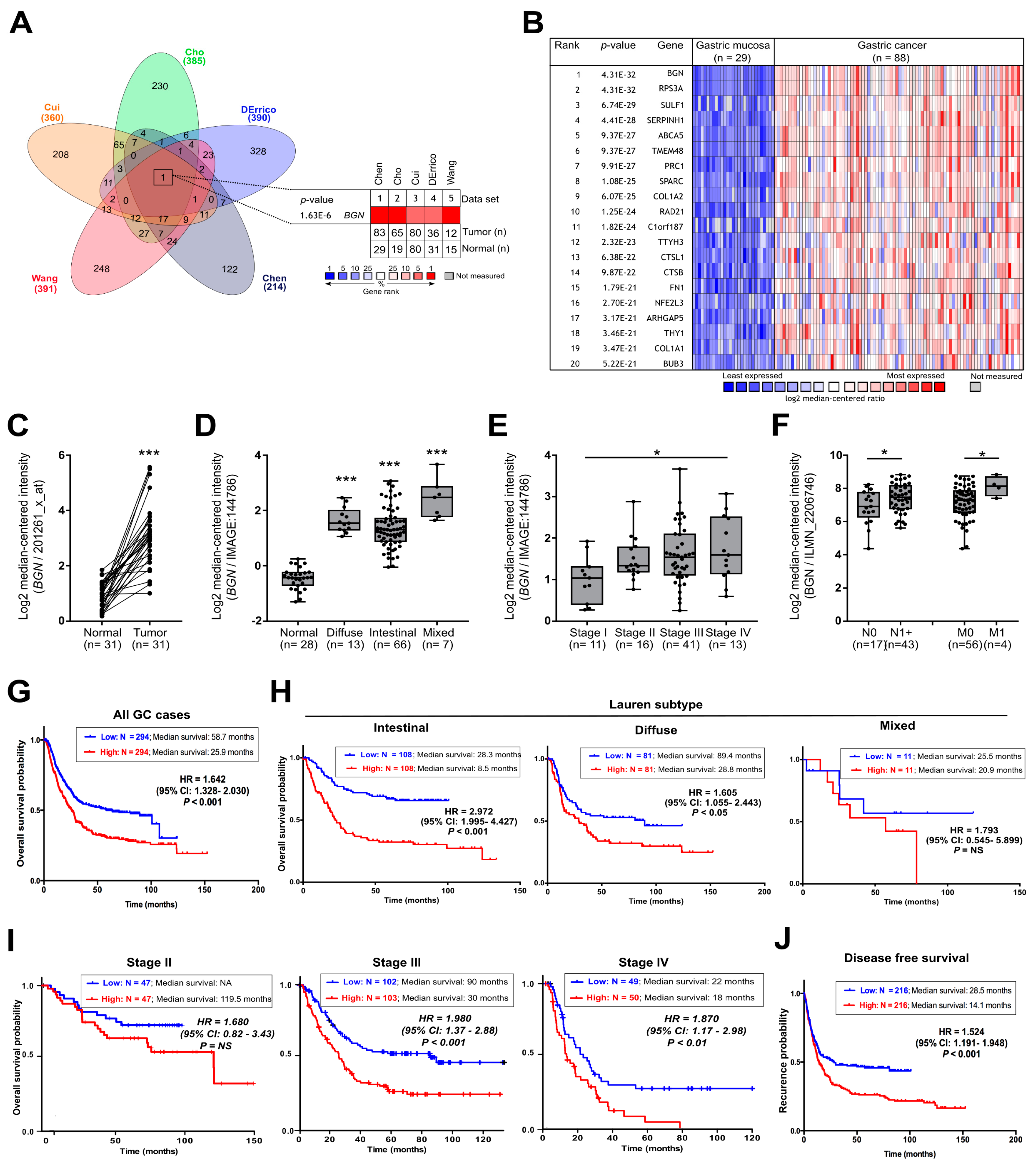
2.2. Clinical and Prognostic Impact of Biglycan in Gastric Cancer
2.3. Cell Culture
2.4. Quantitative Reverse Transcriptase PCR (qPCR)
2.5. Biglycan CRISPR/Cas9 Knock-Out
2.6. Total Protein Extraction and Secretome Protein Enrichment
2.7. Enzymatic Chondroitinase ABC Deglycosylation Digestion
2.8. Western Blot
2.9. Immunofluorescence
2.10. Trypan Blue Exclusion Assay
2.11. Colony Formation Assay
2.12. Wound-Healing Migration Assay
2.13. In Vivo Angiogenic Chicken Embryo Choriallantoic Membrane (CAM) Assay
2.14. Immunohsitochemical Analysis
2.15. Statistical Analysis
3. Results
3.1. BGN Is Commonly Over-Expressed in GC and Predict Poor Prognosis in Advanced GC Patients
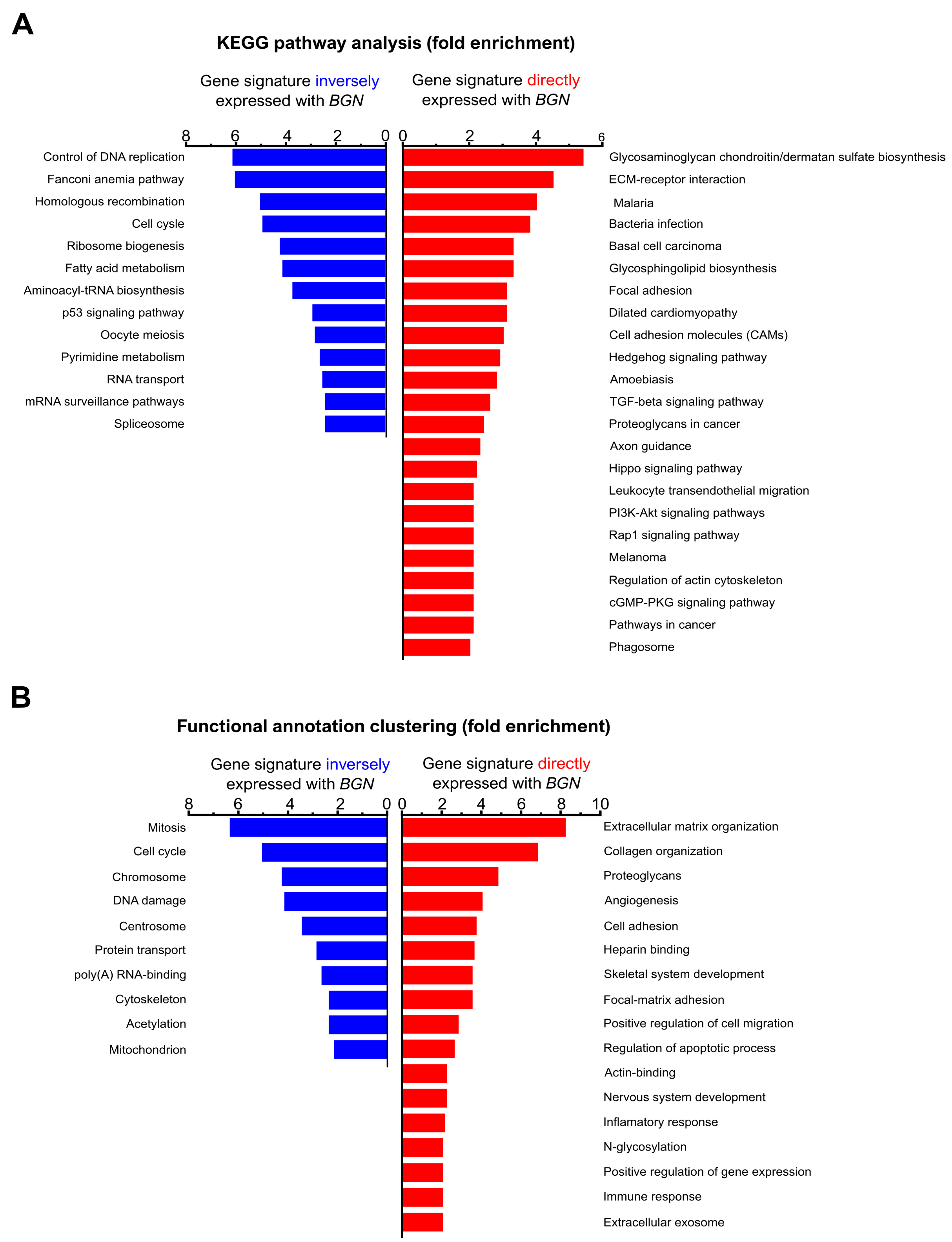
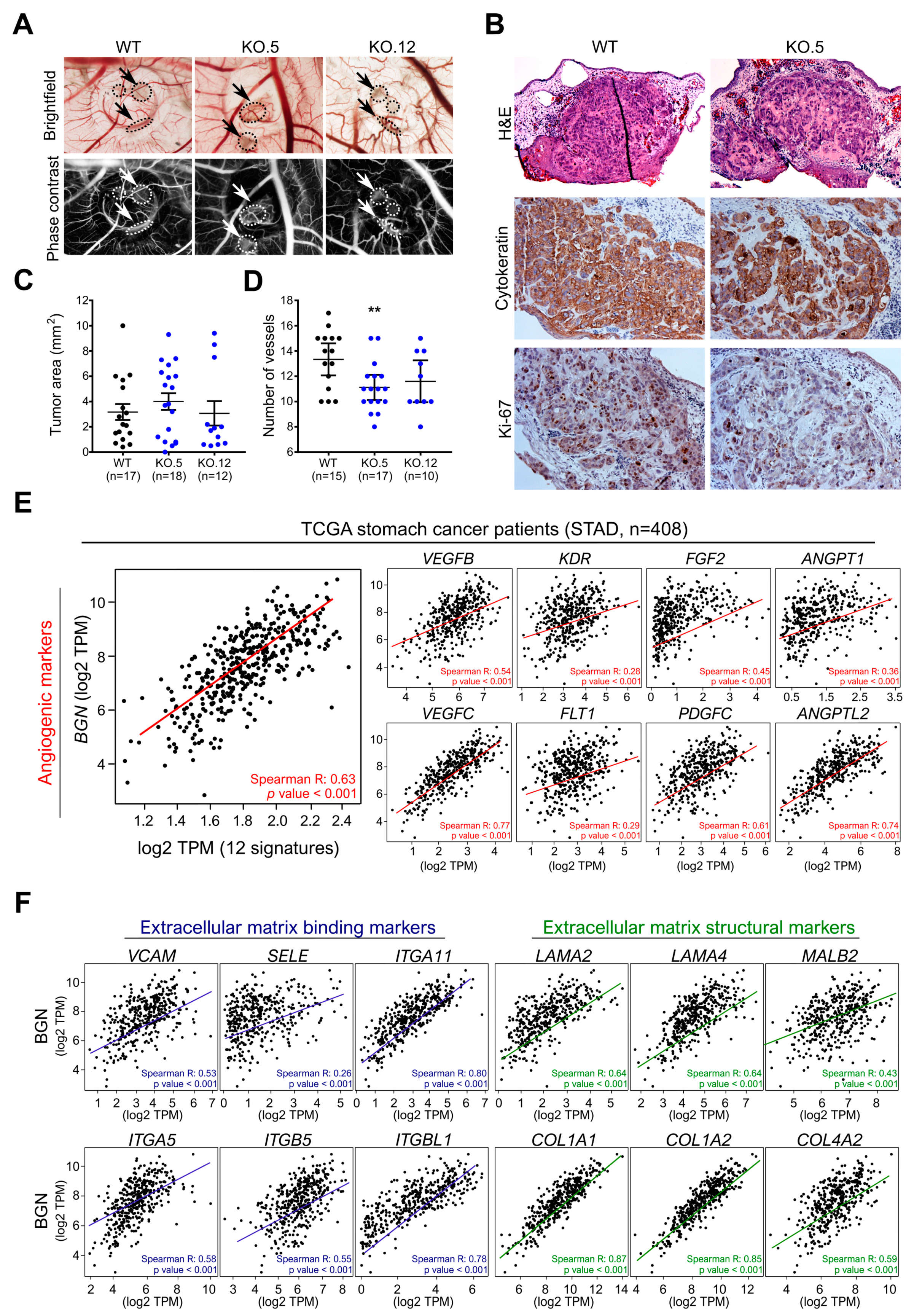
3.2. BGN Gene Profiling in GC Patients: Functional Annotation
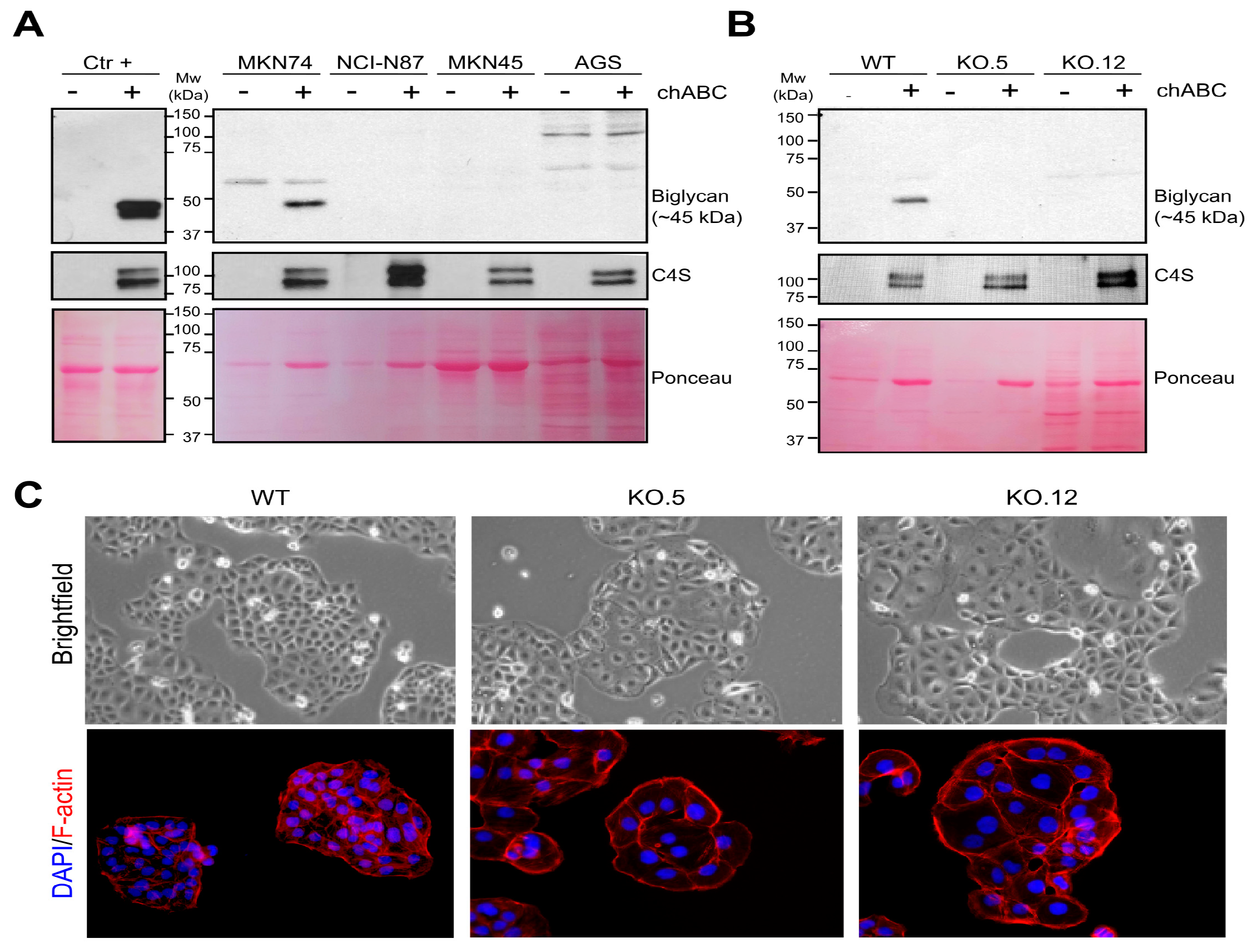
3.3. GC Cell Line Characterization of Secreted Biglycan and Establishment of Biglycan Knock Out Cell Models
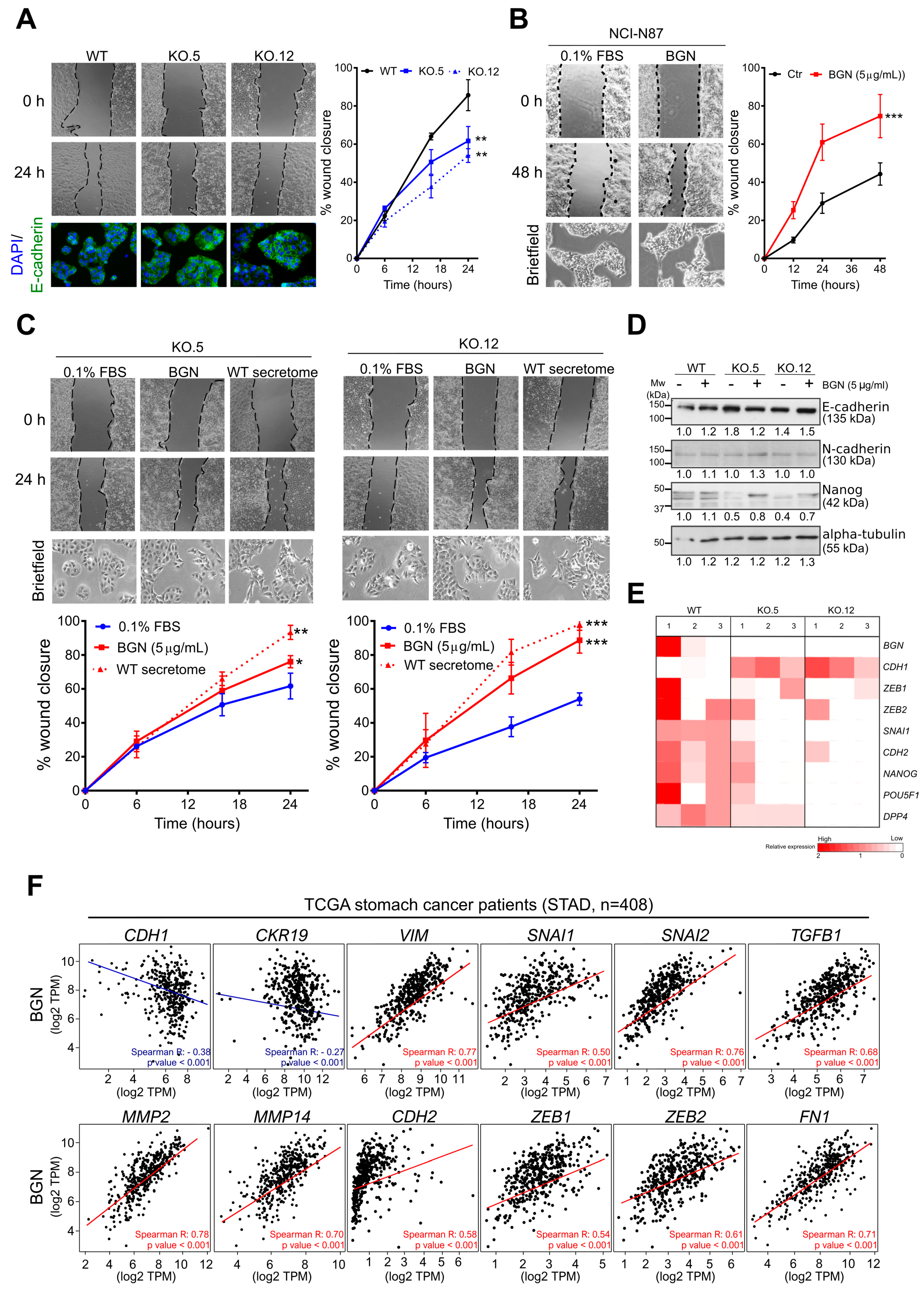
3.4. Biglycan Modulates GC Cell Migration through the Regulation of Epithelial-to-Mesenchymal (EMT) Markers
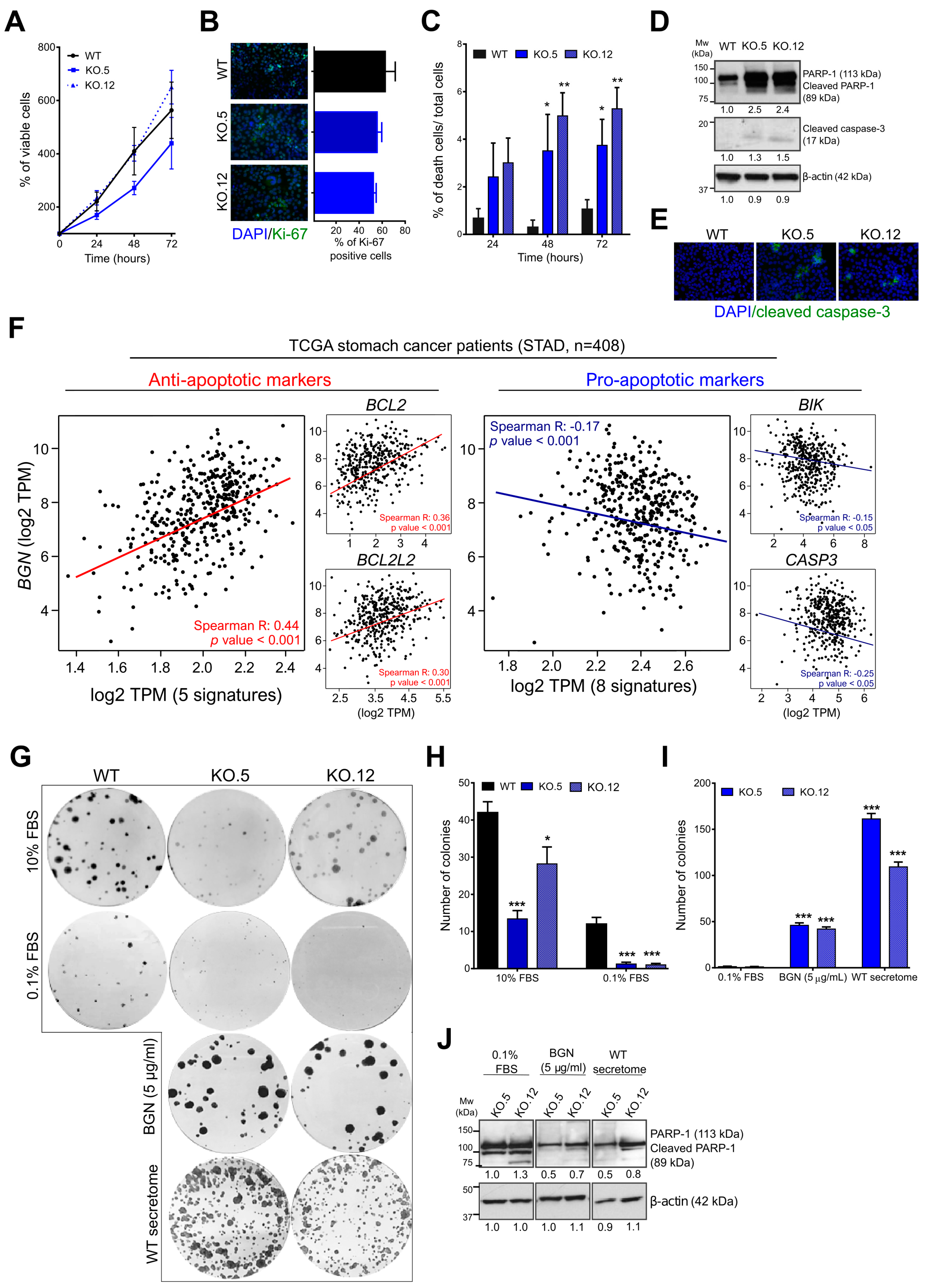
3.5. Loss of Biglycan Promotes Cell Death by Apoptosis, and BGN Expression Interferes with Anti- and Pro-Apoptotic Gene Regulation in GC Patients
3.6. Exogenous Biglycan Protein Stimuli is Able to Restore GC Cell Survival Capacity
3.7. In Vivo Biglycan Angiogenic Potential of Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Sagaert, X.; Topal, B.; Haustermans, K.; Prenen, H. Gastric Cancer. Lancet 2016, 388, 2654–2664. [Google Scholar] [CrossRef]
