
Genetic Events Inhibiting Apoptosis in Diffuse Large B Cell Lymphoma

1
Faculty of Medicine, McGill University, Montreal, QC H3G 2M1, Canada
2
Department of Medicine, McGill University, Montreal, QC H4A 3J1, Canada
3
Departments of Medicine and Oncology, Jewish General Hospital, Montreal, QC H3T 1E2, Canada
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(9), 2167; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13092167
Submission received: 13 April 2021
/
Revised: 27 April 2021
/
Accepted: 27 April 2021
/
Published: 30 April 2021
(This article belongs to the Section Molecular Cancer Biology)
Abstract
:Simple Summary
Diffuse large B cell lymphoma (DLBCL) is the most common type of non-Hodgkin lymphoma (NHL). Despite the genetic heterogeneity of the disease, most patients are initially treated with a combination of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP), but relapse occurs in ~50% of patients. One of the hallmarks of DLBCL is the occurrence of genetic events that inhibit apoptosis, which contributes to disease development and resistance to therapy. These events can affect the intrinsic or extrinsic apoptotic pathways, or their modulators. Understanding the factors that contribute to inhibition of apoptosis in DLBCL is crucial in order to be able to develop targeted therapies and improve outcomes, particularly in relapsed and refractory DLBCL (rrDLBCL). This review provides a description of the genetic events inhibiting apoptosis in DLBCL, their contribution to lymphomagenesis and chemoresistance, and their implication for the future of DLBCL therapy.
Abstract
Diffuse large B cell lymphoma (DLBCL) is curable with chemoimmunotherapy in ~65% of patients. One of the hallmarks of the pathogenesis and resistance to therapy in DLBCL is inhibition of apoptosis, which allows malignant cells to survive and acquire further alterations. Inhibition of apoptosis can be the result of genetic events inhibiting the intrinsic or extrinsic apoptotic pathways, as well as their modulators, such as the inhibitor of apoptosis proteins, P53, and components of the NF-kB pathway. Mechanisms of dysregulation include upregulation of anti-apoptotic proteins and downregulation of pro-apoptotic proteins via point mutations, amplifications, deletions, translocations, and influences of other proteins. Understanding the factors contributing to resistance to apoptosis in DLBCL is crucial in order to be able to develop targeted therapies that could improve outcomes by restoring apoptosis in malignant cells. This review describes the genetic events inhibiting apoptosis in DLBCL, provides a perspective of their interactions in lymphomagenesis, and discusses their implication for the future of DLBCL therapy.
1. Introduction
Diffuse large B cell lymphoma (DLBCL) is the most common type of lymphoma and comprises about 25–40% of all non-Hodgkin lymphomas (NHL) in the Western world [1]. Patients are initially treated with chemoimmunotherapy, most commonly rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) [2,3]. Unfortunately, more than 50% of patients experience disease progression which, in young and fit patients, is treated with salvage chemotherapy followed by autologous stem cell transplant. Ultimately, only 10% of patients with relapsed or refractory DLBCL (rrDLBCL) are cured with this approach [4]. Work over the past decade has led to important insights into the disease biology. DLBCL is a heterogenous disease that evolves to evade apoptosis [5]. Many novel targeted therapies have been tested and are active in a subset of patients with rrDLBCL that is otherwise resistant to conventional chemotherapy [6]. Some of these have the potential to activate the intrinsic apoptotic pathway, independently of DNA-damage response pathways or P53, or engage the extrinsic apoptotic pathway through cell-mediated cytotoxicity. Furthermore, the genomic characterization of DLBCL at diagnosis and relapse allows for non-invasive monitoring of disease progression and clonal evolution over time using circulating tumor DNA (ctDNA) in the plasma [7,8]. This review focuses on the genomic alterations that affect critical survival and apoptotic pathways in DLBCL. An improved understanding of mechanisms that impair apoptosis in DLBCL may reveal vulnerabilities that could be exploited therapeutically in the future.
1.1. DLBCL Classification
DLBCL is classified into molecular subtypes that share common survival pathways and mechanisms that inhibit apoptosis. The most widely used classification stratifies DLBCL according to cell-of-origin (COO) molecular signatures, determined by gene expression profiling (GEP). These include the germinal center B cell-like (GCB) and activated B cell-like (ABC) subtypes, with 10–20% of cases having an intermediate or “unclassifiable” profile. GCB and ABC subtypes have distinct genomic alterations and clinical outcomes, with the latter being associated with an inferior overall survival [9,10,11,12]. The exact mechanisms of lymphomagenesis differ in GCB and ABC DLBCL, as they have distinct patterns of GEP and are, respectively derived from germinal center centroblasts and post-germinal center plasmablasts [13]. For instance, activation of the NF-kB pathway is mostly seen in ABC DLBCL, while BCL2 translocations are almost exclusive to GCB DLBCL. Other genetic events, such as TP53 mutations, are observed in both types of DLBCL [14]. An alternative gene expression classifier segregates DLBCL into three clusters that, for the purposes of this review, may have different mechanisms involved in survival and apoptosis. The OxPhos cluster is enriched in genes involved in mitochondrial function, oxidative phosphorylation, and the electron transport chain. The BCR/proliferation cluster expresses genes encoding components of the B cell receptor (BCR) signaling cascade, cell-cycle regulators, and DNA repair proteins. Finally, the host response (HR) cluster is characterized by increased expression of inflammatory mediators as well as components of the T cell receptor pathway and complement cascade [15]. The classification of DLCBL continues to evolve to include new information gained by recent whole exome or genome sequencing. The main genetic alterations identified in newer DLBCL classification systems are summarized in Table 1. These classification systems better represent the genetic heterogeneity of DLBCL and their distinct pathogenic pathways that could be modulated with targeted therapies. Although they overlap, significant differences are observed between those classification systems (reviewed in [16]), and a novel consensus classification remains to be determined [14,16,17].
1.2. Apoptosis in Normal Germinal Center B Cells
Apoptosis in the normal germinal center reaction is regulated by several pro- and anti-apoptotic proteins that have been previously reviewed [18]. In the normal maturation process of B cells in the germinal center, the intrinsic and extrinsic apoptotic pathways are essential to the proper functioning of positive and negative B cell selection [19]. The intrinsic pathway is triggered by cellular stress, including DNA alteration, that leads to mitochondrial permeabilization and release of pro-apoptotic factors, while the extrinsic pathway is triggered by ligands binding to death receptors and subsequent activation of the death-inducing signaling complex (DISC). Both pathways converge into the activation of effector caspases, the main apoptotic proteases (Figure 1) [20].
1.3. Inhibition of Apoptosis in DLBCL
A hallmark of cancer, including DLBCL, is the occurrence and accumulation of genetic alterations that promote malignant cell survival via inhibition of apoptosis [21]. This survival advantage can be exacerbated by the selective pressure imposed by therapies such as R-CHOP and is a major factor in the development of resistance to chemotherapy [22,23]. Accordingly, relapsed and refractory DLBCL (rrDLBCL) have a distinct genetic landscape that further inhibits apoptosis, contributing to resistance to additional chemotherapy and a poor outcome [4,7,24,25]. Understanding apoptotic pathways in DLBCL and the genetic factors contributing to their inhibition has the potential to dramatically improve the prognosis of DLBCL, notably by facilitating the development of targeted therapies that could selectively relieve apoptotic blockade and therefore restore response to treatment. Herein, the genetic events leading to inhibition of apoptosis in DLBCL are described (Table 2). The intrinsic and extrinsic apoptotic pathways, as well as their modulators, will be discussed.
2. Intrinsic Apoptotic Pathway
The intrinsic apoptotic pathway, also known as the mitochondrial or BCL2-regulated apoptotic pathway, is predominantly under the control of the BCL2 protein family [20,26]. These proteins are classified based on their influence on apoptosis and the number of BCL2 homology (BH) regions they contain. The anti-apoptotic proteins, which bind and sequester proapoptotic proteins, contain 4 BH domains (BH1–4) and include BCL2, MCL1, BCL-XL, and BCLW. The proapoptotic BH3-only proteins can be further divided into apoptotic sensitizers (BAD, NOXA, and HRK) that bind and inhibit anti-apoptotic proteins, and apoptotic activators (BID, BIM, and, to a lesser extent, PUMA) that can also directly activate the BH1–3 apoptotic effectors BAX and BAK [20]. Various cellular stresses, including oncogenes, DNA damage, and chemotherapy can trigger the intrinsic apoptotic pathway by activating the BH3-only proteins [27]. BAX and BAK are subsequently activated and permeabilize the outer mitochondrial membrane, allowing the release of cytochrome c in the cytosol, which is considered to be an irreversible commitment to apoptosis. Cytochrome c activates caspase 9, with APAF1 acting as a scaffold in the process. SMAC is also released from the mitochondrial outer membrane and leads to activation of caspase 9 by inhibiting XIAP, a caspase inhibitor. Activated caspase 9 in turn engages effector caspases, namely caspase 3, 6, and 7, leading to proteolysis and end stages of apoptosis (Figure 1) [20]. Inhibition of this pathway can be the result of alterations affecting different genes and proteins (Table 2) [28].
2.1. BCL2
BCL2 is the most common and important anti-apoptotic protein inhibiting apoptosis in DLBCL. BCL2 is normally silenced in germinal center (GC) B cells to allow low affinity B cells generated through somatic hypermutation (SHM) to undergo apoptosis. Thus, its expression in DLBCL is pathogenic [19]. BCL2 is located on chromosome 18q21 [29]. In addition to its 4 BH domains, the BCL2 protein structure is notable for the presence of a flexible loop domain that mediates interaction with the P53 tumor suppressor [30]. BCL2 protein expression in DLBCL can be caused by different genetic events such as translocations, mutations, gains, and amplifications as well as transcriptional upregulation from pathways discussed later (BCR signaling and NF-kB) (Table 2) [31]. BCL2 translocations to immunoglobulin genes (IG), t (14;18) (q32; q21), t (2;18) (p11; q21) and t (18;22) (q21; q11), involving the IgH, IgK and IgL loci, respectively, are present in 20–25% of DLBCL, almost exclusively of the GCB subtype [32]. BCL2 translocations are also a dominant feature in the new DLBCL classification systems, occurring in 71% of C3 DLBCL, 89% of BCL2 DLBCL, and 78% of EZB DLBCL [14,16,17]. They lead to constitutive transcription of BCL2 by the enhancer elements within the active IG loci [33]. In addition to leading to BCL2 overexpression, the t (14;18) translocation is associated with a significantly higher rate of SHM-associated BCL2 mutations, which is the most commonly mutated gene in DLBCL [31]. The role of these mutations in the pathogenesis of DLBCL is unclear, as a large proportion are either synonymous or tend to occur outside of the functionally important BH domains [31]. Some of these mutations could still have a functional impact and a role in disease development. BCL2 promoter mutations might increase BCL2 protein expression by preventing binding and transcriptional repression by BCL6 [34]. Mutations in the flexible loop domain might prevent P53 binding, resulting in increased sequestration of BAX by BCL2 and therefore reduced apoptosis [30]. Mutations in the BH4 domain of BCL2 decrease calcium-mediated apoptosis by preventing binding of BCL2 to the inositol 1,4,5-triphosphate receptor (IP3R), a channel that promotes apoptosis by facilitating calcium conductance from the endoplasmic reticulum to the mitochondria [35,36]. However, taken collectively, BCL2 mutations do not seem to affect prognosis [31]. BCL2 gains and amplifications occur in up to 25% of DLBCL, mostly of the ABC subtype. They typically are the result of copy number aberration of chromosome 18q21.33 and are associated with increased BCL2 expression and worse prognosis [37].
