
Hyperglycemia-Induced miR-467 Drives Tumor Inflammation and Growth in Breast Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Protocols
2.2. Induction of Diabetes in Mice
2.3. Injection of Cancer Cells and Tumor Collection
2.4. Tissue Samples
2.5. Immunohistochemical Staining
2.6. RNA Isolation and Real-Time Quantitative RT-PCR
2.7. Statistical Analysis
3. Results
3.1. Hyperglycemia Induces EMT6 Cancer Growth
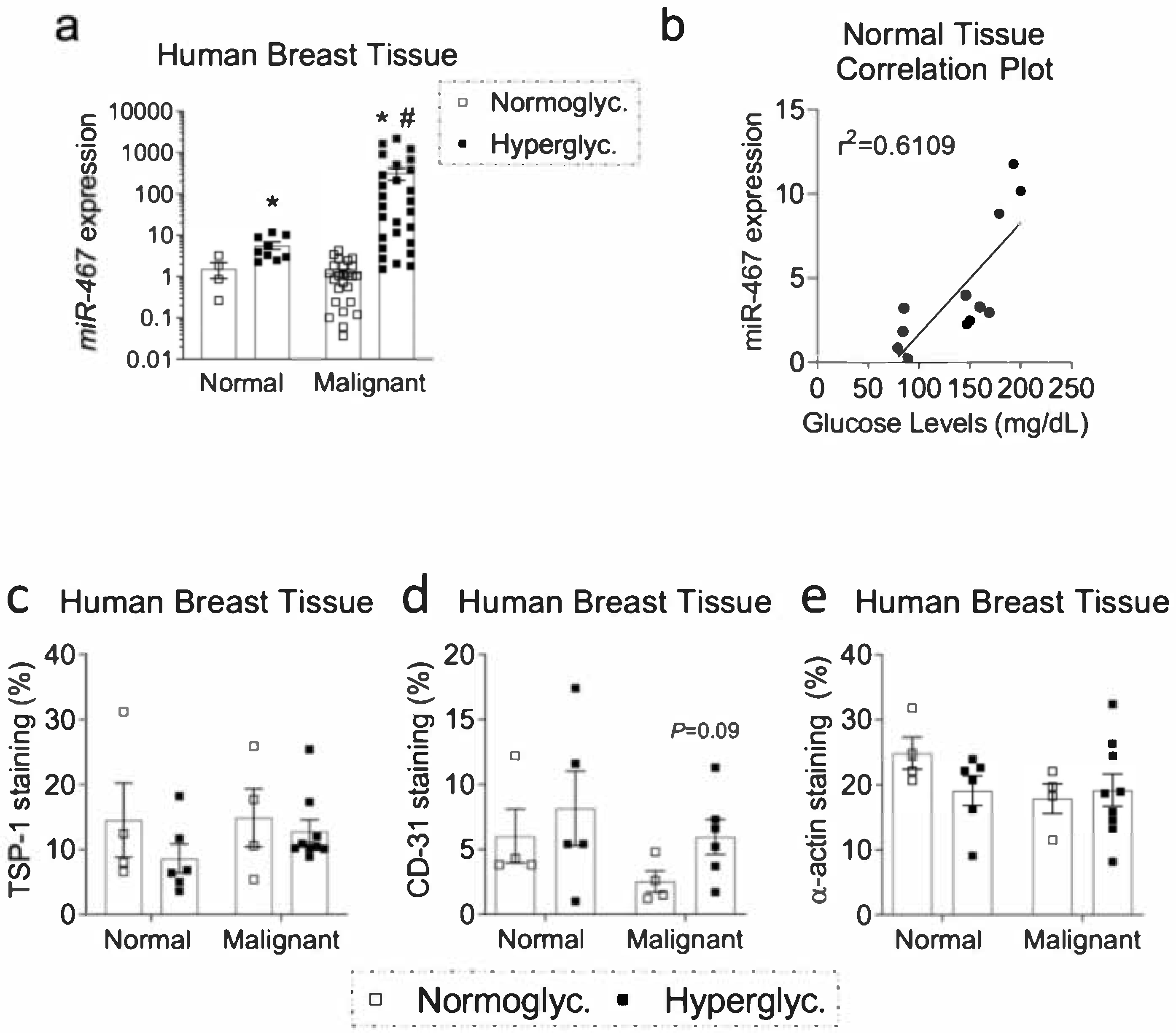
3.2. Expression of miR-467 in Human Breast Tissue Positively Correlates with Glucose Levels
3.3. Increased Macrophage Accumulation and Angiogenesis and Decreased TSP-1 in Human Diabetic BC Samples
3.4. Increased Expression of Pro-Inflammatory Markers and Markers of Macrophages in BC from Hyperglycemic Patients
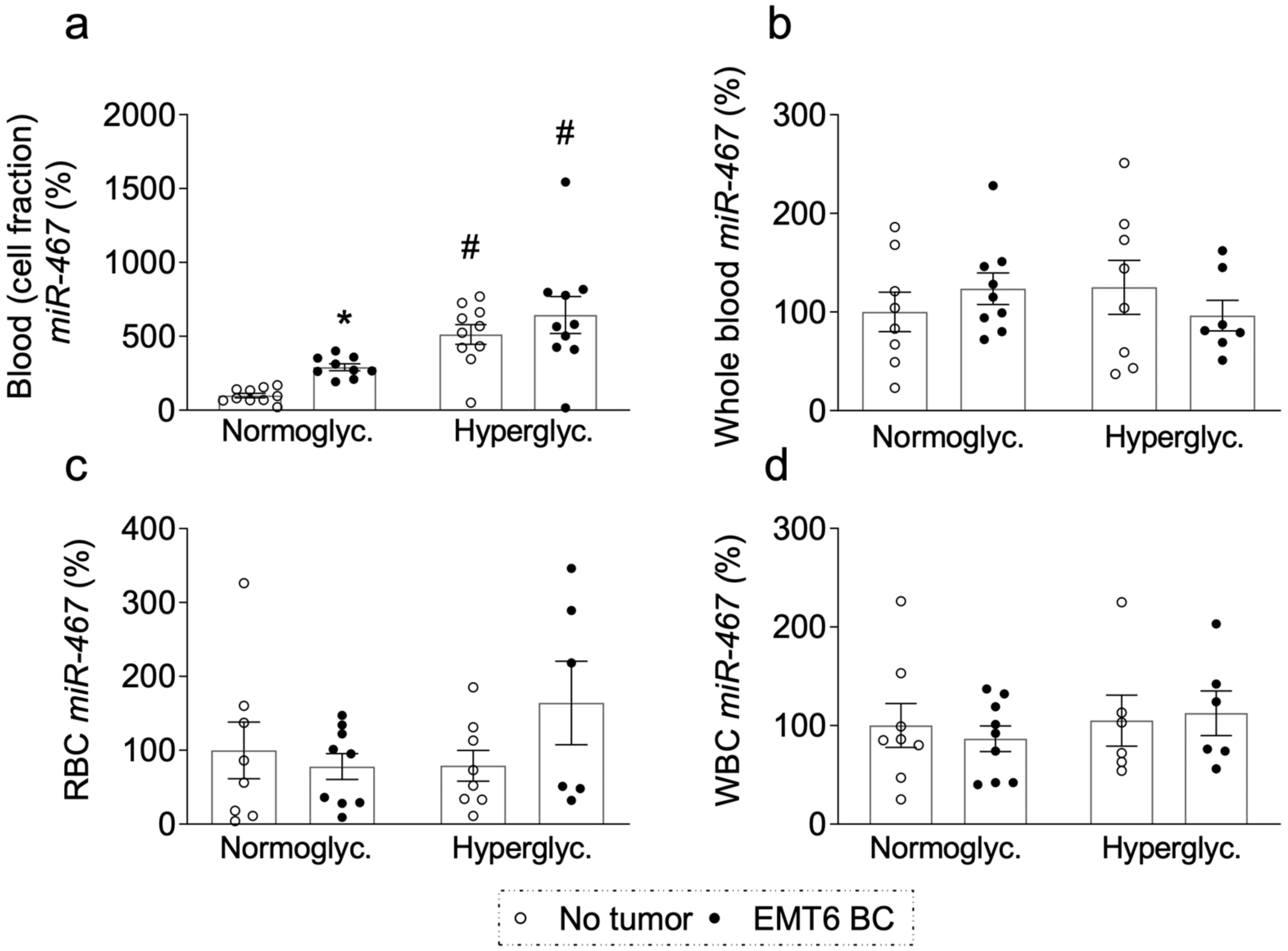
3.5. Level of Circulating Plasma miR-467 Is Higher in Hyperglycemic Patients
3.6. Level of Circulating Plasma miR-467 Is Higher in Hyperglycemic Mice
3.7. Level of Circulating Plasma miR-467 Is Higher in Mice with EMT6 BC Tumors
3.8. miR-467 Levels in Whole Blood, Blood Cells, and Bone Marrow of Mice with BC Tumors
3.9. Increased miR-467 Expression in Bone Marrow (BM) from Hyperglycemic Mice
3.10. Increased miR-467 Expression in Bone Marrow from Mice with EMT6 BC Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TAM | Tumor-Associated Macrophages |
| BC | breast cancer |
| EC | endothelial cells |
| TSP | thropmbospondin |
| miR | microRNA |
| WT | wild type |
| STZ | streptozotocin |
| ATCC | American Type Culture Collection |
| PBS | phosphate buffer saline |
| OCT | optimal cutting temperature compound |
| qPCR | Quantitative polimerase chain reaction |
| SEM | standard error of the mean |
| HbA1c | glycated hemoglobin |
| IL | interleukin |
| EGR | early growth response |
| BM | bone marrow |
References
- U.S. Department of Health and Human Services CfDCaPaNCI; U.S. Cancer Statistics Working Group. U.S. Cancer Statistics Data Visualizations Tool 2018. Available online: https://gis.cdc.gov/Cancer/USCS/DataViz.html (accessed on 15 March 2021).
- Control CfD. Cancers Associated with Overweight and Obesity Make Up 40 Percent of Cancers Diagnosed in the United States 2017. Available online: https://www.cdc.gov/media/releases/2017/p1003-vs-cancer-obesity.html (accessed on 15 March 2021).