- Matsuoka, T.; Yashiro, M. Biomarkers of Gastric Cancer: Current Topics and Future Perspective. World J. Gastroenterol. 2018, 24, 2818–2832. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Reis, C.A. Glycosylation in Cancer: Mechanisms and Clinical Implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Reis, C.A.; Osorio, H.; Silva, L.; Gomes, C.; David, L. Alterations in glycosylation as biomarkers for cancer detection. J. Clin. Pathol. 2010, 63, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Orditura, M. Treatment of Gastric Cancer. World J. Gastroenterol. 2014, 20, 1635. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.-M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive Molecular Characterization of Clinical Responses to PD-1 Inhibition in Metastatic Gastric Cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Gerarduzzi, C.; Hartmann, U.; Leask, A.; Drobetsky, E. The Matrix Revolution: Matricellular Proteins and Restructuring of the Cancer Microenvironment. Cancer Res. 2020, 80, 2705–2717. [Google Scholar] [CrossRef] [Green Version]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Faria-Ramos, I.; Poças, J.; Marques, C.; Santos-Antunes, J.; Macedo, G.; Reis, A.C.; Magalhães, A. Heparan Sulfate Glycosaminoglycans: (Un)Expected Allies in Cancer Clinical Management. Biomolecules 2021, 11, 136. [Google Scholar] [CrossRef]
- Li, W.; Ng, J.M.-K.; Wong, C.C.; Ng, E.K.W.; Yu, J. Molecular Alterations of Cancer Cell and Tumour Microenvironment in Metastatic Gastric Cancer. Oncogene 2018, 37, 4903–4920. [Google Scholar] [CrossRef]