2.2. MCL1
MCL1 is an anti-apoptotic protein from the BCL2 family that is essential in B cell development and germinal center formation. It is therefore normally expressed in GC B cells [38,39]. It directly binds and sequesters BAX and BAK and prevents their activation by BH3-only proteins [40]. MCL1 is deregulated in several types of lymphoma and contributes to lymphomagenesis in cell lines and mouse models [41,42,43]. In a recent study, strong MCL1 expression was predominantly seen in ABC DLBCL, and apoptosis was induced in MCL1-positive DLBCL cell lines after knockdown of MCL1 or treatment with the BH3 mimetic obatoclax [44]. In the same study, gains and amplifications of MCL1 were observed in 26% of ABC DLBCL. However, MCL1 mutations were identified in less than 1% of cases [44]. Other potential mechanisms for deregulation of MCL1 include upregulation by the proto-oncogene STAT3, which is expressed in ABC DLBCL [45], and decreased MCL1 degradation due to upregulation of the USP9X deubiquitinase [46].
2.3. BCLX and BCLW
BCLX produces two proteins via alternative splicing: BCL-XL, an anti-apoptotic protein, and BCL-XS, a BCL2 inhibitor [47]. BCL-XL is expressed in DLBCL, although its contribution to inhibition of apoptosis is unclear [44,48,49,50]. It remains a potential therapeutic target, as pharmacologic inhibition of BCL-XL leads to apoptosis in some DLBCL cell lines [44,49]. A major pitfall of BCL-XL inhibition is thrombocytopenia, as it is the primary survival factor in platelets [51]. BCLW is a relatively understudied anti-apoptotic gene, as it was initially shown to be only essential in spermatogenesis [52,53]. Overexpression of BCLW has been observed in DLBCL cell lines and has been associated with resistance to apoptosis and decreased patient survival [54,55]. However, these results have not been observed in a recent study [56]. Overall, there are no notable genetic alteration in BCLX and BCLW in DLBCL, and the role of the proteins encoded by these two genes is minor in comparison to BCL2 and MCL1 [44].
2.4. Pro-Apoptotic Proteins
Although genetic events amplifying the effect of anti-apoptotic proteins are well characterized, pro-apoptotic defects in DLBCL remain a relatively understudied phenomenon. Functional assessment of the intrinsic apoptotic pathway through BH3 profiling in DLBCL cell lines and primary samples has revealed defects in BH3-only pro-apoptotic proteins (class A apoptotic block) and in apoptotic effectors (class B block). Such pro-apoptotic defects are a factor contributing to resistance to the BCL2 inhibitor venetoclax [50,57]. However, they are unlikely to be the result of alterations at the genomic level, as mutations or other genetic events directly affecting pro-apoptotic proteins are rarely seen in DLBCL [24,28]. The exact causes of these pro-apoptotic defects remain to be determined, but could notably involve epigenetic silencing, transcriptional repression, or interaction with other proteins. There is also a possibility that genetic alteration of pro-apoptotic proteins is more common in rrDLBCL and a contributing factor to chemoresistance, as observed in other hematological malignancies [58,59,60].
In summary, deregulation of the intrinsic apoptotic pathway is a clear contributor to the pathogenesis of DLBCL. Translocations and mutations affecting BCL2 are characteristic of GCB DLBCL, while BCL2 and MCL1 gains and amplifications are more common in the ABC subtype. These proteins can also be upregulated through other mechanisms that do not require direct alteration of the gene loci. The therapeutic relevance of these alterations is discussed at the end of this review.
3. Extrinsic Apoptotic Pathway
The extrinsic apoptotic pathway is important in the regulation of the germinal center reaction and prevention of lymphomagenesis [19]. It is also known as the death receptor-mediated pathway, as it is initiated by the binding of a death receptor ligand to its corresponding death receptor. These receptor-ligand pairs include FAS (CD95, APO-1) and FAS ligand (FasL), as well as TRAIL (APO2-L) and its receptors [61]. Activation of the death receptor leads to binding of its intracellular death domain to FADD (for FAS) or TRADD (for TRAIL receptors). The death effector domain of FADD/TRADD then binds to cFLIP, pro-caspase 8 (FLICE), and pro-caspase 10. The protein complex formed by FADD/TRADD, cFLIP, and pro-caspases is termed the death-inducing signaling complex (DISC) and allows conversion of pro-caspases into caspases. In the absence of death receptor ligand binding, formation of the DISC is inhibited by the anti-apoptotic regulator cFLIP [61]. The intrinsic and extrinsic apoptotic pathways converge when initiator caspases 8 and 10 activate effector caspases, resulting in proteolysis and apoptosis (Figure 1) [61]. Genetic events affecting proteins involved in different death receptor classes from the extrinsic apoptotic pathway have been reported in DLBCL (Table 2).
3.1. FAS Pathway
FAS, located on chromosome 10q23 and containing 9 exons, encodes the FAS cell surface death receptor [62,63]. The last exon of the gene encodes the death domain of the receptor, which is essential for initiation of FAS-mediated apoptosis [64]. FAS is highly expressed in germinal center B cells and is critical for the negative selection of suboptimal or self-reactive B cells during the germinal center reaction [65,66]. The mechanism by which FAS-mediated apoptosis is induced in the germinal center is not entirely understood, but might involve CD4+ T helper cells, which express FasL, and autonomous FAS-FasL signaling by B cells [19,67,68,69,70]. The FAS pathway is also one of the mechanisms by which cytotoxic T cells and natural killer (NK) cells kill their cellular targets, along with the perforin/granzyme pathway [71]. Deregulation of the extrinsic apoptotic pathway contributes to lymphomagenesis, notably by making NHL cells resistant to FAS-mediated apoptosis [72,73,74]. Heterozygous germline mutations in FAS are associated with autoimmune lymphoproliferative syndrome, a rare disorder characterized by lymphadenopathy, splenomegaly, autoimmune cytopenias, and a significantly increased risk of B cell lymphoma [75]. Somatic FAS mutations are consistently identified in GCB and ABC DLBCL, with reported frequencies of 5–15% [16,17,24]. The majority of mutations are located either in exon 9, encoding the death domain [64], or are frameshift or nonsense mutations that lead to loss of the death domain [76]. These mutations exert a dominant negative effect, as they prevent formation of the DISC and initiation of apoptosis via the extrinsic pathway [75]. Mutations are also seen in the 5′ region of the gene, which is also the case in healthy germinal center B cells, likely as a consequence of aberrant SHM [77]. In addition, FAS deletions have been observed in 7% of DLBCL [78]. Another potential mechanism leading to resistance to FAS-mediated apoptosis is an increased concentration of soluble FAS receptor (sFAS), a FasL sequestrant that results from alternative splicing out of exon 6 of FAS [79]. Prognostic information regarding FAS mutations in DLBCL is limited, but decreased FAS or FasL expression has been associated with decreased survival [80]. Preclinical studies with mouse models and lymphoma cell lines have explored different mechanisms of modulation of FAS-mediated apoptosis such as local administration of FasL, bispecific antibodies, and fusion proteins [81]. However, FAS-directed therapy is notably limited by severe hepatoxicity that precludes it from being used in clinical practice, and by the fact that FAS has other functions that are pro-oncogenic [82,83].
3.2. TRAIL Pathway
TRAIL (APO2-L) is a ligand from the tumor necrosis factor (TNF) family that can trigger extrinsic apoptosis by binding to either TRAIL-R1 (DR4) or TRAIL-R2 (DR5) [84,85]. The genes encoding these receptors, TRAIL-R1 and TRAIL-R2, are both located on chromosome 8p21 [86,87]. Three other receptors can bind TRAIL and inhibit apoptosis by acting as decoys: TRAIL-R3 (DcR1), TRAIL-R4 (DcR2), and osteoprotegerin [84]. TRAIL-mediated apoptosis is one of the main effector mechanism of NK cells and is also used by cytotoxic T cells [88,89,90]. Highest levels of TRAIL receptors expression in B cells are seen in GC B cells, and evidence suggests that TRAIL-mediated apoptosis is an important regulator of B cell selection and germinal center homeostasis [91,92,93]. In addition, TRAIL and its receptors are important in immune surveillance against tumor development [88]. Resistance to TRAIL-mediated apoptosis has been reported in DLBCL [94]. A study that included 46 DLBCL has identified TRAIL-R1 or TRAIL-R2 mutations in 5 of them (10.9%), all of which were inside or in close proximity to the region encoding the death domain [95]. 8p21 deletions comprising TRAIL-R1 and TRAIL-R2 are also common in DLBCL [96]. TRAIL is an interesting therapeutic target, as it preferentially targets tumor cells and shows low levels of toxicity in animal models [84]. TRAIL agonists such as recombinant TRAIL, TRAIL-R antibodies, fusion proteins, and small molecules are being investigated preclinically in various malignancies, including DLBCL [97,98]. In addition, the TRAIL receptor agonist ABBV-621 is currently being investigated in a phase I clinical trial in solid tumors and hematological malignancies (NCT03082209; Supplementary Table S1).
4. Inhibitor of Apoptosis Proteins
The inhibitor of apoptosis proteins (IAPs) are a family of antiapoptotic proteins that inhibit the intrinsic and extrinsic apoptotic pathways, mainly via inhibition of caspases 3,7, and 9. Eight IAPs have been identified including XIAP, cIAP1, cIAP2, and survivin [99]. XIAP is a direct caspase inhibitor, while the other IAPs act by inhibiting SMAC, a XIAP inhibitor (Figure 1) [100]. Several of these IAPs can be upregulated in DLBCL [101,102,103,104]. As IAPs inhibit both the intrinsic and extrinsic apoptotic pathways due to their downstream action, they likely contribute to the pathogenesis of DLBCL and resistance to chemotherapy [99]. However, mutations in genes encoding IAPs are rare in DLBCL. Downregulation of IAPs is being investigated as a potential therapeutic strategy that could be combined with other apoptotic modulators in DLBCL [105,106,107].
5. P53 Pathway
TP53, located on chromosome 17p13.1 and containing 11 exons, encodes the tumor suppressor P53 and is the most frequently mutated gene in human cancers [108,109,110]. Germline mutations in TP53 cause Li-Fraumeni syndrome, a cancer predisposition syndrome associated with breast cancer, brain tumors, adrenocortical carcinoma, leukemias, and many other malignancies [111]. P53 is a crucial regulator of cell cycle, cell proliferation, DNA repair, cellular senescence, and apoptosis [112]. Its structure is notable for a central DNA-binding domain that is necessary for the transcriptional activation of target genes. This domain contains several residues that are frequently mutated in different malignancies [112]. Under normal circumstances, MDM2 inhibits P53-mediated transcriptional activation by transporting P53 to the cytoplasm, binding its DNA-binding domain, and promoting its degradation. P53 also self-regulates in a negative feedback manner by inducing expression of MDM2 [113,114,115]. Cellular stresses such as DNA damage, oncogene activation, hypoxia, and loss of normal cell contact can lead to P53 activation by disrupting the binding of MDM2 to P53 [116]. Activated P53 can then exert its proapoptotic functions by modulating the transcription of several proteins. This results in an increased proportion of pro-apoptotic BCL2 proteins, increased expression of extrinsic pathway proteins FAS, FasL and TRAIL-R2, and upregulation of effector caspases 9 (via coactivator Apaf-1) and 6 (Figure 1) [117].
Genomic alterations in TP53 and its associated proteins are common in DLBCL. TP53 mutations are seen in more than 20% of GCB and ABC DLBCL and are associated with poor prognosis in the GCB subtype [118,119,120]. Approximately 90% of mutations lead to loss of P53 function, and most of them are located in the DNA-binding domain (exons 5-8), thus preventing P53-mediated transcriptional activation [119]. Mutant P53 can act as an oncogenic transcription factor and could therefore further contribute to lymphomagenesis if its expression is increased [121]. Chromosome 17p13.1 deletion occurs in approximately 10% of DLBCL, but does not seem to be correlated with survival [119]. The P53 inhibitor MDM2 is overexpressed in 40% of DLBCL, although gene amplifications are rare and expression is not affected by a common polymorphism (SNP309) [122]. MDM4 and RFWD2 encode two other P53 inhibitors and can both be amplified with gains of chromosome 1q23.3, reported in 15% of DLBCL [78]. Amplification of BCL2L12, an atypical BCL2 protein that inhibits P53 and caspases 3/7, have been observed in 10% of cases [78]. CDKN2A encodes the ARF (p14) and INK4a (p16) tumor suppressors [123]. ARF promotes activation of the P53 pathway by binding and inhibiting MDM2 [124]. CDKN2A deletions are present in 19–35% of DLBCL. They are associated with an ABC subtype and a decreased survival [14,125,126]. Alterations of the P53 pathway also include P53 target genes such as PERP (caspase 8 activator) and SCOTIN (pro-caspase 3/7 activator), which are deleted in 27% and 8% of DLBCL, respectively [78].
