
Recruitment and Expansion of Tregs Cells in the Tumor Environment—How to Target Them?

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Natural and Peripherally—Induced Tregs
1.2. Mechanisms Developed by Tregs to Suppress T Cells
1.3. Tregs Infiltrate Tumors and Participate to Cancer Progression
1.4. Origin of Tregs Present in the TME
2. Chemokines: A Key Role in the Active Recruitment of Tregs in Tumors
2.1. Major Chemokine Receptors and Ligands Involved in Tregs Recruitment in the TME
2.1.1. CCR4 and Its Cognate Ligands
2.1.2. CCR8 and Its Cognate Ligands
2.2. Chemokine/Chemokine Receptors Favoring Recruitment and Treg/Myeloid Cell Interactions
2.2.1. CCR2/CCL2
2.2.2. CCR5/CCL5
2.3. CCR6, CCR10 and CXCR3: Receptors Favoring Co-Localisation of Tregs with CD4+ or CD8+ Effector T Cells with Specific Homing Tropisms
2.3.1. CCR6/CCL20
2.3.2. CCR10 and Its Ligands
2.3.3. CXCR3 and Its Ligands
2.3.4. Conclusions
3. Expansion in the Tumor Environment
3.1. Role of ICOS-ICOS-L
3.2. Role of TNFR2
3.3. Role of Other TNF Receptor Superfamily Costimulatory Molecules (4-1BB, OX40 and GITR)
3.4. Negative Regulators
3.5. Metabolism
4. Treg Stabilization
4.1. Helios
4.2. STAT-5
4.3. Von Hippel-Lindau E3-Ubiquitin Ligase
4.4. Neuropilin 1 (NRP1)
4.5. PI3K/AKT/mTOR
4.6. CCL1/CCR8
4.7. IL-1 Receptors
4.8. ST2/IL-33 Pathway
4.9. Conclusions
5. Therapeutic Strategies to Block Treg Recruitment and Expansion or Limit Their Stability
5.1. Inhibit Tregs Recruitment
5.1.1. CCR4 Inhibitors
5.1.2. CCR8 Inhibitors
5.1.3. Other Chemokine Receptor Antagonists
5.1.4. TLR9 Ligand and Type-I Interferon
5.2. Inhibit Tregs Amplification in the TME
5.2.1. Anti-ICOS Antagonists
5.2.2. Fully Human Anti-TNFR2 Antibodies
5.2.3. Restore Type-I Interferon to Inhibit Treg Amplification in TME
5.3. Destabilize TA-Tregs
5.4. Favor Tregs Depletion
5.4.1. CTLA-4 Blockade
5.4.2. Humanized Anti-CCR4 Antibody
5.4.3. Humanized Anti-CCR8 Antibodies
5.4.4. Antibodies Targeting ICOS
5.4.5. TCR-Mimic Antibody Recognizing a FOXP3-Derived Epitope
5.5. Risk to Reactivate TA-Tregs through Anti-ICP mAbs
5.5.1. Antibodies Neutralizing PD-1/PD-L1 Interaction
5.5.2. Other Antibodies Targeting Activating Receptors
- OX40 and 4-1BB agonists antibodies
- ICOS agonist antibodies
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome (IPEX) Is Caused by Mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Morikawa, H.; Sakaguchi, S. Genetic and Epigenetic Basis of Treg Cell Development and Function: From a FoxP3-centered View to an Epigenome-defined View of Natural Treg Cells. Immunol. Rev. 2014, 259, 192–205. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of Peripheral CD4+CD25− Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-β Induction of Transcription Factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Kretschmer, K.; Apostolou, I.; Hawiger, D.; Khazaie, K.; Nussenzweig, M.C. Inducing and Expanding Regulatory T Cell Populations by Foreign Antigen. Nat. Immunol. 2005, 6, 1219–1227. [Google Scholar] [CrossRef]
- Selvaraj, R.K.; Geiger, T.L. A Kinetic and Dynamic Analysis of Foxp3 Induced in T Cells by TGF-β. J. Immunol. 2007, 179, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siewert, C.; Lauer, U.; Cording, S.; Schmitt, E.; Hamann, A.; Huehn, J. Experience-Driven Development: Effector/Memory-Like AE+Foxp3+ Regulatory T Cells Originate from Both Naive T Cells and Naturally Occurring Naive-Like Regulatory T Cells. J. Immunol. 2008, 180, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.G.; Wang, J.H.; Stohl, W.; Kim, K.S.; Gray, J.D.; Horwitz, D.A. TGF-β Requires CTLA-4 Early after T Cell Activation to Induce FoxP3 and Generate Adaptive CD4+ CD25+ Regulatory Cells. J. Immunol. 2006, 176, 3321–3329. [Google Scholar] [CrossRef] [Green Version]
- Roncarolo, M.G.; Gregori, S.; Bacchetta, R.; Battaglia, M. Tr1 Cells and the Counter-Regulation of Immunity: Natural Mechanisms and Therapeutic Applications. Curr. Top. Microbiol. Immunol. 2014, 380, 39–68. [Google Scholar] [CrossRef]
- De Lafaille, M.A.C. Natural and Adaptive Foxp3+ Regulatory T Cells: More of the Same or a Division of Labor? Immunity 2009, 30, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Baron, U.; Floess, S.; Wieczorek, G.; Baumann, K.; Grützkau, A.; Dong, J.; Thiel, A.; Boeld, T.J.; Hoffmann, P.; Edinger, M.; et al. DNA Demethylation in the Human FOXP3 Locus Discriminates Regulatory T Cells from Activated FOXP3+ Conventional T Cells. Eur. J. Immunol. 2007, 37, 2378–2389. [Google Scholar] [CrossRef]
- Thornton, A.M.; Korty, P.E.; Tran, D.Q.; Wohlfert, E.A.; Murray, P.E.; Belkaid, Y.; Shevach, E.M. Expression of Helios, an Ikaros Transcription Factor Family Member, Differentiates Thymic-Derived from Peripherally Induced Foxp3+ T Regulatory Cells. J. Immunol. 2010, 184, 3433–3441. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Chen, M.; Liu, Y.; Guo, Z.; He, X.; Brand, D.; Zheng, S.G. Advances in Distinguishing Natural from Induced Foxp3+ Regulatory T Cells. Int. J. Clin. Exp. Pathol. 2013, 6, 116–123. [Google Scholar]
- Ahmadzadeh, M.; Pasetto, A.; Jia, L.; Deniger, D.C.; Stevanović, S.; Robbins, P.F.; Rosenberg, S.A. Tumor-Infiltrating Human CD4+ Regulatory T Cells Display a Distinct TCR Repertoire and Exhibit Tumor and Neoantigen Reactivity. Sci. Immunol. 2019, 4, eaao4310. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H. CD4+ CD25+ Regulatory T Cells Control the Induction of Antigen-Specific CD4+ Helper T Cell Responses in Cancer Patients. Blood 2005, 106, 1008–1011. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Sakaguchi, S. Regulatory T Cells in Cancer Immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.-F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine Generation Catalyzed by CD39 and CD73 Expressed on Regulatory T Cells Mediates Immune Suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- Bynoe, M.S.; Viret, C. Foxp3+CD4+ T Cell-Mediated Immunosuppression Involves Extracellular Nucleotide Catabolism. Trends Immunol. 2008, 29, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Mastelic-Gavillet, B.; Navarro Rodrigo, B.; Décombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derré, L.; Valerio, M.; et al. Adenosine Mediates Functional and Metabolic Suppression of Peripheral and Tumor-Infiltrating CD8+ T Cells. J. Immunother. Cancer 2019, 7, 257. [Google Scholar] [CrossRef]
- Gourdin, N.; Bossennec, M.; Rodriguez, C.; Vigano, S.; Machon, C.; Jandus, C.; Bauché, D.; Faget, J.; Durand, I.; Chopin, N.; et al. Autocrine Adenosine Regulates Tumor Polyfunctional CD73+CD4+ Effector T Cells Devoid of Immune Checkpoints. Cancer Res. 2018, 78, 3604–3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, G.J.; Fox, S.B.; Han, C.; Leek, R.D.; Garcia, J.F.; Harris, A.L.; Banham, A.H. Quantification of Regulatory T Cells Enables the Identification of High-Risk Breast Cancer Patients and Those at Risk of Late Relapse. J. Clin. Oncol. 2006, 24, 5373–5380. [Google Scholar] [CrossRef] [PubMed]
- Gobert, M.; Treilleux, I.; Bendriss-Vermare, N.; Bachelot, T.; Goddard-Leon, S.; Arfi, V.; Biota, C.; Doffin, A.C.; Durand, I.; Olive, D.; et al. Regulatory T Cells Recruited through CCL22/CCR4 Are Selectively Activated in Lymphoid Infiltrates Surrounding Primary Breast Tumors and Lead to an Adverse Clinical Outcome. Cancer Res. 2009, 69, 2000–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Kim, K.M.; Bae, J.S.; Park, H.S.; Lee, H.; Chung, M.J.; Moon, W.S.; Lee, D.G.; Jang, K.Y. Tumor-Infiltrating PD1-Positive Lymphocytes and FoxP3-Positive Regulatory T Cells Predict Distant Metastatic Relapse and Survival of Clear Cell Renal Cell Carcinoma. Transl. Oncol. 2013, 6, 282–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liotta, F.; Gacci, M.; Frosali, F.; Querci, V.; Vittori, G.; Lapini, A.; Santarlasci, V.; Serni, S.; Cosmi, L.; Maggi, L.; et al. Frequency of Regulatory T Cells in Peripheral Blood and in Tumour-Infiltrating Lymphocytes Correlates with Poor Prognosis in Renal Cell Carcinoma. BJU Int. 2011, 107, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Schmoeckel, E.; Kost, B.P.; Kuhn, C.; Vattai, A.; Vilsmaier, T.; Mahner, S.; Mayr, D.; Jeschke, U.; Heidegger, H.H. Higher CCL22+ Cell Infiltration Is Associated with Poor Prognosis in Cervical Cancer Patients. Cancers 2019, 11, 2004. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Kanao, K.; Suzuki, S.; Muramatsu, H.; Morinaga, S.; Kajikawa, K.; Kobayashi, I.; Nishikawa, G.; Kato, Y.; Zennami, K.; et al. Increased Infiltration of CCR4-positive Regulatory T Cells in Prostate Cancer Tissue Is Associated with a Poor Prognosis. Prostate 2019, 79, 1658–1665. [Google Scholar] [CrossRef]
- Oh, D.Y. Intratumoral CD4+ T Cells Mediate Anti-Tumor Cytotoxicity in Human Bladder Cancer. Cell 2020, 181, 1612–1625. [Google Scholar] [CrossRef]
- Tao, H.; Mimura, Y.; Aoe, K.; Kobayashi, S.; Yamamoto, H.; Matsuda, E.; Okabe, K.; Matsumoto, T.; Sugi, K.; Ueoka, H. Prognostic Potential of FOXP3 Expression in Non-Small Cell Lung Cancer Cells Combined with Tumor-Infiltrating Regulatory T Cells. Lung Cancer 2012, 75, 95–101. [Google Scholar] [CrossRef]
- Zhou, J.; Ding, T.; Pan, W.; Zhu, L.; Li, L.; Zheng, L. Increased Intratumoral Regulatory T Cells Are Related to Intratumoral Macrophages and Poor Prognosis in Hepatocellular Carcinoma Patients. Int. J. Cancer 2009, 125, 1640–1648. [Google Scholar] [CrossRef]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ Regulatory T Cells Increases During the Progression of Pancreatic Ductal Adenocarcinoma and Its Premalignant Lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, J.F.M.; Idema, A.J.; Bol, K.F.; Grotenhuis, J.A.; de Vries, I.J.M.; Wesseling, P.; Adema, G.J. Prognostic Significance and Mechanism of Treg Infiltration in Human Brain Tumors. J. Neuroimmunol. 2010, 225, 195–199. [Google Scholar] [CrossRef]
- Badoual, C. Prognostic Value of Tumor-Infiltrating CD4+ T-Cell Subpopulations in Head and Neck Cancers. Clin. Cancer Res. 2006, 12, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Hillen, F.; Baeten, C.I.M.; van de Winkel, A.; Creytens, D.; Winnepenninckx, V.; Gri, A.W. Leukocyte Infiltration and Tumor Cell Plasticity Are Parameters of Aggressiveness in Primary Cutaneous Melanoma. Cancer Immunol. Immunother. 2008, 57, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Mourmouras, V.; Biagioli, M.; Miracco, C.; Luzi, P.; Tosi, P.; Cosci, E.; Monciatti, I.; Mannucci, S.; Rubegni, P. Utility of Tumour-Infiltrating CD25+FOXP3+ Regulatory T Cell Evaluation in Predicting Local Recurrence in Vertical Growth Phase Cutaneous Melanoma. Oncol. Rep. 2007, 18, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Ménétrier-Caux, C.; Curiel, T.; Faget, J.; Manuel, M.; Caux, C.; Zou, W. Targeting Regulatory T Cells. Target. Oncol. 2012, 7, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Carreras, J.; Lopez-Guillermo, A.; Fox, B.C.; Colomo, L.; Martinez, A.; Roncador, G.; Montserrat, E.; Campo, E.; Banham, A.H. High Numbers of Tumor-Infiltrating FOXP3-Positive Regulatory T Cells Are Associated with Improved Overall Survival in Follicular Lymphoma. Blood 2006, 108, 2957–2964. [Google Scholar] [CrossRef] [PubMed]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-Infiltrating FOXP3+ T Regulatory Cells Show Strong Prognostic Significance in Colorectal Cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef]
- Tzankov, A.; Meier, C.; Hirschmann, P.; Went, P.; Pileri, S.A.; Dirnhofer, S. Correlation of High Numbers of Intratumoral FOXP3+ Regulatory T Cells with Improved Survival in Germinal Center-like Diffuse Large B-Cell Lymphoma, Follicular Lymphoma and Classical Hodgkin’s Lymphoma. Haematologica 2008, 93, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Kryczek, I.; Wu, K.; Zhao, E.; Wei, S.; Vatan, L.; Szeliga, W.; Huang, E.; Greenson, J.; Chang, A.; Roliński, J.; et al. IL-17+ Regulatory T Cells in the Microenvironments of Chronic Inflammation and Cancer. J. Immunol. 2011, 186, 4388–4395. [Google Scholar] [CrossRef] [Green Version]
- Kryczek, I.; Wei, S.; Szeliga, W.; Vatan, L.; Zou, W. Endogenous IL-17 Contributes to Reduced Tumor Growth and Metastasis. Blood 2009, 114, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3+CD4+ T Cell Subpopulations Distinctly Control the Prognosis of Colorectal Cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Panduro, M.; Benoist, C.; Mathis, D. Tissue Tregs. Annu. Rev. Immunol. 2016, 34, 609–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Simone, M.; Arrigoni, A.; Rossetti, G.; Gruarin, P.; Ranzani, V.; Politano, C.; Bonnal, R.J.P.; Provasi, E.; Sarnicola, M.L.; Panzeri, I.; et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity 2016, 45, 1135–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global Characterization of T Cells in Non-Small-Cell Lung Cancer by Single-Cell Sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef]
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-Cell Profiling of Breast Cancer T Cells Reveals a Tissue-Resident Memory Subset Associated with Improved Prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Plitas, G.; Konopacki, C.; Wu, K.; Bos, P.D.; Morrow, M.; Putintseva, E.V.; Chudakov, D.M.; Rudensky, A.Y. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 2016, 45, 1122–1134. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20. [Google Scholar] [CrossRef]
- Niedzielska, M.; Israelsson, E.; Angermann, B.; Sidders, B.S.; Clausen, M.; Catley, M.; Malhotra, R.; Dumont, C. Differential Gene Expression in Human Tissue Resident Regulatory T Cells from Lung, Colon, and Blood. Oncotarget 2018, 9, 36166–36184. [Google Scholar] [CrossRef] [Green Version]
- Núñez, N.G.; Tosello Boari, J.; Ramos, R.N.; Richer, W.; Cagnard, N.; Anderfuhren, C.D.; Niborski, L.L.; Bigot, J.; Meseure, D.; De La Rochere, P.; et al. Tumor Invasion in Draining Lymph Nodes Is Associated with Treg Accumulation in Breast Cancer Patients. Nat. Commun. 2020, 11, 3272. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the Cancer Microenvironment and Their Relevance in Cancer Immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wing, J.B.; Sakaguchi, S. Multiple Treg Suppressive Modules and Their Adaptability. Front. Immun. 2012, 3, 178. [Google Scholar] [CrossRef] [Green Version]
