
The TGF-β Pathway: A Pharmacological Target in Hepatocellular Carcinoma?

 ,
,  , ,
, ,  , ,
, ,
Abstract
:Simple Summary
Abstract
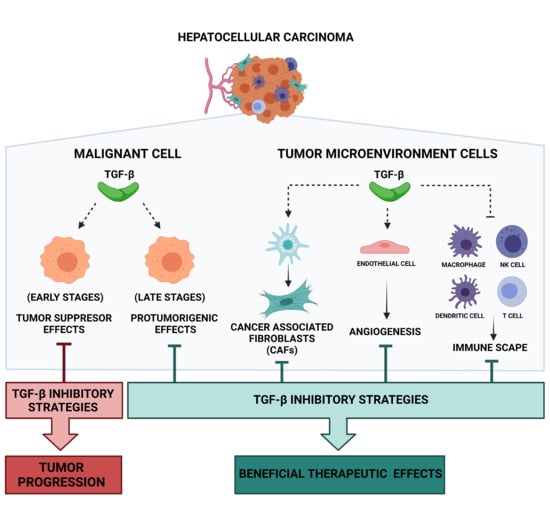
1. Introduction
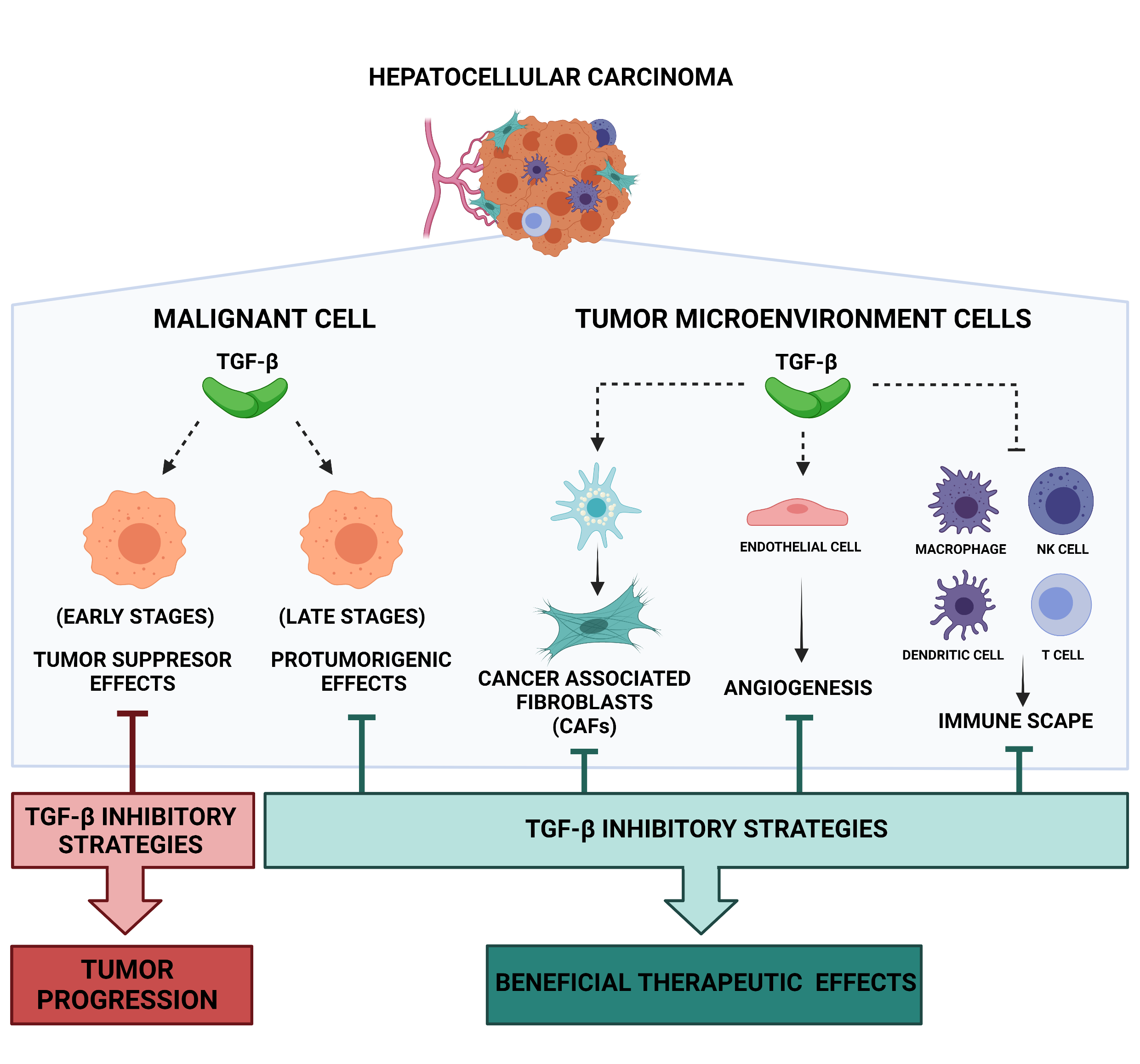
2. TGF-β Signaling
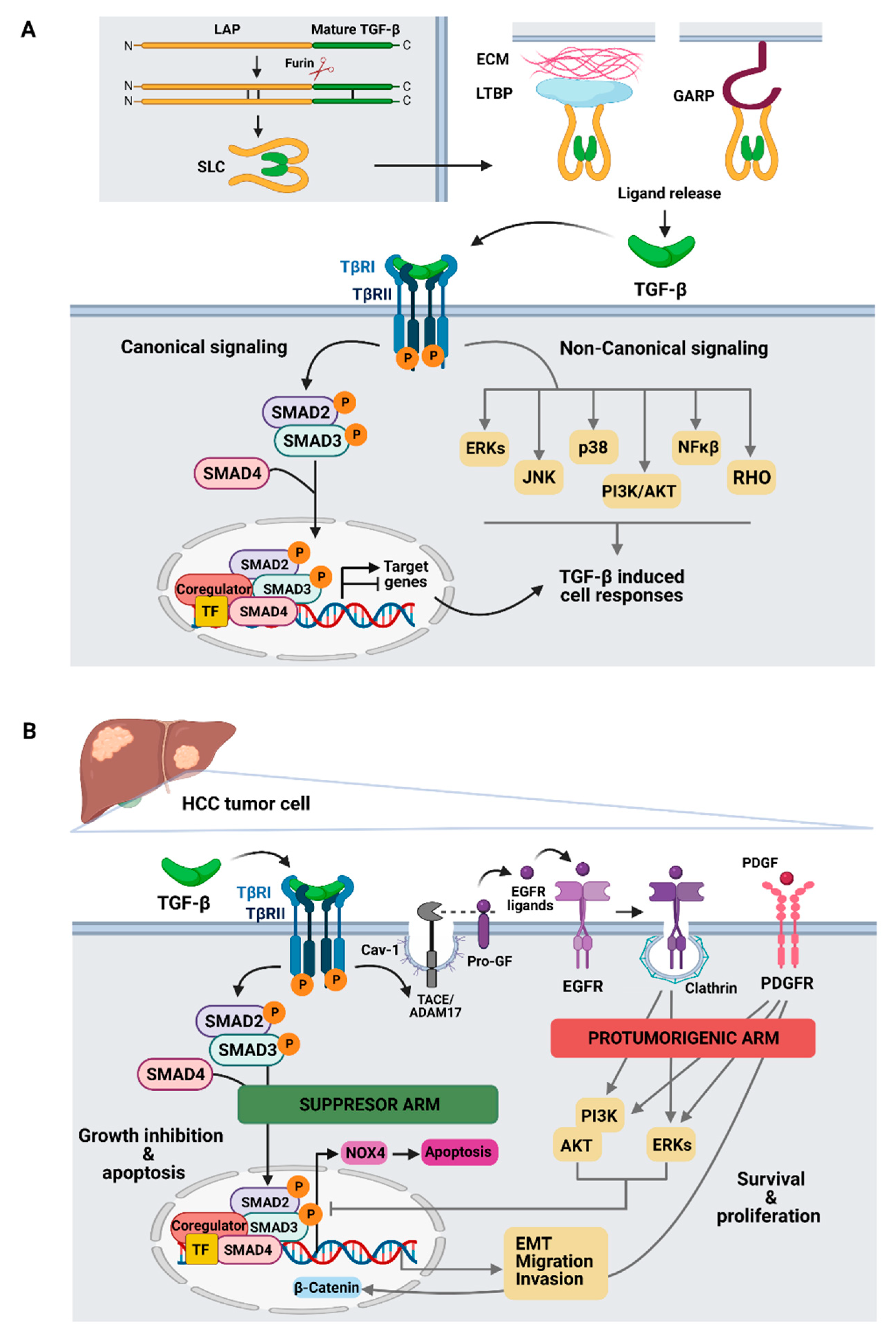
3. Role of TGF-β in HCC Cells
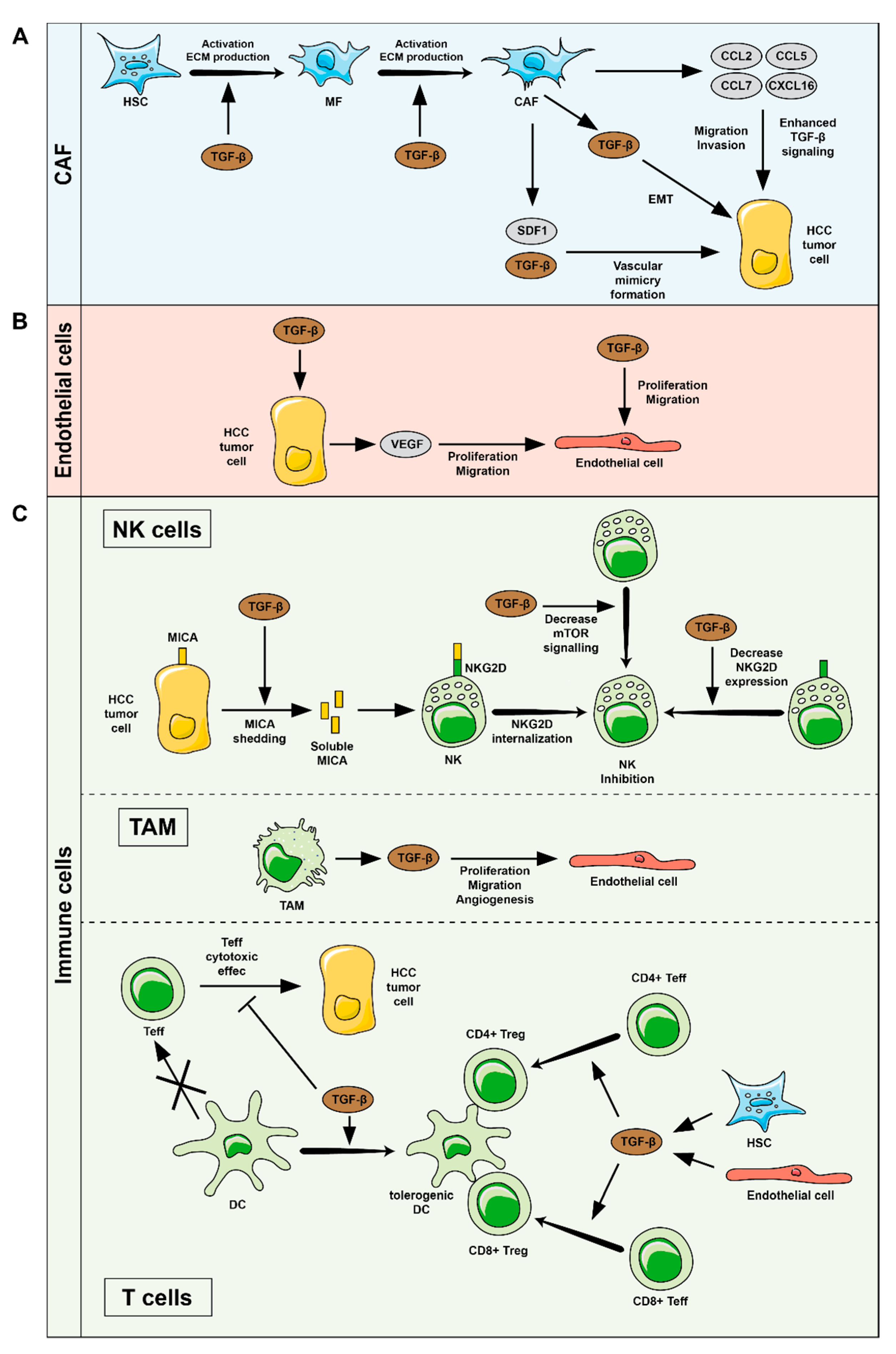
4. TGFβ-Related Functions in HCC Tumor Microenvironment (TME)
4.1. HSC and CAF
4.2. Endothelial Cells
4.3. Immune System
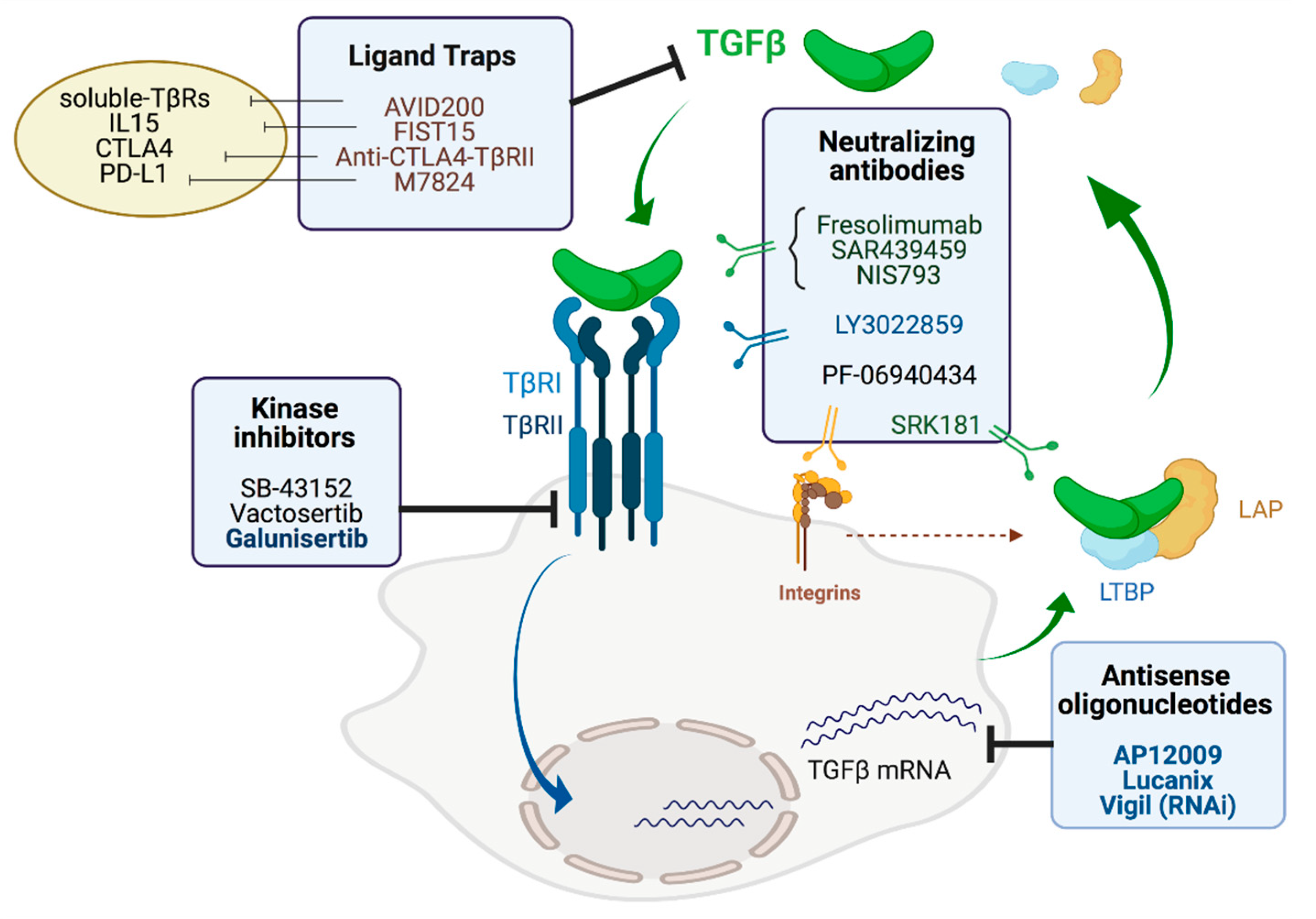
5. TGF-β Inhibitors
5.1. Antisense Oligonucleotides
5.2. Neutralizing Antibodies
5.3. Ligand Traps
5.4. Small Molecule Inhibitors
6. Current Therapies in HCC and New Perspectives for TGF-β Inhibitors
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Bruix, J.; Da Fonseca, L.G.; Reig, M. Insights into the success and failure of systemic therapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 617–630. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 1–28. [Google Scholar] [CrossRef]
- Caruso, S.; O’Brien, D.R.; Cleary, S.P.; Roberts, L.R.; Zucman-Rossi, J. Genetics of Hepatocellular Carcinoma: Approaches to Explore Molecular Diversity. Hepatology 2021, 73, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Chan, S.L.; Meyer, T.; Reig, M.; El-Khoueiry, A.; Galle, P.R. Diagnosis and management of toxicities of immune checkpoint inhibitors in hepatocellular carcinoma. J. Hepatol. 2020, 72, 320–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casak, S.J.; Donoghue, M.; Fashoyin-Aje, L.; Jiang, X.; Rodriguez, L.; Shen, Y.-L.; Xu, Y.; Jiang, X.; Liu, J.; Zhao, H.; et al. FDA Approval Summary: Atezolizumab Plus Bevacizumab for the Treatment of Patients with Advanced Unresectable or Metastatic Hepatocellular Carcinoma. Clin. Cancer Res. 2021, 27, 1836–1841. [Google Scholar] [CrossRef]
- Moses, H.L.; Roberts, A.B.; Derynck, R. The Discovery and Early Days of TGF-β: A Historical Perspective. Cold Spring Harb. Perspect. Biol. 2016, 8, a021865. [Google Scholar] [CrossRef] [Green Version]
- Schon, H.-T.; Weiskirchen, R. Immunomodulatory effects of transforming growth factor-β in the liver. Hepatobiliary Surg. Nutr. 2014, 3, 386–406. [Google Scholar] [CrossRef]
- Robertson, I.B.; Rifkin, D.B. Unchaining the beast; insights from structural and evolutionary studies on TGFβ secretion, sequestration, and activation. Cytokine Growth Factor Rev. 2013, 24, 355–372. [Google Scholar] [CrossRef] [Green Version]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming Growth Factor-β Receptors and Smads: Regulatory Complexity and Functional Versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Ren, J.; Dijke, P.T. Targeting TGFβ signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 1–20. [Google Scholar] [CrossRef]
- Fabregat, I.; Moreno-Caceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Dijke, P.T.; The IT-LIVER Consortium. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, I.; Caballero-Díaz, D. Transforming Growth Factor-β-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [Green Version]
- Giannelli, G.; Mikulits, W.; Dooley, S.; Fabregat, I.; Moustakas, A.; Dijke, P.T.; Portincasa, P.; Winter, P.; Janssen, R.; Leporatti, S.; et al. The rationale for targeting TGF-β in chronic liver diseases. Eur. J. Clin. Investig. 2016, 46, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancarella, S.; Cigliano, A.; Chieti, A.; Giannelli, G.; Dituri, F. TGF-β as Multifaceted Orchestrator in HCC Progression: Signaling, EMT, Immune Microenvironment, and Novel Therapeutic Perspectives. Semin. Liver Dis. 2019, 39, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.E.; Coffey, R.J.; Ouellette, A.J.; Moses, H.L. Type beta transforming growth factor reversibly inhibits the early proliferative response to partial hepatectomy in the rat. Proc. Natl. Acad. Sci. USA 1988, 85, 5126–5130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberhammer, F.A.; Pavelka, M.; Sharma, S.; Tiefenbacher, R.; Purchio, A.F.; Bursch, W.; Schulte-Hermann, R. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc. Natl. Acad. Sci. USA 1992, 89, 5408–5412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, A.; Alvarez-Barrientos, A.; Benito, M.; Fabregat, I. Apoptosis Induced by Transforming Growth Factor-β in Fetal Hepatocyte Primary Cultures. J. Biol. Chem. 1996, 271, 7416–7422. [Google Scholar] [CrossRef] [Green Version]
- Laiho, M.; DeCaprio, J.A.; Ludlow, J.W.; Livingston, D.M.; Massague, J. Growth inhibition by TGF-β linked to suppression of retinoblastoma protein phosphorylation. Cell 1990, 62, 175–185. [Google Scholar] [CrossRef]
- Polyak, K.; Kato, J.Y.; Solomon, M.J.; Sherr, C.J.; Massague, J.; Roberts, J.M.; Koff, A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994, 8, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Warner, B.J.; Blain, S.W.; Seoane, J.; Massagué, J. Myc Downregulation by Transforming Growth Factor β Required for Activation of the p15 Ink4b G 1 Arrest Pathway. Mol. Cell. Biol. 1999, 19, 5913–5922. [Google Scholar] [CrossRef] [Green Version]
- Herrera, B.; Alvarez-Barrientos, A.; Sánchez, A.; Fernández, M.; Roncero, C.; Benito, M.; Fabregat, I. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor ß in fetal hepatocytes. FASEB J. 2001, 15, 741–751. [Google Scholar] [CrossRef]
- Herrera, B.; Gil, J.; Fernández, M.; Álvarez, A.M.; Roncero, C.; Benito, M.; Fabregat, I. Activation of caspases occurs downstream from radical oxygen species production, Bcl-xL down-regulation, and early cytochrome C release in apoptosis induced by transforming growth factor β in rat fetal hepatocytes. Hepatology 2001, 34, 548–556. [Google Scholar] [CrossRef]