TP53 is the most commonly mutated gene in rrDLBCL, with mutations observed in up to half of cases [7,24]. Clonal evolution studies have shown that most of these mutations are present in primary DLBCL subclones that are selected for during chemotherapy [7,127,128]. For this reason, tracking of TP53 mutations in the plasma ctDNA in patients undergoing chemotherapy has the potential to detect and monitor early resistant clones [129,130].
6. Transcriptional Regulation of Apoptotic Pathways
In addition to alterations in the apoptotic pathways themselves, resistance to apoptosis in DLBCL is driven by the constitutive activation of transcriptional regulators that decrease apoptotic signals and potentiate anti-apoptotic proteins. Inhibition of apoptosis by the nuclear factor kappa beta (NF-kB) pathway is the most notable example of such transcriptional regulation in DLBCL [131]. The role of the NF-kB pathway in DLBCL and other hematological malignancies has been previously reviewed [131]. In B cells, activation of the NF-kB pathway is triggered by B cell receptor (BCR) signaling. This leads to formation of the CARD11-BCL10-MALT1 (CBM) complex, which allows translocation of cytoplasmic NF-kB to the nucleus to facilitate target gene transcription [132,133,134]. Regulation of apoptosis by the NF-kB pathway affects both the intrinsic and extrinsic apoptotic pathways by targeting and upregulating cFLIP [135,136], BCL2 [137], BCL-XL [138], cIAP1 [139,140], cIAP2 [139,140], XIAP [141], and survivin [142] (Figure 1). Constitutive NF-kB activation is a hallmark of ABC DLBCL and can involve different components of the pathway (Table 2) [143].
6.1. B Cell Receptor Signaling
In ABC DLBCL, the NF-kB pathway is constitutively activated by chronic active BCR signaling [144]. This involves BCR signal transduction via phosphorylation of the immunoreceptor tyrosine-based activation motifs (ITAMs) of CD79A and CD79B. Phosphorylated ITAMs trigger a signaling cascade that sequentially activate the SYK and BTK tyrosine kinases and results in activation of the CBM complex [144,145]. Gain-of-function mutations in the ITAM of CD79B occur in 10–25% of ABC DLBCL and 3% of GCB DLBCL, and lead to increased BCR signaling [14,144,146]. CD79A mutations can also be observed but are rare [144]. Potential loss-of-function mutations in genes encoding negative regulators of BCR signaling, such as LYN, LAPTM5, PTPN6, GRB2, PRKCD, DGKZ, SLA, and MAP4K1 have collectively been identified in almost 40% of all DLBCL [14]. Of note, in addition to NF-kB, BCR signaling upregulates several pathways that contribute to cellular proliferation and oncogenesis, notably the MEK/ERK and AKT/PI3K pathways [5].
6.2. CARD11-BCL10-MALT1 Complex
As described above, the CBM complex is essential for NF-kB activation in lymphocytes. In resting cells, CARD11, a scaffold protein from the membrane-associated guanylate kinase family, is inactivated by its autoinhibitory domain. B cell activation leads to phosphorylation and activation of CARD11, recruitment of BCL10 and MALT1, formation of the CBM complex, and NF-kB activation [147,148]. Missense mutations in CARD11 occur in approximately 10% of ABC DLBCL and in a smaller proportion of GCB DLBCL. These mutations occur almost exclusively in the coiled-coil domain of CARD11 and result in a gain of function by preventing functioning of the autoinhibitory domain [147]. Chromosomal rearrangements involving BCL10 are identified in up to 20% of DLBCL and are more common in the GCB subtype [149,150]. BCL10 amplifications can also occur but are rare [14]. In addition to its role in the NF-kB pathway, wild-type BCL10 also has pro-apoptotic functions [151]. BCL10 mutations occur in about 5% of DLBCL [14,17] and result in a loss of BCL10 pro-apoptotic function while preserving its ability to activate NF-kB [151]. As for BCL2, MALT1 is located on chromosome 18q21 and can therefore be gained in the same copy number alteration events [152].
6.3. NF-kB Genes and Regulators
NF-kB is a family of five dimeric transcription factors (RelA, RelB, c-Rel, p50, and p52) that are involved in numerous processes such as cellular development and proliferation, immune cell activation, and regulation of apoptosis. Two other proteins, p100 and p105, are precursors of p52 and p50, respectively [133]. These 7 proteins share a Rel Homology Domain (RHD) that mediates their dimerization, interactions with inhibitors, and DNA binding [133]. Despite the prominent role of the NF-kB pathway in ABC DLBCL, mutations affecting the NF-kB genes themselves are rare [153]. Other rare genetic events directly involving NF-kB include chromosome 10q24 rearrangements that lead to loss of the 3′-end of NFKB2 (encoding p100) and constitutive protein activation [154], and amplifications of REL (encoding c-Rel), which are mostly seen in GCB DLBCL [9]. However, the significance of REL amplifications is unclear, as they do not correlate with NF-kB target gene expression [154]. Mutations involving positive and negative NF-kB regulators are more common and are collectively seen in more than half of ABC DLBCL, and in more than 20% of GCB DLBCL [153]. A20 is located on chromosome 6q23.3 and encodes a ubiquitin-modifying enzyme that can downregulate the NF-kB response [155]. Loss-of-function mutations in A20 have been identified in 24% of ABC DLBCL [153]. Inactivation of A20 can also be the result of 6q23 deletions [156,157]. Mutations in TNIP1, encoding an A20-binding inhibitor of NF-kB, can also be seen [14]. MYD88 encodes an adaptor protein that upregulates the NF-kB pathway by mediating toll and interleukin-1 signaling. The MYD88L265P mutation, a gain-of-function mutation that leads to constitutive NF-kB activation, is observed in 18–30% of ABC DLBCL and is associated with poor prognosis [14,16,158]. In addition, this mutation is particularly prevalent in primary extranodal DLBCL and is notably present in most cases of primary testicular lymphoma and primary central nervous system lymphoma [14,16,17].
Inhibition of apoptosis is the main mechanism by which NF-kB contributes to the pathogenesis of DLBCL, particularly of the ABC subtype [131]. This anti-apoptotic effect affects the intrinsic and extrinsic apoptotic pathways as well as the inhibitor of apoptosis proteins (Figure 1, Table 2). Numerous steps of the NF-kB can be altered, which may lead to different responses in attempts to pharmacologically downregulate this pathway.
7. Therapeutic Targeting of Apoptosis
The central role of inhibition of apoptosis in DLBCL and other malignancies has led to increasing investigation of apoptotic pathways and proteins as potential therapeutic targets [159]. Venetoclax is a BH3 mimetic and BCL2 inhibitor studied in multiple hematological malignancies. It is approved in chronic lymphocytic leukemia (CLL) and in older acute myeloid leukemia (AML) patients deemed unfit for conventional chemotherapy [160]. Initial trials in DLBCL have demonstrated modest activity [161,162,163], but a recent phase II study adding venetoclax to R-CHOP for previously untreated DLBCL has shown promising results, particularly in BCL2-positive cases [164]. One of the main factors contributing to venetoclax resistance in DLBCL is upregulation of MCL1, and inhibition of this protein can induce apoptosis and increase venetoclax-induced apoptosis in DLBCL cell lines [41,50,165,166,167]. DLBCL where resistance to apoptosis is driven by an increase in anti-apoptotic proteins (class C apoptotic block) might therefore respond to venetoclax with the possible addition of an MCL1 inhibitor. Doxorubicin and vincristine might also be beneficial in such patients, as these agents are associated with decreased levels of MCL1 and increased venetoclax-induced apoptosis [50]. Another factor contributing to inhibition of intrinsic apoptosis is pro-apoptotic functional defects (class A and B blocks). DLBCL with such defects might benefit from therapies that trigger cell death independently of the mitochondrial apoptotic pathway, for example by inducing cell-mediated cytotoxicity [57].
Extrinsic apoptosis is of particular importance in the use of therapies that involve cytotoxic T cells, namely immune checkpoint inhibitors, chimeric antigen receptor T (CAR T) cells, and bispecific T cell engagers (BiTEs) [168,169,170]. Immune checkpoint inhibitors have not been studied extensively in DLBCL, but have shown relatively low response rates in rrDLBCL [171,172]. This could be explained by the fact that most DLBCL are not characterized by robust T cell infiltration and activation. However, the immune landscape of DLBCL is heterogeneous and certain subtypes, such as those with constitutive NF-kB activation, could still benefit from immune checkpoint inhibition [173]. Anti-CD19 CAR T cell therapy has shown good response rates in selected cases of rrDLBCL and is approved as third-line therapy in multiple countries [174]. Blinatumomab, a BiTE that links CD3-positive T cells and CD19-positive B cells, has shown a 43% overall response rate and a 19% complete remission rate in a phase 2 trial including 25 patients with rrDLBCL [175]. It can be hypothesized that response to these therapies requires a functional extrinsic apoptotic pathway, which could explain the lack of response in a significant proportion of patients. Recent evidence using CRISPR knockout screens suggests that defects in the extrinsic apoptotic pathway contribute to resistance to CAR T cell and BiTE therapy in DLBCL and other hematological malignancies [176,177]. Such defects seem to exert a dominant negative effect and also prevent perforin/granzyme cytotoxicity by leading to prolonged antigen exposure and subsequent T cell exhaustion and dysfunction [177]. However, the nature of the extrinsic apoptosis defects contributing to T cell-based therapy resistance remains to be characterized. This has several potential clinical implications: for instance, DLBCL with mutations in genes that encode components of the extrinsic apoptotic pathway might have suboptimal responses or higher rates of resistance to CAR T cell or BiTE therapy. Patients receiving these therapies might also benefit from combination therapy that simultaneously targets the intrinsic apoptotic pathway, as shown in B cell malignancies cell lines where CAR T cell therapy was combined with the BH3 mimetic ABT-737 [178]. Currently, therapies involving cytotoxic T cells remain mostly used and investigated in rrDLBCL (Table S1).
An important consideration in the choice of therapy for rrDLBCL is a high rate of TP53 mutations [7]. Loss of P53 function notably drives the development of chemoresistance by blunting the DNA-damage response and downregulating the initiation of apoptosis [179]. This implies that agents depending on P53 to trigger apoptosis by inducing DNA damage, such as doxorubicin, are unlikely to be effective in a large proportion of rrDLBCL [179]. BH3 mimetics, T cell-based therapies, IAPs inhibitors, and other therapeutic strategies that kill cells independently of P53 might therefore be more beneficial in those cases. Pharmacologic inhibition of the P53 inhibitor MDM2 has also shown potential in preclinical studies [180]. However, two recent phase 1 trials combining the MDM2 inhibitor idasanutlin with rituximab (plus venetoclax in one of the two trials) in rrDLBCL were terminated because of the overall modest benefits observed (NCT03135262 and NCT02624986).