- Duhen, T.; Geiger, R.; Jarrossay, D.; Lanzavecchia, A.; Sallusto, F. Production of Interleukin 22 but Not Interleukin 17 by a Subset of Human Skin-Homing Memory T Cells. Nat. Immunol. 2009, 10, 857–863. [Google Scholar] [CrossRef]
- Imai, T.; Nagira, M.; Takagi, S.; Kakizaki, M.; Nishimura, M.; Wang, J.; Gray, P.W.; Matsushima, K.; Yoshie, O. Selective Recruitment of CCR4-Bearing Th2 Cells toward Antigen-Presenting Cells by the CC Chemokines Thymus and Activation-Regulated Chemokine and Macrophage-Derived Chemokine. Int. Immunol. 1999, 11, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.W.; Lee, J.; Hillsamer, P. Human Th17 Cells Share Major Trafficking Receptors with Both Polarized Effector T Cells and FOXP3+ Regulatory T Cells. J. Immunol. 2008, 180, 122–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iellem, A.; Mariani, M.; Lang, R.; Recalde, H.; Panina-Bordignon, P.; Sinigaglia, F.; D’Ambrosio, D. Unique Chemotactic Response Profile and Specific Expression of Chemokine Receptors CCR4 and CCR8 by CD4+CD25+ Regulatory T Cells. J. Exp. Med. 2001, 194, 847–853. [Google Scholar] [CrossRef] [Green Version]
- Sather, B.D.; Treuting, P.; Perdue, N.; Miazgowicz, M.; Fontenot, J.D.; Rudensky, A.Y.; Campbell, D.J. Altering the Distribution of Foxp3+ Regulatory T Cells Results in Tissue-Specific Inflammatory Disease. J. Exp. Med. 2007, 204, 1335–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oo, Y.H.; Weston, C.J.; Lalor, P.F.; Curbishley, S.M.; Withers, D.R.; Reynolds, G.M.; Shetty, S.; Harki, J.; Shaw, J.C.; Eksteen, B.; et al. Distinct Roles for CCR4 and CXCR3 in the Recruitment and Positioning of Regulatory T Cells in the Inflamed Human Liver. J. Immunol. 2010, 184, 2886–2898. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [Green Version]
- Faget, J.; Biota, C.; Bachelot, T.; Gobert, M.; Treilleux, I.; Goutagny, N.; Durand, I.; Leon-Goddard, S.; Blay, J.Y.; Caux, C.; et al. Early Detection of Tumor Cells by Innate Immune Cells Leads to Treg Recruitment through CCL22 Production by Tumor Cells. Cancer Res. 2011, 71, 6143–6152. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, T.; Matsuo, K.; Hashida, Y.; Kitahata, K.; Ujihara, T.; Taniguchi, A.; Yoshie, O.; Nakayama, T.; Daibata, M. Epstein–Barr Virus-Positive Pyothorax-Associated Lymphoma Expresses CCL17 and CCL22 Chemokines That Attract CCR4-Expressing Regulatory T Cells. Cancer Lett. 2019, 453, 184–192. [Google Scholar] [CrossRef]
- Ishida, T.; Ishii, T.; Inagaki, A.; Yano, H.; Komatsu, H.; Iida, S.; Inagaki, H.; Ueda, R. Specific Recruitment of CC Chemokine Receptor 4–Positive Regulatory T Cells in Hodgkin Lymphoma Fosters Immune Privilege. Cancer Res. 2006, 66, 5716–5722. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Wei, X.; Li, L.; Wu, X.; Yan, J.; Yang, H.; Song, F. CCR4 Mediated Chemotaxis of Regulatory T Cells Suppress the Activation of T Cells and NK Cells via TGF-β Pathway in Human Non-Small Cell Lung Cancer. Biochem. Biophys. Res. Commun. 2017, 488, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Svensson, H.; Olofsson, V.; Lundin, S.; Yakkala, C. Accumulation of CCR4+ CTLA-4hi FOXP3+CD25hi Regulatory T Cells in Colon Adenocarcinomas Correlate to Reduced Activation of Conventional T Cells. PLoS ONE 2012, 7, e30695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Chen, J.; Xiao, L.; Tang, F.; Zhang, Z.; Zhang, Y.; Feng, Z.; Jiang, Y.; Shao, C. Accumulation Mechanisms of CD4+CD25+FOXP3+ Regulatory T Cells in EBV-Associated Gastric Carcinoma. Sci. Rep. 2015, 5, 18057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zheng, L.; Zhang, L.; Hu, X.; Ren, X.; Zhang, Z. Deep Single-Cell RNA Sequencing Data of Individual T Cells from Treatment-Naïve Colorectal Cancer Patients. Sci. Data 2019, 6, 131. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Nanbu, U.; Noguchi, H.; Harada, Y.; Kumamoto, K.; Sasaguri, Y.; Nakayama, T. Macrophage CCL22 Expression in the Tumor Microenvironment and Implications for Survival in Patients with Squamous Cell Carcinoma of the Tongue. J. Oral. Pathol. Med. 2019, 48, 677–685. [Google Scholar] [CrossRef]
- Kuehnemuth, B.; Piseddu, I.; Wiedemann, G.M.; Lauseker, M.; Kuhn, C.; Hofmann, S.; Schmoeckel, E.; Endres, S.; Mayr, D.; Jeschke, U.; et al. CCL1 Is a Major Regulatory T Cell Attracting Factor in Human Breast Cancer. BMC Cancer 2018, 18, 1278. [Google Scholar] [CrossRef]
- Li, Y.-Q.; Liu, F.-F.; Zhang, X.-M.; Guo, X.-J.; Ren, M.-J.; Fu, L. Tumor Secretion of CCL22 Activates Intratumoral Treg Infiltration and Is Independent Prognostic Predictor of Breast Cancer. PLoS ONE 2013, 8, e76379. [Google Scholar] [CrossRef]
- Takegawa, S.; Jin, Z.; Nakayama, T.; Oyama, T.; Hieshima, K.; Nagakubo, D.; Shirakawa, A.-K.; Tsuzuki, T.; Nakamura, S.; Yoshie, O. Expression of CCL17 and CCL22 by Latent Membrane Protein 1-Positive Tumor Cells in Age-Related Epstein–Barr Virus-Associated B-Cell Lymphoproliferative Disorder. Cancer Sci. 2008, 99, 296–302. [Google Scholar] [CrossRef]
- Wiedemann, G.M.; Knott, M.M.L.; Vetter, V.K.; Rapp, M.; Haubner, S.; Fesseler, J.; Kühnemuth, B.; Layritz, P.; Thaler, R.; Kruger, S.; et al. Cancer Cell-Derived IL-1α Induces CCL22 and the Recruitment of Regulatory T Cells. OncoImmunology 2016, 5, e1175794. [Google Scholar] [CrossRef] [Green Version]
- Mizukami, Y.; Kono, K.; Kawaguchi, Y.; Akaike, H.; Kamimura, K.; Sugai, H.; Fujii, H. CCL17 and CCL22 Chemokines within Tumor Microenvironment Are Related to Accumulation of Foxp3+ Regulatory T Cells in Gastric Cancer. Int. J. Cancer 2008, 122, 2286–2293. [Google Scholar] [CrossRef]
- Tsujikawa, T.; Yaguchi, T.; Ohmura, G.; Ohta, S.; Kobayashi, A.; Kawamura, N.; Fujita, T.; Nakano, H.; Shimada, T.; Takahashi, T.; et al. Autocrine and Paracrine Loops between Cancer Cells and Macrophages Promote Lymph Node Metastasis via CCR4/CCL22 in Head and Neck Squamous Cell Carcinoma. Int. J. Cancer 2013, 132, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chang, Y.; Kuo, Z.; Fang, Y.; Kao, Y.; Tsai, T.; Wu, W. Cancer-associated Fibroblast-derived Interleukin-1β Activates Protumor C-C Motif Chemokine Ligand 22 Signaling in Head and Neck Cancer. Cancer Sci. 2019, 110, 2783–2793. [Google Scholar] [CrossRef] [Green Version]
- Cremonesi, E.; Governa, V.; Garzon, J.F.G.; Mele, V.; Amicarella, F.; Muraro, M.G.; Trella, E.; Galati-Fournier, V.; Oertli, D.; Däster, S.R.; et al. Gut Microbiota Modulate T Cell Trafficking into Human Colorectal Cancer. Gut 2018, 67, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Chensue, S.W.; Lukacs, N.W.; Yang, T.-Y.; Shang, X.; Frait, K.A.; Kunkel, S.L.; Kung, T.; Wiekowski, M.T.; Hedrick, J.A.; Cook, D.N.; et al. Aberrant In Vivo T Helper Type 2 Cell Response and Impaired Eosinophil Recruitment in CC Chemokine Receptor 8 Knockout Mice. J. Exp. Med. 2001, 193, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Soler, D.; Chapman, T.R.; Poisson, L.R.; Cote-Sierra, J.; Ryan, M.; McDonald, A.; Badola, S.; Fedyk, E.; Coyle, A.J.; Kolbeck, R. CCR8 Expression Identifies CD4 Memory T Cells Enriched for FOXP3 + Regulatory and Th2 Effector Lymphocytes. J. Immunol. 2006, 177, 6940–6951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coghill, J.M.; Fowler, K.A.; West, M.L.; Fulton, L.M.; van Deventer, H.; McKinnon, K.P.; Vincent, B.G.; Lin, K.; Panoskaltsis-Mortari, A.; Cook, D.N.; et al. CC Chemokine Receptor 8 Potentiates Donor Treg Survival and Is Critical for the Prevention of Murine Graft-versus-Host Disease. Blood 2013, 122, 825–836. [Google Scholar] [CrossRef] [Green Version]
- Borcherding, N.; Ahmed, K.K.; Voigt, A.P.; Vishwakarma, A.; Kolb, R.; Kluz, P.; Pandey, G.; Gibson-Corley, K.N.; Klesney-Tait, J.; Zhu, Y.; et al. Transcriptional Heterogeneity in Cancer-Associated Regulatory T Cells Is Predictive of Survival. bioRxiv 2018. [Google Scholar] [CrossRef]
- Magnuson, A.M.; Kiner, E.; Ergun, A.; Park, J.S.; Asinovski, N.; Ortiz-Lopez, A.; Kilcoyne, A.; Paoluzzi-Tomada, E.; Weissleder, R.; Mathis, D.; et al. Identification and Validation of a Tumor-Infiltrating Treg Transcriptional Signature Conserved across Species and Tumor Types. Proc. Natl. Acad. Sci. USA 2018, 115, E10672–E10681. [Google Scholar] [CrossRef] [Green Version]
- Miragaia, R.J.; Gomes, T.; Chomka, A.; Jardine, L.; Riedel, A.; Hegazy, A.N.; Whibley, N.; Tucci, A.; Chen, X.; Lindeman, I.; et al. Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity 2019, 50, 493–504.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhou, Q.; Zeng, H.; Zhang, H.; Liu, Z.; Shao, J.; Wang, Z.; Xiong, Y.; Wang, J.; Bai, Q.; et al. CCR8 Blockade Primes Anti-Tumor Immunity through Intratumoral Regulatory T Cells Destabilization in Muscle-Invasive Bladder Cancer. Cancer Immunol. Immunother. 2020, 69, 1855–1867. [Google Scholar] [CrossRef]
- Sallusto, F.; Kremmer, E.; Palermo, B.; Hoy, A.; Ponath, P.; Qin, S.; Förster, R.; Lipp, M.; Lanzavecchia, A. Switch in Chemokine Receptor Expression upon TCR Stimulation Reveals Novel Homing Potential for Recently Activated T Cells. Eur. J. Immunol. 1999, 29, 2037–2045. [Google Scholar] [CrossRef]
- Alvisi, G.; Brummelman, J.; Puccio, S.; Mazza, E.M.C.; Tomada, E.P.; Losurdo, A.; Zanon, V.; Peano, C.; Colombo, F.S.; Scarpa, A.; et al. IRF4 Instructs Effector Treg Differentiation and Immune Suppression in Human Cancer. J. Clin. Investig. 2020, 130, 3137–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yao, Y.; Gong, C.; Yu, F.; Su, S.; Liu, B.; Deng, H.; Wang, F.; Lin, L.; Yao, H.; et al. CCL18 from Tumor-Associated Macrophages Promotes Breast Cancer Metastasis via PITPNM3. Cancer Cell 2011, 19, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Zhao, L.; Hou, Y.; Sun, Y.; Wang, L.; Luo, H.; Peng, H.; Liu, M. Biological Characteristics and Genetic Heterogeneity between Carcinoma-Associated Fibroblasts and Their Paired Normal Fibroblasts in Human Breast Cancer. PLoS ONE 2013, 8, e60321. [Google Scholar] [CrossRef] [PubMed]
- Ma, L. Chemokine (C-C Motif) Ligand 18 Is Highly Expressed in Glioma Tissues and Promotes Invasion of Glioblastoma Cells. J. Cancer Res. Ther. 2019, 15, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Gombert, M.; Dieu-Nosjean, M.-C.; Bünemann, E.; Kubitza, R.C.; Cunha, D.; Haahtela, A.; Lehtimäki, S.; Müller, A.; Rieker, J.; Meller, S.; et al. CCL1-CCR8 Interactions: An Axis Mediating the Recruitment of T Cells and Langerhans-Type Dendritic Cells to Sites of Atopic Skin Inflammation. J. Immunol. 2005, 174, 5082–5091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenivesse, C.; Chang, Y.; Azzaoui, I.; Ait Yahia, S.; Morales, O.; Plé, C.; Foussat, A.; Tonnel, A.-B.; Delhem, N.; Yssel, H.; et al. Pulmonary CCL18 Recruits Human Regulatory T Cells. J. Immunol. 2012, 189, 128–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Chen, Y.-S.; Yao, Y.-D.; Chen, J.-Q.; Chen, J.-N.; Huang, S.-Y.; Zeng, Y.-J.; Yao, H.-R.; Zeng, S.-H.; Fu, Y.-S.; et al. CCL18 from Tumor-Associated Macrophages Promotes Angiogenesis in Breast Cancer. Oncotarget 2015, 6, 34758–34773. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Du, C.; Xu, P.; Miao, C. Surgical Trauma-Induced CCL18 Promotes Recruitment of Regulatory T Cells and Colon Cancer Progression. J. Cell Physiol. 2019, 2334, 4608–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Liao, J.; Liu, J.; Huang, D.; He, C.; Chen, F.; Yang, L.; Wu, W.; Chen, J.; Lin, L.; et al. Blocking the Recruitment of Naive CD4+ T Cells Reverses Immunosuppression in Breast Cancer. Cell Res. 2017, 27, 461–482. [Google Scholar] [CrossRef] [PubMed]
- Barsheshet, Y.; Wildbaum, G.; Levy, E.; Vitenshtein, A.; Akinseye, C.; Griggs, J.; Lira, S.A.; Karin, N. CCR8+ FOXp3+ Treg Cells as Master Drivers of Immune Regulation. Proc. Natl. Acad. Sci. USA 2017, 114, 6086–6091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelzinger, D.B.; Smith, S.E.; Mirza, N.; Dominguez, A.L.; Manrique, S.Z.; Lustgarten, J. Blockade of CCL1 Inhibits T Regulatory Cell Suppressive Function Enhancing Tumor Immunity without Affecting T Effector Responses. J. Immunol. 2010, 184, 6833–6842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarreal, D.O.; L’Huillier, A.; Armington, S.; Mottershead, C.; Filippova, E.V.; Coder, B.D.; Petit, R.G.; Princiotta, M.F. Targeting CCR8 Induces Protective Antitumor Immunity and Enhances Vaccine-Induced Responses in Colon Cancer. Cancer Res. 2018, 78, 5340–5348. [Google Scholar] [CrossRef] [Green Version]
- Eruslanov, E.; Stoffs, T.; Kim, W.-J.; Daurkin, I.; Gilbert, S.M.; Su, L.-M.; Vieweg, J.; Daaka, Y.; Kusmartsev, S. Expansion of CCR8+ Inflammatory Myeloid Cells in Cancer Patients with Urothelial and Renal Carcinomas. Clin. Cancer Res. 2013, 19, 1670–1680. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, K. The Chemokine CCL2 Increases Prostate Tumor Growth and Bone Metastasis through Macrophage and Osteoclast Recruitment. Neoplasia 2009, 11, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 Recruits Inflammatory Monocytes to Facilitate Breast-Tumour Metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2+ Myeloid-Derived Suppressor Cells Promote Immune Escape by Limiting Activated CD8 T-Cell Infiltration into the Tumor Microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef] [Green Version]
- Loyher, P.-L.; Rochefort, J.; Baudesson de Chanville, C.; Hamon, P.; Lescaille, G.; Bertolus, C.; Guillot-Delost, M.; Krummel, M.F.; Lemoine, F.M.; Combadière, C.; et al. CCR2 Influences T Regulatory Cell Migration to Tumors and Serves as a Biomarker of Cyclophosphamide Sensitivity. Cancer Res. 2016, 76, 6483–6494. [Google Scholar] [CrossRef] [Green Version]
- Craig, M.J.; Loberg, R.D. CCL2 (Monocyte Chemoattractant Protein-1) in Cancer Bone Metastases. Cancer Metastasis Rev. 2006, 25, 611–619. [Google Scholar] [CrossRef]
- Wang, Z.; Xie, H.; Zhou, L.; Liu, Z.; Fu, H.; Zhu, Y.; Xu, L.; Xu, J. CCL2/CCR2 Axis Is Associated with Postoperative Survival and Recurrence of Patients with Non-Metastatic Clear-Cell Renal Cell Carcinoma. Oncotarget 2016, 7, 51525–51534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaszczyk, J.; Coillie, E.V.; Proost, P.; Damme, J.V.; Opdenakker, G.; Bujacz, G.D.; Wang, J.M.; Ji, X. Complete Crystal Structure of Monocyte Chemotactic Protein-2, a CC Chemokine That Interacts with Multiple Receptors. Biochemistry 2000, 39, 14075–14081. [Google Scholar] [CrossRef]
- Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers 2020, 12, 1765. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.; Cihak, J.; Simonis, C.; Luckow, B.; Proudfoot, A.E.I.; Plachý, J.; Brühl, H.; Frink, M.; Anders, H.-J.; Vielhauer, V.; et al. Expression and Characterization of the Chemokine Receptors CCR2 and CCR5 in Mice. J. Immunol. 2001, 166, 4697–4704. [Google Scholar] [CrossRef] [Green Version]
- Ward, S.T.; Li, K.K.; Hepburn, E.; Weston, C.J.; Curbishley, S.M.; Reynolds, G.M.; Hejmadi, R.K.; Bicknell, R.; Eksteen, B.; Ismail, T.; et al. The Effects of CCR5 Inhibition on Regulatory T-Cell Recruitment to Colorectal Cancer. Br. J. Cancer 2015, 112, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, C.E.; Gasparoto, T.H.; Pinheiro, C.R. CCR5-Dependent Homing of T Regulatory Cells to the Tumor Microenvironment Contributes to Skin Squamous Cell Carcinoma Development. Mol. Cancer Ther. 2017, 16, 2871–2880. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.-Y.; Lin, Y.-C.; Kang, C.-W.; Hsu, C.-Y.; Chu, Y.-Y.; Huang, C.-T.; Day, Y.-J.; Chen, T.-C.; Yeh, C.-T.; Lin, C.-Y. The Indispensable Role of CCR5 for In Vivo Suppressor Function of Tumor-Derived CD103+ Effector/Memory Regulatory T Cells. J. Immunol. 2012, 189, 567–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.C.B.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.-S.; Linehan, D.C. Disruption of CCR5-Dependent Homing of Regulatory T Cells Inhibits Tumor Growth in a Murine Model of Pancreatic Cancer. J. Immunol. 2009, 182, 1746–1755. [Google Scholar] [CrossRef]
- Korbecki, J.; Grochans, S.; Gutowska, I.; Barczak, K.; Baranowska-Bosiacka, I. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of Receptors CCR5, CCR6, CCR7, CCR8, CCR9, and CCR10 Ligands. Int. J. Mol. Sci. 2020, 21, 7619. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Imai, T.; Nishimura, M.; Kakizaki, M.; Takagi, S.; Hieshima, K.; Nomiyama, H.; Yoshie, O. Identification of CCR6, the Specific Receptor for a Novel Lymphocyte-Directed CC Chemokine LARC. J. Biol. Chem. 1997, 272, 14893–14898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, T.; Yang, X.O.; Chung, Y.; Fukunaga, A.; Nurieva, R.; Pappu, B.; Kang, H.S.; Ma, L.; Craig, S.; Watowich, S.S.; et al. CCR6 Regulates the Migration of Inflammatory and Regulatory T Cells. J. Immunol. 2008, 181, 8391–8401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hromas, R.; Gray, P.W.; Chantry, D.; Godiska, R.; Krathwohl, M.; Fife, K.; Bell, G.I.; Takeda, J.; Aronica, S.; Gordon, M.; et al. Cloning and Characterization of Exodus, a Novel b-Chemokine. Blood 1997, 89, 3315–3322. [Google Scholar] [PubMed]
- Kadomoto, S.; Izumi, K.; Mizokami, A. The CCL20-CCR6 Axis in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 5186. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, N.; Li, Q.; Zhang, W.; Ke, F.; Leng, Q.; Wang, H.; Chen, J.; Wang, H. Tumor-Associated Macrophages Recruit CCR6+ Regulatory T Cells and Promote the Development of Colorectal Cancer via Enhancing CCL20 Production in Mice. PLoS ONE 2011, 6, e19495. [Google Scholar] [CrossRef]
- Wang, D.; Yang, L.; Yu, W.; Wu, Q.; Lian, J.; Li, F.; Liu, S.; Li, A.; He, Z.; Liu, J.; et al. Colorectal Cancer Cell-Derived CCL20 Recruits Regulatory T Cells to Promote Chemoresistance via FOXO1/CEBPB/NF-ΚB Signaling. J. Immunother. Cancer 2019, 7, 215. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Qi, Y.; Li, X.; Yang, Y.; Liu, D.; Zhao, J.; Zhu, D.; Wu, K.; Zhou, X.; Zhao, S. The Role of CCL20/CCR6 Axis in Recruiting Treg Cells to Tumor Sites of NSCLC Patients. Biomed. Pharmacother. 2015, 69, 242–248. [Google Scholar] [CrossRef]
- Lee, J.-J.; Kao, K.-C.; Chiu, Y.-L.; Jung, C.-J.; Liu, C.-J.; Cheng, S.-J.; Chang, Y.-L.; Ko, J.-Y.; Chia, J.-S. Enrichment of Human CCR6+ Regulatory T Cells with Superior Suppressive Activity in Oral Cancer. J. Immunol. 2017, 199, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Zhang, D.; Zhou, J.; Li, Q.; Zhou, L.; Li, S.-M.; Zhu, L.; Chou, K.-Y.; Zhou, L.; Tao, L.; et al. High CCR6/CCR7 Expression and Foxp3+ Treg Cell Number Are Positively Related to the Progression of Laryngeal Squamous Cell Carcinoma. Oncol. Rep. 2013, 30, 1380–1390. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Y.; Li, F.; Wang, L.P.; Chen, X.F.; Wang, D.; Cao, L.; Ping, Y.; Zhao, S.; Li, B.; Thorne, S.H.; et al. CTL- vs Treg Lymphocyte-Attracting Chemokines, CCL4 and CCL20, Are Strong Reciprocal Predictive Markers for Survival of Patients with Oesophageal Squamous Cell Carcinoma. Br. J. Cancer 2015, 113, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Qin, Y.; Wang, D.; Zhou, L.; Liu, Y.; Chen, S.; Yin, L.; Xiao, Y.; Yao, X.-H.; Yang, X.; et al. CCL20 Triggered by Chemotherapy Hinders the Therapeutic Efficacy of Breast Cancer. PLoS Biol. 2018, 16, e2005869. [Google Scholar] [CrossRef]
- Marsigliante CCL20 Induces Migration and Proliferation on Breast Epithelial Cells. J. Cell Physiol. 2013, 228, 1873–1883. [CrossRef]
- Chen, D. Chemokine/Chemokine Receptor Interactions Contribute to the Accumulation of Th17 Cells in Patients with Esophageal Squamous Cell Carcinoma. Hum. Immunol. 2012, 73, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- Kirshberg, S.; Izhar, U.; Amir, G.; Demma, J.; Vernea, F.; Beider, K.; Shlomai, Z.; Wald, H.; Zamir, G.; Shapira, O.M.; et al. Involvement of CCR6/CCL20/IL-17 Axis in NSCLC Disease Progression. PLoS ONE 2011, 6, e24856. [Google Scholar] [CrossRef] [PubMed]
- Kesselring, R.; Thiel, A.; Pries, R.; Trenkle, T.; Wollenberg, B. Human Th17 Cells Can Be Induced through Head and Neck Cancer and Have a Functional Impact on HNSCC Development. Br. J. Cancer 2010, 103, 1245–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Soto, H.; Oldham, E.R.; Buchanan, M.E.; Homey, B.; Catron, D.; Jenkins, N.; Copeland, N.G.; Gilbert, D.J.; Nguyen, N.; et al. Identification of a Novel Chemokine (CCL28), Which Binds CCR10 (GPR2). J. Biol. Chem. 2000, 275, 22313–22323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler, D.; Humphreys, T.L.; Spinola, S.M.; Campbell, J.J. CCR4 versus CCR10 in Human Cutaneous TH Lymphocyte Trafficking. Blood 2003, 101, 1677–1682. [Google Scholar] [CrossRef] [Green Version]
- Eksteen, B.; Miles, A.; Curbishley, S.M.; Tselepis, C.; Grant, A.J.; Walker, L.S.K.; Adams, D.H. Epithelial Inflammation Is Associated with CCL28 Production and the Recruitment of Regulatory T Cells Expressing CCR10. J. Immunol. 2006, 177, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Facciabene, A. Tumour Hypoxia Promotes Tolerance and Angiogenesis via CCL28 and Treg Cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Yu, Y.; Wang, L.; Zhu, Z.; Lu, R.; Yao, Z. Hypoxia-Induced CCL28 Promotes Recruitment of Regulatory T Cells and Tumor Growth in Liver Cancer. Oncotarget 2016, 7, 75763–75773. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shoshan, J.; Maysel-Auslender, S.; Mor, A.; Keren, G.; George, J. Hypoxia Controls CD4+CD25+ Regulatory T-Cell Homeostasis via Hypoxia-Inducible Factor-1α. Eur. J. Immunol. 2008, 38, 2412–2418. [Google Scholar] [CrossRef] [PubMed]
- Westendorf, A.M.; Skibbe, K.; Adamczyk, A.; Buer, J.; Geffers, R.; Hansen, W.; Pastille, E.; Jendrossek, V. Hypoxia Enhances Immunosuppression by Inhibiting CD4+ Effector T Cell Function and Promoting Treg Activity. Cell Physiol. Biochem. 2017, 41, 1271–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Jene, N.; Byrne, D.; Millar, E.K.; Bates, G.J.; Harris, A.L.; Banham, A.H.; Sutherland, R.L.; Fox, S.B. Recruitment of Regulatory T Cells Is Correlated with Hypoxia-Induced CXCR4 Expression, and Is Associated with Poor Prognosis in Basal-like Breast Cancers. Breast Cancer Res. 2011, 13, R47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wald, O.; Izhar, U.; Amir, G.; Avniel, S.; Bar-Shavit, Y.; Wald, H.; Weiss, I.D.; Galun, E.; Peled, A. CD4+ CXCR4high CD69+ T Cells Accumulate in Lung Adenocarcinoma. J. Immunol. 2006, 177, 6983–6990. [Google Scholar] [CrossRef] [Green Version]
- Jaafar, F. Correlation of CXCL12 Expression and FoxP3+ Cell Infiltration with Human Papillomavirus Infection and Clinicopathological Progression of Cervical Cancer. Am. J. Pathol. 2009, 175, 1525–1535. [Google Scholar] [CrossRef] [Green Version]
- Metzemaekers, M.; Vanheule, V.; Janssens, R.; Struyf, S.; Proost, P. Overview of the Mechanisms That May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Front. Immunol. 2018, 8, 1970. [Google Scholar] [CrossRef]
- Reynders, N.; Abboud, D.; Baragli, A.; Noman, M.Z.; Rogister, B.; Niclou, S.P.; Heveker, N.; Janji, B.; Hanson, J.; Szpakowska, M.; et al. The Distinct Roles of CXCR3 Variants and Their Ligands in the Tumor Microenvironment. Cells 2019, 8, 613. [Google Scholar] [CrossRef] [Green Version]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-Redundant Requirement for CXCR3 Signalling during Tumoricidal T-Cell Trafficking across Tumour Vascular Checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e4. [Google Scholar] [CrossRef] [Green Version]
- Pekarek, V.; Srinivas, S.; Eskdale, J.; Gallagher, G. Interferon Lambda-1 (IFN-L1/IL-29) Induces ELR- CXC Chemokine MRNA in Human Peripheral Blood Mononuclear Cells, in an IFN-g-Independent Manner. Genes Immun. 2007, 8, 177–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, M.A.; Tucker-Heard, G.; Perdue, N.R.; Killebrew, J.R.; Urdahl, K.B.; Campbell, D.J. The Transcription Factor T-Bet Controls Regulatory T Cell Homeostasis and Function during Type 1 Inflammation. Nat. Immunol. 2009, 10, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Hoerning, A.; Koss, K.; Datta, D.; Boneschansker, L.; Jones, C.N.; Wong, I.Y.; Irimia, D.; Calzadilla, K.; Benitez, F.; Hoyer, P.F.; et al. Subsets of Human CD4+ Regulatory T Cells Express the Peripheral Homing Receptor CXCR3. Eur. J. Immunol. 2011, 41, 2291–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClymont, S.A.; Putnam, A.L.; Lee, M.R.; Esensten, J.H.; Liu, W.; Hulme, M.A.; Hoffmüller, U.; Baron, U.; Olek, S.; Bluestone, J.A.; et al. Plasticity of Human Regulatory T Cells in Healthy Subjects and Patients with Type 1 Diabetes. J. Immunol. 2011, 186, 3918–3926. [Google Scholar] [CrossRef]
- Ferreira, C.; Barros, L.; Baptista, M.; Blankenhaus, B.; Barros, A.; Figueiredo-Campos, P.; Konjar, Š.; Lainé, A.; Kamenjarin, N.; Stojanovic, A.; et al. Type 1 Treg Cells Promote the Generation of CD8+ Tissue-Resident Memory T Cells. Nat. Immunol. 2020, 21, 766–776. [Google Scholar] [CrossRef]
- Mami-Chouaib, F.; Blanc, C.; Corgnac, S.; Hans, S.; Malenica, I.; Granier, C.; Tihy, I.; Tartour, E. Resident Memory T Cells, Critical Components in Tumor Immunology. J. Immunother. Cancer 2018, 6, 87. [Google Scholar] [CrossRef]
- Redjimi, N.; Raffin, C.; Raimbaud, I.; Pignon, P.; Matsuzaki, J.; Odunsi, K.; Valmori, D.; Ayyoub, M. CXCR3+ T Regulatory Cells Selectively Accumulate in Human Ovarian Carcinomas to Limit Type I Immunity. Cancer Res. 2012, 72, 4351–4360. [Google Scholar] [CrossRef] [Green Version]
- Li, C.X.; Ling, C.C.; Shao, Y.; Xu, A.; Li, X.C.; Ng, K.T.-P.; Liu, X.B.; Ma, Y.Y.; Qi, X.; Liu, H.; et al. CXCL10/CXCR3 Signaling Mobilized-Regulatory T Cells Promote Liver Tumor Recurrence after Transplantation. J. Hepatol. 2016, 65, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Winkler, A.E.; Brotman, J.J.; Pittman, M.E.; Judd, N.P.; Lewis, J.S.; Schreiber, R.D.; Uppaluri, R. CXCR3 Enhances a T-Cell-Dependent Epidermal Proliferative Response and Promotes Skin Tumorigenesis. Cancer Res. 2011, 71, 5707–5716. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, D.A.; Sengupta, S.; Han, Y.; Lesniak, M.S. Thymus-Derived Rather than Tumor-Induced Regulatory T Cells Predominate in Brain Tumors. Neuro Oncol. 2011, 13, 1308–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Juedes, A.E.; Temann, U.-A.; Shresta, S.; Allison, J.P.; Ruddle, N.H.; Flavell, R.A. ICOS Co-Stimulatory Receptor Is Essential for T-Cell Activation and Function. Nature 2001, 409, 97–101. [Google Scholar] [CrossRef]
- Faget, J.; Bendriss-Vermare, N.; Gobert, M.; Durand, I.; Olive, D.; Biota, C.; Bachelot, T.; Treilleux, I.; Goddard-Leon, S.; Lavergne, E.; et al. ICOS-Ligand Expression on Plasmacytoid Dendritic Cells Supports Breast Cancer Progression by Promoting the Accumulation of Immunosuppressive CD4+ T Cells. Cancer Res. 2012, 72, 6130–6141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, C.; Gregorio, J.; Wang, Y.-H.; Ito, T.; Meller, S.; Hanabuchi, S.; Anderson, S.; Atkinson, N.; Ramirez, P.T.; Liu, Y.-J.; et al. Plasmacytoid Dendritic Cells Promote Immunosuppression in Ovarian Cancer via ICOS Costimulation of Foxp3+ T-Regulatory Cells. Cancer Res. 2012, 72, 5240–5249. [Google Scholar] [CrossRef] [Green Version]
- Toker, A.; Nguyen, L.T.; Stone, S.C.; Yang, S.Y.C.; Katz, S.R.; Shaw, P.A.; Clarke, B.A.; Ghazarian, D.; Al-Habeeb, A.; Easson, A.; et al. Regulatory T Cells in Ovarian Cancer Are Characterized by a Highly Activated Phenotype Distinct from That in Melanoma. Clin. Cancer Res. 2018, 24, 5685–5696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, K.-S.; Thibult, M.-L.; Just-Landi, S.; Pastor, S.; Gondois-Rey, F.; Granjeaud, S.; Broussais, F.; Bouabdallah, R.; Colisson, R.; Caux, C.; et al. Follicular B Lymphomas Generate Regulatory T Cells via the ICOS/ICOSL Pathway and Are Susceptible to Treatment by Anti-ICOS/ICOSL Therapy. Cancer Res. 2016, 76, 4648–4660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Orozco, N.; Li, Y.; Wang, Y.; Liu, S.; Hwu, P.; Liu, Y.-J.; Dong, C.; Radvanyi, L. Melanoma Cells Express ICOS Ligand to Promote the Activation and Expansion of T-Regulatory Cells. Cancer Res. 2010, 70, 9581–9590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, L.; Bergmann, C.; Szczepanski, M.J.; Lang, S.; Kirkwood, J.M.; Whiteside, T.L. Expression of ICOS on Human Melanoma-Infiltrating CD4+ CD25high Foxp3+ T Regulatory Cells: Implications and Impact on Tumor-Mediated Immune Suppression. J. Immunol. 2008, 180, 2967–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraldo, N.A.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A.; et al. Tumor-Infiltrating and Peripheral Blood T-Cell Immunophenotypes Predict Early Relapse in Localized Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, H.; Takeoka, T.; Urakawa, S.; Morimoto-Okazawa, A.; Kawashima, A.; Iwahori, K.; Takiguchi, S.; Nishikawa, H.; Sato, E.; Sakaguchi, S.; et al. ICOS+ Foxp3+ TILs in Gastric Cancer Are Prognostic Markers and Effector Regulatory T Cells Associated with H Elicobacter Pylori: Unique Expression of ICOS on TREGS in Gastric Cancer. Int. J. Cancer 2017, 140, 686–695. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [Green Version]