- Carmona-Cuenca, I.; Roncero, C.; Sancho, P.; Caja, L.; Fausto, N.; Fernández, M.; Fabregat, I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J. Hepatol. 2008, 49, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Gotzmann, J.; Fischer, A.N.M.; Zojer, M.; Mikula, M.; Proell, V.; Huber, H.; Jechlinger, M.; Waerner, T.; Weith, A.; Beug, H.; et al. A crucial function of PDGF in TGF-β-mediated cancer progression of hepatocytes. Oncogene 2006, 25, 3170–3185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caja, L.; Sancho, P.; Bertran, E.; Fabregat, I. Dissecting the effect of targeting the epidermal growth factor receptor on TGF-β-induced-apoptosis in human hepatocellular carcinoma cells. J. Hepatol. 2011, 55, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Caceres, J.; Caja, L.; Mainez, J.; Mayoral, R.; Martín-Sanz, P.; Moreno-Vicente, R.; Del Pozo, M.Á.; Dooley, S.; Egea, G.; Fabregat, I. Caveolin-1 is required for TGF-β-induced transactivation of the EGF receptor pathway in hepatocytes through the activation of the metalloprotease TACE/ADAM17. Cell Death Dis. 2014, 5, e1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero-Díaz, D.; Bertran, E.; Peñuelas-Haro, I.; Moreno-Caceres, J.; Malfettone, A.; Luque, J.L.; Addante, A.; Herrera, B.; Sánchez, A.; Alay, A.; et al. Clathrin switches transforming growth factor-β role to pro-tumorigenic in liver cancer. J. Hepatol. 2020, 72, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Giannelli, G.; Bergamini, C.; Fransvea, E.; Sgarra, C.; Antonaci, S. Laminin-5 with Transforming Growth Factor-β1 Induces Epithelial to Mesenchymal Transition in Hepatocellular Carcinoma. Gastroenterology 2005, 129, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Malfettone, A.; Soukupova, J.; Bertran, E.; Molist, E.C.; Lastra, R.; Fernando, J.; Koudelkova, P.; Rani, B.; Fabra, Á.; Serrano, T.; et al. Transforming growth factor-β-induced plasticity causes a migratory stemness phenotype in hepatocellular carcinoma. Cancer Lett. 2017, 392, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Transforming growth factor-β gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology 2008, 47, 2059–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zaidi, S.; Rao, S.; Chen, J.-S.; Phan, L.; Farci, P.; Su, X.; Shetty, K.; White, J.; Zamboni, F.; et al. Analysis of Genomes and Transcriptomes of Hepatocellular Carcinomas Identifies Mutations and Gene Expression Changes in the Transforming Growth Factor-β Pathway. Gastroenterology 2018, 154, 195–210. [Google Scholar] [CrossRef]
- Rebouissou, S.; Nault, J.-C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J. Hepatol. 2020, 72, 215–229. [Google Scholar] [CrossRef] [Green Version]
- Bertran, E.; Molist, E.C.; Sancho, P.; Caja, L.; Luque, J.L.; Navarro, E.; Egea, G.; Lastra, R.; Serrano, T.; Ramos, E.; et al. Overactivation of the TGF-β pathway confers a mesenchymal-like phenotype and CXCR4-dependent migratory properties to liver tumor cells. Hepatology 2013, 58, 2032–2044. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Dzieran, J.; Gu, X.; Marhenke, S.; Vogel, A.; Machida, K.; Weiss, T.; Ruemmele, P.; Kollmar, O.; Hoffmann, P.; et al. Smad7 regulates compensatory hepatocyte proliferation in damaged mouse liver and positively relates to better clinical outcome in human hepatocellular carcinoma. Clin. Sci. 2015, 128, 761–774. [Google Scholar] [CrossRef]
- Rani, B.; Malfettone, A.; Dituri, F.; Soukupova, J.; Lupo, L.; Mancarella, S.; Fabregat, I.; Giannelli, G. Galunisertib suppresses the staminal phenotype in hepatocellular carcinoma by modulating CD44 expression. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawi, M.; Kim, J.; Dauki, A.; Sutaria, D.; Motiwala, T.; Reyes, R.; Wani, N.; Kolli, S.; Jiang, J.; Coss, C.C.; et al. CD44 positive and sorafenib insensitive hepatocellular carcinomas respond to the ATP-competitive mTOR inhibitor INK128. Oncotarget 2018, 9, 26032–26045. [Google Scholar] [CrossRef] [Green Version]
- Dzieran, J.; Fabian, J.; Feng, T.; Coulouarn, C.; Ilkavets, I.; Kyselova, A.; Breuhahn, K.; Dooley, S.; Meindl-Beinker, N.M. Comparative Analysis of TGF-β/Smad Signaling Dependent Cytostasis in Human Hepatocellular Carcinoma Cell Lines. PLoS ONE 2013, 8, e72252. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Agarwal, R.; Dituri, F.; Lupo, L.; Trerotoli, P.; Mancarella, S.; Winter, P.; Giannelli, G. NGS-based transcriptome profiling reveals biomarkers for companion diagnostics of the TGF-β receptor blocker galunisertib in HCC. Cell Death Dis. 2017, 8, e2634. [Google Scholar] [CrossRef] [Green Version]
- Barry, A.E.; Baldeosingh, R.; Lamm, R.; Patel, K.; Zhang, K.; Dominguez, D.A.; Kirton, K.J.; Shah, A.P.; Dang, H. Hepatic Stellate Cells and Hepatocarcinogenesis. Front. Cell Dev. Biol. 2020, 8, 709. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradère, J.-P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [Green Version]
- Dooley, S.; Delvoux, B.; Lahme, B.; Mangasser-Stephan, K.; Gressner, A.M. Modulation of transforming growth factorβ response and signaling during transdifferentiation of rat hepatic stellate cells to myofibroblasts. Hepatology 2000, 31, 1094–1106. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Choi, I.; Kim, S.S. Progression of hepatic stellate cell activation is associated with the level of oxidative stress rather than cytokines during CCl4-induced fibrogenesis. Mol. Cells 2000, 10, 289–300. [Google Scholar]