Another potential therapeutic strategy in DLBCL is to modulate transcriptional regulators of apoptosis. The predominant effect of NF-kB on inhibition of apoptosis in DLBCL, particularly of the ABC subtype, makes it a therapeutic target of interest. Inhibition of BCR signaling with the BTK tyrosine kinase inhibitor ibrutinib has initially shown good tolerability but modest response rates in ABC DLBCL [181,182]. Phase 1 trials using ibrutinib in combination with either lenalidomide or R-ICE (rituximab, ifosfamide, carboplatin, and etoposide) have shown good response rates in non-GCB rrDLBCL [183,184]. However, the addition of ibrutinib to R-CHOP did not improve progression-free or overall survival in patients with de novo non-GCB DLBCL [185]. Lenalidomide is an immunomodulatory agent that downregulates BCR signaling [186]. Its addition to R-CHOP in de novo ABC DLBCL has shown promising results in phase II trials [187,188], but a recent phase III trial has shown no improvement in progression-free or overall survival in previously untreated ABC DLBCL [189]. Bortezomib, a proteasome inhibitor that downregulates NF-kB by decreasing the degradation of inhibitory kB proteins [190], has shown no benefit in phase II and III trials when combined with R-CHOP in de novo ABC DLBCL [191,192,193]. The disappointing results seen with modulation of NF-kB do not exclude this pathway as a potential therapeutic target, as the events leading to NF-kB upregulation in DLBCL are heterogeneous. For instance, it can be hypothesized that BTK inhibition with ibrutinib could have a limited effect in a DLBCL with a mutation that affects a downstream step in the NF-kB pathway. Modulation of the NF-kB pathway could therefore be of therapeutic benefit in selected patients, particularly if used in conjunction with other apoptotic modulators that are chosen based on apoptotic profiling. The new DLBCL classification systems might contribute to better identification of patients that would best respond to NF-kB downregulation or other therapies that aim to restore apoptosis. Therapeutic targeting of apoptosis is currently being investigated in newly diagnosed DLBCL and rrDLBCL in numerous clinical trials (Table 3; Table S1).
8. Conclusions
DLBCL genetically reprograms itself to inhibit apoptosis. This inhibition of apoptosis is a consequence of genetic events leading to dysregulation of several interacting pathways that modulate intrinsic and extrinsic apoptosis (Table 2). Overall, this results in an increase in anti-apoptotic BCL2 proteins, dysfunction of death receptor and DISC signaling, and blunting of the P53-mediated DNA-damage response. In addition, this anti-apoptotic state is upregulated by BCR signaling and the NF-kB pathway (Figure 1). Ultimately, inhibition of apoptosis confers a survival advantage to malignant cells, especially under the selective pressure of chemotherapy. As a result, inhibition of apoptosis is even more predominant in rrDLBCL, which harbor very high rates of TP53 mutations [7,8,194,195].
In addition to improving the understanding of the pathogenesis of DLBCL, characterizing the genetic events inhibiting apoptosis have several important clinical implications. Indeed, newer DLBCL classification systems, which are based on genetic defects, recapitulate the different mechanisms of inhibition of apoptosis more accurately than the COO classification. Once a consensus classification system is established, it would likely replace the current COO classification in genetic studies and clinical trials. Sequencing lymphomas in clinical practice may reveal mutation patterns that would help in establishing the diagnosis of DLBCL. Monitoring these mutations in the plasma ctDNA over time would be a non-invasive approach to identify the emergence of resistant subclones. This could then lead to the timely implementation of targeted therapies. For instance, ibrutinib could be beneficial in MCD and BN2 DLBCL, which are characterized by predominant B cell receptor-dependent NF-kB activation, while BH3 mimetics might be particularly beneficial in the EZB subtype, which shows frequent BCL2 translocations [14,16,17].
A better understanding of the genetic events contributing to the inhibition of apoptosis in DLBCL is essential. It gives insight into the mechanisms involved in disease development and progression. The identification of potential therapeutic targets in the plasma could help guide physicians to select effective therapies based on DLBCL subtype or apoptotic profile. This precision medicine may ultimately improve the survival of patients with rrDLBCL.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/cancers13092167/s1, Table S1: Ongoing clinical trials investigating therapies modulating apoptosis in DLBCL.
Author Contributions
Conceptualization—E.L. and N.A.J.; Writing—Original draft preparation, E.L.; Writing—Review and editing, E.L. and N.A.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
N.A.J. has received research funding from Roche Canada and consulting fees/honoraria from Roche, Abbvie, Lundbeck, Seattle Genetics, Janssen, and Gilead. E.L. has no conflict of interest.
References
- Teras, L.R.; DeSantis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US lymphoid malignancy statistics by world health organization subtypes. CA Cancer J. Clin. 2016, 66, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat. Rev. Clin. Oncol. 2014, 11, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Lepage, E.; Briere, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Van Den Neste, E.; Salles, G.; Gaulard, P.; et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Van Den Neste, E.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 2017, 130, 1800–1808. [Google Scholar] [CrossRef]
- Lossos, I.S. Molecular pathogenesis of diffuse large B-cell lymphoma. J. Clin. Oncol. 2005, 23, 6351–6357. [Google Scholar] [CrossRef] [Green Version]
- Di Rocco, A.; De Angelis, F.; Ansuinelli, M.; Foà, R.; Martelli, M. Is now the time for molecular driven therapy for diffuse large B-cell lymphoma? Expert Rev. Hematol. 2017, 10, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Rushton, C.K.; Arthur, S.E.; Alcaide, M.; Cheung, M.; Jiang, A.; Coyle, K.M.; Cleary, K.L.S.; Thomas, N.; Hilton, L.K.; Michaud, N.; et al. Genetic and evolutionary patterns of treatment resistance in relapsed B-cell lymphoma. Blood Adv. 2020, 4, 2886–2898. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Scherer, F.; Jin, M.C.; Soo, J.; Craig, A.F.M.; Esfahani, M.S.; Chabon, J.J.; Stehr, H.; Liu, C.L.; Tibshirani, R.; et al. Circulating Tumor DNA measurements as early outcome predictors in diffuse large b-cell lymphoma. J. Clin. Oncol. 2018, 36, 2845–2853. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef] [Green Version]
- Wright, G.; Tan, B.; Rosenwald, A.; Hurt, E.H.; Wiestner, A.; Staudt, L.M. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2003, 100, 9991–9996. [Google Scholar] [CrossRef] [Green Version]
- Lenz, G.; Staudt, L.M. Aggressive lymphomas. N. Engl. J. Med. 2010, 362, 1417–1429. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and pathogenesis of diffuse large B-Cell lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Monti, S.; Savage, K.J.; Kutok, J.L.; Feuerhake, F.; Kurtin, P.; Mihm, M.; Wu, B.; Pasqualucci, L.; Neuberg, D.; Aguiar, R.C.; et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861. [Google Scholar] [CrossRef] [Green Version]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A haematological malignancy research network report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]
- Klein, U.; Dalla-Favera, R. Germinal centres: Role in B-cell physiology and malignancy. Nat. Rev. Immunol. 2008, 8, 22–33. [Google Scholar] [CrossRef]
- Kondo, E.; Yoshino, T. Expression of apoptosis regulators in germinal centers and germinal center-derived B-cell lymphomas: Insight into B-cell lymphomagenesis. Pathol. Int. 2007, 57, 391–397. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indran, I.R.; Tufo, G.; Pervaiz, S.; Brenner, C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim. Biophys. Acta 2011, 1807, 735–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muris, J.J.; Meijer, C.J.; Ossenkoppele, G.J.; Vos, W.; Oudejans, J.J. Apoptosis resistance and response to chemotherapy in primary nodal diffuse large B-cell lymphoma. Hematol. Oncol. 2006, 24, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Assouline, S.; Alcaide, M.; Mohajeri, A.; Johnston, R.L.; Chong, L.; Grewal, J.; Yu, S.; Fornika, D.; Bushell, K.; et al. Genetic landscapes of relapsed and refractory diffuse large B-Cell Lymphomas. Clin. Cancer Res. 2016, 22, 2290–2300. [Google Scholar] [CrossRef] [Green Version]
- Rovira, J.; Valera, A.; Colomo, L.; Setoain, X.; Rodriguez, S.; Martinez-Trillos, A.; Gine, E.; Dlouhy, I.; Magnano, L.; Gaya, A.; et al. Prognosis of patients with diffuse large B cell lymphoma not reaching complete response or relapsing after frontline chemotherapy or immunochemotherapy. Ann. Hematol. 2015, 94, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Klanova, M.; Klener, P. BCL-2 Proteins in pathogenesis and therapy of B-Cell non-hodgkin lymphomas. Cancers 2020, 12, 938. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.M.; Clark-Garvey, S.; Porcu, P.; Eischen, C.M. Targeting the Bcl-2 Family in B Cell Lymphoma. Front. Oncol. 2018, 8, 636. [Google Scholar] [CrossRef]
- Seto, M.; Jaeger, U.; Hockett, R.D.; Graninger, W.; Bennett, S.; Goldman, P.; Korsmeyer, S.J. Alternative promoters and exons, somatic mutation and deregulation of the Bcl-2-Ig fusion gene in lymphoma. EMBO J. 1988, 7, 123–131. [Google Scholar] [CrossRef]
- Deng, X.; Gao, F.; Flagg, T.; Anderson, J.; May, W.S. Bcl2’s flexible loop domain regulates p53 binding and survival. Mol. Cell Biol. 2006, 26, 4421–4434. [Google Scholar] [CrossRef] [Green Version]
- Schuetz, J.M.; Johnson, N.A.; Morin, R.D.; Scott, D.W.; Tan, K.; Ben-Nierah, S.; Boyle, M.; Slack, G.W.; Marra, M.A.; Connors, J.M.; et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia 2012, 26, 1383–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, J.; Sanger, W.G.; Horsman, D.E.; Rosenwald, A.; Pickering, D.L.; Dave, B.; Dave, S.; Xiao, L.; Cao, K.; Zhu, Q.; et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am. J. Pathol. 2004, 165, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Kuppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Novak, U.; Piovan, E.; Basso, K.; Sumazin, P.; Schneider, C.; Crespo, M.; Shen, Q.; Bhagat, G.; Califano, A.; et al. BCL6 suppression of BCL2 via Miz1 and its disruption in diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2009, 106, 11294–11299. [Google Scholar] [CrossRef] [Green Version]
- Deniaud, A.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Monaco, G.; Decrock, E.; Akl, H.; Ponsaerts, R.; Vervliet, T.; Luyten, T.; De Maeyer, M.; Missiaen, L.; Distelhorst, C.W.; De Smedt, H.; et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012, 19, 295–309. [Google Scholar] [CrossRef]
- Ennishi, D.; Mottok, A.; Ben-Neriah, S.; Shulha, H.P.; Farinha, P.; Chan, F.; Meissner, B.; Boyle, M.; Hother, C.; Kridel, R.; et al. Genetic profiling of MYC and BCL2 in diffuse large B-cell lymphoma determines cell-of-origin-specific clinical impact. Blood 2017, 129, 2760–2770. [Google Scholar] [CrossRef] [Green Version]