- Moreira, A.M.; Pereira, J.; Melo, S.; Fernandes, M.S.; Carneiro, P.; Seruca, R.; Figueiredo, J. The Extracellular Matrix: An Accomplice in Gastric Cancer Development and Progression. Cells 2020, 9, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbaszadegan, M.R.; Mojarrad, M.; Moghbeli, M. Role of Extra Cellular Proteins in Gastric Cancer Progression and Metastasis: An Update. Genes Environ. 2020, 42, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, X.; Hu, X.; Jiang, F.; Shen, Y.; Xu, R.; Wu, L.; Wei, P.; Shen, X. LUM Expression and Its Prognostic Significance in Gastric Cancer. Front. Oncol. 2020, 10, 605. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Han, F.; Fu, M.; Wang, Z. High Expression of VCAN Is an Independent Predictor of Poor Prognosis in Gastric Cancer. J. Int. Med. Res. 2020, 48, 030006051989127. [Google Scholar] [CrossRef]
- Bianco, P.; Fisher, L.W.; Young, M.F.; Termine, J.D.; Robey, P.G. Expression and Localization of the Two Small Proteoglycans Biglycan and Decorin in Developing Human Skeletal and Non-Skeletal Tissues. J. Histochem. Cytochem. 1990, 38, 1549–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tufvesson, E.; Malmström, J.; Marko-Varga, G.; Westergren-Thorsson, G. Biglycan Isoforms with Differences in Polysaccharide Substitution and Core Protein in Human Lung Fibroblasts. Eur. J. Biochem. 2002, 269, 3688–3696. [Google Scholar] [CrossRef]
- Nastase, M.V.; Young, M.F.; Schaefer, L. Biglycan: A Multivalent Proteoglycan Providing Structure and Signals. J. Histochem. Cytochem. 2012, 60, 963–975. [Google Scholar] [CrossRef]
- Schulz, G.B.; Grimm, T.; Sers, C.; Riemer, P.; Elmasry, M.; Kirchner, T.; Stief, C.G.; Karl, A.; Horst, D. Prognostic Value and Association with Epithelial-Mesenchymal Transition and Molecular Subtypes of the Proteoglycan Biglycan in Advanced Bladder Cancer. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 530.e9–530.e18. [Google Scholar] [CrossRef] [PubMed]