- Wu, J.; Zern, M.A. Hepatic stellate cells: A target for the treatment of liver fibrosis. J. Gastroenterol. 2000, 35, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, A.; Di Sario, A.; Casini, A.; Ridolfi, F.; Bendia, E.; Pigini, P.; Tonnini, C.; D’Ambrosio, L.; Feliciangeli, G.; Macarri, G.; et al. Inhibition of the Na+/H+ exchanger reduces rat hepatic stellate cell activity and liver fibrosis: An in vitro and in vivo study. Gastroenterology 2001, 120, 545–556. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, J.L.; Gorin, Y. Myofibroblast differentiation during fibrosis: Role of NAD(P)H oxidases. Kidney Int. 2011, 79, 944–956. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernández-Rodríguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. NADPH Oxidase NOX4 Mediates Stellate Cell Activation and Hepatocyte Cell Death during Liver Fibrosis Development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Mak, K.M.; Lieber, C.S. DLPC decreases TGF-β1-induced collagen mRNA by inhibiting p38 MAPK in hepatic stellate cells. Am. J. Physiol. Liver Physiol. 2002, 283, G1051–G1061. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, F. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology 2003, 38, 879–889. [Google Scholar] [CrossRef]
- Dooley, S.; Dijke, P.T. TGF-β in progression of liver disease. Cell Tissue Res. 2011, 347, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.-Y.; Jin, G.-N.; Liang, H.-F.; Wang, W.; Chen, W.-X.; Datta, P.K.; Zhang, M.-Z.; Zhang, B.; Chen, X.-P. Transforming growth factor β induces expression of connective tissue growth factor in hepatic progenitor cells through Smad independent signaling. Cell. Signal. 2013, 25, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Königsrainer, A.; Weng, H.; Dooley, S.; Dijke, P.T. Transforming Growth Factor-β (TGF-β)-mediated Connective Tissue Growth Factor (CTGF) Expression in Hepatic Stellate Cells Requires Stat3 Signaling Activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proell, V.; Carmona-Cuenca, I.; Murillo, M.M.; Huber, H.; Fabregat, I.; Mikulits, W. TGF-β dependent regulation of oxygen radicals during transdifferentiation of activated hepatic stellate cells to myofibroblastoid cells. Comp. Hepatol. 2007, 6, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Young, C.D.; Zhou, H.; Wang, X.-J. Transforming Growth Factor-β Signaling in Fibrotic Diseases and Cancer-Associated Fibroblasts. Biomolecules 2020, 10, 1666. [Google Scholar] [CrossRef] [PubMed]
- Morén, A.; Bellomo, C.; Tsubakihara, Y.; Kardassis, D.; Mikulits, W.; Heldin, C.-H.; Moustakas, A. LXRα limits TGFβ-dependent hepatocellular carcinoma associated fibroblast differentiation. Oncogenesis 2019, 8, 1–14. [Google Scholar] [CrossRef]
- Yang, J.; Lu, Y.; Lin, Y.-Y.; Zheng, Z.-Y.; Fang, J.-H.; He, S.; Zhuang, S.-M. Vascular mimicry formation is promoted by paracrine TGF-β and SDF1 of cancer-associated fibroblasts and inhibited by miR-101 in hepatocellular carcinoma. Cancer Lett. 2016, 383, 18–27. [Google Scholar] [CrossRef]
- Liu, J.; Chen, S.; Wang, W.; Ning, B.-F.; Chen, F.; Shen, W.; Ding, J.; Chen, W.; Xie, W.-F.; Zhang, X. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-β pathways. Cancer Lett. 2016, 379, 49–59. [Google Scholar] [CrossRef]
- Mikula, M.; Proell, V.; Fischer, A.; Mikulits, W. Activated hepatic stellate cells induce tumor progression of neoplastic hepatocytes in a TGF-β dependent fashion. J. Cell. Physiol. 2006, 209, 560–567. [Google Scholar] [CrossRef] [Green Version]
- Ronnov-Jessen, L.; Petersen, O.W. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab. Investig. 1993, 68, 696–707. [Google Scholar]
- Postlethwaite, A.E.; Keski-Oja, J.; Moses, H.L.; Kang, A.H. Stimulation of the chemotactic migration of human fibroblasts by transforming growth factor beta. J. Exp. Med. 1987, 165, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Yang, H.; Wan, L.; Wang, Z.; Wang, H.; Ge, C.; Liu, Y.; Hao, Y.; Zhang, D.; Shi, G.; et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-C.; Zhang, H.-W.; Zhao, Q.-C.; Sun, L.; Yang, J.-J.; Hong, L.; Feng, F.; Cai, L. Mesenchymal stem cells promote tumor angiogenesis via the action of transforming growth factor β1. Oncol. Lett. 2015, 11, 1089–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benetti, A.; Berenzi, A.; Gambarotti, M.; Garrafa, E.; Gelati, M.; Dessy, E.; Portolani, N.; Piardi, T.; Giulini, S.M.; Caruso, A.; et al. Transforming Growth Factor-β1 and CD105 Promote the Migration of Hepatocellular Carcinoma–Derived Endothelium. Cancer Res. 2008, 68, 8626–8634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.; Wang, X.; Liu, Q.; Shen, J.; Li, Z.; Li, Y.; Zhang, J. Inhibition of TGF-β/SMAD3/NF-κB signaling by microRNA-491 is involved in arsenic trioxide-induced anti-angiogenesis in hepatocellular carcinoma cells. Toxicol. Lett. 2014, 231, 55–61. [Google Scholar] [CrossRef]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Gingold, J.A.; Su, X. Immunomodulatory TGF-β Signaling in Hepatocellular Carcinoma. Trends Mol. Med. 2019, 25, 1010–1023. [Google Scholar] [CrossRef] [Green Version]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; de Moura, M.C.; Putra, J.; Campreciós, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [Green Version]