- Opferman, J.T.; Letai, A.; Beard, C.; Sorcinelli, M.D.; Ong, C.C.; Korsmeyer, S.J. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 2003, 426, 671–676. [Google Scholar] [CrossRef]
- Vikstrom, I.; Carotta, S.; Luthje, K.; Peperzak, V.; Jost, P.J.; Glaser, S.; Busslinger, M.; Bouillet, P.; Strasser, A.; Nutt, S.L.; et al. Mcl-1 is essential for germinal center formation and B cell memory. Science 2010, 330, 1095–1099. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1, the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, S.S.; Grau, M.; Mavis, C.; Hailfinger, S.; Wolf, A.; Madle, H.; Deeb, G.; Dorken, B.; Thome, M.; Lenz, P.; et al. MCL1 is deregulated in subgroups of diffuse large B-cell lymphoma. Leukemia 2013, 27, 1381–1390. [Google Scholar] [CrossRef]
- Fernandez-Marrero, Y.; Spinner, S.; Kaufmann, T.; Jost, P.J. Survival control of malignant lymphocytes by anti-apoptotic MCL-1. Leukemia 2016, 30, 2152–2159. [Google Scholar] [CrossRef]
- Cho-Vega, J.H.; Rassidakis, G.Z.; Admirand, J.H.; Oyarzo, M.; Ramalingam, P.; Paraguya, A.; McDonnell, T.J.; Amin, H.M.; Medeiros, L.J. MCL-1 expression in B-cell non-Hodgkin’s lymphomas. Human Pathol. 2004, 35, 1095–1100. [Google Scholar] [CrossRef]
- Smith, V.M.; Dietz, A.; Henz, K.; Bruecher, D.; Jackson, R.; Kowald, L.; van Wijk, S.J.L.; Jayne, S.; Macip, S.; Fulda, S.; et al. Specific interactions of BCL-2 family proteins mediate sensitivity to BH3-mimetics in diffuse large B-cell lymphoma. Haematologica 2020, 105, 2150–2163. [Google Scholar] [CrossRef]
- Ding, B.B.; Yu, J.J.; Yu, R.Y.; Mendez, L.M.; Shaknovich, R.; Zhang, Y.; Cattoretti, G.; Ye, B.H. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood 2008, 111, 1515–1523. [Google Scholar] [CrossRef] [Green Version]
- Schwickart, M.; Huang, X.; Lill, J.R.; Liu, J.; Ferrando, R.; French, D.M.; Maecker, H.; O’Rourke, K.; Bazan, F.; Eastham-Anderson, J.; et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010, 463, 103–107. [Google Scholar] [CrossRef]
- Boise, L.H.; Gonzalez-Garcia, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Bcl, L.A.; Mao, X.; Nunez, G.; Thompson, C.B. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Xerri, L.; Parc, P.; Brousset, P.; Schlaifer, D.; Hassoun, J.; Reed, J.C.; Krajewski, S.; Birnbaum, D. Predominant expression of the long isoform of Bcl-x (Bcl-xL) in human lymphomas. Br. J. Haematol. 1996, 92, 900–906. [Google Scholar] [CrossRef]
- Klanova, M.; Andera, L.; Brazina, J.; Svadlenka, J.; Benesova, S.; Soukup, J.; Prukova, D.; Vejmelkova, D.; Jaksa, R.; Helman, K.; et al. Targeting of BCL2 family proteins with ABT-199 and homoharringtonine Reveals BCL2- and MCL1-dependent subgroups of diffuse large B-Cell lymphoma. Clin. Cancer Res. 2016, 22, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Rys, R.N.; Wever, C.M.; Geoffrion, D.; Goncalves, C.; Ghassemian, A.; Brailovski, E.; Ryan, J.; Stoica, L.; Hebert, J.; Petrogiannis-Haliotis, T.; et al. Apoptotic blocks in primary non-hodgkin B cell lymphomas identified by BH3 profiling. Cancers 2021, 13, 1002. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Print, C.G.; Loveland, K.L.; Gibson, L.; Meehan, T.; Stylianou, A.; Wreford, N.; de Kretser, D.; Metcalf, D.; Kontgen, F.; Adams, J.M.; et al. Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc. Natl. Acad. Sci. USA 1998, 95, 12424–12431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, A.J.; Waymire, K.G.; Moss, J.E.; Parlow, A.F.; Skinner, M.K.; Russell, L.D.; MacGregor, G.R. Testicular degeneration in Bclw-deficient mice. Nat. Genet. 1998, 18, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M.; Mitra, R.; Gong, J.Z.; Eischen, C.M. Non-hodgkin and hodgkin lymphomas select for overexpression of BCLW. Clin. Cancer Res. 2017, 23, 7119–7129. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.M.; Kim, A.S.; Mitra, R.; Choi, J.K.; Gong, J.Z.; Eischen, C.M. BCL-W has a fundamental role in B cell survival and lymphomagenesis. J. Clin. Investig. 2017, 127, 635–650. [Google Scholar] [CrossRef] [Green Version]
- Diepstraten, S.T.; Chang, C.; Tai, L.; Gong, J.N.; Lan, P.; Dowell, A.C.; Taylor, G.S.; Strasser, A.; Kelly, G.L. BCL-W is dispensable for the sustained survival of select Burkitt lymphoma and diffuse large B-cell lymphoma cell lines. Blood Adv. 2020, 4, 356–366. [Google Scholar] [CrossRef]
- Deng, J.; Carlson, N.; Takeyama, K.; Dal Cin, P.; Shipp, M.; Letai, A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 2007, 12, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Brimmell, M.; Mendiola, R.; Mangion, J.; Packham, G. BAX frameshift mutations in cell lines derived from human haemopoietic malignancies are associated with resistance to apoptosis and microsatellite instability. Oncogene 1998, 16, 1803–1812. [Google Scholar] [CrossRef] [Green Version]
- Fresquet, V.; Rieger, M.; Carolis, C.; Garcia-Barchino, M.J.; Martinez-Climent, J.A. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 2014, 123, 4111–4119. [Google Scholar] [CrossRef] [Green Version]
- Wever, C.M.; Geoffrion, D.; Grande, B.M.; Yu, S.; Alcaide, M.; Lemaire, M.; Riazalhosseini, Y.; Hebert, J.; Gavino, C.; Vinh, D.C.; et al. The genomic landscape of two Burkitt lymphoma cases and derived cell lines: Comparison between primary and relapse samples. Leuk. Lymphoma 2018, 59, 2159–2174. [Google Scholar] [CrossRef]
- Safa, A.R. c-FLIP, a master anti-apoptotic regulator. Exp. Oncol. 2012, 34, 176–184. [Google Scholar]
- Cheng, J.; Liu, C.; Koopman, W.J.; Mountz, J.D. Characterization of human Fas gene. Exon/intron organization and promoter region. J. Immunol. 1995, 154, 1239–1245. [Google Scholar]
- Lichter, P.; Walczak, H.; Weitz, S.; Behrmann, I.; Krammer, P.H. The human APO-1 (APT) antigen maps to 10q23, a region that is syntenic with mouse chromosome 19. Genomics 1992, 14, 179–180. [Google Scholar] [CrossRef]
- Behrmann, I.; Walczak, H.; Krammer, P.H. Structure of the human APO-1 gene. Eur. J. Immunol. 1994, 24, 3057–3062. [Google Scholar] [CrossRef]
- Hao, Z.; Duncan, G.S.; Seagal, J.; Su, Y.W.; Hong, C.; Haight, J.; Chen, N.J.; Elia, A.; Wakeham, A.; Li, W.Y.; et al. Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity 2008, 29, 615–627. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Ohta, H.; Takemori, T. Fas is required for clonal selection in germinal centers and the subsequent establishment of the memory B cell repertoire. Immunity 2001, 14, 181–192. [Google Scholar] [CrossRef]
- Guzman-Rojas, L.; Sims-Mourtada, J.C.; Rangel, R.; Martinez-Valdez, H. Life and death within germinal centres: A double-edged sword. Immunology 2002, 107, 167–175. [Google Scholar] [CrossRef]
- Mintz, M.A.; Cyster, J.G. T follicular helper cells in germinal center B cell selection and lymphomagenesis. Immunol. Rev. 2020, 296, 48–61. [Google Scholar] [CrossRef]
- Koncz, G.; Hueber, A.O. The Fas/CD95 Receptor Regulates the Death of Autoreactive B cells and the selection of antigen-specific B cells. Front. Immunol. 2012, 3, 207. [Google Scholar] [CrossRef] [Green Version]
- Afshar-Sterle, S.; Zotos, D.; Bernard, N.J.; Scherger, A.K.; Rodling, L.; Alsop, A.E.; Walker, J.; Masson, F.; Belz, G.T.; Corcoran, L.M.; et al. Fas ligand-mediated immune surveillance by T cells is essential for the control of spontaneous B cell lymphomas. Nat. Med. 2014, 20, 283–290. [Google Scholar] [CrossRef]
- Trapani, J.A.; Smyth, M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002, 2, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Plumas, J.; Jacob, M.C.; Chaperot, L.; Molens, J.P.; Sotto, J.J.; Bensa, J.C. Tumor B cells from non-Hodgkin’s lymphoma are resistant to CD95 (Fas/Apo-1)-mediated apoptosis. Blood 1998, 91, 2875–2885. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Manley, T.J.; Pichert, G.; Cameron, C.; Cochran, K.J.; Levine, H.; Ritz, J. Functional consequences of APO-1/Fas (CD95) antigen expression by normal and neoplastic hematopoietic cells. Leuk. Lymphoma 1995, 17, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Kondo, E.; Yoshino, T.; Yamadori, I.; Matsuo, Y.; Kawasaki, N.; Minowada, J.; Akagi, T. Expression of Bcl-2 protein and Fas antigen in non-Hodgkin’s lymphomas. Am. J. Pathol. 1994, 145, 330–337. [Google Scholar] [PubMed]
- Fisher, G.H.; Rosenberg, F.J.; Straus, S.E.; Dale, J.K.; Middleton, L.A.; Lin, A.Y.; Strober, W.; Lenardo, M.J.; Puck, J.M. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 1995, 81, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Feuerhake, F.; Kutok, J.L.; Monti, S.; Dal Cin, P.; Neuberg, D.; Aster, J.C.; Shipp, M.A. FAS death domain deletions and cellular FADD-like interleukin 1beta converting enzyme inhibitory protein (long) overexpression: Alternative mechanisms for deregulating the extrinsic apoptotic pathway in diffuse large B-cell lymphoma subtypes. Clin. Cancer Res. 2006, 12 Pt. 1, 3265–3271. [Google Scholar] [CrossRef] [Green Version]
- Muschen, M.; Re, D.; Jungnickel, B.; Diehl, V.; Rajewsky, K.; Kuppers, R. Somatic mutation of the CD95 gene in human B cells as a side-effect of the germinal center reaction. J. Exp. Med. 2000, 192, 1833–1840. [Google Scholar] [CrossRef]
- Monti, S.; Chapuy, B.; Takeyama, K.; Rodig, S.J.; Hao, Y.; Yeda, K.; Inguilizian, H.; Mermel, C.; Currie, T.; Dogan, A.; et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 2012, 22, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, L.; Mathur, R.; Braun, F.K.; Wise, J.F.; Berkova, Z.; Neelapu, S.; Kwak, L.W.; Samaniego, F. FAS-antisense 1 lncRNA and production of soluble versus membrane Fas in B-cell lymphoma. Leukemia 2014, 28, 2376–2387. [Google Scholar] [CrossRef]
- Kojima, Y.; Tsurumi, H.; Goto, N.; Shimizu, M.; Kasahara, S.; Yamada, T.; Kanemura, N.; Hara, T.; Sawada, M.; Saio, M.; et al. Fas and Fas ligand expression on germinal center type-diffuse large B-cell lymphoma is associated with the clinical outcome. Eur. J. Haematol. 2006, 76, 465–472. [Google Scholar] [CrossRef]
- Villa-Morales, M.; Fernandez-Piqueras, J. Targeting the Fas/FasL signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 85–101. [Google Scholar] [CrossRef]
- Peter, M.E.; Hadji, A.; Murmann, A.E.; Brockway, S.; Putzbach, W.; Pattanayak, A.; Ceppi, P. The role of CD95 and CD95 ligand in cancer. Cell Death Differ. 2015, 22, 549–559. [Google Scholar] [CrossRef]
- Ichim, G.; Tait, S.W. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; El-Deiry, W.S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 2003, 22, 8628–8633. [Google Scholar] [CrossRef] [Green Version]
- Sessler, T.; Healy, S.; Samali, A.; Szegezdi, E. Structural determinants of DISC function: New insights into death receptor-mediated apoptosis signalling. Pharmacol. Ther. 2013, 140, 186–199. [Google Scholar] [CrossRef]
- Walczak, H.; Degli-Esposti, M.A.; Johnson, R.S.; Smolak, P.J.; Waugh, J.Y.; Boiani, N.; Timour, M.S.; Gerhart, M.J.; Schooley, K.A.; Smith, C.A.; et al. TRAIL-R2: A novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997, 16, 5386–5397. [Google Scholar] [CrossRef]