- Niedworok, C.; Röck, K.; Kretschmer, I.; Freudenberger, T.; Nagy, N.; Szarvas, T.; vom Dorp, F.; Reis, H.; Rübben, H.; Fischer, J.W. Inhibitory Role of the Small Leucine-Rich Proteoglycan Biglycan in Bladder Cancer. PLoS ONE 2013, 8, e80084. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Ma, Y.; Xiao, J.; Zheng, H.; Song, C.; Gong, Y.; Xing, X. Up-Regulated Biglycan Expression Correlates with the Malignancy in Human Colorectal Cancers. Clin. Exp. Med. 2012, 12, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, X.; Zhang, Y.; Che, X.; Liu, Z.; Zhang, L.; Qiu, C.; Lv, Q.; Jiang, J. Biglycan Enhances the Ability of Migration and Invasion in Endometrial Cancer. Arch. Gynecol. Obstet. 2016, 293, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Gu, X.; Ma, T.; Ye, H. Biglycan Up-Regulated Vascular Endothelial Growth Factor (VEGF) Expression and Promoted Angiogenesis in Colon Cancer. Tumor Biol. 2015, 36, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Andrlová, H.; Mastroianni, J.; Madl, J.; Kern, J.S.; Melchinger, W.; Dierbach, H.; Wernet, F.; Follo, M.; Technau-Hafsi, K.; Has, C.; et al. Biglycan Expression in the Melanoma Microenvironment Promotes Invasiveness via Increased Tissue Stiffness Inducing Integrin-Β1 Expression. Oncotarget 2017, 8, 42901–42916. [Google Scholar] [CrossRef] [Green Version]
- Maishi, N.; Ohba, Y.; Akiyama, K.; Ohga, N.; Hamada, J.; Nagao-Kitamoto, H.; Alam, M.T.; Yamamoto, K.; Kawamoto, T.; Inoue, N.; et al. Tumour Endothelial Cells in High Metastatic Tumours Promote Metastasis via Epigenetic Dysregulation of Biglycan. Sci. Rep. 2016, 6, 28039. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ohga, N.; Hida, Y.; Maishi, N.; Kawamoto, T.; Kitayama, K.; Akiyama, K.; Osawa, T.; Kondoh, M.; Matsuda, K.; et al. Biglycan Is a Specific Marker and an Autocrine Angiogenic Factor of Tumour Endothelial Cells. Br. J. Cancer 2012, 106, 1214–1223. [Google Scholar] [CrossRef]