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.B.; Ren, G.S. Diabetes mellitus and prognosis in women with breast cancer: A systematic review and meta-analysis. Medicine 2016, 95, e5602. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Ryu, T.Y.; Park, J.; Scherer, P.E. Hyperglycemia as a risk factor for cancer progression. Diabetes Metab. J. 2014, 38, 330–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Casbas-Hernandez, P.; Bigelow, C.; Makowski, L.; Jerry, D.J.; Schneider, S.S.; Troester, M.A. Normal breast tissue of obese women is enriched for macrophage markers and macrophage-associated gene expression. Breast Cancer Res. Treat. 2012, 131, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Morris, P.G.; Hudis, C.A.; Giri, D.; Morrow, M.; Falcone, D.J.; Zhou, X.K.; Du, B.; Brogi, E.; Crawford, C.B.; Kopelovich, L.; et al. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev. Res. 2011, 4, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Iyengar, N.M.; Zhou, X.K.; Gucalp, A.; Morris, P.G.; Howe, L.R.; Giri, D.D.; Morrow, M.; Wang, H.; Pollak, M.; Jones, L.W.; et al. Systemic Correlates of White Adipose Tissue Inflammation in Early-Stage Breast Cancer. Clin. Cancer Res. 2016, 22, 2283–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbaramaiah, K.; Howe, L.R.; Bhardwaj, P.; Du, B.; Gravaghi, C.; Yantiss, R.K.; Zhou, X.K.; Blaho, V.A.; Hla, T.; Yang, P.; et al. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer Prev. Res. 2011, 4, 329–346. [Google Scholar] [CrossRef] [Green Version]
- Iyengar, N.M.; Hudis, C.A.; Dannenberg, A.J. Obesity and cancer: Local and systemic mechanisms. Annu. Rev. Med. 2015, 66, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Yeh, E.S.; Soloff, A.C. Tumor-associated macrophages: Unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2016, 2, 15025. [Google Scholar] [CrossRef] [Green Version]
- Murano, I.; Barbatelli, G.; Parisani, V.; Latini, C.; Muzzonigro, G.; Castellucci, M.; Cinti, S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J. Lipid Res. 2008, 49, 1562–1568. [Google Scholar] [CrossRef] [Green Version]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [Green Version]
- Leek, R.D.; Harris, A.L. Tumor-associated macrophages in breast cancer. J. Mammary Gland Biol. Neoplasia 2002, 7, 177–189. [Google Scholar] [CrossRef]
- O’Sullivan, C.; Lewis, C.E. Tumour-associated leucocytes: Friends or foes in breast carcinoma. J. Pathol. 1994, 172, 229–235. [Google Scholar] [CrossRef]