- Marsters, S.A.; Sheridan, J.P.; Pitti, R.M.; Huang, A.; Skubatch, M.; Baldwin, D.; Yuan, J.; Gurney, A.; Goddard, A.D.; Godowski, P.; et al. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr. Biol. 1997, 7, 1003–1006. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Smyth, M.J.; Cretney, E.; Hayakawa, Y.; Kayagaki, N.; Yagita, H.; Okumura, K. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J. Exp. Med. 2002, 195, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Hayakawa, Y.; Smyth, M.J.; Kayagaki, N.; Yamaguchi, N.; Kakuta, S.; Iwakura, Y.; Yagita, H.; Okumura, K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat. Med. 2001, 7, 94–100. [Google Scholar] [CrossRef]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Eto, H.; Okumura, K.; Yagita, H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 1999, 189, 1451–1460. [Google Scholar] [CrossRef]
- Staniek, J.; Lorenzetti, R.; Heller, B.; Janowska, I.; Schneider, P.; Unger, S.; Warnatz, K.; Seidl, M.; Venhoff, N.; Thiel, J.; et al. TRAIL-R1 and TRAIL-R2 Mediate TRAIL-dependent apoptosis in activated primary human B lymphocytes. Front. Immunol. 2019, 10, 951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerreiro-Cacais, A.O.; Levitskaya, J.; Levitsky, V. B cell receptor triggering sensitizes human B cells to TRAIL-induced apoptosis. J. Leukoc Biol. 2010, 88, 937–945. [Google Scholar] [CrossRef]
- Sedger, L.M.; Katewa, A.; Pettersen, A.K.; Osvath, S.R.; Farrell, G.C.; Stewart, G.J.; Bendall, L.J.; Alexander, S.I. Extreme lymphoproliferative disease and fatal autoimmune thrombocytopenia in FasL and TRAIL double-deficient mice. Blood 2010, 115, 3258–3268. [Google Scholar] [CrossRef] [Green Version]
- Testa, U. TRAIL/TRAIL-R in hematologic malignancies. J. Cell Biochem. 2010, 110, 21–34. [Google Scholar] [CrossRef]
- Lee, S.H.; Shin, M.S.; Kim, H.S.; Lee, H.K.; Park, W.S.; Kim, S.Y.; Lee, J.H.; Han, S.Y.; Park, J.Y.; Oh, R.R.; et al. Somatic mutations of TRAIL-receptor 1 and TRAIL-receptor 2 genes in non-Hodgkin’s lymphoma. Oncogene 2001, 20, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Moscardo, F.; Blesa, D.; Mestre, C.; Siebert, R.; Balasas, T.; Benito, A.; Rosenwald, A.; Climent, J.; Martinez, J.I.; Schilhabel, M.; et al. Characterization of 8p21.3 chromosomal deletions in B-cell lymphoma: TRAIL-R1 and TRAIL-R2 as candidate dosage-dependent tumor suppressor genes. Blood 2005, 106, 3214–3222. [Google Scholar] [CrossRef] [Green Version]
- Ion, G.N.D.; Nitulescu, G.M.; Popescu, C.I. Targeting TRAIL. Bioorg. Med. Chem. Lett. 2019, 29, 2527–2534. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.E. Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Schimmer, A.D. Inhibitor of apoptosis proteins: Translating basic knowledge into clinical practice. Cancer Res. 2004, 64, 7183–7190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef]
- Hussain, A.R.; Uddin, S.; Ahmed, M.; Bu, R.; Ahmed, S.O.; Abubaker, J.; Sultana, M.; Ajarim, D.; Al-Dayel, F.; Bavi, P.P.; et al. Prognostic significance of XIAP expression in DLBCL and effect of its inhibition on AKT signalling. J. Pathol. 2010, 222, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Engel, K.; Rudelius, M.; Slawska, J.; Jacobs, L.; Ahangarian Abhari, B.; Altmann, B.; Kurutz, J.; Rathakrishnan, A.; Fernandez-Saiz, V.; Brunner, A.; et al. USP9X stabilizes XIAP to regulate mitotic cell death and chemoresistance in aggressive B-cell lymphoma. EMBO Mol. Med. 2016, 8, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Akyurek, N.; Ren, Y.; Rassidakis, G.Z.; Schlette, E.J.; Medeiros, L.J. Expression of inhibitor of apoptosis proteins in B-cell non-Hodgkin and Hodgkin lymphomas. Cancer 2006, 107, 1844–1851. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Sui, X.; Li, Y.; Lu, K.; Fang, X.; Jiang, Y.; Wang, X. Prognostic and Clinicopathological Value of Survivin in Diffuse Large B-cell Lymphoma: A Meta-Analysis. Medicine 2015, 94, e1432. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14, 18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Cheson, B.D.; Bartlett, N.L.; Vose, J.M.; Lopez-Hernandez, A.; Seiz, A.L.; Keating, A.T.; Shamsili, S.; Papadopoulos, K.P. A phase II study of the survivin suppressant YM155 in patients with refractory diffuse large B-cell lymphoma. Cancer 2012, 118, 3128–3134. [Google Scholar] [CrossRef]
- Kita, A.; Mitsuoka, K.; Kaneko, N.; Nakata, M.; Yamanaka, K.; Jitsuoka, M.; Miyoshi, S.; Noda, A.; Mori, M.; Nakahara, T.; et al. Sepantronium bromide (YM155) enhances response of human B-cell non-Hodgkin lymphoma to rituximab. J. Pharmacol. Exp. Ther. 2012, 343, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Isobe, M.; Emanuel, B.S.; Givol, D.; Oren, M.; Croce, C.M. Localization of gene for human p53 tumour antigen to band 17p13. Nature 1986, 320, 84–85. [Google Scholar] [CrossRef]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [Green Version]
- Varley, J.M. Germline TP53 mutations and Li-Fraumeni syndrome. Hum. Mutat. 2003, 21, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the MDM2 oncoprotein. Cell Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [Green Version]
- Young, K.H.; Leroy, K.; Moller, M.B.; Colleoni, G.W.; Sanchez-Beato, M.; Kerbauy, F.R.; Haioun, C.; Eickhoff, J.C.; Young, A.H.; Gaulard, P.; et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: An international collaborative study. Blood 2008, 112, 3088–3098. [Google Scholar] [CrossRef] [Green Version]
- Xu-Monette, Z.Y.; Wu, L.; Visco, C.; Tai, Y.C.; Tzankov, A.; Liu, W.M.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A.; Orazi, A.; et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: Report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012, 120, 3986–3996. [Google Scholar] [CrossRef]
- Young, K.H.; Weisenburger, D.D.; Dave, B.J.; Smith, L.; Sanger, W.; Iqbal, J.; Campo, E.; Delabie, J.; Gascoyne, R.D.; Ott, G.; et al. Mutations in the DNA-binding codons of TP53, which are associated with decreased expression of TRAILreceptor-2, predict for poor survival in diffuse large B-cell lymphoma. Blood 2007, 110, 4396–4405. [Google Scholar] [CrossRef]
- Strano, S.; Dell’Orso, S.; Di Agostino, S.; Fontemaggi, G.; Sacchi, A.; Blandino, G. Mutant p53: An oncogenic transcription factor. Oncogene 2007, 26, 2212–2219. [Google Scholar] [CrossRef] [Green Version]
- Xu-Monette, Z.Y.; Moller, M.B.; Tzankov, A.; Montes-Moreno, S.; Hu, W.; Manyam, G.C.; Kristensen, L.; Fan, L.; Visco, C.; Dybkaer, K.; et al. MDM2 phenotypic and genotypic profiling, respective to TP53 genetic status, in diffuse large B-cell lymphoma patients treated with rituximab-CHOP immunochemotherapy: A report from the International DLBCL Rituximab-CHOP Consortium Program. Blood 2013, 122, 2630–2640. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J.; Weber, J.D. The ARF/p53 pathway. Curr. Opin. Genet. Dev. 2000, 10, 94–99. [Google Scholar] [CrossRef]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef] [Green Version]
- Jardin, F.; Jais, J.P.; Molina, T.J.; Parmentier, F.; Picquenot, J.M.; Ruminy, P.; Tilly, H.; Bastard, C.; Salles, G.A.; Feugier, P.; et al. Diffuse large B-cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R-CHOP treatment: A GELA study. Blood 2010, 116, 1092–1104. [Google Scholar] [CrossRef] [Green Version]
- Moller, M.B.; Ino, Y.; Gerdes, A.M.; Skjodt, K.; Louis, D.N.; Pedersen, N.T. Aberrations of the p53 pathway components p53, MDM2 and CDKN2A appear independent in diffuse large B cell lymphoma. Leukemia 1999, 13, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, M.R.; Stevens, W.B.C.; van den Brand, M.; van Krieken, J.H.; Scheijen, B. Molecular genetics of relapsed diffuse large B-Cell Lymphoma: Insight into mechanisms of therapy resistance. Cancers 2020, 12, 3553. [Google Scholar] [CrossRef]
- Jiang, Y.; Redmond, D.; Nie, K.; Eng, K.W.; Clozel, T.; Martin, P.; Tan, L.H.; Melnick, A.M.; Tam, W.; Elemento, O. Deep sequencing reveals clonal evolution patterns and mutation events associated with relapse in B-cell lymphomas. Genome Biol. 2014, 15, 432. [Google Scholar]
- Murray, G.F.; Guest, D.; Mikheykin, A.; Toor, A.; Reed, J. Single cell biomass tracking allows identification and isolation of rare targeted therapy-resistant DLBCL cells within a mixed population. Analyst 2021, 146, 1157–1162. [Google Scholar] [CrossRef]
- Rossi, D.; Diop, F.; Spaccarotella, E.; Monti, S.; Zanni, M.; Rasi, S.; Deambrogi, C.; Spina, V.; Bruscaggin, A.; Favini, C.; et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood 2017, 129, 1947–1957. [Google Scholar] [CrossRef]
- Staudt, L.M. Oncogenic activation of NF-kappaB. Cold Spring Harb. Perspect Biol. 2010, 2, a000109. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Thome, M.; Charton, J.E.; Pelzer, C.; Hailfinger, S. Antigen receptor signaling to NF-kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harb. Perspect Biol. 2010, 2, a003004. [Google Scholar] [CrossRef]
- Kreuz, S.; Siegmund, D.; Scheurich, P.; Wajant, H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell Biol. 2001, 21, 3964–3973. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Le, K.; Cheng, H.; Aplin, A.E. NF-kappaB Regulation of c-FLIP Promotes TNFalpha-Mediated RAF Inhibitor Resistance in Melanoma. J. Investig. Dermatol. 2015, 135, 1839–1848. [Google Scholar] [CrossRef] [Green Version]
- Catz, S.D.; Johnson, J.L. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene 2001, 20, 7342–7351. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Edelstein, L.C.; Gelinas, C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol. Cell Biol. 2000, 20, 2687–2695. [Google Scholar] [CrossRef]
- Stehlik, C.; de Martin, R.; Binder, B.R.; Lipp, J. Cytokine induced expression of porcine inhibitor of apoptosis protein (iap) family member is regulated by NF-kappa B. Biochem. Biophys. Res. Commun. 1998, 243, 827–832. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S., Jr. NF-kappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef]
- Stehlik, C.; de Martin, R.; Kumabashiri, I.; Schmid, J.A.; Binder, B.R.; Lipp, J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J. Exp. Med. 1998, 188, 211–216. [Google Scholar] [CrossRef]
- Kawakami, H.; Tomita, M.; Matsuda, T.; Ohta, T.; Tanaka, Y.; Fujii, M.; Hatano, M.; Tokuhisa, T.; Mori, N. Transcriptional activation of survivin through the NF-kappaB pathway by human T-cell leukemia virus type I tax. Int. J. Cancer 2005, 115, 967–974. [Google Scholar] [CrossRef]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Rickert, R.C. New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat. Rev. Immunol. 2013, 13, 578–591. [Google Scholar] [CrossRef]
- Kim, Y.; Ju, H.; Kim, D.H.; Yoo, H.Y.; Kim, S.J.; Kim, W.S.; Ko, Y.H. CD79B and MYD88 mutations in diffuse large B-cell lymphoma. Hum. Pathol. 2014, 45, 556–564. [Google Scholar] [CrossRef]
- Lamason, R.L.; McCully, R.R.; Lew, S.M.; Pomerantz, J.L. Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC-responsive inhibitory domain. Biochemistry 2010, 49, 8240–8250. [Google Scholar] [CrossRef] [Green Version]
- Knies, N.; Alankus, B.; Weilemann, A.; Tzankov, A.; Brunner, K.; Ruff, T.; Kremer, M.; Keller, U.B.; Lenz, G.; Ruland, J. Lymphomagenic CARD11/BCL10/MALT1 signaling drives malignant B-cell proliferation via cooperative NF-kappaB and JNK activation. Proc. Natl. Acad. Sci. USA 2015, 112, E7230–E7238. [Google Scholar] [CrossRef] [Green Version]
- Tibiletti, M.G.; Martin, V.; Bernasconi, B.; Del Curto, B.; Pecciarini, L.; Uccella, S.; Pruneri, G.; Ponzoni, M.; Mazzucchelli, L.; Martinelli, G.; et al. BCL2, BCL6, MYC, MALT 1, and BCL10 rearrangements in nodal diffuse large B-cell lymphomas: A multicenter evaluation of a new set of fluorescent in situ hybridization probes and correlation with clinical outcome. Hum. Pathol. 2009, 40, 645–652. [Google Scholar] [CrossRef]