- Saleh, R.; Taha, R.Z.; Toor, S.M.; Sasidharan Nair, V.; Murshed, K.; Khawar, M.; Al-Dhaheri, M.; Petkar, M.A.; Abu Nada, M.; Elkord, E. Expression of Immune Checkpoints and T Cell Exhaustion Markers in Early and Advanced Stages of Colorectal Cancer. Cancer Immunol. Immunother. 2020, 69, 1989–1999. [Google Scholar] [CrossRef]
- Huang, X.; Liu, X.; Lin, X.; Yu, H.; Sun, J.; Liu, X.; Chen, C.; Jin, H.; Zhang, G.; Shi, X.; et al. Role of Plasmacytoid Dendritic Cells and Inducible Costimulator-positive Regulatory T Cells in the Immunosuppression Microenvironment of Gastric Cancer. Cancer Sci. 2014, 105, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Dong, Y.; Yang, Q.; Xu, W.; Jiang, S.; Yu, Z.; Yu, K.; Zhang, S. Acute Myeloid Leukemia Cells Express ICOS Ligand to Promote the Expansion of Regulatory T Cells. Front. Immunol. 2018, 9, 2227. [Google Scholar] [CrossRef] [Green Version]
- Burlion, A.; Ramos, R.N.; Kc, P.; Sendeyo, K.; Corneau, A.; Ménétrier-Caux, C.; Piaggio, E.; Olive, D.; Caux, C.; Marodon, G. A Novel Combination of Chemotherapy and Immunotherapy Controls Tumor Growth in Mice with a Human Immune System. OncoImmunology 2019, 8, e1596005. [Google Scholar] [CrossRef]
- Ito, T.; Hanabuchi, S.; Wang, Y.-H.; Park, W.R.; Arima, K.; Bover, L.; Qin, F.X.-F.; Gilliet, M.; Liu, Y.-J. Two Functional Subsets of FOXP3+ Regulatory T Cells in Human Thymus and Periphery. Immunity 2008, 28, 870–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Mo, L.; Cai, X.; Wei, L.; Xie, Z.; Li, H.; Li, J.; Hu, Z. ICOS Signal Facilitates Foxp3 Transcription to Favor Suppressive Function of Regulatory T Cells. Int. J. Med. Sci. 2018, 15, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landuyt, A.E.; Klocke, B.J.; Colvin, T.B.; Schoeb, T.R.; Maynard, C.L. Cutting Edge: ICOS-Deficient Regulatory T Cells Display Normal Induction of Il10 but Readily Downregulate Expression of Foxp3. J. Immunol. 2019, 202, 1039–1044. [Google Scholar] [CrossRef] [Green Version]
- Burmeister, Y.; Lischke, T.; Dahler, A.C.; Mages, H.W.; Lam, K.-P.; Coyle, A.J.; Kroczek, R.A.; Hutloff, A. ICOS Controls the Pool Size of Effector-Memory and Regulatory T Cells. J. Immunol. 2008, 180, 774–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, A.E.; Freeman, G.J.; Mathis, D.; Benoist, C. CD4+CD25+ T Regulatory Cells Dependent on ICOS Promote Regulation of Effector Cells in the Prediabetic Lesion. J. Exp. Med. 2004, 199, 1479–1489. [Google Scholar] [CrossRef]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF Receptor Superfamilies. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Xie, P. TRAF Molecules in Cell Signaling and in Human Diseases. J. Mol. Signal. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Chee, J.; Angstetra, E.; Mariana, L.; Graham, K.L.; Carrington, E.M.; Bluethmann, H.; Santamaria, P.; Allison, J.; Kay, T.W.H.; Krishnamurthy, B.; et al. TNF Receptor 1 Deficiency Increases Regulatory T Cell Function in Nonobese Diabetic Mice. J. Immunol. 2011, 187, 1702–1712. [Google Scholar] [CrossRef]
- Chopra, M.; Biehl, M.; Steinfatt, T.; Brandl, A.; Kums, J.; Amich, J.; Vaeth, M.; Kuen, J.; Holtappels, R.; Podlech, J.; et al. Exogenous TNFR2 Activation Protects from Acute GvHD via Host TReg Cell Expansion. J. Exp. Med. 2016, 213, 1881–1900. [Google Scholar] [CrossRef]
- Lubrano di Ricco, M.; Ronin, E.; Collares, D.; Divoux, J.; Grégoire, S.; Wajant, H.; Gomes, T.; Grinberg-Bleyer, Y.; Baud, V.; Marodon, G.; et al. Tumor Necrosis Factor Receptor Family Costimulation Increases Regulatory T-cell Activation and Function via NF-κB. Eur. J. Immunol. 2020, 50, 972–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Subleski, J.J.; Kopf, H.; Howard, O.M.Z.; Männel, D.N.; Oppenheim, J.J. Expression of TNFR2 Defines a Maximally Suppressive Subset of Mouse CD4+CD25+FoxP3+ T Regulatory Cells: Applicability to Tumor Infiltrating T Regulatory Cells. J. Immunol. 2008, 180, 6467–6471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Landman, S.; Bauland, S.C.G.; van den Dolder, J.; Koenen, H.J.P.M.; Joosten, I. A TNFR2-Agonist Facilitates High Purity Expansion of Human Low Purity Treg Cells. PLoS ONE 2016, 11, e0156311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamontain, V.; Schmid, T.; Weber-Steffens, D.; Zeller, D.; Jenei-Lanzl, Z.; Wajant, H.; Straub, R.H.; Männel, D.N. Stimulation of TNF Receptor Type 2 Expands Regulatory T Cells and Ameliorates Established Collagen-Induced Arthritis in Mice. Cell Mol. Immunol. 2019, 16, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Okubo, Y.; Mera, T.; Wang, L.; Faustman, D.L. Homogeneous Expansion of Human T-Regulatory Cells Via Tumor Necrosis Factor Receptor 2. Sci. Rep. 2013, 3, 3153. [Google Scholar] [CrossRef] [Green Version]
- Atretkhany, K.-S.N.; Mufazalov, I.A.; Dunst, J.; Kuchmiy, A.; Gogoleva, V.S.; Andruszewski, D.; Drutskaya, M.S.; Faustman, D.L.; Schwabenland, M.; Prinz, M.; et al. Intrinsic TNFR2 Signaling in T Regulatory Cells Provides Protection in CNS Autoimmunity. Proc. Natl. Acad. Sci. USA 2018, 115, 13051–13056. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.W.; Chen, Q.Q.; Cao, J.; Xu, L.Q.; Tang, X.; Wang, J.; Zhang, J.; Dong, L.X. Expression of Tumor Necrosis Factor Receptor 2 in Human Non-small Cell Lung Cancer and Its Role as a Potential Prognostic Biomarker. Thorac. Cancer 2019, 10, 437–444. [Google Scholar] [CrossRef]
- Zhang, T.; Jiao, J.; Jiao, X.; Zhao, L.; Tian, X.; Zhang, Q.; Ma, D.; Cui, B. Aberrant Frequency of TNFR2+ Treg and Related Cytokines in Patients with CIN and Cervical Cancer. Oncotarget 2017, 9, 5073–5083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamano, R.; Huang, J.; Yoshimura, T.; Oppenheim, J.J.; Chen, X. TNF Optimally Activatives Regulatory T Cells by Inducing TNF Receptor Superfamily Members TNFR2, 4-1BB and OX40: Immunomodulation. Eur. J. Immunol. 2011, 41, 2010–2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindaraj, C.; Scalzo-Inguanti, K.; Madondo, M.; Hallo, J.; Flanagan, K.; Quinn, M.; Plebanski, M. Impaired Th1 Immunity in Ovarian Cancer Patients Is Mediated by TNFR2+ Tregs within the Tumor Microenvironment. Clin. Immunol. 2013, 149, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Kampan, N.C.; Madondo, M.T.; McNally, O.M.; Stephens, A.N.; Quinn, M.A.; Plebanski, M. Interleukin 6 Present in Inflammatory Ascites from Advanced Epithelial Ovarian Cancer Patients Promotes Tumor Necrosis Factor Receptor 2-Expressing Regulatory T Cells. Front. Immunol. 2017, 8, 1482. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.A.; Lin, Y.; Yeku, O.; LaFramboise, W.A.; Ashraf, M.; Sander, C.; Kirkwood, J.M.; Lee, S. A Four-Marker Signature of TNF-RII, TGF-α, TIMP-1 and CRP Is Prognostic of Worse Survival in High-Risk Surgically Resected Melanoma. J. Transl. Med. 2014, 12, 19. [Google Scholar] [CrossRef] [Green Version]
- Chopra, M.; Riedel, S.S.; Biehl, M.; Krieger, S.; von Krosigk, V.; Bäuerlein, C.A.; Brede, C.; Jordan Garrote, A.-L.; Kraus, S.; Schäfer, V.; et al. Tumor Necrosis Factor Receptor 2-Dependent Homeostasis of Regulatory T Cells as a Player in TNF-Induced Experimental Metastasis. Carcinogenesis 2013, 34, 1296–1303. [Google Scholar] [CrossRef]
- Chang, L.-Y.; Lin, Y.-C.; Chiang, J.-M.; Mahalingam, J.; Su, S.-H.; Huang, C.-T.; Chen, W.-T.; Huang, C.-H.; Jeng, W.-J.; Chen, Y.-C.; et al. Blockade of TNF-α Signaling Benefits Cancer Therapy by Suppressing Effector Regulatory T Cell Expansion. OncoImmunology 2015, 4, e1040215. [Google Scholar] [CrossRef]
- Torrey, H.; Butterworth, J.; Mera, T.; Okubo, Y.; Wang, L.; Baum, D.; Defusco, A.; Plager, S.; Warden, S.; Huang, D.; et al. Targeting TNFR2 with Antagonistic Antibodies Inhibits Proliferation of Ovarian Cancer Cells and Tumor-Associated Tregs. Sci. Signal. 2017, 10, eaaf8608. [Google Scholar] [CrossRef] [PubMed]
- Torrey, H.; Khodadoust, M.; Tran, L.; Baum, D.; Defusco, A.; Kim, Y.H.; Faustman, D.L. Targeted Killing of TNFR2-Expressing Tumor Cells and Tregs by TNFR2 Antagonistic Antibodies in Advanced Sézary Syndrome. Leukemia 2019, 33, 1206–1218. [Google Scholar] [CrossRef] [Green Version]
- McHugh, R.S.; Whitters, M.J.; Piccirillo, C.A.; Young, D.A.; Shevach, E.M.; Collins, M.; Byrne, M.C. CD4+CD25+ Immunoregulatory T Cells: Gene Expression Analysis Reveals a Functional Role for the Glucocorticoid-Induced TNF Receptor. Immunity 2002, 16, 311–323. [Google Scholar] [CrossRef] [Green Version]
- So, T.; Croft, M. Cutting Edge: OX40 Inhibits TGF-β- and Antigen-Driven Conversion of Naive CD4 T Cells into CD25+ Foxp3+ T Cells. J. Immunol. 2007, 179, 1427–1430. [Google Scholar] [CrossRef] [Green Version]
- Sukumar, S.; Wilson, D.C.; Yu, Y.; Wong, J.; Naravula, S.; Ermakov, G.; Riener, R.; Bhagwat, B.; Necheva, A.S.; Grein, J.; et al. Characterization of MK-4166, a Clinical Agonistic Antibody That Targets Human GITR and Inhibits the Generation and Suppressive Effects of T Regulatory Cells. Cancer Res. 2017, 77, 4378–4388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, M.D.; Xiao, X.; Gao, W.; Degauque, N.; Chen, M.; Kroemer, A.; Killeen, N.; Ishii, N.; Chang Li, X. OX40 Costimulation Turns off Foxp3+ Tregs. Blood 2007, 110, 2501–2510. [Google Scholar] [CrossRef] [Green Version]
- De Kleer, I.M.; Wedderburn, L.R.; Taams, L.S.; Patel, A.; Varsani, H.; Klein, M.; de Jager, W.; Pugayung, G.; Giannoni, F.; Rijkers, G.; et al. CD4+ CD25bright Regulatory T Cells Actively Regulate Inflammation in the Joints of Patients with the Remitting Form of Juvenile Idiopathic Arthritis. J. Immunol. 2004, 172, 6435–6443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenbrunn, A.; Frentsch, M.; Kohler, S.; Keye, J.; Dooms, H.; Moewes, B.; Dong, J.; Loddenkemper, C.; Sieper, J.; Wu, P.; et al. A Converse 4-1BB and CD40 Ligand Expression Pattern Delineates Activated Regulatory T Cells (Treg) and Conventional T Cells Enabling Direct Isolation of Alloantigen-Reactive Natural Foxp3+ Treg. J. Immunol. 2012, 189, 5985–5994. [Google Scholar] [CrossRef] [Green Version]
- Voo, K.S.; Bover, L.; Harline, M.L.; Vien, L.T.; Facchinetti, V.; Arima, K.; Kwak, L.W.; Liu, Y.J. Antibodies Targeting Human OX40 Expand Effector T Cells and Block Inducible and Natural Regulatory T Cell Function. J. Immunol. 2013, 191, 3641–3650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ephrem, A.; Epstein, A.L.; Stephens, G.L.; Thornton, A.M.; Glass, D.; Shevach, E.M. Modulation of Treg Cells/T Effector Function by GITR Signaling Is Context-Dependent: Immunomodulation. Eur. J. Immunol. 2013, 43, 2421–2429. [Google Scholar] [CrossRef]
- Liao, G.; Nayak, S.; Regueiro, J.R.; Berger, S.B.; Detre, C.; Romero, X.; de Waal Malefyt, R.; Chatila, T.A.; Herzog, R.W.; Terhorst, C. GITR Engagement Preferentially Enhances Proliferation of Functionally Competent CD4+CD25+FoxP3+ Regulatory T Cells. Int. Immunol. 2010, 22, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Mahmud, S.A.; Manlove, L.S.; Schmitz, H.M.; Xing, Y.; Wang, Y.; Owen, D.L.; Schenkel, J.M.; Boomer, J.S.; Green, J.M.; Yagita, H.; et al. Costimulation via the Tumor-Necrosis Factor Receptor Superfamily Couples TCR Signal Strength to the Thymic Differentiation of Regulatory T Cells. Nat. Immunol. 2014, 15, 473–481. [Google Scholar] [CrossRef]
- Ni, X.Y.; Sui, H.X.; Liu, Y.; Ke, S.Z.; Wang, Y.N.; Gao, F.G. TGF-β of Lung Cancer Microenvironment Upregulates B7H1 and GITRL Expression in Dendritic Cells and Is Associated with Regulatory T Cell Generation. Oncol. Rep. 2012, 28, 615–621. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Shin, S.M.; Choi, B.K.; Oh, H.S.; Kim, C.H.; Lee, S.J.; Kim, K.H.; Lee, D.G.; Park, S.H.; Kwon, B.S. Authentic GITR Signaling Fails To Induce Tumor Regression Unless Foxp3+ Regulatory T Cells Are Depleted. J. Immunol. 2015, 195, 4721–4729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Olffen, R.W.; Koning, N.; van Gisbergen, K.P.J.M.; Wensveen, F.M.; Hoek, R.M.; Boon, L.; Hamann, J.; van Lier, R.A.W.; Nolte, M.A. GITR Triggering Induces Expansion of Both Effector and Regulatory CD4+ T Cells In Vivo. J. Immunol. 2009, 182, 7490–7500. [Google Scholar] [CrossRef] [Green Version]
- Heinhuis, K.M.; Carlino, M.; Joerger, M.; Di Nicola, M.; Meniawy, T.; Rottey, S.; Moreno, V.; Gazzah, A.; Delord, J.-P.; Paz-Ares, L.; et al. Safety, Tolerability, and Potential Clinical Activity of a Glucocorticoid-Induced TNF Receptor–Related Protein Agonist Alone or in Combination with Nivolumab for Patients With Advanced Solid Tumors: A Phase 1/2a Dose-Escalation and Cohort-Expansion Clinical Trial. JAMA Oncol. 2019, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Aspeslagh, S.; Postel-Vinay, S.; Rusakiewicz, S.; Soria, J.-C.; Zitvogel, L.; Marabelle, A. Rationale for Anti-OX40 Cancer Immunotherapy. Eur. J. Cancer 2016, 52, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Kohrt, H.; Sagiv-Barfi, I.; Ajami, B.; Axtell, R.C.; Zhou, G.; Rajapaksa, R.; Green, M.R.; Torchia, J.; Brody, J.; et al. Depleting Tumor-Specific Tregs at a Single Site Eradicates Disseminated Tumors. J. Clin. Investig. 2013, 123, 2447–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulliard, Y.; Jolicoeur, R.; Zhang, J.; Dranoff, G.; Wilson, N.S.; Brogdon, J.L. OX40 Engagement Depletes Intratumoral Tregs via Activating FcγRs, Leading to Antitumor Efficacy. Immunol. Cell Biol. 2014, 92, 475–480. [Google Scholar] [CrossRef]
- Piconese, S.; Timperi, E.; Pacella, I.; Schinzari, V.; Tripodo, C.; Rossi, M.; Guglielmo, N.; Mennini, G.; Grazi, G.L.; Filippo, S.D.; et al. Human OX40 Tunes the Function of Regulatory T Cells in Tumor and Nontumor Areas of Hepatitis C Virus–Infected Liver Tissue. Hepatology 2014, 60, 1494–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chen, Y.; Miao, L.; Wang, Y.; Yu, M.; Yan, X.; Zhao, Q.; Cai, H.; Xiao, Y.; Huang, G. Stress-Induced Upregulation of TNFSF4 in Cancer-Associated Fibroblast Facilitates Chemoresistance of Lung Adenocarcinoma through Inhibiting Apoptosis of Tumor Cells. Cancer Lett. 2021, 497, 212–220. [Google Scholar] [CrossRef]