- Carpizo, D.; Iruela-Arispe, M.L. Endogenous regulators of angiogenesis—Emphasis on proteins with thrombospondin—Type I motifs. Cancer Metastasis Rev. 2000, 19, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Sheibani, N.; Sorenson, C.M.; Cornelius, L.A.; Frazier, W.A. Thrombospondin-1, a natural inhibitor of angiogenesis, is present in vitreous and aqueous humor and is modulated by hyperglycemia. Biochem. Biophys. Res. Commun. 2000, 267, 257–261. [Google Scholar] [CrossRef] [Green Version]
- Lawler, J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J. Cell. Mol. Med. 2002, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.C.; Bornstein, P. Thrombospondins 1 and 2 function as inhibitors of angiogenesis. Matrix Biol. 2003, 22, 63–71. [Google Scholar] [CrossRef]
- Lopez-Dee, Z.; Pidcock, K.; Gutierrez, L.S. Thrombospondin-1: Multiple paths to inflammation. Mediat. Inflamm. 2011, 2011, 296069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almog, N.; Henke, V.; Flores, L.; Hlatky, L.; Kung, A.L.; Wright, R.D.; Berger, R.; Hutchinson, L.; Naumov, G.N.; Bender, E.; et al. Prolonged dormancy of human liposarcoma is associated with impaired tumor angiogenesis. FASEB J. 2006, 20, 947–949. [Google Scholar] [CrossRef] [PubMed]
- Zaslavsky, A.; Chen, C.; Grillo, J.; Baek, K.-H.; Holmgren, L.; Yoon, S.S.; Folkman, J.; Ryeom, S. Regional control of tumor growth. Mol. Cancer Res. 2010, 8, 1198–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Sul, K.; Krukovets, I.; Nestor, C.; Li, J.; Adognravi, O.S. Novel tissue-specific mechanism of regulation of angiogenesis and cancer growth in response to hyperglycemia. J. Am. Heart Assoc. 2012, 1, e005967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krukovets, I.; Legerski, M.; Sul, P.; Stenina-Adognravi, O. Inhibition of hyperglycemia-induced angiogenesis and breast cancer tumor growth by systemic injection of microRNA-467 antagonist. FASEB J. 2015, 29, 3726–3736. [Google Scholar] [CrossRef] [Green Version]
- Gajeton, J.; Krukovets, I.; Yendamuri, R.; Verbovetskiy, D.; Vasanji, A.; Sul, L.; Stenina-Adognravi, O. miR-467 regulates inflammation and blood insulin and glucose. J. Cell. Mol. Med. 2021, 25, 2549–2562. [Google Scholar] [CrossRef]
- Muppala, S.; Xiao, R.; Gajeton, J.; Krukovets, I.; Verbovetskiy, D.; Stenina-Adognravi, O. Thrombospondin-4 mediates hyperglycemia- and TGF-beta-induced inflammation in breast cancer. Int. J. Cancer 2021, 148, 2010–2022. [Google Scholar] [CrossRef]
- Abe, H.; Aida, Y.; Ishiguro, H.; Yoshizawa, K.; Miyazaki, T.; Itagaki, M.; Sutoh, S.; Aizawa, Y. Alcohol, postprandial plasma glucose, and prognosis of hepatocellular carcinoma. World J. Gastroenterol. 2013, 19, 78–85. [Google Scholar] [CrossRef]