- Gabrielson, A.; Wu, Y.; Wang, H.; Jiang, J.; Kallakury, B.; Gatalica, Z.; Reddy, S.; Kleiner, D.; Fishbein, T.; Johnson, L.; et al. Intratumoral CD3 and CD8 T-cell Densities Associated with Relapse-Free Survival in HCC. Cancer Immunol. Res. 2016, 4, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.-Z.; Zhang, Z.; Zheng, B.-H.; Shi, Y.; Duan, M.; Ma, L.-J.; Wang, Z.-C.; Dong, L.-Q.; Dong, P.-P.; Shi, J.-Y.; et al. CCL15 Recruits Suppressive Monocytes to Facilitate Immune Escape and Disease Progression in Hepatocellular Carcinoma. Hepatology 2019, 69, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Kuang, D.-M.; Pan, W.-D.; Wan, Y.-L.; Lao, X.-M.; Wang, D.; Li, X.-F.; Zheng, L. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology 2013, 57, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.L.; Takami, A.; Yoshioka, K.; Nakata, K.; Sato, T.; Kasahara, Y.; Nakao, S. Human microRNA-1245 down-regulates the NKG2D receptor in natural killer cells and impairs NKG2D-mediated functions. Haematologica 2012, 97, 1295–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easom, N.J.W.; Stegmann, K.A.; Swadling, L.; Pallett, L.J.; Burton, A.R.; Odera, D.; Schmidt, N.; Huang, W.-C.; Fusai, G.; Davidson, B.; et al. IL-15 Overcomes Hepatocellular Carcinoma-Induced NK Cell Dysfunction. Front. Immunol. 2018, 9, 1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, J.; Goto, K.; Tanoue, Y.; Ito, S.; Muroyama, R.; Matsubara, Y.; Nakagawa, R.; Kaise, Y.; Lim, L.A.; Yoshida, H.; et al. Enzymatic inhibition of MICA sheddase ADAM17 by lomofungin in hepatocellular carcinoma cells. Int. J. Cancer 2018, 143, 2575–2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Kim, Y.; Park, G.; Kim, Y.-S.; Kim, S.; Lee, H.-K.; Chung, W.Y.; Park, S.J.; Han, S.-Y.; Cho, D.; et al. Transforming growth factor-β1 regulates human renal proximal tubular epithelial cell susceptibility to natural killer cells via modulation of the NKG2D ligands. Int. J. Mol. Med. 2015, 36, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Hasmim, M.; Messai, Y.; Ziani, L.; Thiery, J.; Bouhris, J.-H.; Noman, M.Z.; Chouaib, S. Critical Role of Tumor Microenvironment in Shaping NK Cell Functions: Implication of Hypoxic Stress. Front. Immunol. 2015, 6, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiringhelli, F.; Ménard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor–β–dependent manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Gara, S.K.; Achyut, B.R.; Li, Z.; Yan, H.H.; Day, C.-P.; Weiss, J.M.; Trinchieri, G.; Morris, J.C.; Yang, L. TGF-β Signaling in Myeloid Cells Is Required for Tumor Metastasis. Cancer Discov. 2013, 3, 936–951. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Chen, Y.; Chen, Y.; Yang, Y.; Bing, W.; Qi, J. CD4+ CD25+ regulatory T cells promote hepatocellular carcinoma invasion via TGF-β1-induced epithelial–mesenchymal transition. OncoTargets Ther. 2018, 12, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.R.; Mantovani, A. Cancer-related inflammation: Common themes and therapeutic opportunities. Semin. Cancer Biol. 2012, 22, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhu, X.-D.; Sun, H.-C.; Xiong, Y.-Q.; Zhuang, P.-Y.; Xu, H.-X.; Kong, L.-Q.; Wang, L.; Wu, W.-Z.; Tang, Z.-Y. Depletion of Tumor-Associated Macrophages Enhances the Effect of Sorafenib in Metastatic Liver Cancer Models by Antimetastatic and Antiangiogenic Effects. Clin. Cancer Res. 2010, 16, 3420–3430. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Zhong, C.; Cui, W.; Wang, G.; Zheng, G.; Li, L.; Zhang, J.; Ren, R.; Gao, H.; Wang, T.; et al. Induction of tolerogenic dendritic cells by activated TGF-β/Akt/Smad2 signaling in RIG-I-deficient stemness-high human liver cancer cells. BMC Cancer 2019, 19, 439. [Google Scholar] [CrossRef]
- Nandan, D.; Reiner, N.E. TGF-beta attenuates the class II transactivator and reveals an accessory pathway of IFN-gamma action. J. Immunol. 1997, 158, 1095–1101. [Google Scholar] [PubMed]
- Harimoto, H.; Shimizu, M.; Nakagawa, Y.; Nakatsuka, K.; Wakabayashi, A.; Sakamoto, C.; Takahashi, H. Inactivation of tumor-specific CD8+ CTLs by tumor-infiltrating tolerogenic dendritic cells. Immunol. Cell Biol. 2013, 91, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Steinman, R.M. Antigen-bearing immature dendritic cells induce peptide-specific CD8+ regulatory T cells in vivo in humans. Blood 2002, 100, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Gorelik, L.; Constant, S.L.; Flavell, R.A. Mechanism of Transforming Growth Factor β–induced Inhibition of T Helper Type 1 Differentiation. J. Exp. Med. 2002, 195, 1499–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.T.; Martin, S.L.; Xia, L.; Gorham, J.D. TGF-β1 Uses Distinct Mechanisms to Inhibit IFN-γ Expression in CD4+ T Cells at Priming and at Recall: Differential Involvement of Stat4 and T-bet. J. Immunol. 2005, 174, 5950–5958. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, M.; Yamashita, M.; Shinoda, K.; Tofukuji, S.; Onodera, A.; Shinnakasu, R.; Motohashi, S.; Hosokawa, H.; Tumes, D.; Iwamura, C.; et al. The transcription factor Sox4 is a downstream target of signaling by the cytokine TGF-β and suppresses TH2 differentiation. Nat. Immunol. 2012, 13, 778–786. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFβ. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Pfeuffer, I.; Schorr, E.; Siebelt, F.; Wirth, T.; Serfling, E. Transforming growth factor beta and cyclosporin A inhibit the inducible activity of the interleukin-2 gene in T cells through a noncanonical octamer-binding site. Mol. Cell. Biol. 1993, 13, 1155–1162. [Google Scholar] [CrossRef] [Green Version]