- Kitamura, N.; Murata, S.; Ueki, T.; Mekata, E.; Reilly, R.T.; Jaffee, E.M.; Tani, T. OX40 Costimulation Can Abrogate Foxp3+ Regulatory T Cell-Mediated Suppression of Antitumor Immunity. Int. J. Cancer 2009, 125, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Oberst, M.D.; Auge, C.; Morris, C.; Kentner, S.; Mulgrew, K.; McGlinchey, K.; Hair, J.; Hanabuchi, S.; Du, Q.; Damschroder, M.; et al. An Engineered Human OX40 Ligand IgG4P Fc Fusion Protein. Mol. Cancer Ther. 2018, 17, 1024–1038. [Google Scholar] [CrossRef] [Green Version]
- Nowak, A.; Lock, D.; Bacher, P.; Hohnstein, T.; Vogt, K.; Gottfreund, J.; Giehr, P.; Polansky, J.K.; Sawitzki, B.; Kaiser, A.; et al. CD137+CD154− Expression As a Regulatory T Cell (Treg)-Specific Activation Signature for Identification and Sorting of Stable Human Tregs from In Vitro Expansion Cultures. Front. Immunol. 2018, 9, 199. [Google Scholar] [CrossRef] [Green Version]
- Elpek, K.G.; Yolcu, E.S.; Franke, D.D.H.; Lacelle, C.; Schabowsky, R.-H.; Shirwan, H. Ex Vivo Expansion of CD4+ CD25+ FoxP3+ T Regulatory Cells Based on Synergy between IL-2 and 4-1BB Signaling. J. Immunol. 2007, 179, 7295–7304. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Gao, F.; Wang, Q.; Wang, X.; Zhu, F.; Ma, C.; Sun, W.; Zhang, L. Agonistic Anti-4-1BB Antibody Promotes the Expansion of Natural Regulatory T Cells While Maintaining Foxp3 Expression. Scand. J. Immunol. 2007, 66, 435–440. [Google Scholar] [CrossRef]
- Choi, B.K.; Bae, J.S.; Choi, E.M.; Kang, W.J.; Sakaguchi, S.; Vinay, D.S.; Kwon, B.S. 4-1BB-Dependent Inhibition of Immunosuppression by Activated CD4+ CD25+ T Cells. J. Leukoc. Biol. 2004, 75, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Madireddi, S.; Schabowsky, R.-H.; Srivastava, A.K.; Sharma, R.K.; Yolcu, E.S.; Shirwan, H. SA-4-1BBL Costimulation Inhibits Conversion of Conventional CD4+ T Cells into CD4+FoxP3+ T Regulatory Cells by Production of IFN-γ. PLoS ONE 2012, 7, e42459. [Google Scholar] [CrossRef]
- Wang, L.; Simons, D.L.; Lu, X.; Tu, T.Y.; Solomon, S.; Wang, R.; Rosario, A.; Avalos, C.; Schmolze, D.; Yim, J.; et al. Connecting Blood and Intratumoral Treg Cell Activity in Predicting Future Relapse in Breast Cancer. Nat. Immunol. 2019, 20, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, Y.; Zhang, J.-P.; Liang, J.; Li, L.; Zheng, L. Tim-3 Expression Defines Regulatory T Cells in Human Tumors. PLoS ONE 2013, 8, e58006. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Khaja, A.S.S.; Toor, S.M.; El Salhat, H.; Faour, I.; Ul Haq, N.; Ali, B.R.; Elkord, E. Preferential Accumulation of Regulatory T Cells with Highly Immunosuppressive Characteristics in Breast Tumor Microenvironment. Oncotarget 2017, 8, 33159–33171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.R.; Park, H.J.; Son, J.; Lee, J.G.; Chung, K.Y.; Cho, N.H.; Shim, H.S.; Park, S.; Kim, G.; In Yoon, H.; et al. Tumor Microenvironment Dictates Regulatory T Cell Phenotype: Upregulated Immune Checkpoints Reinforce Suppressive Function. J. Immunother. Cancer 2019, 7, 339. [Google Scholar] [CrossRef]
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1+ Regulatory T Cells Amplified by PD-1 Blockade Promote Hyperprogression of Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 9999–10008. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory Effect of Tumor Cell–Derived Lactic Acid on Human T Cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Procaccini, C.; De Rosa, V.; Galgani, M.; Abanni, L.; Calì, G.; Porcellini, A.; Carbone, F.; Fontana, S.; Horvath, T.L.; La Cava, A.; et al. An Oscillatory Switch in MTOR Kinase Activity Sets Regulatory T Cell Responsiveness. Immunity 2010, 33, 929–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishore, M.; Cheung, K.C.P.; Fu, H.; Bonacina, F.; Wang, G.; Coe, D.; Ward, E.J.; Colamatteo, A.; Jangani, M.; Baragetti, A.; et al. Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity 2017, 47, 875–889.e10. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, D.; Bhattacharya, R.; Sinha, B.P.; Liu, C.S.C.; Ghosh, A.R.; Rahaman, O.; Bandopadhyay, P.; Sarif, J.; D’Rozario, R.; Paul, S.; et al. Lactate Induces Pro-Tumor Reprogramming in Intratumoral Plasmacytoid Dendritic Cells. Front. Immunol. 2019, 10, 1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, S.E.; Singer, B.D.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martínez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial Complex III Is Essential for Suppressive Function of Regulatory T Cells. Nature 2019, 565, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Franco, F.; Tsui, Y.-C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.-H.; Schulze, I.; et al. CD36-Mediated Metabolic Adaptation Supports Regulatory T Cell Survival and Function in Tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J.; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [Green Version]
- Muroski, M.E.; Miska, J.; Chang, A.L.; Zhang, P.; Rashidi, A.; Moore, H.; Lopez-Rosas, A.; Han, Y.; Lesniak, M.S. Fatty Acid Uptake in T Cell Subsets Using a Quantum Dot Fatty Acid Conjugate. Sci. Rep. 2017, 7, 5790. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, S.; Togashi, Y.; Sakai, C.; Kawazoe, A.; Kawazu, M.; Ueno, T.; Sato, E.; Kuwata, T.; Kinoshita, T.; Yamamoto, M.; et al. An Oncogenic Alteration Creates a Microenvironment That Promotes Tumor Progression by Conferring a Metabolic Advantage to Regulatory T Cells. Immunity 2020, 53, 187–203.e8. [Google Scholar] [CrossRef]
- Miska, J.; Lee-Chang, C.; Rashidi, A.; Muroski, M.E.; Chang, A.L.; Lopez-Rosas, A.; Zhang, P.; Panek, W.K.; Cordero, A.; Han, Y.; et al. HIF-1α Is a Metabolic Switch between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep. 2019, 27, 226–237.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floess, S.; Freyer, J.; Siewert, C.; Baron, U.; Olek, S.; Polansky, J.; Schlawe, K.; Chang, H.-D.; Bopp, T.; Schmitt, E.; et al. Epigenetic Control of the Foxp3 Locus in Regulatory T Cells. PLoS Biol. 2007, 5, e38. [Google Scholar] [CrossRef]
- Bailey-Bucktrout, S.L.; Martinez-Llordella, M.; Zhou, X.; Anthony, B.; Rosenthal, W.; Luche, H.; Fehling, H.J.; Bluestone, J.A. Self-Antigen-Driven Activation Induces Instability of Regulatory T Cells during an Inflammatory Autoimmune Response. Immunity 2013, 39, 949–962. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic Conversion of Foxp3+ T Cells into TH17 Cells in Autoimmune Arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef]
- Zhou, X.; Bailey-Bucktrout, S.L.; Jeker, L.T.; Penaranda, C.; Martínez-Llordella, M.; Ashby, M.; Nakayama, M.; Rosenthal, W.; Bluestone, J.A. Instability of the Transcription Factor Foxp3 Leads to the Generation of Pathogenic Memory T Cells in Vivo. Nat. Immunol. 2009, 10, 1000–1007. [Google Scholar] [CrossRef]
- Kim, H.-J.; Barnitz, R.A.; Kreslavsky, T.; Brown, F.D.; Moffett, H.; Lemieux, M.E.; Kaygusuz, Y.; Meissner, T.; Holderried, T.A.W.; Chan, S.; et al. Stable Inhibitory Activity of Regulatory T Cells Requires the Transcription Factor Helios. Science 2015, 350, 334–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, M.; Lopez-Ocasio, M.; Metidji, A.; Rieder, S.A.; Shevach, E.M.; Thornton, A.M. Helios Controls a Limited Subset of Regulatory T Cell Functions. J. Immunol. 2016, 196, 144–155. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Sido, J.M.; Reyes, E.E.; Kiers, V.; Cantor, H.; Kim, H.-J. Instability of Helios-Deficient Tregs Is Associated with Conversion to a T-Effector Phenotype and Enhanced Antitumor Immunity. Proc. Natl. Acad. Sci. USA 2016, 113, 6248–6253. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Lin, J.-X.; Leonard, W.J. Interleukin-2 at the Crossroads of Effector Responses, Tolerance, and Immunotherapy. Immunity 2013, 38, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, T.R.; Castro, I. Interleukin-2 Receptor Signaling: At the Interface between Tolerance and Immunity. Immunity 2010, 33, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, P.T. PTEN Inhibits IL-2 Receptor-Mediated Expansion of CD4+CD25+ Tregs. J. Clin. Investig. 2006, 116, 2521–2531. [Google Scholar] [CrossRef] [Green Version]
- Burchill, M.A.; Yang, J.; Vogtenhuber, C.; Blazar, B.R.; Farrar, M.A. IL-2 Receptor β-Dependent STAT5 Activation Is Required for the Development of Foxp3+ Regulatory T Cells. J. Immunol. 2007, 178, 280–290. [Google Scholar] [CrossRef] [Green Version]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y. An Essential Role for the IL-2 Receptor in Treg Cell Function. Nat. Immunol. 2016, 17, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Arvey, A.; Chinen, T.; van der Veeken, J.; Gasteiger, G.; Rudensky, A.Y. Control of the Inheritance of Regulatory T Cell Identity by a Cis Element in the Foxp3 Locus. Cell 2014, 158, 749–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontenot, J.D.; Rasmussen, J.P.; Gavin, M.A.; Rudensky, A.Y. A Function for Interleukin 2 in Foxp3-Expressing Regulatory T Cells. Nat. Immunol. 2005, 6, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Liu, C.; Tan, H.; Li, Y.; Nguyen, T.-L.M.; Dhungana, Y.; Guy, C.; Vogel, P.; Neale, G.; Rankin, S.; et al. Hippo Kinases Mst1 and Mst2 Sense and Amplify IL-2R-STAT5 Signaling in Regulatory T Cells to Establish Stable Regulatory Activity. Immunity 2018, 49, 899–914.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Shi, H.; Li, J.; Dong, Y.; Liang, J.; Ye, J.; Kong, S.; Zhang, S.; Zhong, T.; Yuan, Z.; et al. Mst1/Mst2 Regulate Development and Function of Regulatory T Cells through Modulation of Foxo1/Foxo3 Stability in Autoimmune Disease. J. Immunol. 2014, 192, 1525–1535. [Google Scholar] [CrossRef] [Green Version]
- Abdollahpour, H.; Appaswamy, G.; Kotlarz, D.; Diestelhorst, J.; Beier, R. The Phenotype of Human STK4 Deficiency. Blood 2012, 119, 3450–3457. [Google Scholar] [CrossRef] [PubMed]
- Nehme, N.T.; Schmid, J.P.; Debeurme, F.; André-Schmutz, I.; Lim, A.; Nitschke, P.; Rieux-Laucat, F.; Lutz, P.; Picard, C.; Mahlaoui, N.; et al. MST1 Mutations in Autosomal Recessive Primary Immunodeficiency Characterized by Defective Naive T-Cell Survival. Blood 2012, 119, 3458–3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Elly, C.; Park, Y.; Liu, Y.-C. E3 Ubiquitin Ligase VHL Regulates Hypoxia-Inducible Factor-1α to Maintain Regulatory T Cell Stability and Suppressive Capacity. Immunity 2015, 42, 1062–1074. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-Inducible Factor 1 (HIF-1) Pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef]
- Zhu, F.; Yi, G.; Liu, X.; Zhu, F.; Zhao, A.; Wang, A.; Zhu, R.; Chen, Z.; Zhao, B.; Fang, S.; et al. Ring Finger Protein 31–Mediated Atypical Ubiquitination Stabilizes Forkhead Box P3 and Thereby Stimulates Regulatory T-Cell Function. J. Biol. Chem. 2018, 293, 20099–20111. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Melo-Cardenas, J.; Zhang, Y.; Gau, I.; Wei, J.; Montauti, E.; Zhang, Y.; Gao, B.; Jin, H.; Sun, Z.; et al. The E3 Ligase Hrd1 Stabilizes Tregs by Antagonizing Inflammatory Cytokine–Induced ER Stress Response. JCI Insight 2019, 4, e121887. [Google Scholar] [CrossRef]
- Cortez, J.T.; Montauti, E.; Shifrut, E.; Gatchalian, J.; Zhang, Y.; Shaked, O.; Xu, Y.; Roth, T.L.; Simeonov, D.R.; Zhang, Y.; et al. CRISPR Screen in Regulatory T Cells Reveals Modulators of Foxp3. Nature 2020, 582, 416–420. [Google Scholar] [CrossRef]
- Hill, J.A.; Feuerer, M.; Tash, K.; Haxhinasto, S.; Perez, J.; Melamed, R.; Mathis, D.; Benoist, C. Foxp3 Transcription-Factor-Dependent and -Independent Regulation of the Regulatory T Cell Transcriptional Signature. Immunity 2007, 27, 786–800. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Josefowicz, S.Z.; Kas, A.; Chu, T.-T.; Gavin, M.A.; Rudensky, A.Y. Genome-Wide Analysis of Foxp3 Target Genes in Developing and Mature Regulatory T Cells. Nature 2007, 445, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Bilate, A.M.; Gobert, M.; Ding, Y.; Curotto de Lafaille, M.A.; Parkhurst, C.N.; Xiong, H.; Dolpady, J.; Frey, A.B.; Ruocco, M.G.; et al. Neuropilin 1 Is Expressed on Thymus-Derived Natural Regulatory T Cells, but Not Mucosa-Generated Induced Foxp3+ T Reg Cells. J. Exp. Med. 2012, 209, 1723–1742. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Louvet, C.; Davini, D.; Gardner, J.M.; Martinez-Llordella, M.; Bailey-Bucktrout, S.; Anthony, B.A.; Sverdrup, F.M.; Head, R.; Kuster, D.J.; et al. Neuropilin-1 Distinguishes Natural and Inducible Regulatory T Cells among Regulatory T Cell Subsets in Vivo. J. Exp. Med. 2012, 209, 1713–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaglia, A.; Buzzonetti, A.; Monego, G.; Peri, L.; Ferrandina, G.; Fanfani, F.; Scambia, G.; Fattorossi, A. Neuropilin-1 Expression Identifies a Subset of Regulatory T Cells in Human Lymph Nodes That Is Modulated by Preoperative Chemoradiation Therapy in Cervical Cancer. Immunology 2008, 123, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, B.; Elkord, E. Novel Expression of Neuropilin 1 on Human Tumor-Infiltrating Lymphocytes in Colorectal Cancer Liver Metastases. Expert Opin. Ther. Targets 2015, 19, 147–161. [Google Scholar] [CrossRef]
- Overacre-Delgoffe, A.E.; Chikina, M.; Dadey, R.E.; Yano, H.; Brunazzi, E.A.; Shayan, G.; Horne, W.; Moskovitz, J.M.; Kolls, J.K.; Sander, C.; et al. Interferon-γ Drives T Reg Fragility to Promote Anti-Tumor Immunity. Cell 2017, 169, 1130–1141.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soker, S.; Miao, H.-Q.; Nomi, M.; Takashima, S.; Klagsbrun, M. VEGF165 Mediates Formation of Complexes Containing VEGFR-2 and Neuropilin-1 That Enhance VEGF165-receptor Binding. J. Cell Biochem. 2002, 85, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Woo, S.-R.; Turnis, M.E.; Gravano, D.M.; Guy, C.; Overacre, A.E.; Bettini, M.L.; Vogel, P.; Finkelstein, D.; Bonnevier, J.; et al. Stability and Function of Regulatory T Cells Is Maintained by a Neuropilin-1–Semaphorin-4a Axis. Nature 2013, 501, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Kim, J.-A.; Kim, Y.-J.; Lee, H.W.; Kim, C.-H.; Haam, S.; Kim, Y.-S. A Neuropilin-1 Antagonist Exerts Antitumor Immunity by Inhibiting the Suppressive Function of Intratumoral Regulatory T Cells. Cancer Immunol. Res. 2020, 8, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Merkenschlager, M.; von Boehmer, H. PI3 Kinase Signalling Blocks Foxp3 Expression by Sequestering Foxo Factors. J. Exp. Med. 2010, 207, 1347–1350. [Google Scholar] [CrossRef] [Green Version]