- Augustin, L.S.; Kendall, C.W.; Jenkins, D.J.; Willett, W.C.; Astrup, A.; Barclay, A.W.; Björck, I.; Brand-Miller, J.C.; Brighenti, F.; Buyken, A.E.; et al. Glycemic index, glycemic load and glycemic response: An International Scientific Consensus Summit from the International Carbohydrate Quality Consortium (ICQC). Nutr. Metab. Cardiovasc. Dis. 2015, 25, 795–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustin, L.S.; Libra, M.; Crispo, A.; Grimaldi, M.; De Laurentiis, M.; Rinaldo, M.; D’Aiuto, M.; Catalano, F.; Banna, G.; Rossello, R.; et al. Low glycemic index diet, exercise and vitamin D to reduce breast cancer recurrence (DEDiCa): Design of a clinical trial. BMC Cancer 2017, 17, 69. [Google Scholar] [CrossRef] [Green Version]
- Blaser, M.J.; Perez-Perez, G.I.; Kleanthous, H.; Cover, T.L.; Peek, R.M.; Chyou, P.H.; Stemmermann, G.N.; Nomura, A. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995, 55, 2111–2115. [Google Scholar] [PubMed]
- Dehghan, M.; Mente, A.; Zhang, X.; Swaminathan, S.; Li, W.; Mohan, V.; Iqbal, R.; Kumar, R.; Wentzel-Viljoen, E.; Rosengren, A.; et al. Associations of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): A prospective cohort study. Lancet 2017, 390, 2050–2062. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.Y.; Qin, L.Q. Dietary glycemic index, glycemic load, and risk of breast cancer: Meta-analysis of prospective cohort studies. Breast Cancer Res. Treat. 2011, 126, 287–294. [Google Scholar] [CrossRef]
- Hardin, J.; Cheng, I.; Witte, J.S. Impact of consumption of vegetable, fruit, grain, and high glycemic index foods on aggressive prostate cancer risk. Nutr. Cancer 2011, 63, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; La Vecchia, C.; Augustin, L.; Negri, E.; de Groh, M.; Morrison, H.; Mery, L. Glycemic index, glycemic load and cancer risk. Ann. Oncol. 2013, 24, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.Y.; Cho, S.W.; Kim, Y.A.; Kim, D.; Oh, B.-C.; Park, D.J.; Park, Y.J. Cancers with Higher Density of Tumor-Associated Macrophages Were Associated with Poor Survival Rates. J. Pathol. Transl. Med. 2015, 49, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Keum, N.; Yuan, C.; Nishihara, R.; Zoltick, E.; Hamada, T.; Fernandez, A.M.; Zhang, X.; Hanyuda, A.; Liu, L.; Kosumi, K.; et al. Dietary glycemic and insulin scores and colorectal cancer survival by tumor molecular biomarkers. Int. J. Cancer 2017, 140, 2648–2656. [Google Scholar] [CrossRef] [PubMed]