- Wang, B.; Li, G.-X.; Zhang, S.-G.; Wang, Q.; Wen, Y.-G.; Tang, H.-M.; Zhou, C.-Z.; Xing, A.-Y.; Fan, J.-W.; Yan, D.-W.; et al. Biglycan Expression Correlates with Aggressiveness and Poor Prognosis of Gastric Cancer. Exp. Biol. Med. 2011, 236, 1247–1253. [Google Scholar] [CrossRef]
- Hu, L.; Duan, Y.; Li, J.; Su, L.; Yan, M.; Zhu, Z.; Liu, B.; Yang, Q. Biglycan Enhances Gastric Cancer Invasion by Activating FAK Signaling Pathway. Oncotarget 2014, 5, 1885–1896. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Zang, M.; Wang, H.; Li, J.; Su, L.; Yan, M.; Li, C.; Yang, Q.; Liu, B.; Zhu, Z. Biglycan Stimulates VEGF Expression in Endothelial Cells by Activating the TLR Signaling Pathway. Mol. Oncol. 2016, 10, 1473–1484. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Yoshida, E.; Shinkai, Y.; Yamamoto, C.; Fujiwara, Y.; Kumagai, Y.; Kaji, T. Biglycan Intensifies ALK5-Smad2/3 Signaling by TGF-β1 and Downregulates Syndecan-4 in Cultured Vascular Endothelial Cells. J. Cell. Biochem. 2017, 118, 1087–1096. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pander, A.; Chinnaiyan, A.M. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Pinto, F.; Costa, Â.M.; Andrade, R.P.; Reis, R.M. Brachyury Is Associated with Glioma Differentiation and Response to Temozolomide. Neurotherapeutics 2020. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Á.; Lánczky, A.; Menyhárt, O.; Győrffy, B. Validation of MiRNA Prognostic Power in Hepatocellular Carcinoma Using Expression Data of Independent Datasets. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Lonowski, L.; Narimatsu, Y.; Riaz, A.; Delay, C.E.; Yang, Z.; Niola, F.; Katarzyna, D.; Over, E.A.; Clausen, H.; Wandall, H.H.; et al. Genome editing using FACS enrichment of nuclease-expressing cells and indel detection by amplicon analysis. Nat. Protoc. 2017, 12, 581–603. [Google Scholar] [CrossRef]
- Gomes, C.; Almeida, A.; Barreira, A.; Calheiros, J.; Pinto, F.; Abrantes, R.; Costa, A.; Polonia, A.; Campos, D.; Osório, H.; et al. Carcinoembryonic Antigen Carrying SLeX as a New Biomarker of More Aggressive Gastric Carcinomas. Theranostics 2019, 9, 7431–7446. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Arao, T.; Tamura, D.; Aomatsu, K.; Furuta, K.; Matsumoto, K.; Kaneda, H.; Kudo, K.; Fujita, Y.; Kimura, H.; et al. SRPX2 Is a Novel Chondroitin Sulfate Proteoglycan That Is Overexpressed in Gastrointestinal Cancer. PLoS ONE 2012, 7, e27922. [Google Scholar] [CrossRef] [Green Version]
- Pinto, F.; Pértega-Gomes, N.; Pereira, M.S.; Vizcaíno, J.R.; Monteiro, P.; Henrique, R.M.; Baltazar, F.; Andrade, R.P.; Reis, R.M. T-Box Transcription Factor Brachyury Is Associated with Prostate Cancer Progression and Aggressiveness. Clin. Cancer Res. 2014, 20, 4949–4961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, F.; Costa, Â.M.; Santos, G.C.; Matsushita, M.M.; Costa, S.; Silva, V.A.; Miranda-Gonçalves, V.; Lopes, C.M.; Clara, C.A.; Becker, A.P.; et al. The T-box Transcription Factor Brachyury Behaves as a Tumor Suppressor in Gliomas. J. Pathol. 2020, 251, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.; Marques, M.S.; Melo, J.; Pinto, M.T.; Cavadas, B.; Aroso, M.; Gomez-Lazaro, M.; Seruca, R.; Figueiredo, C. Helicobacter Pylori Targets the EPHA2 Receptor Tyrosine Kinase in Gastric Cells Modulating Key Cellular Functions. Cells 2020, 