- Sauer, S.; Bruno, L.; Hertweck, A.; Finlay, D.; Leleu, M.; Spivakov, M.; Knight, Z.A.; Cobb, B.S.; Cantrell, D.; O’Connor, E.; et al. T Cell Receptor Signaling Controls Foxp3 Expression via PI3K, Akt, and MTOR. Proc. Natl. Acad. Sci. USA 2008, 105, 7797–7802. [Google Scholar] [CrossRef] [Green Version]
- Huynh, A.; DuPage, M.; Priyadharshini, B.; Sage, P.T.; Quiros, J.; Borges, C.M.; Townamchai, N.; Gerriets, V.A.; Rathmell, J.C.; Sharpe, A.H.; et al. Control of PI(3) Kinase in Treg Cells Maintains Homeostasis and Lineage Stability. Nat. Immunol. 2015, 16, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Yang, K.; Guy, C.; Vogel, P.; Neale, G.; Chi, H. Treg Cells Require the Phosphatase PTEN to Restrain TH1 and TFH Cell Responses. Nat. Immunol. 2015, 16, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Yang, K.; Cloer, C.; Neale, G.; Vogel, P.; Chi, H. MTORC1 Couples Immune Signals and Metabolic Programming to Establish Treg-Cell Function. Nature 2013, 499, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Haxhinasto, S.; Mathis, D.; Benoist, C. The AKT–MTOR Axis Regulates de Novo Differentiation of CD4+Foxp3+ Cells. J. Exp. Med. 2008, 205, 565–574. [Google Scholar] [CrossRef]
- Liu, G.; Burns, S.; Huang, G.; Boyd, K.; Proia, R.L.; Flavell, R.A.; Chi, H. The Receptor S1P1 Overrides Regulatory T Cell–Mediated Immune Suppression through Akt-MTOR. Nat. Immunol. 2009, 10, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, M.; Stabilini, A.; Roncarolo, M.-G. Rapamycin Selectively Expands CD4+CD25+FoxP3+ Regulatory T Cells. Blood 2005, 105, 4743–4748. [Google Scholar] [CrossRef]
- Lampson, B.L.; Kasar, S.N.; Matos, T.R.; Morgan, E.A.; Rassenti, L.; Davids, M.S.; Fisher, D.C.; Freedman, A.S.; Jacobson, C.A.; Armand, P.; et al. Idelalisib given Front-Line for Treatment of Chronic Lymphocytic Leukemia Causes Frequent Immune-Mediated Hepatotoxicity. Blood 2016, 128, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chellappa, S.; Kushekhar, K.; Munthe, L.A.; Tjønnfjord, G.E.; Aandahl, E.M.; Okkenhaug, K.; Taskén, K. The PI3K P110δ Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. J. Immunol. 2019, 202, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Abu-Eid, R.; Shrimali, R.; Webb, M.; Verma, V.; Doroodchi, A.; Berrong, Z.; Samara, R.; Rodriguez, P.C.; Mkrtichyan, M.; et al. Differential PI3Kδ Signaling in CD4+ T-Cell Subsets Enables Selective Targeting of T Regulatory Cells to Enhance Cancer Immunotherapy. Cancer Res. 2017, 77, 1892–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soond, D.R.; Slack, E.C.M.; Garden, O.A.; Patton, D.T.; Okkenhaug, K. Does the PI3K Pathway Promote or Antagonize Regulatory T Cell Development and Function? Front. Immun. 2012, 3, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A. Interleukin-1, Interleukin-1 Receptors and Interleukin-1 Receptor Antagonist. Int. Rev. Immunol. 1998, 16, 457–499. [Google Scholar] [CrossRef]
- Kuhn, P.-H.; Marjaux, E.; Imhof, A.; De Strooper, B.; Haass, C.; Lichtenthaler, S.F. Regulated Intramembrane Proteolysis of the Interleukin-1 Receptor II by α-, β-, and γ-Secretase. J. Biol. Chem. 2007, 282, 11982–11995. [Google Scholar] [CrossRef] [Green Version]
- Orlando, S.; Matteucci, C.; Fadlon, E.J.; Buurman, W.A.; Bardella, M.T.; Colotta, F.; Introna, M.; Mantovani, A. TNF-Alpha, Unlike Other pro- and Anti-Inflammatory Cytokines, Induces Rapid Release of the IL-1 Type II Decoy Receptor in Human Myelomonocytic Cells. J. Immunol. 1997, 158, 3861–3868. [Google Scholar]
- Tran, D.Q.; Andersson, J.; Hardwick, D.; Bebris, L.; Illei, G.G.; Shevach, E.M. Selective Expression of Latency-Associated Peptide (LAP) and IL-1 Receptor Type I/II (CD121a/CD121b) on Activated Human FOXP3+ Regulatory T Cells Allows for Their Purification from Expansion Cultures. Blood 2009, 113, 5125–5133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; Herrath, J.; Malmström, V. IL-1R1 Is Expressed on Both Helios+ and Helios− FoxP3+ CD4+ T Cells in the Rheumatic Joint: Helios and IL-1R1 Expression in Joint-Derived FoxP3+ T Cells. Clin. Exp. Immunol. 2015, 182, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, F.; Istomine, R.; Shourian, M.; Pavey, N.; Al-Aubodah, T.A.-F.; Qureshi, S.; Fritz, J.H.; Piccirillo, C.A. The Alarmins IL-1 and IL-33 Differentially Regulate the Functional Specialisation of Foxp3+ Regulatory T Cells during Mucosal Inflammation. Mucosal. Immunol. 2019, 12, 746–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Humphry, M.; Maguire, J.J.; Bennett, M.R.; Clarke, M.C.H. Intracellular Interleukin-1 Receptor 2 Binding Prevents Cleavage and Activity of Interleukin-1a, Controlling Necrosis-Induced Sterile Inflammation. Immunity 2013, 38, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Nikolouli, E.; Elfaki, Y.; Herppich, S.; Schelmbauer, C.; Delacher, M.; Falk, C.; Mufazalov, I.A.; Waisman, A.; Feuerer, M.; Huehn, J. Recirculating IL-1R2+ Tregs Fine-Tune Intrathymic Treg Development under Inflammatory Conditions. Cell Mol. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary Lymphoid Structures Improve Immunotherapy and Survival in Melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B Cells and Tertiary Lymphoid Structures Promote Immunotherapy Response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.-W.; Sun, C.-M.; Calderaro, J.; Jeng, Y.-M.; Hsiao, L.-P.; Lacroix, L.; Bougoüin, A.; et al. B Cells Are Associated with Survival and Immunotherapy Response in Sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qiang, J.; Yang, X.; Wang, D.; Rehman, A.; He, X.; Chen, W.; Sheng, D.; Zhou, L.; Jiang, Y.; et al. Breast Cancer: IL1R2 Blockade Suppresses Breast Tumorigenesis and Progression by Impairing USP15-Dependent BMI1 Stability. Adv. Sci. 2019, 7, 1901728. [Google Scholar] [CrossRef] [Green Version]
- Mercer, F.; Kozhaya, L.; Unutmaz, D. Expression and Function of TNF and IL-1 Receptors on Human Regulatory T Cells. PLoS ONE 2010, 5, e8639. [Google Scholar] [CrossRef] [Green Version]
- Vasanthakumar, A.; Moro, K.; Xin, A.; Liao, Y.; Gloury, R.; Kawamoto, S.; Fagarasan, S.; Mielke, L.A.; Afshar-Sterle, S.; Masters, S.L.; et al. The Transcriptional Regulators IRF4, BATF and IL-33 Orchestrate Development and Maintenance of Adipose Tissue–Resident Regulatory T Cells. Nat. Immunol. 2015, 16, 276–285. [Google Scholar] [CrossRef]
- Arpaia, N.; Green, J.A.; Moltedo, B.; Arvey, A.; Hemmers, S.; Yuan, S.; Treuting, P.M.; Rudensky, A.Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 2015, 162, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Schiering, C.; Krausgruber, T.; Chomka, A.; Fröhlich, A.; Adelmann, K.; Wohlfert, E.A.; Pott, J.; Griseri, T.; Bollrath, J.; Hegazy, A.N.; et al. The Alarmin IL-33 Promotes Regulatory T-Cell Function in the Intestine. Nature 2014, 513, 564–568. [Google Scholar] [CrossRef] [Green Version]
- Pastille, E.; Wasmer, M.-H.; Adamczyk, A.; Vu, V.P.; Mager, L.F.; Phuong, N.N.T.; Palmieri, V.; Simillion, C.; Hansen, W.; Kasper, S.; et al. The IL-33/ST2 Pathway Shapes the Regulatory T Cell Phenotype to Promote Intestinal Cancer. Mucosal. Immunol. 2019, 12, 990–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. A Special Population of Regulatory T Cells Potentiates Muscle Repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuswanto, W.; Burzyn, D.; Panduro, M.; Wang, K.K.; Jang, Y.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity 2016, 44, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delacher, M.; Imbusch, C.D.; Weichenhan, D.; Breiling, A.; Hotz-Wagenblatt, A.; Träger, U.; Hofer, A.-C.; Kägebein, D.; Wang, Q.; Frauhammer, F.; et al. Genome-Wide DNA-Methylation Landscape Defines Specialization of Regulatory T Cells in Tissues. Nat. Immunol. 2017, 18, 1160–1172. [Google Scholar] [CrossRef]
- Kolodin, D.; van Panhuys, N.; Li, C.; Magnuson, A.M.; Cipolletta, D.; Miller, C.M.; Wagers, A.; Germain, R.N.; Benoist, C.; Mathis, D. Antigen- and Cytokine-Driven Accumulation of Regulatory T Cells in Visceral Adipose Tissue of Lean Mice. Cell Metab. 2015, 21, 543–557. [Google Scholar] [CrossRef] [Green Version]
- Moussion, C.; Ortega, N.; Girard, J.-P. The IL-1-Like Cytokine IL-33 Is Constitutively Expressed in the Nucleus of Endothelial Cells and Epithelial Cells In Vivo: A Novel ‘Alarmin’? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [Green Version]
- Pichery, M.; Mirey, E.; Mercier, P.; Lefrancais, E.; Dujardin, A.; Ortega, N.; Girard, J.-P. Endogenous IL-33 Is Highly Expressed in Mouse Epithelial Barrier Tissues, Lymphoid Organs, Brain, Embryos, and Inflamed Tissues: In Situ Analysis Using a Novel Il-33–LacZ Gene Trap Reporter Strain. J. Immunol. 2012, 188, 3488–3495. [Google Scholar] [CrossRef] [Green Version]
- Préfontaine, D.; Nadigel, J.; Chouiali, F.; Audusseau, S.; Semlali, A.; Chakir, J.; Martin, J.G.; Hamid, Q. Increased IL-33 Expression by Epithelial Cells in Bronchial Asthma. J. Allergy Clin. Immunol. 2010, 125, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an Interleukin-1-like Cytokine That Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Manetti, M.; Ibba-Manneschi, L.; Liakouli, V.; Guiducci, S.; Milia, A.F.; Benelli, G.; Marrelli, A.; Conforti, M.L.; Romano, E.; Giacomelli, R.; et al. The IL1-like Cytokine IL33 and Its Receptor ST2 Are Abnormally Expressed in the Affected Skin and Visceral Organs of Patients with Systemic Sclerosis. Ann. Rheum. Dis. 2010, 69, 598–605. [Google Scholar] [CrossRef]
- Masamune, A.; Watanabe, T.; Kikuta, K.; Satoh, K.; Kanno, A.; Shimosegawa, T. Nuclear Expression of Interleukin-33 in Pancreatic Stellate Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G821–G832. [Google Scholar] [CrossRef] [Green Version]
- Sponheim, J.; Pollheimer, J.; Olsen, T.; Balogh, J.; Hammarström, C.; Loos, T.; Kasprzycka, M.; Sørensen, D.R.; Nilsen, H.R.; Küchler, A.M.; et al. Inflammatory Bowel Disease-Associated Interleukin-33 Is Preferentially Expressed in Ulceration-Associated Myofibroblasts. Am. J. Pathol. 2010, 177, 2804–2815. [Google Scholar] [CrossRef]
- Zhang, C.; Li, L.; Feng, K.; Fan, D.; Xue, W.; Lu, J. ‘Repair’ Treg Cells in Tissue Injury. Cell Physiol. Biochem. 2017, 43, 2155–2169. [Google Scholar] [CrossRef] [Green Version]
- Ameri, A.H.; Moradi Tuchayi, S.; Zaalberg, A.; Park, J.H.; Ngo, K.H.; Li, T.; Lopez, E.; Colonna, M.; Lee, R.T.; Mino-Kenudson, M.; et al. IL-33/Regulatory T Cell Axis Triggers the Development of a Tumor-Promoting Immune Environment in Chronic Inflammation. Proc. Natl. Acad. Sci. USA 2019, 116, 2646–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzioannou, A.; Banos, A.; Sakelaropoulos, T.; Fedonidis, C.; Vidali, M.-S.; Köhne, M.; Händler, K.; Boon, L.; Henriques, A.; Koliaraki, V.; et al. An Intrinsic Role of IL-33 in Treg Cell–Mediated Tumor Immunoevasion. Nat. Immunol. 2020, 21, 75–85. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The Kinase MTOR Regulates the Differentiation of Helper T Cells through the Selective Activation of Signaling by MTORC1 and MTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindon, N.; Andrews, G.; Baxter, A.; Cheshire, D.; Hemsley, P.; Johnson, T.; Liu, Y.-Z.; McGinnity, D.; McHale, M.; Mete, A.; et al. Discovery of AZD-2098 and AZD-1678, Two Potent and Bioavailable CCR4 Receptor Antagonists. ACS Med. Chem. Lett. 2017, 8, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Cahn, A.; Hodgson, S.; Wilson, R.; Robertson, J.; Watson, J.; Beerahee, M.; Hughes, S.C.; Young, G.; Graves, R.; Hall, D.; et al. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of GSK2239633, a CC-Chemokine Receptor 4 Antagonist, in Healthy Male Subjects: Results from an Open-Label and from a Randomised Study. BMC Pharmacol. Toxicol. 2013, 14, 14. [Google Scholar] [CrossRef] [Green Version]
- Toogood, P.L. Small Molecule Immuno-Oncology Therapeutic Agents. Bioorg. Med. Chem. Lett. 2018, 28, 319–329. [Google Scholar] [CrossRef]
- Bayry, J.; Tchilian, E.Z.; Davies, M.N.; Forbes, E.K.; Draper, S.J.; Kaveri, S.V.; Hill, A.V.S.; Kazatchkine, M.D.; Beverley, P.C.L.; Flower, D.R.; et al. In Silico Identified CCR4 Antagonists Target Regulatory T Cells and Exert Adjuvant Activity in Vaccination. Proc. Natl. Acad. Sci. USA 2008, 105, 10221–10226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pere, H.; Montier, Y.; Bayry, J.; Quintin-Colonna, F.; Merillon, N.; Dransart, E.; Badoual, C.; Gey, A.; Ravel, P.; Marcheteau, E.; et al. A CCR4 Antagonist Combined with Vaccines Induces Antigen-Specific CD8+ T Cells and Tumor Immunity against Self Antigens. Blood 2011, 118, 4853–4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, J.J.; Ketcham, J.M.; Younai, A.; Abraham, B.; Biannic, B.; Beck, H.P.; Bui, M.H.T.; Chian, D.; Cutler, G.; Diokno, R.; et al. Discovery of a Potent and Selective CCR4 Antagonist That Inhibits Treg Trafficking into the Tumor Microenvironment. J. Med. Chem. 2019, 62, 6190–6213. [Google Scholar] [CrossRef]
- Robles, O.; Jackson, J.J.; Marshall, L.; Talay, O.; Chian, D.; Cutler, G.; Diokno, R.; Hu, D.X.; Jacobson, S.; Karbarz, E.; et al. Novel Piperidinyl-Azetidines as Potent and Selective CCR4 Antagonists Elicit Antitumor Response as a Single Agent and in Combination with Checkpoint Inhibitors. J. Med. Chem. 2020, 63, 8584–8607. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Matsuo, K.; Nagakubo, D.; Higashiyama, S.; Nishiwaki, K.; Oiso, N.; Kawada, A.; Yoshie, O.; Nakayama, T. A CCR4 Antagonist Enhances DC Activation and Homing to the Regional Lymph Node and Shows Potent Vaccine Adjuvant Activity through the Inhibition of Regulatory T-Cell Recruitment. J. Pharmacol. Sci. 2018, 136, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Mutalithas, K.; Guillen, C.; Raport, C.; Kolbeck, R.; Soler, D.; Brightling, C.E.; Pavord, I.D.; Wardlaw, A.J. Expression of CCR8 Is Increased in Asthma: CCR8 in Asthma. Clin. Exp. Allergy 2010, 40, 1175–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Jenkins, T.J.; Dai, M.; Yin, W.; Pulido, J.C.; LaMantia-Martin, E.; Hodge, M.R.; Ocain, T.; Kolbeck, R. Antagonism of Chemokine Receptor CCR8 Is Ineffective in a Primate Model of Asthma. Thorax 2013, 68, 506–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, S.; Skrinjar, M.; Rosendahl, A. Orally Bioavailable Allosteric CCR8 Antagonists Inhibit Dendritic Cell, T Cell and Eosinophil Migration. Biochem. Pharmacol. 2012, 83, 778–787. [Google Scholar] [CrossRef]