- Kuper, H.; Adami, H.O.; Trichopoulos, D. Infections as a major preventable cause of human cancer. J. Intern. Med. 2000, 248, 171–183. [Google Scholar] [CrossRef]
- Larsson, S.C.; Bergkvist, L.; Wolk, A. Glycemic load, glycemic index and breast cancer risk in a prospective cohort of Swedish women. Int. J. Cancer 2009, 125, 153–157. [Google Scholar] [CrossRef]
- Larsson, S.C.; Giovannucci, E.L.; Wolk, A. Prospective Study of Glycemic Load, Glycemic Index, and Carbohydrate Intake in Relation to Risk of Biliary Tract Cancer. Am. J. Gastroenterol. 2016, 111, 891–896. [Google Scholar] [CrossRef]
- Lega, I.C.; Austin, P.C.; Fischer, H.D.; Fung, K.; Krzyzanowska, M.K.; Amir, E.; Lipscombe, L.L. The Impact of Diabetes on Breast Cancer Treatments and Outcomes: A Population-Based Study. Diabetes Care 2018, 41, 755–761. [Google Scholar] [CrossRef] [Green Version]
- Makarem, N.; Bandera, E.V.; Lin, Y.; Jacques, P.F.; Hayes, R.B.; Parekh, N. Carbohydrate nutrition and risk of adiposity-related cancers: Results from the Framingham Offspring cohort (1991–2013). Br. J. Nutr. 2017, 117, 1603–1614. [Google Scholar] [CrossRef] [Green Version]
- Marinac, C.R.; Nelson, S.H.; Breen, C.I.; Hartman, S.J.; Natarajan, L.; Pierce, J.P.; Flatt, S.W.; Sears, D.D.; Patterson, R.E. Prolonged Nightly Fasting and Breast Cancer Prognosis. JAMA Oncol. 2016, 2, 1049–1055. [Google Scholar] [CrossRef]
- Melkonian, S.C.; Daniel, C.R.; Ye, Y.; Pierzynski, J.A.; Roth, J.A.; Wu, X. Glycemic Index, Glycemic Load, and Lung Cancer Risk in Non-Hispanic Whites. Cancer Epidemiol. Biomark. Prev. 2016, 25, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Mullie, P.; Koechlin, A.; Boniol, M.; Autier, P.; Boyle, P. Relation between Breast Cancer and High Glycemic Index or Glycemic Load: A Meta-analysis of Prospective Cohort Studies. Crit. Rev. Food Sci. Nutr. 2016, 56, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Nagle, C.M.; Olsen, C.M.; Ibiebele, T.I.; Spurdle, A.B.; Webb, P.M.; The Australian National Endometrial Cancer Study Group; The Australian Ovarian Cancer Study Group. Glycemic index, glycemic load and endometrial cancer risk: Results from the Australian National Endometrial Cancer study and an updated systematic review and meta-analysis. Eur. J. Nutr. 2013, 52, 705–715. [Google Scholar] [CrossRef]