- Tinoco, R.; Alcalde, V.; Yang, Y.; Sauer, K.; Zuniga, E.I. Cell-Intrinsic Transforming Growth Factor-β Signaling Mediates Virus-Specific CD8+ T Cell Deletion and Viral Persistence In Vivo. Immunity 2009, 31, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Wolfraim, L.A.; Walz, T.M.; James, Z.; Fernandez, T.; Letterio, J.J. p21Cip1 and p27Kip1 Act in Synergy to Alter the Sensitivity of Naive T Cells to TGF-β-Mediated G1 Arrest through Modulation of IL-2 Responsiveness. J. Immunol. 2004, 173, 3093–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Roh, Y.S.; Song, J.; Zhang, B.; Liu, C.; Loomba, R.; Seki, E. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology 2014, 59, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- Rau, M.; Schilling, A.-K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2015, 196, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Doisne, J.-M.; Bartholin, L.; Yan, K.-P.; Garcia, C.N.; Duarte, N.; Le Luduec, J.-B.; Vincent, D.; Cyprian, F.; Horvat, B.; Martel, S.; et al. iNKT cell development is orchestrated by different branches of TGF-β signaling. J. Exp. Med. 2009, 206, 1365–1378. [Google Scholar] [CrossRef]
- Liew, P.X.; Lee, W.-Y.; Kubes, P. iNKT Cells Orchestrate a Switch from Inflammation to Resolution of Sterile Liver Injury. Immunity 2017, 47, 752–765.e5. [Google Scholar] [CrossRef] [Green Version]
- Cariani, E.; Pilli, M.; Zerbini, A.; Rota, C.; Olivani, A.; Pelosi, G.; Schianchi, C.; Soliani, P.; Campanini, N.; Silini, E.M.; et al. Immunological and Molecular Correlates of Disease Recurrence after Liver Resection for Hepatocellular Carcinoma. PLoS ONE 2012, 7, e32493. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.-S.; Gao, Q.; Xu, X.-N.; Li, Y.-W.; Ju, M.-J.; Cai, M.-Y.; Dai, C.-X.; Hu, J.; Qiu, S.-J.; Zhou, J.; et al. Combination of Intratumoral Invariant Natural Killer T Cells and Interferon-Gamma Is Associated with Prognosis of Hepatocellular Carcinoma after Curative Resection. PLoS ONE 2013, 8, e70345. [Google Scholar] [CrossRef]
- Shen, Y.; Wei, Y.; Wang, Z.; Jing, Y.; He, H.; Yuan, J.; Li, R.; Zhao, Q.; Wei, L.; Yang, T.; et al. TGF-β Regulates Hepatocellular Carcinoma Progression by Inducing Treg Cell Polarization. Cell. Physiol. Biochem. 2015, 35, 1623–1632. [Google Scholar] [CrossRef]
- Carambia, A.; Freund, B.; Schwinge, D.; Heine, M.; Laschtowitz, A.; Huber, S.; Wraith, D.C.; Korn, T.; Schramm, C.; Lohse, A.W.; et al. TGF-β-dependent induction of CD4+CD25+Foxp3+ Tregs by liver sinusoidal endothelial cells. J. Hepatol. 2014, 61, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Kakita, N.; Kanto, T.; Itose, I.; Kuroda, S.; Inoue, M.; Matsubara, T.; Higashitani, K.; Miyazaki, M.; Sakakibara, M.; Hiramatsu, N.; et al. Comparative analyses of regulatory T cell subsets in patients with hepatocellular carcinoma: A crucial role of CD25−FOXP3−T cells. Int. J. Cancer 2012, 131, 2573–2583. [Google Scholar] [CrossRef]
- Yu, S.; Wang, Y.; Hou, J.; Li, W.; Wang, X.; Xiang, L.; Tan, D.; Wang, W.; Jiang, L.; Claret, F.X.; et al. Tumor-infiltrating immune cells in hepatocellular carcinoma: Tregs is correlated with poor overall survival. PLoS ONE 2020, 15, e0231003. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Yang, W.; Huang, X.; Cao, A.T.; Bilotta, A.J.; Xiao, Y.; Sun, M.; Chen, L.; Ma, C.; Liu, X.; et al. Neutrophils Promote Amphiregulin Production in Intestinal Epithelial Cells through TGF-β and Contribute to Intestinal Homeostasis. J. Immunol. 2018, 201, 2492–2501. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Y.; Wang, Y.; Ye, P.; Li, J.; Li, H.; Ding, Q.; Xia, J. Amphiregulin Confers Regulatory T Cell Suppressive Function and Tumor Invasion via the EGFR/GSK-3β/Foxp3 Axis. J. Biol. Chem. 2016, 291, 21085–21095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaiss, D.M.; van Loosdregt, J.; Gorlani, A.; Bekker, C.P.; Gröne, A.; Sibilia, M.; Henegouwen, P.M.V.B.E.; Roovers, R.C.; Coffer, P.J.; Sijts, A.J. Amphiregulin Enhances Regulatory T Cell-Suppressive Function via the Epidermal Growth Factor Receptor. Immunity 2013, 38, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Park, B.V.; Freeman, Z.T.; Ghasemzadeh, A.; Chattergoon, M.A.; Rutebemberwa, A.; Steigner, J.; Winter, M.E.; Huynh, T.V.; Sebald, S.M.; Lee, S.-J.; et al. TGFβ1-Mediated SMAD3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov. 2016, 6, 1366–1381. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ding, Z.-Y.; Li, S.; Liu, S.; Xiao, C.; Li, Z.; Zhang, B.-X.; Chen, X.-P.; Yang, X. Targeting transforming growth factor-β signaling for enhanced cancer chemotherapy. Theranostics 2021, 11, 1345–1363. [Google Scholar] [CrossRef]
- Lee, H.-J. Recent Advances in the Development of TGF-β Signaling Inhibitors for Anticancer Therapy. J. Cancer Prev. 2020, 25, 213–222. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Chung, C.-L.; Hu, T.-H.; Chen, J.-J.; Liu, P.-F.; Chen, C.-L. Recent progress in TGF-β inhibitors for cancer therapy. Biomed. Pharmacother. 2021, 134, 111046. [Google Scholar] [CrossRef]
- Tu, Y.; Han, J.; Dong, Q.; Chai, R.; Li, N.; Lu, Q.; Xiao, Z.; Guo, Y.; Wan, Z.; Xu, Q. TGF-β2 is a Prognostic Biomarker Correlated with Immune Cell Infiltration in Colorectal Cancer. Medicine 2020, 99, e23024. [Google Scholar] [CrossRef]