- Berenguer, J.; Lagerweij, T.; Zhao, X.W.; Dusoswa, S.; van der Stoop, P.; Westerman, B.; de Gooijer, M.C.; Zoetemelk, M.; Zomer, A.; Crommentuijn, M.H.W.; et al. Glycosylated Extracellular Vesicles Released by Glioblastoma Cells Are Decorated by CCL18 Allowing for Cellular Uptake via Chemokine Receptor CCR8. J. Extracell. Vesicles 2018, 7, 1446660. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.; Nasrah, N.; Johnson, D. Phase I/II Dose Escalation and Expansion Study of FLX475 Alone and in Combination with Pembrolizumab in Advanced Cancer. JCO 2019, 37, TPS24. [Google Scholar] [CrossRef] [Green Version]
- Powderly, J.D.; Chmielowski, B.; Brahmer, J.R.; Piha-Paul, S.A.; Bowyer, S.E.; LoRusso, P.; Catenacci, D.V.T.; Wu, C.; Barve, M.A.; Chisamore, M.J.; et al. Phase I/II Dose-Escalation and Expansion Study of FLX475 Alone and in Combination with Pembrolizumab in Advanced Cancer. JCO 2020, 38, TPS3163. [Google Scholar] [CrossRef]
- Kurose, K.; Ohue, Y.; Wada, H.; Iida, S.; Ishida, T.; Kojima, T.; Doi, T.; Suzuki, S.; Isobe, M.; Funakoshi, T.; et al. Phase Ia Study of FoxP3+ CD4 Treg Depletion by Infusion of a Humanized Anti-CCR4 Antibody, KW-0761, in Cancer Patients. Clin. Cancer Res. 2015, 21, 4327–4336. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Joh, T.; Uike, N.; Yamamoto, K.; Utsunomiya, A.; Yoshida, S.; Saburi, Y.; Miyamoto, T.; Takemoto, S.; Suzushima, H.; et al. Defucosylated Anti-CCR4 Monoclonal Antibody (KW-0761) for Relapsed Adult T-Cell Leukemia-Lymphoma: A Multicenter Phase II Study. JCO 2012, 30, 837–842. [Google Scholar] [CrossRef]
- Joffe, E.; Vardhana, S.A.; Kumar, A.; Abedi, M.; Hoeg, R.; Sharon, E.; Younes, A. A Phase I and Randomized Phase II Etctn Study of KW-0761 (Mogamulizumab) and MK-3475 (Pembrolizumab) in Relapsed and Refractory Diffuse Large B-Cell Lymphoma. JCO 2020, 38, TPS8072. [Google Scholar] [CrossRef]
- Cohen, E.E.W. A Phase Ib Study of Utomilumab (PF-05082566) in Combination with Mogamulizumab in Patients with Advanced Solid Tumors. J. Immunother. Cancer 2019, 7, 342. [Google Scholar] [CrossRef]
- Zamarin, D.; Hamid, O.; Nayak-Kapoor, A.; Sahebjam, S.; Sznol, M.; Collaku, A.; Fox, F.E.; Marshall, M.A.; Hong, D.S. Mogamulizumab in Combination with Durvalumab or Tremelimumab in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2020, 26, 4531–4541. [Google Scholar] [CrossRef]
- Doi, T.; Muro, K.; Ishii, H.; Kato, T.; Tsushima, T.; Takenoyama, M.; Oizumi, S.; Gemmoto, K.; Suna, H.; Enokitani, K.; et al. A Phase I Study of the Anti-CC Chemokine Receptor 4 Antibody, Mogamulizumab, in Combination with Nivolumab in Patients with Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2019, 25, 6614–6622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wei, M.; Zhang, H.; Chen, H.; Germana, S.; Huang, C.A.; Madsen, J.C.; Sachs, D.H.; Wang, Z. Diphtheria-Toxin Based Anti-Human CCR4 Immunotoxin for Targeting Human CCR4+ Cells in Vivo. Mol. Oncol. 2015, 9, 1458–1470. [Google Scholar] [CrossRef]
- Wang, Z.; Pratts, S.G.; Zhang, H.; Spencer, P.J.; Yu, R.; Tonsho, M.; Shah, J.A.; Tanabe, T.; Powell, H.R.; Huang, C.A.; et al. Treg Depletion in Non-Human Primates Using a Novel Diphtheria Toxin-Based Anti-Human CCR4 Immunotoxin. Mol. Oncol. 2016, 10, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Louras, N.J.; Lellouch, A.G.; Pratts, S.G.; Zhang, H.; Wang, H.; Huang, C.A.; Cetrulo, C.L.; Madsen, J.C.; Sachs, D.H.; et al. Dosing Optimization of CCR 4 Immunotoxin for Improved Depletion of CCR 4+ Treg in Nonhuman Primates. Mol. Oncol. 2018, 12, 1374–1382. [Google Scholar] [CrossRef] [Green Version]
- Dépis, F.; Hu, C.; Weaver, J.; McGrath, L.; Klebanov, B.; Buggé, J.; Umiker, B.; Fregeau, C.; Upadhyay, D.; Singh, A.; et al. Abstract 4532: Preclinical Evaluation of JTX-1811, an Anti-CCR8 Antibody with Enhanced ADCC Activity, for Preferential Depletion of Tumor-Infiltrating Regulatory T Cells. Cancer Res. 2020, 4532. [Google Scholar] [CrossRef]
- Lake, A.; Warren, M.; Das, S.; Wells, C.; Scrivens, M.; Smith, E.; Palombella, V.; Etemad-Gilbertson, B.; Holland, P.; Dulak, A. 726 SRF114 Is a Fully Human, CCR8 Selective IgG1 Antibody That Induces Destruction of Tumor Tregs through ADCC. J. Immunother. Cancer 2020, 8, A656–A959. [Google Scholar] [CrossRef]
- Lu, S.; Hu, S.; Gan, X.; Wang, Y.; Zhao, C.; Ding, Y.; He, J.; Du, Q.; Lv, X.; Qin, B.; et al. 711 HBM1022, a Novel Anti-CCR8 Antibody Depletes Tumor-Infiltrating Regulatory T Cells via Enhanced ADCC Activity, Mediates Potent Anti-Tumor Activity with Keytruda. J. Immunother. Cancer 2020, 8, A753. [Google Scholar] [CrossRef]
- Rankin, A.; Naik, E. 861 Development of FPA157, an Anti-CCR8 Depleting Antibody Engineered to Preferentially Eliminate Tumor-Infiltrating T Regulatory Cells. J. Immunother. Cancer 2020, 8, A914. [Google Scholar] [CrossRef]
- Campbell, J.R.; McDonald, B.R.; Mesko, P.B.; Siemers, N.O.; Singh, P.B.; Selby, M.; Sproul, T.W.; Korman, A.J.; Vlach, L.M.; Houser, J.; et al. Fc-Optimized Anti-CCR8 Antibody Depletes Regulatory T Cells in Human Tumor Models. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Le, D.; Gutierrez, M.E.; Saleh, M.; Chen, E.; Mallick, A.B.; Pishvaian, M.J.; Krauss, J.; O’Dwyer, P.; Garrido-Laguna, I.; Zhao, Q.; et al. Abstract CT124: A Phase Ib/II Study of BMS-813160, a CC Chemokine Receptor (CCR) 2/5 Dual Antagonist, in Combination with Chemotherapy or Nivolumab in Patients (Pts) with Advanced Pancreatic or Colorectal Cancer. Cancer Res. 2018, 78, CT124. [Google Scholar] [CrossRef]
- Noel, M.S.; Hezel, A.F.; Linehan, D.; Wang-Gillam, A.; Eskens, F.; Sleijfer, S.; Desar, I.; Erdkamp, F.; Wilmink, J.; Diehl, J.; et al. Orally Administered CCR2 Selective Inhibitor CCX872-b Clinical Trial in Pancreatic Cancer. JCO 2017, 35, 276. [Google Scholar] [CrossRef]
- Quaratino, S.; Sainson, R.; Thotakura, A.; Henderson, S.J.; Pryke, K.; Newton, A.; Brennan, N.; Oelmann, E. A First-in-Human Study of KY1044, a Fully Human Anti-ICOS IgG1 Antibody as Monotherapy and in Combination with Atezolizumab in Patients with Selected Advanced Malignancies. JCO 2019, 37, TPS2644. [Google Scholar] [CrossRef]
- Maio, M.; Groenland, S.L.; Bauer, T.; Rischin, D.; Gardeazabal, I.; Moreno, V.; Trigo Perez, J.M.; Chisamore, M.; Sadik Shaik, J.; Rigat, F.; et al. Pharmacokinetic/ Pharmacodynamic (PK/PD) Exposure-Response Characterization of GSK3359609 (GSK609) from INDUCE-1, a Phase I Open-Label Study. Ann. Oncol. 2019, 30, v793. [Google Scholar] [CrossRef]
- Angevin, E.; Groenland, S.L.; Lim, A.M.L.; Martin-Liberal, J.; Moreno, V.; Trigo, J.M.; Le Tourneau, C.; Mathew, M.; Cho, D.C.; Hansen, A.R.; et al. Updated Analysis of the Inducible T-Cell Co-Stimulatory Receptor (ICOS) Agonist, GSK3359609 (GSK609), Combination with Pembrolizumab (PE) in Patients (Pts) with Anti-PD-1/L1 Treatment-Naïve Head and Neck Squamous Cell Carcinoma (HNSCC). JCO 2020, 38, 6517. [Google Scholar] [CrossRef]
- Burris, H.A.; Callahan, M.K.; Tolcher, A.W.; Kummar, S.; Falchook, G.S.; Pachynski, R.K.; Tykodi, S.S.; Gibney, G.T.; Seiwert, T.Y.; Gainor, J.F.; et al. Phase 1 Safety of ICOS Agonist Antibody JTX-2011 Alone and with Nivolumab (Nivo) in Advanced Solid Tumors; Predicted vs Observed Pharmacokinetics (PK) in ICONIC. J. Clin. Oncol. 2017, 35, 3033. [Google Scholar] [CrossRef]
- Chen, X.; Oppenheim, J.J. Targeting TNFR2, an Immune Checkpoint Stimulator and Oncoprotein, Is a Promising Treatment for Cancer. Sci. Signal. 2017, 10, eaal2328. [Google Scholar] [CrossRef] [PubMed]
- Mårtensson, L.; Kovacek, M.; Holmkvist, P.; Semmrich, M.; Svensson, C.; Blidberg, T.; Roos, C.; McAllister, A.; Demiri, M.; Borggren, M.; et al. 725 Pre-Clinical Development of TNFR2 Ligand-Blocking BI-1808 for Cancer Immunotherapy. J. Immunother. Cancer 2020, 8, A768. [Google Scholar] [CrossRef]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 Is a Potent Immune-Stimulating Target in Late-Stage Cancer Patients. Cancer Res. 2013, 73, 7189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crittenden, M.; Conlin, A.K.; Moxon, N.; Curti, B.D. Phase I/II Study of Stereotactic Body Radiation Therapy (SBRT) to Metastatic Lesions in the Liver or Lung in Combination with Monoclonal Antibody to OX40 in Patients with Progressive Metastatic Breast Cancer (MBC) after Systemic Therapy. JCO 2015, 33, TPS3103. [Google Scholar] [CrossRef]
- Segal, N.H.; He, A.R.; Doi, T.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 1816–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, C.; Revenko, A.S.; Johnson, R.B.; Peter, A.; Taylor, M.A.; Hettrick, L.A.; Klein, S.; Solanki, A.; Chapman, M.; Yates, J.; et al. Abstract 2713: Discovery and Characterization of AZD8701, a High Affinity Antisense Oligonucleotide Targeting FOXP3 to Relieve Immunosuppression in Cancer. Cancer Res. 2019, 79, 2713. [Google Scholar] [CrossRef]
- Yoo, C.; Oh, D.-Y.; Choi, H.J.; Kudo, M.; Ueno, M.; Kondo, S.; Chen, L.-T.; Osada, M.; Helwig, C.; Dussault, I.; et al. Phase I Study of Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Patients with Pretreated Biliary Tract Cancer. J. Immunother. Cancer 2020, 8, e000564. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Kim, T.M.; Vicente, D.; Felip, E.; Lee, D.H.; Lee, K.H.; Lin, C.-C.; Flor, M.J.; Di Nicola, M.; Alvarez, R.M.; et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Second-Line Treatment of Patients With NSCLC: Results From an Expansion Cohort of a Phase 1 Trial. J. Thorac. Oncol. 2020, 15, 1210–1222. [Google Scholar] [CrossRef]
- Barlesi, F.; Hiret, S.; Rolfo, C.; Grenga, I.; Köenig, A.; Vugmeyster, Y.; Horn, L. Abstract CT267: A Phase 1b/2, Open-Label Study of Bintrafusp Alfa with Chemotherapy in Patients with Stage IV Non-Small Cell Lung Cancer. Cancer Res. 2020, 80, CT267. [Google Scholar] [CrossRef]
- Strauss, J.; Heery, C.R.; Schlom, J.; Madan, R.A.; Cao, L.; Kang, Z.; Lamping, E.; Marté, J.L.; Donahue, R.N.; Grenga, I.; et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFβ, in Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coward, J.; Ganju, V.; Behzadigohar, R.; Kwong, K.; Xu, J.; Van, H.; Kong, P.; Yang, F.; Chen, L.; Guo, K.; et al. Preliminary Safety, Efficacy, and Pharmacokinetics (PK) Results of KN046 (Bispecific Anti-PD-L1/CTLA4) from a First-in-Human Study in Subjects with Advanced Solid Tumors. J. Clin. Oncol. 2019, 37, 2554. [Google Scholar] [CrossRef]
- Kvarnhammar, A.M.; Veitonmäki, N.; Hägerbrand, K.; Dahlman, A.; Smith, K.E.; Fritzell, S.; von Schantz, L.; Thagesson, M.; Werchau, D.; Smedenfors, K.; et al. The CTLA-4 x OX40 Bispecific Antibody ATOR-1015 Induces Anti-Tumor Effects through Tumor-Directed Immune Activation. J. Immunother. Cancer 2019, 7, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. CCR2 Inhibition Reduces Tumor Myeloid Cells and Unmasks a Checkpoint Inhibitor Effect to Slow Progression of Resistant Murine Gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Anz, D.; Rapp, M.; Eiber, S.; Koelzer, V.H.; Thaler, R.; Haubner, S.; Knott, M.; Nagel, S.; Golic, M.; Wiedemann, G.M.; et al. Suppression of Intratumoral CCL22 by Type I Interferon Inhibits Migration of Regulatory T Cells and Blocks Cancer Progression. Cancer Res. 2015, 75, 4483–4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, E.; Wollenberg, B.; Rothenfusser, S.; Wagner, M.; Wellisch, D.; Mack, B.; Giese, T.; Gires, O.; Endres, S.; Hartmann, G. Identification and Functional Analysis of Tumor-Infiltrating Plasmacytoid Dendritic Cells in Head and Neck Cancer. Cancer Res. 2003, 63, 6478–6487. [Google Scholar] [PubMed]
- Labidi-Galy, S.I.; Sisirak, V.; Meeus, P.; Gobert, M.; Treilleux, I.; Bajard, A.; Combes, J.-D.; Faget, J.; Tredan, O.; Durand, I.; et al. Quantitative and Functional Alterations of Plasmacytoid Dendritic Cells Contribute to Immune Tolerance in Ovarian Cancer. Cancer Res. 2011, 71, 5423–5434. [Google Scholar] [CrossRef] [Green Version]
- Sisirak, V.; Vey, N.; Goutagny, N.; Renaudineau, S.; Malfroy, M.; Thys, S.; Treilleux, I.; Labidi-Galy, S.I.; Bachelot, T.; Dezutter-Dambuyant, C.; et al. Breast Cancer-Derived Transforming Growth Factor-β and Tumor Necrosis Factor-α Compromise Interferon-α Production by Tumor-Associated Plasmacytoid Dendritic Cells: TGF-β and TNF-α Inhibit PDC-Derived IFN-α. Int. J. Cancer 2013, 133, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S.; et al. Impaired IFN-a Production by Plasmacytoid Dendritic Cells Favors Regulatory T-Cell Expansion That May Contribute to Breast Cancer Progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Mercier, I.; Poujol, D.; Sanlaville, A.; Sisirak, V.; Gobert, M.; Durand, I.; Dubois, B.; Treilleux, I.; Marvel, J.; Vlach, J.; et al. Tumor Promotion by Intratumoral Plasmacytoid Dendritic Cells Is Reversed by TLR7 Ligand Treatment. Cancer Res. 2013, 73, 4629–4640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terra, M.; Oberkampf, M.; Fayolle, C.; Rosenbaum, P.; Guillerey, C.; Dadaglio, G.; Leclerc, C. Tumor-Derived TGFb Alters the Ability of Plasmacytoid Dendritic Cells to Respond to Innate Immune Signaling. Cancer Res. 2018, 78, 3014–3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, S.M.; Carlesso, G.; Cheng, L.I.; Cook, H.; DaCosta, K.; Leininger, J.; McKeever, K.; Scott, S. (Weasel); Taylor, D.; Streicher, K.; et al. Effects of ICOS+ T Cell Depletion via Afucosylated Monoclonal Antibody MEDI-570 on Pregnant Cynomolgus Monkeys and the Developing Offspring. Reprod. Toxicol. 2017, 74, 116–133. [Google Scholar] [CrossRef]
- Mo, L.; Chen, Q.; Zhang, X.; Shi, X.; Wei, L.; Zheng, D.; Li, H.; Gao, J.; Li, J.; Hu, Z. Depletion of Regulatory T Cells by Anti-ICOS Antibody Enhances Anti-Tumor Immunity of Tumor Cell Vaccine in Prostate Cancer. Vaccine 2017, 35, 5932–5938. [Google Scholar] [CrossRef] [PubMed]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS Is an Inducible T-Cell Co-Stimulator Structurally and Functionally Related to CD28. Nature 1999, 397, 263–266. [Google Scholar] [CrossRef]
- Chen, H.; Liakou, C.I.; Kamat, A.; Pettaway, C.; Ward, J.F.; Tang, D.N.; Sun, J.; Jungbluth, A.A.; Troncoso, P.; Logothetis, C.; et al. Anti-CTLA-4 Therapy Results in Higher CD4+ICOShi T Cell Frequency and IFN-g Levels in Both Nonmalignant and Malignant Prostate Tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 2729–2734. [Google Scholar] [CrossRef] [Green Version]