- Dave, B.J.; Nelson, M.; Pickering, D.L.; Chan, W.C.; Greiner, T.C.; Weisenburger, D.D.; Armitage, J.O.; Sanger, W.G. Cytogenetic characterization of diffuse large cell lymphoma using multi-color fluorescence in situ hybridization. Cancer Genet. Cytogenet. 2002, 132, 125–132. [Google Scholar] [CrossRef]
- Willis, T.G.; Jadayel, D.M.; Du, M.Q.; Peng, H.; Perry, A.R.; Abdul-Rauf, M.; Price, H.; Karran, L.; Majekodunmi, O.; Wlodarska, I.; et al. Bcl10 is involved in t(1, 14)(p22, q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell 1999, 96, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Dierlamm, J.; Murga Penas, E.M.; Bentink, S.; Wessendorf, S.; Berger, H.; Hummel, M.; Klapper, W.; Lenze, D.; Rosenwald, A.; Haralambieva, E.; et al. Gain of chromosome region 18q21 including the MALT1 gene is associated with the activated B-cell-like gene expression subtype and increased BCL2 gene dosage and protein expression in diffuse large B-cell lymphoma. Haematologica 2008, 93, 688–696. [Google Scholar] [CrossRef]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [Green Version]
- Jost, P.J.; Ruland, J. Aberrant NF-kappaB signaling in lymphoma: Mechanisms, consequences, and therapeutic implications. Blood 2007, 109, 2700–2707. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef]
- Gaidano, G.; Hauptschein, R.S.; Parsa, N.Z.; Offit, K.; Rao, P.H.; Lenoir, G.; Knowles, D.M.; Chaganti, R.S.; Dalla-Favera, R. Deletions involving two distinct regions of 6q in B-cell non-Hodgkin lymphoma. Blood 1992, 80, 1781–1787. [Google Scholar] [CrossRef] [Green Version]
- Thelander, E.F.; Ichimura, K.; Corcoran, M.; Barbany, G.; Nordgren, A.; Heyman, M.; Berglund, M.; Mungall, A.; Rosenquist, R.; Collins, V.P.; et al. Characterization of 6q deletions in mature B cell lymphomas and childhood acute lymphoblastic leukemia. Leuk. Lymphoma 2008, 49, 477–487. [Google Scholar] [CrossRef]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Valentin, R.; Grabow, S.; Davids, M.S. The rise of apoptosis: Targeting apoptosis in hematologic malignancies. Blood 2018, 132, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Pollyea, D.A.; Amaya, M.; Strati, P.; Konopleva, M.Y. Venetoclax for AML: Changing the treatment paradigm. Blood Adv. 2019, 3, 4326–4335. [Google Scholar] [CrossRef] [Green Version]
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients With Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vos, S.; Swinnen, L.J.; Wang, D.; Reid, E.; Fowler, N.; Cordero, J.; Dunbar, M.; Enschede, S.H.; Nolan, C.; Petrich, A.M.; et al. Venetoclax, bendamustine, and rituximab in patients with relapsed or refractory NHL: A phase Ib dose-finding study. Ann. Oncol. 2018, 29, 1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Gerecitano, J.F.; Roberts, A.W.; Seymour, J.F.; Wierda, W.G.; Kahl, B.S.; Pagel, J.M.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Dunbar, M.; et al. A phase 1 study of venetoclax (ABT-199/GDC-0199) monotherapy in patients with relapsed/refractory non-hodgkin lymphoma. Blood 2015, 126, 254. [Google Scholar] [CrossRef]
- Morschhauser, F.; Feugier, P.; Flinn, I.W.; Gasiorowski, R.; Greil, R.; Illes, A.; Johnson, N.A.; Larouche, J.F.; Lugtenburg, P.J.; Patti, C.; et al. A phase 2 study of venetoclax plus R-CHOP as first-line treatment for patients with diffuse large B-cell lymphoma. Blood 2021, 137, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pongtornpipat, P.; Tiutan, T.; Kendrick, S.L.; Park, S.; Persky, D.O.; Rimsza, L.M.; Puvvada, S.D.; Schatz, J.H. Synergistic induction of apoptosis in high-risk DLBCL by BCL2 inhibition with ABT-199 combined with pharmacologic loss of MCL1. Leukemia 2015, 29, 1702–1712. [Google Scholar] [CrossRef] [Green Version]
- Brem, E.A.; Thudium, K.; Khubchandani, S.; Tsai, P.C.; Olejniczak, S.H.; Bhat, S.; Riaz, W.; Gu, J.; Iqbal, A.; Campagna, R.; et al. Distinct cellular and therapeutic effects of obatoclax in rituximab-sensitive and -resistant lymphomas. Br. J. Haematol. 2011, 153, 599–611. [Google Scholar] [CrossRef]
- Phillips, D.C.; Xiao, Y.; Lam, L.T.; Litvinovich, E.; Roberts-Rapp, L.; Souers, A.J.; Leverson, J.D. Loss in MCL-1 function sensitizes non-Hodgkin’s lymphoma cell lines to the BCL-2-selective inhibitor venetoclax (ABT-199). Blood Cancer J. 2015, 5, e368. [Google Scholar] [CrossRef] [Green Version]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.A.; Dotti, G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin. Biol. Ther. 2011, 11, 855–873. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients with relapsed or refractory hematologic malignancy: Preliminary results of a phase Ib study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Armand, P.; Lesokhin, A.; Borrello, I.; Timmerman, J.; Gutierrez, M.; Zhu, L.; Popa McKiver, M.; Ansell, S.M. A phase 1b study of dual PD-1 and CTLA-4 or KIR blockade in patients with relapsed/refractory lymphoid malignancies. Leukemia 2020, 35, 777–786. [Google Scholar] [CrossRef]
- Kline, J.; Godfrey, J.; Ansell, S.M. The immune landscape and response to immune checkpoint blockade therapy in lymphoma. Blood 2020, 135, 523–533. [Google Scholar] [CrossRef]
- Chavez, J.C.; Bachmeier, C.; Kharfan-Dabaja, M.A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 2019, 10, 2040620719841581. [Google Scholar] [CrossRef] [Green Version]
- Viardot, A.; Goebeler, M.E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Liu, S.Q.; Grantham, A.; Landry, C.; Granda, B.; Chopra, R.; Chakravarthy, S.; Deutsch, S.; Vogel, M.; Russo, K.; Seiss, K.; et al. A CRISPR Screen Reveals Resistance Mechanisms to CD3-Bispecific Antibody Therapy. Cancer Immunol. Res. 2021, 9, 34–49. [Google Scholar] [CrossRef]
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.J.; Kuramitsu, S.; et al. Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-cell Dysfunction. Cancer Discov. 2020, 10, 552–567. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, H.; Lindqvist, A.C.; Fransson, M.; Paul-Wetterberg, G.; Nilsson, B.; Essand, M.; Nilsson, K.; Frisk, P.; Jernberg-Wiklund, H.; Loskog, A. Combining CAR T cells and the Bcl-2 family apoptosis inhibitor ABT-737 for treating B-cell malignancy. Cancer Gene Ther. 2013, 20, 386–393. [Google Scholar] [CrossRef] [Green Version]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [Green Version]
- Drakos, E.; Singh, R.R.; Rassidakis, G.Z.; Schlette, E.; Li, J.; Claret, F.X.; Ford, R.J., Jr.; Vega, F.X.; Medeiros, L.J. Activation of the p53 pathway by the MDM2 inhibitor nutlin-3a overcomes BCL2 overexpression in a preclinical model of diffuse large B-cell lymphoma associated with t(14, 18)(q32, q21). Leukemia 2011, 25, 856–867. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.M.; Landsburg, D.J.; Mato, A.R.; Isaac, K.; Hernandez-Ilizaliturri, F.J.; Reddy, N.; Smith, S.; Shadman, M.; Smith, M.R.; Caimi, P.; et al. A multi-institutional outcomes analysis of patients with relapsed or refractory DLBCL treated with ibrutinib. Blood 2017, 130, 1676–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goy, A.; Ramchandren, R.; Ghosh, N.; Munoz, J.; Morgan, D.S.; Dang, N.H.; Knapp, M.; Delioukina, M.; Kingsley, E.; Ping, J.; et al. Ibrutinib plus lenalidomide and rituximab has promising activity in relapsed/refractory non-germinal center B-cell-like DLBCL. Blood 2019, 134, 1024–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauter, C.S.; Matasar, M.J.; Schoder, H.; Devlin, S.M.; Drullinsky, P.; Gerecitano, J.; Kumar, A.; Noy, A.; Palomba, M.L.; Portlock, C.S.; et al. A phase 1 study of ibrutinib in combination with R-ICE in patients with relapsed or primary refractory DLBCL. Blood 2018, 131, 1805–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younes, A.; Sehn, L.H.; Johnson, P.; Zinzani, P.L.; Hong, X.; Zhu, J.; Patti, C.; Belada, D.; Samoilova, O.; Suh, C.; et al. Randomized Phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-cell diffuse large B-Cell lymphoma. J. Clin. Oncol. 2019, 37, 1285–1295. [Google Scholar] [CrossRef]
- Zhang, L.H.; Kosek, J.; Wang, M.; Heise, C.; Schafer, P.H.; Chopra, R. Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. Br. J. Haematol. 2013, 160, 487–502. [Google Scholar] [CrossRef]
- Castellino, A.; Chiappella, A.; LaPlant, B.R.; Pederson, L.D.; Gaidano, G.; Macon, W.R.; Inghirami, G.; Reeder, C.B.; Tucci, A.; King, R.L.; et al. Lenalidomide plus R-CHOP21 in newly diagnosed diffuse large B-cell lymphoma (DLBCL): Long-term follow-up results from a combined analysis from two phase 2 trials. Blood Cancer J. 2018, 8, 108. [Google Scholar] [CrossRef]
- Nowakowski, G.S.; Hong, F.; Scott, D.W.; Macon, W.R.; King, R.L.; Habermann, T.M.; Wagner-Johnston, N.; Casulo, C.; Wade, J.L.; Nagargoje, G.G.; et al. Addition of Lenalidomide to R-CHOP improves outcomes in newly diagnosed diffuse large B-Cell lymphoma in a randomized phase II US intergroup study ECOG-ACRIN E1412. J. Clin. Oncol. 2021, 39, 1329–1338. [Google Scholar] [CrossRef]
- Nowakowski, G.S.; Chiappella, A.; Gascoyne, R.D.; Scott, D.W.; Zhang, Q.; Jurczak, W.; Ozcan, M.; Hong, X.; Zhu, J.; Jin, J.; et al. ROBUST: A phase III study of lenalidomide plus R-CHOP versus placebo plus R-CHOP in previously untreated patients with ABC-type diffuse large B-Cell Lymphoma. J. Clin. Oncol. 2021, 39, 1317–1328. [Google Scholar] [CrossRef]
- Dunleavy, K.; Pittaluga, S.; Czuczman, M.S.; Dave, S.S.; Wright, G.; Grant, N.; Shovlin, M.; Jaffe, E.S.; Janik, J.E.; Staudt, L.M.; et al. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood 2009, 113, 6069–6076. [Google Scholar] [CrossRef]
- Davies, A.; Cummin, T.E.; Barrans, S.; Maishman, T.; Mamot, C.; Novak, U.; Caddy, J.; Stanton, L.; Kazmi-Stokes, S.; McMillan, A.; et al. Gene-expression profiling of bortezomib added to standard chemoimmunotherapy for diffuse large B-cell lymphoma (REMoDL-B): An open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Leonard, J.P.; Kolibaba, K.S.; Reeves, J.A.; Tulpule, A.; Flinn, I.W.; Kolevska, T.; Robles, R.; Flowers, C.R.; Collins, R.; DiBella, N.J.; et al. Randomized Phase II Study of R-CHOP with or Without Bortezomib in Previously Untreated Patients with Non-Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2017, 35, 3538–3546. [Google Scholar] [CrossRef]
- Offner, F.; Samoilova, O.; Osmanov, E.; Eom, H.S.; Topp, M.S.; Raposo, J.; Pavlov, V.; Ricci, D.; Chaturvedi, S.; Zhu, E.; et al. Frontline rituximab, cyclophosphamide, doxorubicin, and prednisone with bortezomib (VR-CAP) or vincristine (R-CHOP) for non-GCB DLBCL. Blood 2015, 126, 1893–1901. [Google Scholar] [CrossRef] [Green Version]
- Mareschal, S.; Dubois, S.; Viailly, P.J.; Bertrand, P.; Bohers, E.; Maingonnat, C.; Jais, J.P.; Tesson, B.; Ruminy, P.; Peyrouze, P.; et al. Whole exome sequencing of relapsed/refractory patients expands the repertoire of somatic mutations in diffuse large B-cell lymphoma. Genes Chromosomes Cancer 2016, 55, 251–267. [Google Scholar] [CrossRef]
- Juskevicius, D.; Lorber, T.; Gsponer, J.; Perrina, V.; Ruiz, C.; Stenner-Liewen, F.; Dirnhofer, S.; Tzankov, A. Distinct genetic evolution patterns of relapsing diffuse large B-cell lymphoma revealed by genome-wide copy number aberration and targeted sequencing analysis. Leukemia 2016, 30, 2385–2395. [Google Scholar] [CrossRef]
Figure 1.