- Scholl, S.M.; Pallud, C.; Beuvon, F.; Hacene, K.; Stanley, E.R.; Rohrschneider, L.; Tang, R.; Pouillart, P.; Lidereau, R. Anti-colony-stimulating factor-1 antibody staining in primary breast adenocarcinomas correlates with marked inflammatory cell infiltrates and prognosis. J. Natl. Cancer Inst. 1994, 86, 120–126. [Google Scholar] [CrossRef]
- Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer. Oncology 2002, 16, 229, discussion 230–232. [Google Scholar]
- Sieri, S.; Agnoli, C.; Pala, V.; Grioni, S.; Brighenti, F.; Pellegrini, N.; Masala, G.; Palli, D.; Mattiello, A.; Panico, S.; et al. Dietary glycemic index, glycemic load, and cancer risk: Results from the EPIC-Italy study. Sci. Rep. 2017, 7, 9757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieri, S.; Krogh, V.; Agnoli, C.; Ricceri, F.; Palli, D.; Masala, G.; Panico, S.; Mattiello, A.; Tumino, R.; Giurdanella, M.; et al. Dietary glycemic index and glycemic load and risk of colorectal cancer: Results from the EPIC-Italy study. Int. J. Cancer 2015, 136, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Sieri, S.; Pala, V.; Brighenti, F.; Agnoli, C.; Grioni, S.; Berrino, F.; Scazzina, F.; Palli, D.; Masala, G.; Vineis, P.; et al. High glycemic diet and breast cancer occurrence in the Italian EPIC cohort. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 628–634. [Google Scholar] [CrossRef]
- Turati, F.; Galeone, C.; Gandini, S.; Augustin, L.S.; Jenkins, D.J.A.; Pelucchi, C.; La Vecchia, C. High glycemic index and glycemic load are associated with moderately increased cancer risk. Mol. Nutr. Food Res. 2015, 59, 1384–1394. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannafon, B.N.; Trigoso, Y.D.; Calloway, C.L.; Zhao, Y.D.; Lum, D.H.; Welm, A.L.; Zhao, Z.J.; Blick, K.E.; Dooley, W.C.; Ding, W.Q. Plasma exosome microRNAs are indicative of breast cancer. Breast Cancer Res. 2016, 18, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, A.; Ferrari, P.; Duffy, M.J. Prognostic and predictive biomarkers in breast cancer: Past, present and future. Semin. Cancer Biol. 2018, 52 Pt 1, 56–73. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Marinic, T.E.; Krukovets, I.; Hoppe, G.; Stenina, O.I. Cell type-specific post-transcriptional regulation of production of the potent antiangiogenic and proatherogenic protein thrombospondin-1 by high glucose. J. Biol. Chem. 2008, 283, 5699–5707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockwell, S.C.; Kallman, R.F.; Fajardo, L.F. Characteristics of a serially transplanted mouse mammary tumor and its tissue-culture-adapted derivative. J. Natl. Cancer Inst. 1972, 49, 735–749. [Google Scholar] [PubMed]