- Biswas, S.; Nyman, J.S.; Alvarez, J.; Chakrabarti, A.; Ayres, A.; Sterling, J.; Edwards, J.; Rana, T.; Johnson, R.; Perrien, D.S.; et al. Anti-Transforming Growth Factor ß Antibody Treatment Rescues Bone Loss and Prevents Breast Cancer Metastasis to Bone. PLoS ONE 2011, 6, e27090. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.-S.; Terabe, M.; Mamura, M.; Kang, M.-J.; Chae, H.; Stuelten, C.; Kohn, E.A.; Tang, B.; Sabzevari, H.; Anver, M.R.; et al. An Anti–Transforming Growth Factor β Antibody Suppresses Metastasis via Cooperative Effects on Multiple Cell Compartments. Cancer Res. 2008, 68, 3835–3843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terabe, M.; Ambrosino, E.; Takaku, S.; O’Konek, J.J.; Venzon, D.; Lonning, S.; McPherson, J.M.; Berzofsky, J.A. Synergistic Enhancement of CD8+ T Cell-Mediated Tumor Vaccine Efficacy by an Anti-Transforming Growth Factor- Monoclonal Antibody. Clin. Cancer Res. 2009, 15, 6560–6569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I Study of GC1008 (Fresolimumab): A Human Anti-Transforming Growth Factor-Beta (TGFβ) Monoclonal Antibody in Patients with Advanced Malignant Melanoma or Renal Cell Carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Stevenson, J.P.; Kindler, H.L.; Papasavvas, E.; Sun, J.; Jacobs-Small, M.; Hull, J.; Schwed, D.; Ranganathan, A.; Newick, K.; Heitjan, D.F.; et al. Immunological effects of the TGFβ-blocking antibody GC1008 in malignant pleural mesothelioma patients. OncoImmunology 2013, 2, e26218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, R.; Qu, H.; Qu, H.; Theilhaber, J.; Shapiro, G.; Gregory, R.; Winter, C.; Malkova, N.; Sun, F.; Jaworski, J.; et al. Pan-TGFβ inhibition by SAR439459 relieves immunosuppression and improves antitumor efficacy of PD-1 blockade. OncoImmunology 2020, 9, 1811605. [Google Scholar] [CrossRef] [PubMed]
- Dodagatta-Marri, E.; Meyer, D.S.; Reeves, M.Q.; Paniagua, R.; To, M.D.; Binnewies, M.; Broz, M.L.; Mori, H.; Wu, D.; Adoumie, M.; et al. α-PD-1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by α-TGFβ antibody to promote durable rejection and immunity in squamous cell carcinomas. J. Immunother. Cancer 2019, 7, 62. [Google Scholar] [CrossRef]
- Zhong, Z.; Carroll, K.D.; Policarpio, D.; Osborn, C.; Gregory, M.; Bassi, R.; Jimenez, X.; Prewett, M.; Liebisch, G.; Persaud, K.; et al. Anti-Transforming Growth Factor Receptor II Antibody Has Therapeutic Efficacy against Primary Tumor Growth and Metastasis through Multieffects on Cancer, Stroma, and Immune Cells. Clin. Cancer Res. 2010, 16, 1191–1205. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Berlin, J.D.; Cosaert, J.; Kauh, J.; Chan, E.; Piha-Paul, S.A.; Amaya, A.; Tang, S.; Driscoll, K.; Kimbung, R.; et al. A phase 1 study of anti-TGFβ receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Khan, Z.; Marshall, J.F. The role of integrins in TGFβ activation in the tumour stroma. Cell Tissue Res. 2016, 365, 657–673. [Google Scholar] [CrossRef] [Green Version]
- Stockis, J.; Liénart, S.; Colau, D.; Collignon, A.; Nishimura, S.L.; Sheppard, D.; Coulie, P.G.; Lucas, S. Blocking immunosuppression by human Tregs in vivo with antibodies targeting integrin αVβ8. Proc. Natl. Acad. Sci. USA 2017, 114, E10161–E10168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasaka, N.; Seed, R.I.; Cormier, A.; Bondesson, A.J.; Lou, J.; Elattma, A.; Ito, S.; Yanagisawa, H.; Hashimoto, M.; Ma, R.; et al. Integrin αvβ8–expressing tumor cells evade host immunity by regulating TGF-β activation in immune cells. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodagatta-Marri, E.; Ma, H.-Y.; Liang, B.; Li, H.; Meyer, D.S.; Sun, K.-H.; Ren, X.; Zivak, B.; Rosenblum, M.D.; Headley, M.B.; et al. Integrin αvβ8 on T cells is responsible for suppression of anti-tumor immunity in multiple syngeneic models and is a promising target for tumor immunotherapy. BioRxiv 2020. [Google Scholar] [CrossRef]
- Martin, C.J.; Datta, A.; Littlefield, C.; Kalra, A.; Chapron, C.; Wawersik, S.; Dagbay, K.B.; Brueckner, C.T.; Nikiforov, A.; Danehy, F.T., Jr.; et al. Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci. Transl. Med. 2020, 12, eaay8456. [Google Scholar] [CrossRef] [PubMed]
- Economides, A.N.; Carpenter, L.R.; Rudge, J.S.; Wong, V.; Koehler-Stec, E.M.; Hartnett, C.; Pyles, E.A.; Xu, X.; Daly, T.J.; Young, M.R.; et al. Cytokine traps: Multi-component, high-affinity blockers of cytokine action. Nat. Med. 2003, 9, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Zhu, Y.; Cibull, M.L.; Bao, L.; Chen, C.; Sun, L. A soluble transforming growth factor beta type III receptor suppresses tumorigenicity and metastasis of human breast cancer MDA-MB-231 cells. Cancer Res. 1999, 59, 5041–5046. [Google Scholar] [PubMed]
- De Crescenzo, G.; Chao, H.; Zwaagstra, J.; Durocher, Y.; O’Connor-McCourt, M.D. Engineering TGF-β Traps: Artificially Dimerized Receptor Ectodomains as High-Affinity Blockers of TGF-β Action. In Transforming Growth Factor-β in Cancer Therapy. Cancer Drug Discovery and Development; Jakowlew, S.B., Ed.; Humana Press: Totowa, NJ, USA, 2008; Volume II, pp. 671–684. [Google Scholar]
- Zwaagstra, J.C.; Sulea, T.; Baardsnes, J.; Lenferink, A.E.; Collins, C.; Cantin, C.; Paul-Roc, B.; Grothe, S.; Hossain, S.; Richer, L.-P.; et al. Engineering and Therapeutic Application of Single-Chain Bivalent TGF-β Family Traps. Mol. Cancer Ther. 2012, 11, 1477–1487. [Google Scholar] [CrossRef] [Green Version]
- Qin, T.; Barron, L.; Xia, L.; Huang, H.; Villarreal, M.M.; Zwaagstra, J.; Collins, C.; Yang, J.; Zwieb, C.; Kodali, R.; et al. A novel highly potent trivalent TGF-β receptor trap inhibits early-stage tumorigenesis and tumor cell invasion in murine Pten-deficient prostate glands. Oncotarget 2016, 7, 86087–86102. [Google Scholar] [CrossRef] [Green Version]