- Liakou, C.I.; Kamat, A.; Tang, D.N.; Chen, H.; Sun, J.; Troncoso, P.; Logothetis, C.; Sharma, P. CTLA-4 Blockade Increases IFN-g Producing CD4+ICOShi Cells to Shift the Ratio of Effector to Regulatory T Cells in Cancer Patients. Proc. Natl. Acad. Sci. USA 2008, 105, 14987–14992. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.-A.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hamano, R.; Subleski, J.J.; Hurwitz, A.A.; Howard, O.M.Z.; Oppenheim, J.J. Expression of Costimulatory TNFR2 Induces Resistance of CD4+FoxP3− Conventional T Cells to Suppression by CD4+ FoxP3+ Regulatory T Cells. J. Immunol. 2010, 185, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Tam, E.M.; Fulton, R.B.; Sampson, J.F.; Muda, M.; Camblin, A.; Richards, J.; Koshkaryev, A.; Tang, J.; Kurella, V.; Jiao, Y.; et al. Antibody-Mediated Targeting of TNFR2 Activates CD8+ T Cells in Mice and Promotes Antitumor Immunity. Sci. Transl. Med. 2019, 11, eaax0720. [Google Scholar] [CrossRef] [PubMed]
- Pace, L.; Vitale, S.; Dettori, B.; Palombi, C.; Sorsa, V.L.; Belardelli, F.; Proietti, E.; Doria, G. APC Activation by IFN-a Decreases Regulatory T Cell and Enhances Th Cell Functions. J. Immunol. 2010, 184, 5969–5979. [Google Scholar] [CrossRef] [Green Version]
- Arce Vargas, F.; Furness, A.J.S.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018, 33, 649–663.e4. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Subudhi, S.K.; Blando, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti–CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers—Response. Clin. Cancer Res. 2019, 25, 3469–3470. [Google Scholar] [CrossRef] [Green Version]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-Dependent Cell-Mediated Cytotoxicity of Regulatory T Cells Ex Vivo by Nonclassical Monocytes in Melanoma Patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurose, K.; Ohue, Y.; Sato, E.; Yamauchi, A.; Eikawa, S.; Isobe, M.; Nishio, Y.; Uenaka, A.; Oka, M.; Nakayama, E. Increase in Activated Treg in TIL in Lung Cancer and In Vitro Depletion of Treg by ADCC Using an Antihuman CCR4 MAb (KM2760). J. Thorac. Oncol. 2015, 10, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, R.; Jhatakia, A.; Campbell, J.; Siemers, N.; Lu, K.; Nasser, M.; Grigoriev, T.; Singh, P.; Vlach, L.; Sproul, T.; et al. Abstract 6694: Highly Selective Anti-CCR8 Antibody-Mediated Depletion of Regulatory T Cells Leads to Potent Antitumor Activity Alone and in Combination with Anti-PD-1 in Preclinical Models. Cancer Res. 2020, 80, 6694. [Google Scholar] [CrossRef]
- Sainson, R.C.A.; Thotakura, A.; Parveen, N.; Kosmac, M.; Borhis, G.; Carvalho, J.; Myers, T.; Rowlands, R.; Ali, H.; Craig, H.; et al. KY1044, a Novel Anti-ICOS Antibody, Elicits Long Term in Vivo Anti-Tumour Efficacy as Monotherapy and in Combination with Immune Checkpoint Inhibitors. In Journal for Immunotherapy of Cancer; BMC CAMPUS: London, UK, 2017. [Google Scholar]
- Carthon, B.C.; Wolchok, J.D.; Yuan, J.; Kamat, A.; Ng Tang, D.S.; Sun, J.; Ku, G.; Troncoso, P.; Logothetis, C.J.; Allison, J.P.; et al. Preoperative CTLA-4 Blockade: Tolerability and Immune Monitoring in the Setting of a Presurgical Clinical Trial. Clin. Cancer Res. 2010, 16, 2861–2871. [Google Scholar] [CrossRef] [Green Version]
- Ng Tang, D.; Shen, Y.; Sun, J.; Wen, S.; Wolchok, J.D.; Yuan, J.; Allison, J.P.; Sharma, P. Increased Frequency of ICOS+ CD4 T Cells as a Pharmacodynamic Biomarker for Anti-CTLA-4 Therapy. Cancer Immunol. Res. 2013, 1, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Fu, T.; He, Q.; Sharma, P. The ICOS/ICOSL Pathway Is Required for Optimal Antitumor Responses Mediated by Anti-CTLA-4 Therapy. Cancer Res. 2011, 71, 5445–5454. [Google Scholar] [CrossRef] [Green Version]
- Dao, T.; Mun, S.S.; Scott, A.C.; Jarvis, C.A.; Korontsvit, T.; Yang, Z.; Liu, L.; Klatt, M.G.; Guerreiro, M.; Brea, E.J.; et al. Depleting T Regulatory Cells by Targeting Intracellular Foxp3 with a TCR Mimic Antibody. Oncoimmunology 2019, 8, 1570778. [Google Scholar] [CrossRef]
- Stathopoulou, C.; Gangaplara, A.; Mallett, G.; Flomerfelt, F.A.; Liniany, L.P.; Knight, D.; Samsel, L.A.; Berlinguer-Palmini, R.; Yim, J.J.; Felizardo, T.C.; et al. PD-1 Inhibitory Receptor Downregulates Asparaginyl Endopeptidase and Maintains Foxp3 Transcription Factor Stability in Induced Regulatory T Cells. Immunity 2018, 49, 247–263.e7. [Google Scholar] [CrossRef] [Green Version]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 Regulates the Development, Maintenance, and Function of Induced Regulatory T Cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan Nair, V.; Toor, S.M.; Taouk, G.; Pfister, G.; Ouararhni, K.; Alajez, N.M.; Elkord, E. Pembrolizumab Interferes with the Differentiation of Human FOXP3+—Induced T Regulatory Cells, but Not with FOXP3 Stability, through Activation of MTOR. J. Immunol. 2020, 204, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Toor, S.M.; Khaja, A.S.S.; Alkurd, I.; Elkord, E. In Vitro Effect of Pembrolizumab on Different T Regulatory Cell Subsets. Clin. Exp. Immunol. 2018, 191, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Toor, S.M.; Sasidharan Nair, V.; Pfister, G.; Elkord, E. Effect of Pembrolizumab on CD4+ CD25+, CD4+ LAP+ and CD4+ TIM-3+ T Cell Subsets. Clin. Exp. Immunol. 2019, 196, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Toor, S.M.; Al-Ali, D.; Sasidharan Nair, V.; Elkord, E. Blockade of PD-1, PD-L1, and TIM-3 Altered Distinct Immune- and Cancer-Related Signaling Pathways in the Transcriptome of Human Breast Cancer Explants. Genes 2020, 11, 703. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.Q. TGF-β: The Sword, the Wand, and the Shield of FOXP3+ Regulatory T Cells. J. Mol. Cell Biol. 2012, 4, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Jochems, C.; Tritsch, S.R.; Pellom, S.T.; Su, Z.; Soon-Shiong, P.; Wong, H.C.; Gulley, J.L.; Schlom, J. Analyses of Functions of an Anti-PD-L1/TGFβR2 Bispecific Fusion Protein (M7824). Oncotarget 2017, 8, 75217–75231. [Google Scholar] [CrossRef] [Green Version]
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The Significance of OX40 and OX40L to T-Cell Biology and Immune Disease. Immunol. Rev. 2009, 229, 173–191. [Google Scholar] [CrossRef] [Green Version]
- Melero, I.; Gangadhar, T.C.; Kohrt, H.E.; Segal, N.H.; Logan, T.; Urba, W.J.; Hodi, F.S.; Ott, P.A.; Perez-Gracia, J.L.; Wolchok, J.D.; et al. A Phase I Study of the Safety, Tolerability, Pharmacokinetics, and Immunoregulatory Activity of Urelumab (BMS-663513) in Subjects with Advanced and/or Metastatic Solid Tumors and Relapsed/Refractory B-Cell Non-Hodgkin’s Lymphoma (B-NHL). J. Clin. Oncol. 2013, 31, TPS3107. [Google Scholar] [CrossRef]
- Zhang, J.; Hou, S.; Gu, J.; Tian, T.; Yuan, Q.; Jia, J.; Qin, Z.; Chen, Z. S100A4 Promotes Colon Inflammation and Colitis-Associated Colon Tumorigenesis. OncoImmunology 2018, 7, e1461301. [Google Scholar] [CrossRef] [Green Version]
- Faget, J.; Sisirak, V.; Blay, J.-Y.; Caux, C.; Bendriss-Vermare, N.; Ménétrier-Caux, C. ICOS Is Associated with Poor Prognosis in Breast Cancer as It Promotes the Amplification of Immunosuppressive CD4+ T Cells by Plasmacytoid Dendritic Cells. OncoImmunology 2013, 2, e23185. [Google Scholar] [CrossRef] [Green Version]





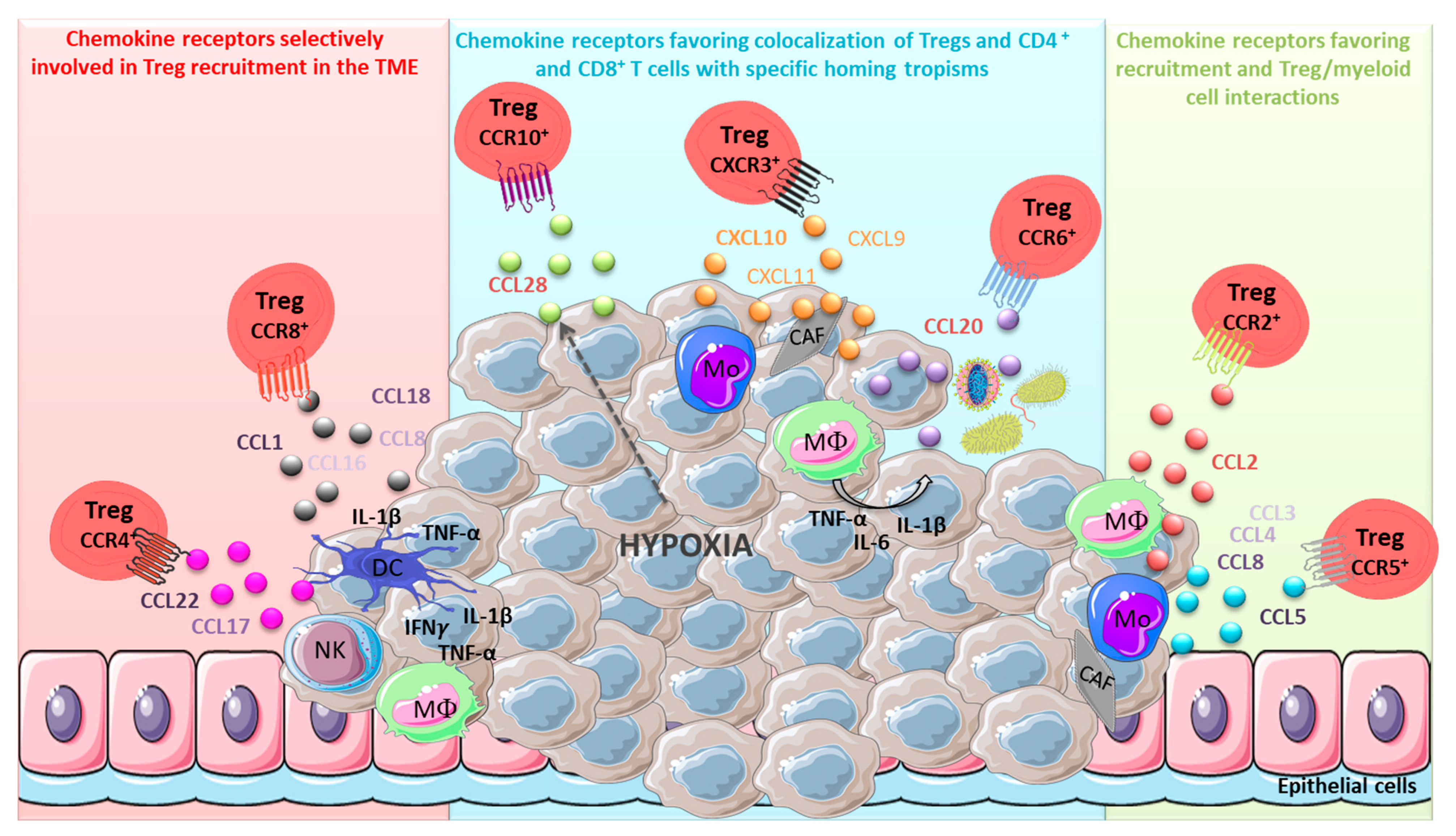{kind=link}
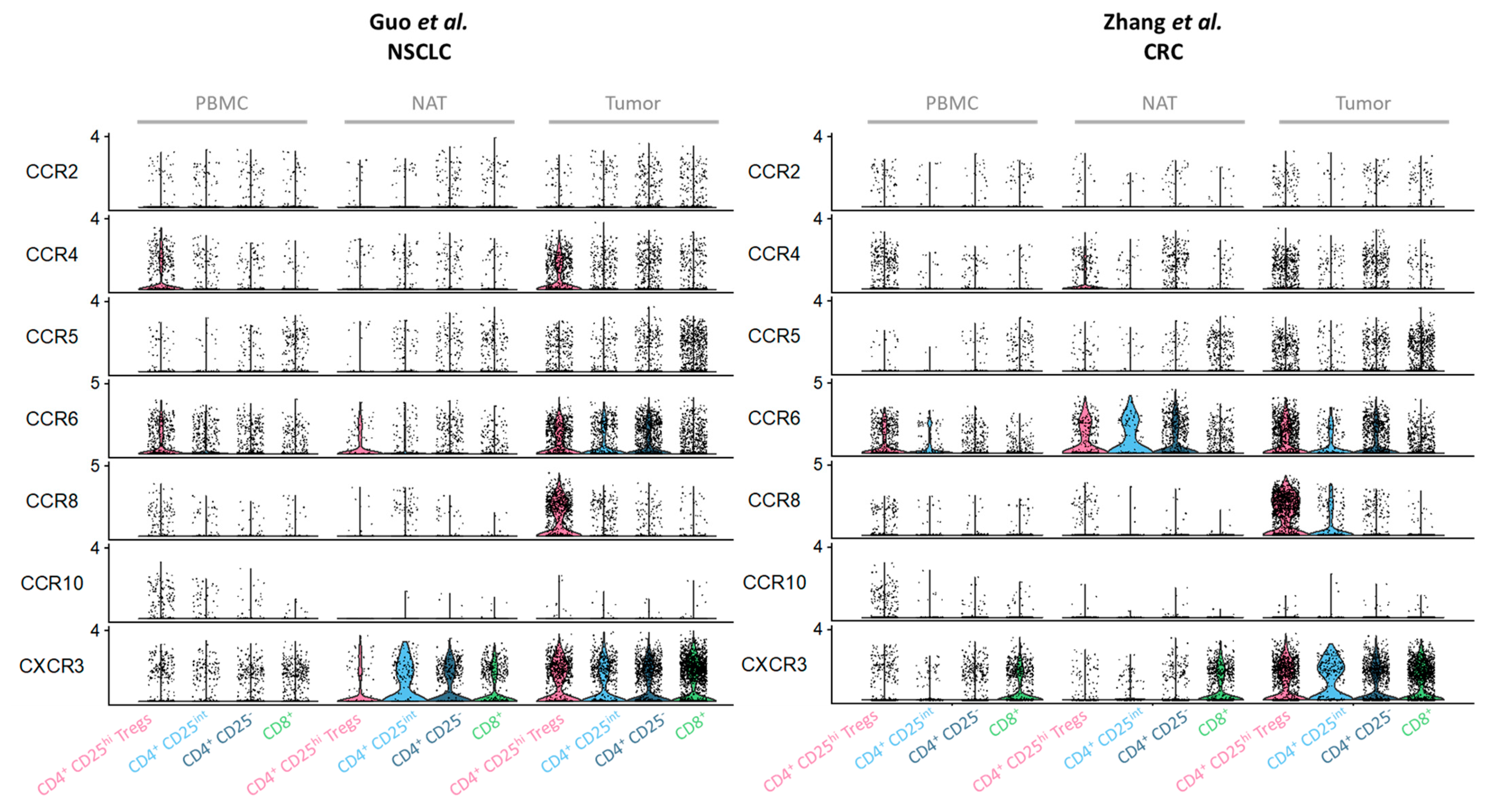{kind=link}
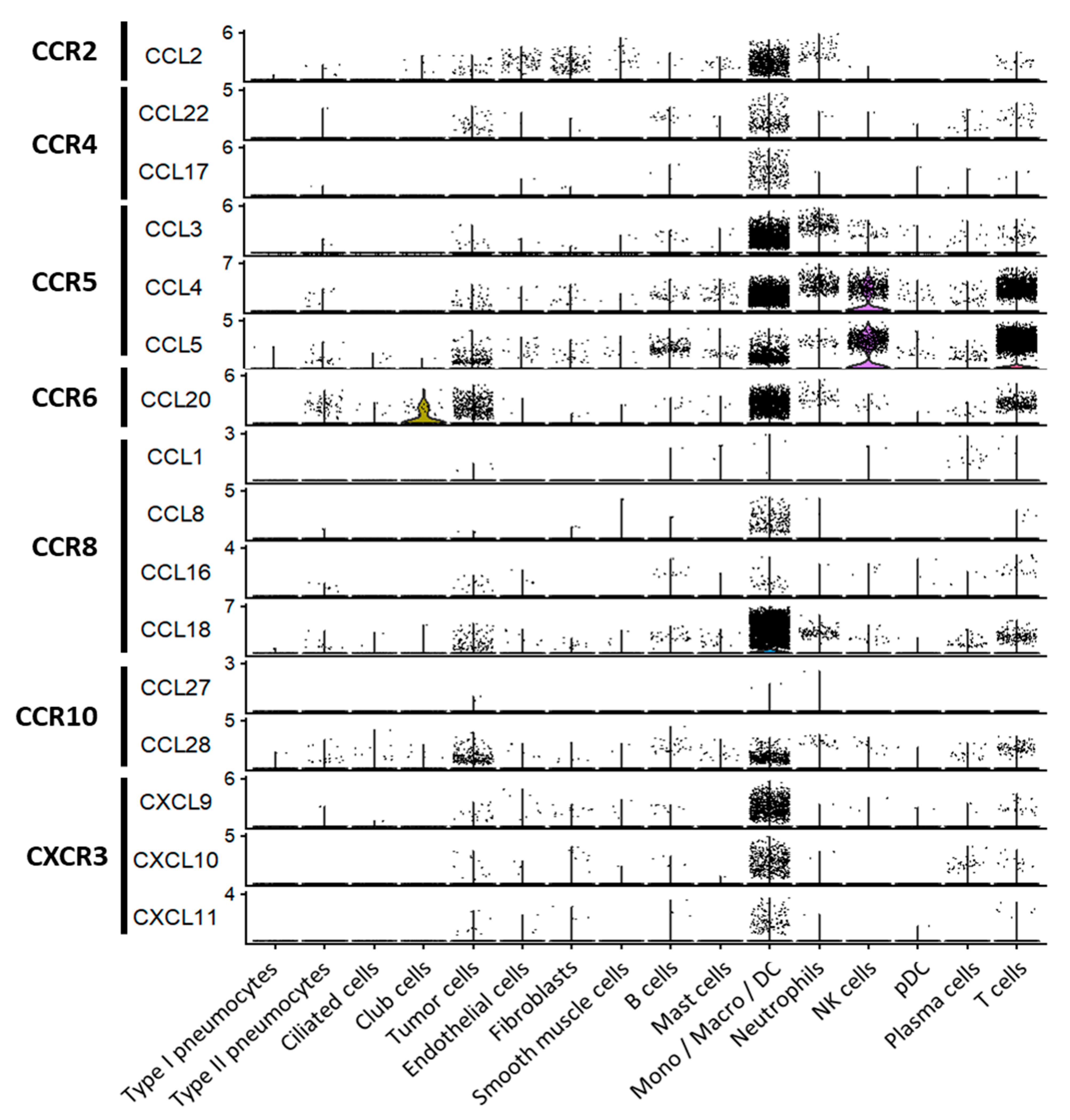{kind=link}
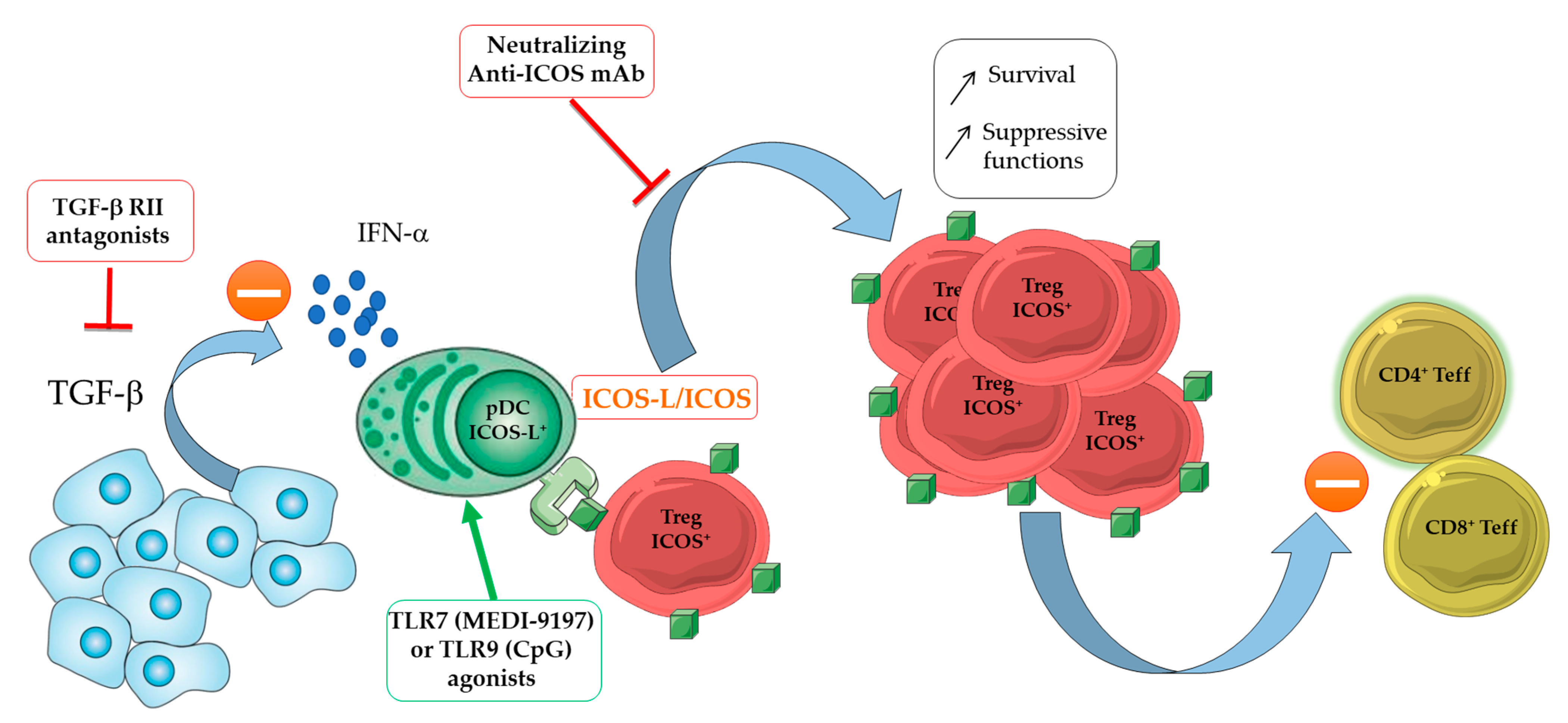{kind=link}
| Target | Name | Producer | Description | Mode of Action | Stage of Development | Clinical Trial Register | Ref. |
|---|---|---|---|---|---|---|---|
| CCR4 | AZD-2098 | AstraZeneca | Small chemical class-II antagonist | Blocks chemokine-induced cellular response (Ca++ influx) Blocks Treg recruitment | Marketed | [310] | |
| AZD-1678 | AstraZeneca | Small chemical class-II antagonist | Blocks Treg recruitment | Preclinical studies | [310] | ||
| FLX475 | FlxBio Inc. | Small chemical class-II antagonist | Prevents link between CCL22 and CCR4 (competition) Blocks Treg migration to the TME | Phase 1/2: Monotherapy or combination with anti-PD-1 (Merck Biopharma (Merck)) (advanced cancers) | NCT03674567 | [322,323] | |
| KW-0761 | Kyowa Hakko Kirin | Humanized mAb (defucosylated hIgG1) | Supports ADCC by linking to CCR4 | Phase 1a: Monotherapy (solid tumors) | NCT01929486 | [324,325] | |
| Phase 1/2: Combination with anti-PD-1 (Merck) (B-cell lymphoma) | NCT03309878 | [326] | |||||
| Phase 1b: Combination with anti-4-1BB (Pfizer) (advanced solid tumors) | NCT02444793 | [327] | |||||
| Phase 1b: combination with anti-PD-L1 or anti-CTLA-4 (AstraZeneca) (advanced solid tumors) | NCT02301130 | [328] | |||||
| Phase 2: combination with anti-PD-1 (BMS) (advanced solid tumors) | NCT02476123 | [329] | |||||
| anti-CCR4-DT immunotoxin | Harvard Medical School | Single-chain fold back diabody anti-hCCR4 coupled to DT | Binds CCR4+ cells Inhibits cell proliferation and protein synthesis ADCC, CDC and ADCP | In vitro studies in human PBMC Preclinical studies in monkey | [330,331,332] | ||
| CCR8 | AZ084 | AstraZeneca | CCR8 allosteric antagonist | Blocks LT, DC and eosinophils migration | Preclinical studies | [320] | |
| JTX-1811 | Jounce Therapeutics | Humanized mAb (hIgG1) | CCR8+ Tregs depletion by ADCC | Preclinical studies | [333] | ||
| SRF114 | Surface Oncology | Humanized mAb (hIgG1) | CCR8+ Tregs depletion by ADCC Blocks CCR8+ Tregs migration | Preclinical studies | [334] | ||
| HBM1022 | Harbour Biomed | Humanized mAb (hIgG1) | CCR8+ Tregs depletion by ADCC Blocks CCR8+ Tregs migration | Preclinical studies | [335] | ||
| FPA157 | Five Prime Therapeutics | Humanized mAb (hIgG1) | CCR8+ Tregs depletion by ADCC Blocks CCR8+ Tregs migration | Preclinical studies | [336] | ||
| 25B3 | BMS | Humanized mAb (non fucosylated hIgG1) | CCR8+ Tregs depletion by ADCC | Preclinical studies | [337] | ||
| CCR2/ CCR5 | BMS-813160 | BMS | Small inhibitor | Reduced Tregs, MDSC and TAM recruitment | Phase 1b/2: Monotherapy or combination with anti-PD-1 (BMS) (CRC/PDAC) | NCT03184870 | [338] |
| CCX872 | ChemoCentryx | Small inhibitor | Reduces the presence of suppressive myeloid cells | Phase 1b: monotherapy (PDAC) | NCT02345408 | [339] | |
| ICOS | KY1044 | KyMab | Human mAb (hIgG1k) | Depletes Tregs by ADCC (in vitro) Enhances production of IFN-γ by CD4+ Teff | Phase 1/2: monotherapy or combination with anti-PD-L1 (Merck) (advanced tumors) | NCT03829501 | [340] |
| GSK3359609 | GSK | Humanized anti-hICOS (hIgG4) | Stimulates ICOS+CD4+ Teff | Phase 1/2: monotherapy or combination with anti-PD-1 (Merck) (neoplasms) | NCT02723955 | [341] | |
| GSK3359609 | GSK | Humanized anti-hICOS (hIgG4) | Stimulates ICOS+CD4+ Teff | Phase 2/3: Combination with anti-PD-1 (Merck) (HNSCC) | NCT04128696 | [342] | |
| JTX-2011 | Jounce Therapeutics | Humanized anti-hICOS (hIgG4) | Stimulates ICOS+CD4+ Teff | Phase 1/2: Combination with anti-PD-1 or anti CTLA-4 (BMS) Phase 2: Combination with anti-PD-1 (Merck/BMS) | NCT02904226 NCT04549025 | [343] | |
| TNFR2 | E4F6 | National Cancer Institute | Fully-human defucosylated mAb (hIgG1) | Induces TNFR2+ Treg killing through ADCC | [344] | ||
| BI-1808 | BioInvent International AB | Fully human mAb (hIgG1) | Combines Treg depletion, CD8+ T cell expansion and modulation of TA-myeloid cells | Phase 1/2a: Monotherapy or combination with anti-PD-1 (Merck) (advanced tumors) | NCT04752826 | [345] | |
| OX40 | 9B12 | Providence Health & Service | Murine anti-hOX40 mAb (mIgG1) | Increases immune cell activity | Phase 1: Monotherapy in advanced cancers | NCT01644968 | [346] |
| MEDI-16469 | MedImmune | Murine anti-hOX40 mAb | Increases Ki67+ ICOS+ CD4+ and CD8+ Teff | Phase 2: Combination with stereotactic radiation and/or chemotherapy | NCT01642290 NCT01303705 | [347] | |
| 4-1BB /CD137 | PF-05082566 | Pfizer | Humanized anti-h4-1BB (hIgG2) | Favors anti-tumor immune response though Teff | Phase 1: Monotherapy or combination with anti-hCD20 (GenenTech) (NHL) | NCT01307267 | [348] |
| FOXP3 | AZD8701 | AstraZeneca | Antisense oligonucleotide | Deletes FOXP3 expression on Tregs | Phase 1: Monotherapy (solid tumors) or combination with anti-PD-L1 (AstraZeneca) (TNBC, RCC) | NCT04504669 | [349] |
| Bi-specific mAbs | M7824 | NIH | Bifunctional fusion protein (hIgG1 anti-PDL-1 fused with 2 extracellular domains of TGFβRII) | Neutralizes and depletes (ADCC) suppressive function of PD-1+ Tregs producing TGFβ | Phase 1: Monotherapy in biliary tract carcinoma | NCT03833661 | [350] |
| Phase 1: Monotherapy in advanced NSCLC | NCT02517398 | [351] | |||||
| Phase 2: Combination with chemotherapy (NSCLC) | NCT03840915 | [352] | |||||
| Phase 2: platinum experienced cervical cancer | NCT04246489 | [353] | |||||
| KN046 | Alphamab Bio Pharmaceuticals | bispecific hIgG1-Fc fused with anti-PD-L1 and anti-CTLA-4 Fab domains | Blocks both PD-L1/PD-1 and CTLA-4/CD80-CD86 interactions | Phase 1b/2: Monotherapy or combination with chemotherapy (TNBC) Phase 2: Combination with chemotherapy and palliative radiotherapy Phase 2: Advanced NSCLC | NCT03872791 NCT03927495 NCT03838848 | [354] | |
| ATOR-1015 | Alligator Biosciences | bispecific hIgG1-Fc [Ig-like-V-type domain of hCD86 + agonist OX40 mAb] | Induces T cell activation and Treg depletion | Phase 1: Monotherapy in solid tumors | NCT03782467 | [355] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cinier, J.; Hubert, M.; Besson, L.; Di Roio, A.; Rodriguez, C.; Lombardi, V.; Caux, C.; Ménétrier-Caux, C. Recruitment and Expansion of Tregs Cells in the Tumor Environment—How to Target Them? Cancers 2021, 13, 1850. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13081850
Cinier J, Hubert M, Besson L, Di Roio A, Rodriguez C, Lombardi V, Caux C, Ménétrier-Caux C. Recruitment and Expansion of Tregs Cells in the Tumor Environment—How to Target Them? Cancers. 2021; 13(8):1850. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13081850
Chicago/Turabian StyleCinier, Justine, Margaux Hubert, Laurie Besson, Anthony Di Roio, Céline Rodriguez, Vincent Lombardi, Christophe Caux, and Christine Ménétrier-Caux. 2021. "Recruitment and Expansion of Tregs Cells in the Tumor Environment—How to Target Them?" Cancers 13, no. 8: 1850. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13081850