- Muller, W.J.; Sinn, E.; Pattengale, P.K.; Wallace, R.; Leder, P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 1988, 54, 105–115. [Google Scholar] [CrossRef]
- McVeigh, T.P.; Kerin, M.J. Clinical use of the Oncotype DX genomic test to guide treatment decisions for patients with invasive breast cancer. Breast Cancer 2017, 9, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [Green Version]
- Pascale, R.M.; Calvisi, D.F.; Simile, M.M.; Feo, C.F.; Feo, F. The Warburg Effect 97 Years after Its Discovery. Cancers 2020, 12, 2819. [Google Scholar] [CrossRef]
- Schwartz, L.; Supuran, C.T.; Alfarouk, K.O. The Warburg Effect and the Hallmarks of Cancer. Anticancer Agents Med. Chem. 2017, 17, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Sona, M.F.; Myung, S.K.; Park, K.; Jargalsaikhan, G. Type 1 diabetes mellitus and risk of cancer: A meta-analysis of observational studies. Jpn. J. Clin. Oncol. 2018, 48, 426–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, P.; Boniol, M.; Koechlin, A.; Robertson, C.; Valentini, F.; Coppens, K.; Fairley, L.L.; Zheng, T.; Zhang, Y.; Pasterk, M.; et al. Diabetes and breast cancer risk: A meta-analysis. Br. J. Cancer 2012, 107, 1608–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carstensen, B.; Read, S.H.; Friis, S.; Sund, R.; Keskimäki, I.; Svensson, A.M.; Ljung, R.; Wild, S.H.; Kerssens, J.J.; Harding, J.L.; et al. Cancer incidence in persons with type 1 diabetes: A five-country study of 9000 cancers in type 1 diabetic individuals. Diabetologia 2016, 59, 980–988. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gajeton, J.; Krukovets, I.; Muppala, S.; Verbovetskiy, D.; Zhang, J.; Stenina-Adognravi, O. Hyperglycemia-Induced miR-467 Drives Tumor Inflammation and Growth in Breast Cancer. Cancers 2021, 13, 1346. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061346
Gajeton J, Krukovets I, Muppala S, Verbovetskiy D, Zhang J, Stenina-Adognravi O. Hyperglycemia-Induced miR-467 Drives Tumor Inflammation and Growth in Breast Cancer. Cancers. 2021; 13(6):1346. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061346
Chicago/Turabian StyleGajeton, Jasmine, Irene Krukovets, Santoshi Muppala, Dmitriy Verbovetskiy, Jessica Zhang, and Olga Stenina-Adognravi. 2021. "Hyperglycemia-Induced miR-467 Drives Tumor Inflammation and Growth in Breast Cancer" Cancers 13, no. 6: 1346. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061346