- Joyce, C.E.; Saadatpour, A.; Ruiz-Gutierrez, M.; Bolukbasi, O.V.; Jiang, L.; Thomas, D.D.; Young, S.; Hofmann, I.; Sieff, C.A.; Myers, K.C.; et al. TGF-β signaling underlies hematopoietic dysfunction and bone marrow failure in Shwachman-Diamond syndrome. J. Clin. Investig. 2019, 129, 3821–3826. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.; Araujo, D.; Wood, D.; Denis, J.-F.; Gruosso, T.; Tremblay, G.; O’Connor-McCourt, M.; Ghosh, R.; Sinclair, S.; Nadler, P.; et al. P856 AVID200, first-in-class TGF-beta1 and beta3 selective inhibitor: Results of a phase 1 monotherapy dose escalation study in solid tumors and evidence of target engagement in patients. J. Immunother. Cancer 2020, 8, A6.2–A7. [Google Scholar] [CrossRef]
- Ng, S.; Deng, J.; Chinnadurai, R.; Yuan, S.; Pennati, A.; Galipeau, J. Stimulation of Natural Killer Cell–Mediated Tumor Immunity by an IL15/TGFβ–Neutralizing Fusion Protein. Cancer Res. 2016, 76, 5683–5695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGF drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018, 10, eaan5488. [Google Scholar] [CrossRef] [Green Version]
- Ravi, R.; Noonan, K.A.; Pham, V.; Bedi, R.; Zhavoronkov, A.; Ozerov, I.V.; Makarev, E.; Artemov, A.V.; Wysocki, P.; Mehra, R.; et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Kim, T.M.; Vicente, D.; Felip, E.; Lee, D.H.; Lee, K.H.; Lin, C.-C.; Flor, M.J.; Di Nicola, M.; Alvarez, R.M.; et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Second-Line Treatment of Patients With NSCLC: Results From an Expansion Cohort of a Phase 1 Trial. J. Thorac. Oncol. 2020, 15, 1210–1222. [Google Scholar] [CrossRef]
- Kang, Y.-K.; Bang, Y.-J.; Kondo, S.; Chung, H.C.; Muro, K.; Dussault, I.; Helwig, C.; Osada, M.; Doi, T. Safety and Tolerability of Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGFβ and PD-L1, in Asian Patients with Pretreated Recurrent or Refractory Gastric Cancer. Clin. Cancer Res. 2020, 26, 3202–3210. [Google Scholar] [CrossRef] [Green Version]
- Yoo, C.; Oh, D.-Y.; Choi, H.J.; Kudo, M.; Ueno, M.; Kondo, S.; Chen, L.-T.; Osada, M.; Helwig, C.; Dussault, I.; et al. Phase I study of bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in patients with pretreated biliary tract cancer. J. Immunother. Cancer 2019, 8, e000564. [Google Scholar] [CrossRef]
- Doi, T.; Fujiwara, Y.; Koyama, T.; Ikeda, M.; Helwig, C.; Watanabe, M.; Vugmeyster, Y.; Kudo, M. Phase I Study of the Bifunctional Fusion Protein Bintrafusp Alfa in Asian Patients with Advanced Solid Tumors, Including a Hepatocellular Carcinoma Safety-Assessment Cohort. Oncologist 2020, 25, e1292–e1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Sun, L.; Jiang, K.; Gao, D.-M.; Kang, X.-N.; Wang, C.; Zhang, S.; Huang, S.; Qin, X.; Li, Y.; et al. NANOG promotes liver cancer cell invasion by inducing epithelial–mesenchymal transition through NODAL/SMAD3 signaling pathway. Int. J. Biochem. Cell Biol. 2013, 45, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Tsukamoto, H.; Liu, J.-C.; Kashiwabara, C.; Feldman, D.; Sher, L.; Dooley, S.; French, S.W.; Mishra, L.; Petrovic, L.; et al. Reciprocal regulation by TLR4 and TGF-β in tumor-initiating stem-like cells. J. Clin. Investig. 2013, 123, 2832–2849. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-A.; Kim, M.-J.; Park, S.-Y.; Kim, J.-S.; Lim, W.; Nam, J.-S.; Sheen, Y.Y. TIMP-1 mediates TGF-β-dependent crosstalk between hepatic stellate and cancer cells via FAK signaling. Sci. Rep. 2015, 5, 16492. [Google Scholar] [CrossRef]
- Bueno, L.; de Alwis, D.P.; Pitou, C.; Yingling, J.; Lahn, M.; Glatt, S.; Trocóniz, I.F. Semi-mechanistic modelling of the tumour growth inhibitory effects of LY2157299, a new type I receptor TGF-β kinase antagonist, in mice. Eur. J. Cancer 2008, 44, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Mazzocca, A.; Peidrò, F.J.; Papappicco, P.; Fabregat, I.; De Santis, F.; Paradiso, A.; Sabba’, C.; Giannelli, G. Differential Inhibition of the TGF-β Signaling Pathway in HCC Cells Using the Small Molecule Inhibitor LY2157299 and the D10 Monoclonal Antibody against TGF-β Receptor Type II. PLoS ONE 2013, 8, e67109. [Google Scholar] [CrossRef]
- Serova, M.; Tijeras-Raballand, A.; Dos Santos, C.; Albuquerque, M.; Paradis, V.; Neuzillet, C.; Benhadji, K.A.; Raymond, E.; Faivre, S.; De Gramont, A. Effects of TGF-beta signalling inhibition with galunisertib (LY2157299) in hepatocellular carcinoma models and inex vivowhole tumor tissue samples from patients. Oncotarget 2015, 6, 21614–21627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungerleider, N.; Han, C.; Zhang, J.; Yao, L.; Wu, T. TGFβ signaling confers sorafenib resistance via induction of multiple RTKs in hepatocellular carcinoma cells. Mol. Carcinog. 2017, 56, 1302–1311. [Google Scholar] [CrossRef] [Green Version]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef] [Green Version]
- Faivre, S.; Santoro, A.; Kelley, R.K.; Gane, E.; Costentin, C.E.; Gueorguieva, I.; Smith, C.; Cleverly, A.; Lahn, M.M.; Raymond, E.; et al. Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int. 2019, 39, 1468–1477. [Google Scholar] [CrossRef]
- Giannelli, G.; Santoro, A.; Kelley, R.K.; Gane, E.; Paradis, V.; Cleverly, A.; Smith, C.; Estrem, S.T.; Man, M.; Wang, S.; et al. Biomarkers and overall survival in patients with advanced hepatocellular carcinoma treated with TGF-βRI inhibitor galunisertib. PLoS ONE 2020, 15, e0222259. [Google Scholar] [CrossRef]