Simplified Overview of Apoptosis in DLBCL, BCR: B cell receptor, Cytc: cytochrome C, DISC: death-inducing signaling complex, IAPs: inhibitor of apoptosis proteins. Created with BioRender.com.
Figure 1.
Simplified Overview of Apoptosis in DLBCL, BCR: B cell receptor, Cytc: cytochrome C, DISC: death-inducing signaling complex, IAPs: inhibitor of apoptosis proteins. Created with BioRender.com.


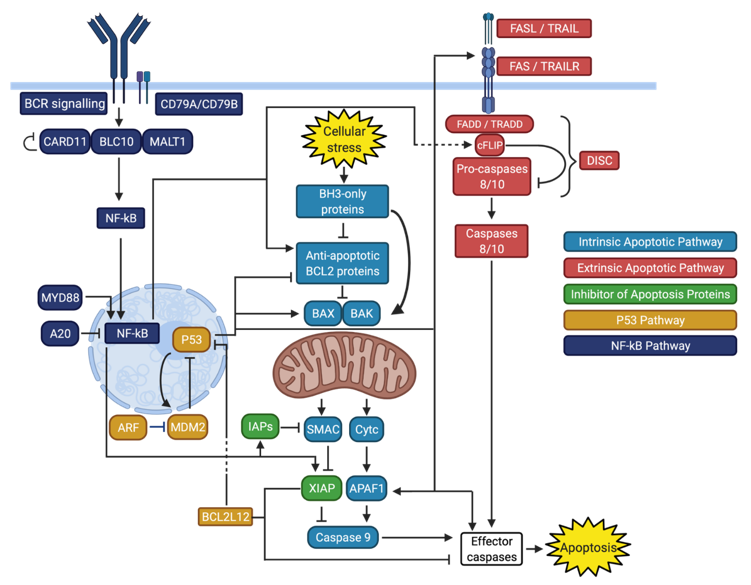{kind=link}
Table 1.
Overview of Recently Proposed Genetic Classifications of DLBCL.
| Lacy et al. [16] | Chapuy et al. [17] | Schmitz et al. [14] | Predominant COO Subtype |
|---|---|---|---|
| MYD88 (16%) MYD88L265P, CD79B, PIM1, and ETV6 mutations 9p21.3/CDKN2A deletions | C5 (21%) 18q gains CD79B and MYD88L265P mutations | MCD (8%) MYD88L265P and CD79B mutations | ABC DLBCL |
| BCL2 (19%) BCL2 mutations and translocations EZH2, CREBBP, TNFRSF14, KMT2D, and MEF2B mutations | C3 (18%) BCL2 mutations and translocations PTEN inactivation Mutations in chromatin modifiers Alterations in BCR and PI3K signaling | EZB (22%) EZH2 mutations and BCL2 translocations | GCB DLBCL |
| SOCS1/SGK1 (12%) SOCS1, SGK1, CD83 NFKBIA, HIST1H1E, and STAT3 mutations | C4 (17%) Mutations in NF-kB modifiers, immune evasion molecules, core histone genes, and RAS/JAK/STAT pathway components | DLBCL NOS | |
| TET2/SGK1 (11%) TET2, SGK1, KLHL6, ZFP36L1, BRAF, MAP2K1, and KRAS mutations | GCB DLBCL | ||
| NOTCH2 (15%) BCL6 rearrangements NOTCH2, BCLL10, TNFAIP3, CCND3, SPEN, TMEM30A, FAS, and CD70 mutations | C1 (19%) BCL6 rearrangements MYD88non-L265P, FAS, NOTCH2 pathway, and NF-kB pathway mutations | BN2 (15%) BCL6 fusions and NOTCH2 mutations | GCB DLBCL, ABC DLBCL, and DLBCL NOS |
| NEC (26%) Not elsewhere classified | Other (54%) | ||
| C2 (21%) TP53 mutations 17p/TP53, 9p21.3/CDKN2A and 13q14.2/RB1 deletions | ABC DLBCL and GCB DLBCL | ||
| C0 (4%) No defining genetic driver | |||
| N1 (2%) NOTCH1 mutations | ABC DLBCL |
Percentage of cases reported is indicated next to each subtype. Reprinted with permission from ref. [16]. 2020 Elsevier.
Table 2.
Main Genetic Events Inhibiting Apoptosis in DLBCL.
| Intrinsic Apoptotic Pathway | Role in Apoptosis | Genetic Alterations |
|---|---|---|
| BCL2 | Inhibition of intrinsic apoptotic pathway via inhibition of pro-apoptotic BCL2 proteins | Increased expression: translocations (mainly t(14;18)), gains and amplifications (18q21.33), mutations (promoter region, BH4 domain, and flexible loop domain)14,16,17, 31−33, 37 |
| MCL1 | Increased expression: gains and amplification43,44 | |
| Extrinsic apoptotic pathway | ||
| FAS | Induction of FAS-mediated apoptosis after binding of FasL | Decreased expression: mutations (death domain), deletions16,17,24,64,76−78 |
| TRAIL-R1/TRAIL-R2 | Induction of TRAIL-mediated apoptosis after binding of TRAIL | Decreased expression: mutations (death domain), deletions (8p21)95−96 |
| CASP10 | Effector caspases activator | Decreased expression: inactivating mutations17 |
| P53 pathway | Promotion of intrinsic and extrinsic apoptosis via upregulation of several pro-apoptotic proteins | |
| TP53 | Decreased expression: mutations (DNA-binding domain), 17p13.1 deletions; polymorphisms in 3’-UTR7,24,118−120 | |
| MDM2, MDM4, and RFWD2 | P53 inhibitors | Increased expression: 1q23.3 gains (MDM4 and RFWD2)78 |
| CDKN2A | Encoding ARF, an MDM2 inhibitor | Decreased expression: deletions14,125,126 |
| BCL2L12 | P53 inhibitor and caspase 3/7 inhibitor | Increased expression: amplifications78 |
| PERP | Target of P53, caspase 8 activator | Decreased expression: deletions78 |
| SCOTIN | Target of P53, pro-caspase 3/7 activator | Decreased expression: deletions78 |
| NF-kB pathway | Upregulation of several anti-apoptotic proteins from the intrinsic and extrinsic pathways | |
| CD79A/CD79B | Activation of NF-kB pathway via transduction of BCR signaling | Increased expression: gain-of-function mutations in ITAM14,144,146 |
| Negative regulators of BCR signaling | Inhibition of NF-kB pathway via downregulation of BCR signaling | Decreased expression: loss-of-function mutations14 |
| CARD11-BCL10-MALT1 complex | Activation of NF-kB pathway | Increased expression: gain-of-function mutations in coiled-coiled domain of CARD11, chromosomal rearrangements, mutations, and gains of BCL10, gains of MALT1 (18q21)14,17,147,149−152 |
| NFKB2 | Encoding p100, which yields the p52 NF-kB transcription factor | Increased expression: 10q24 rearrangements leading to loss of 3’-end and constitutive protein activation154 |
| REL | Encoding c-Rel, an NF-kB transcription factor | Increased expression: amplifications9 |
| A20 | Downregulation of NF-kB response | Decreased expression: loss-of-function mutations, 6q23 deletions, mutations in the A20 binding partner TNIP114,153,156,157 |
| MYD88 | Upregulation of NF-kB response via toll and IL-1 signaling | Increased expression: gain-of-function mutations (L265P)158 |
| Regulators of NF-kB | Modulation of NF-kB response | Increased expression (positive regulators), decreased expression (negative regulators): mutations153 |
BCR: B cell receptor, UTR: untranslated region, ITAM: immunoreceptor tyrosine-based activation motif, IL-1: interleukin-1.
Table 3.
Therapies modulating apoptosis being currently investigated clinically in DLBCL.
| Investigational Therapy | Mechanism of Apoptosis Modulation | Apoptotic Pathway Affected |
|---|---|---|
| Venetoclax | BH3 mimetic and BCL2 inhibitor | Intrinsic apoptotic pathway |
| CAR T cells | Genetically modified cytotoxic T cells | Extrinsic apoptotic pathway |
| Bispecific T cell engagers | Linking of B cells and cytotoxic T cells | Extrinsic apoptotic pathway |
| Immune checkpoint inhibitors | Immune checkpoint inhibition, reactivation of cytotoxic T cells | Extrinsic apoptotic pathway |
| TRAIL agonists | Enhancement of the TRAIL/TRAILR response | Extrinsic apoptotic pathway |
| Ibrutinib | Inhibition of BTK, inhibition of BCR signaling | NF-kB pathway |
| Lenalidomide | Immunomodulation, inhibition of BCR signaling | NF-kB pathway |
| Bortezomib | Proteasome inhibitor, decreased degradation of inhibitory kB proteins | NF-kB pathway |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Leveille, E.; Johnson, N.A. Genetic Events Inhibiting Apoptosis in Diffuse Large B Cell Lymphoma. Cancers 2021, 13, 2167. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13092167
AMA Style
Leveille E, Johnson NA. Genetic Events Inhibiting Apoptosis in Diffuse Large B Cell Lymphoma. Cancers. 2021; 13(9):2167. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13092167
Chicago/Turabian StyleLeveille, Etienne, and Nathalie A. Johnson. 2021. "Genetic Events Inhibiting Apoptosis in Diffuse Large B Cell Lymphoma" Cancers 13, no. 9: 2167. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13092167
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.