9, 513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, M.; Pereira, P.M.; Varanda, A.S.; Carvalho, J.; Azevedo, M.; Mateus, D.D.; Mendes, N.; Oliveira, P.; Trindade, F.; Pinto, M.T.; et al. Codon Misreading TRNAs Promote Tumor Growth in Mice. Rna Biol. 2018, 15, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Freitas, D.; Campos, D.; Gomes, J.; Pinto, F.; Macedo, J.A.; Matos, R.; Mereiter, S.; Pinto, M.T.; Polónia, A.; Gartner, F.; et al. O-Glycans Truncation Modulates Gastric Cancer Cell Signaling and Transcription Leading to a More Aggressive Phenotype. EBioMedicine 2019, 40, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, W.; Li, X.; Tai, Y.; Lü, Q.; Yang, N.; Jiang, J. Expression and significance of biglycan in endometrial cancer. Arch. Gynecol. Obstet. 2014, 289, 649–655. [Google Scholar] [CrossRef]
- Jacobsen, F.; Kraft, J.; Schroeder, C.; Hube-Magg, C.; Kluth, M.; Lang, D.S.; Simon, R.; Sauter, G.; Izbicki, J.R.; Clauditz, T.S.; et al. Up-regulation of Biglycan is Associated with Poor Prognosis and PTEN Deletion in Patients with Prostate Cancer. Neoplasia 2017, 19, 707–715. [Google Scholar] [CrossRef]
- Zhu, Y.H.; Yang, F.; Zhang, S.S.; Zeng, T.T.; Xie, X.; Guan, X.Y. High expression of biglycan is associated with poor prognosis in patients with esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2013, 6, 2497–2505. [Google Scholar]
- Yamamoto, H.; Watanabe, Y.; Sato, Y.; Maehata, T.; Itoh, F. Non-Invasive Early Molecular Detection of Gastric Cancers. Cancers 2020, 12, 2880. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Gu, X.; Ma, T. Knockdown of biglycan expression by RNA interference inhibits the proliferation and invasion of, and induces apoptosis in, the HCT116 colon cancer cell line. Mol. Med. Rep. 2015, 12, 7538–7544. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, T.; Xu, X.; Cui, Y.; Xing, X. Biglycan promotes the chemotherapy resistance of colon cancer by activating NF-κB signal transduction. Mol. Cell. Biochem. 2018, 449, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Kesh, K.; Gupta, V.K.; Durden, B.; Garrido, V.; Mateo-Victoriano, B.; Lavania, S.P.; Banerjee, S. Therapy Resistance, Cancer Stem Cells and ECM in Cancer: The Matrix Reloaded. Cancers 2020, 12, 3067. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Parameter | HR | 95% CI | p Value (Cox) |
|---|---|---|---|
| BGN | 2.219 | 2.056–2.408 | 0.007 |
| Stage II | 1.715 | 0.803–3.665 | 0.164 |
| Stage III | 2.695 | 1.331–5.458 | 0.006 |
| Stage IV | 4.613 | 1.802–11.809 | 0.001 |
| Age | 1.026 | 1.007–1.045 | 0.008 |
| Gender (male) | 1.085 | 0.743–1.585 | 0.673 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, F.; Santos-Ferreira, L.; Pinto, M.T.; Gomes, C.; Reis, C.A. The Extracellular Small Leucine-Rich Proteoglycan Biglycan Is a Key Player in Gastric Cancer Aggressiveness. Cancers 2021, 13, 1330. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061330
Pinto F, Santos-Ferreira L, Pinto MT, Gomes C, Reis CA. The Extracellular Small Leucine-Rich Proteoglycan Biglycan Is a Key Player in Gastric Cancer Aggressiveness. Cancers. 2021; 13(6):1330. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061330
Chicago/Turabian StylePinto, Filipe, Liliana Santos-Ferreira, Marta T. Pinto, Catarina Gomes, and Celso A. Reis. 2021. "The Extracellular Small Leucine-Rich Proteoglycan Biglycan Is a Key Player in Gastric Cancer Aggressiveness" Cancers 13, no. 6: 1330. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061330