- Ikeda, M.; Morimoto, M.; Tajimi, M.; Inoue, K.; Benhadji, K.A.; Lahn, M.M.F.; Sakai, D. A phase 1b study of transforming growth factor-beta receptor I inhibitor galunisertib in combination with sorafenib in Japanese patients with unresectable hepatocellular carcinoma. Investig. New Drugs 2019, 37, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Kelley, R.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.; Merle, P.; Hourmand, I.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients with Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Piñero, F.; Silva, M.; Iavarone, M. Sequencing of systemic treatment for hepatocellular carcinoma: Second line competitors. World J. Gastroenterol. 2020, 26, 1888–1900. [Google Scholar] [CrossRef] [PubMed]
- Kirstein, M.M.; Scheiner, B.; Marwede, T.; Wolf, C.; Voigtländer, T.; Semmler, G.; Wacker, F.; Manns, M.P.; Hinrichs, J.B.; Pinter, M.; et al. Sequential systemic treatment in patients with hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2020, 52, 205–212. [Google Scholar] [CrossRef]
- Reig, M.; Torres, F.; Rodriguez-Lope, C.; Forner, A.; Llarch, N.; Rimola, J.; Darnell, A.; Ríos, J.; Ayuso, C.; Bruix, J. Early dermatologic adverse events predict better outcome in HCC patients treated with sorafenib. J. Hepatol. 2014, 61, 318–324. [Google Scholar] [CrossRef]
- Varghese, J.; Kedarisetty, C.K.; Venkataraman, J.; Srinivasan, V.; Deepashree, T.; Uthappa, M.C.; Ilankumaran, K.; Govil, S.; Reddy, M.S.; Rela, M. Combination of TACE and Sorafenib Improves Outcomes in BCLC Stages B/C of Hepatocellular Carcinoma: A Single Centre Experience. Ann. Hepatol. 2017, 16, 247–254. [Google Scholar] [CrossRef]
- Díaz-González, Á.; Sanduzzi-Zamparelli, M.; Sapena, V.; Torres, F.; Llarch, N.; Iserte, G.; Forner, A.; Da Fonseca, L.; Ríos, J.; Bruix, J.; et al. Systematic review with meta-analysis: The critical role of dermatological events in patients with hepatocellular carcinoma treated with sorafenib. Aliment. Pharmacol. Ther. 2019, 49, 482–491. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.C.C.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients with Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.-H.; Harding, J.J.; Merle, P.; et al. LBA38_PR—CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.A.; Wattenberg, M.M.; Damjanov, N.; Dunphy, E.P.; Jacobs-Small, M.; Lubas, M.J.; Robinson, J.; DiCicco, L.; Garcia-Marcano, L.; Giannone, M.A.; et al. A Pilot Study of Galunisertib plus Stereotactic Body Radiotherapy in Patients with Advanced Hepatocellular Carcinoma. Mol. Cancer Ther. 2021, 20, 389–397. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Tumor Type | Phenotype | TGF-β Signature | Biomarkers (Expression Levels) | |||||
|---|---|---|---|---|---|---|---|---|---|
| CDH1 | VIM | CD44 | CXCR4 | SMAD7 | CLTC | ||||
| HepG2 | Human caucasian HCC | Epithelial | Early | High | Absent | Absent | Low | Low | No data |
| PLC/PRF/5 | Human liver hepatoma | Epithelial | Early | High | Very Low | Low | Low | Low | Low |
| Huh-7 | Human asian HCC | Mixed | Early | Medium | Low | Low | Low | High | Low |
| Hep3B | Human black HCC | Mixed | Early | Medium | Low | Low | High | High | Low |
| SNU-449 | Human asian HCC | Mesenchymal | Late | Absent | High | High | High | No data | High |
| HLE | Human HCC | Mesenchymal | Late | Absent | High | High | High | High | high |
| HLF | Human HCC | Mesenchymal | Late | Absent | High | High | High | High | High |
| No. Clinical Trial | Population | Arms | N | Aim | Status |
|---|---|---|---|---|---|
| NCT01988493 | Advanced | Tepotinib vs. Sorafenib | 117 | Safety and TTP | Active, no Recruiting |
| NCT03970616 | Advanced | Tivozanib + Durvalumab (1st Line) | 42 | Safety | Recruiting |
| NCT04503902 | Advanced | Donafenib Tosilate + Toripalimab | 46 | Safety and ORR | Not yet Recruiting |
| NCT04612504 | Advanced | SynOV1.1 monotherapy vs. SynOV1.1 + Atezolizumab | 45 | Safety | Not yet Recruiting |
| NCT03825705 | Advanced | TQB2450 injection + Anlotinib | 60 | ORR | Recruiting |
| NCT03893695 | Intermediate (not locoregional)/Advanced | GT90001 + Nivolumab (dose escalation and expansion) | 20 | Safety | Active, no Recruiting |
| NCT03864211 | Intermediate (not locoregional)/Advanced | Toriplimab montherapy vs. toriplimab + Ablation | 130 | PFS | Recruiting |
| NCT04502082 | Intermediate (not locoregional)/Advanced | ET140203 autologous T cell product (3rd Line) | 50 | Safety | Recruiting |
| NCT03998033 | Intermediate (not locoregional)/Advanced | ET140202 autologous T cell product | 50 | Safety | Active, no Recruiting |
| NCT04035876 | Intermediate | Camrelizumab + Apatinib (Downstaging for TOH) | 120 | ORR and RFS | Recruiting |
| NCT04069949 | Advanced | Toripalimab + Sorafenib (1st Line) | 39 | Safety and 6-month PFS | Not yet Recruiting |
| NCT03897543 | Intermediate (not locoregional)/Advanced | ABX196 + Nivolumab (2nd Line) | 48 | Safety | Recruiting |
| NCT04212221 | Intermediate (not locoregional)/Advanced | MGD013 monotheray vs. MGD013 + Brivanib (2nd Line) | 300 | Safety | Recruiting |
| NCT04601610 | Advanced | KN046 + Ningetinib (1st or 2nd Line) | 70 | Safety and ORR | Not yet Recruiting |
| NCT04380545 | Intermediate (not locoregional)/Advanced | Fluorouracil + Nivolumab + Recombinant Interferon Alpha 2b-like protein | 15 | Safety | Not yet Recruiting |
| NCT03941873 | Intermediate (not locoregional)/Advanced | Sitravatinib monotherapy vs. Sitravatinib + Tislelizumab (1st o 2nd Line) | 104 | Safety and ORR | Recruiting |
| NCT03812874 | Intermediate | PTX-9908 + TACE vs. PBO + TACE | 50 | Safety | Recruiting |
| NCT04251117 | Intermediate (not locoregional)/Advanced | GNOS-PV02 + INO-9012 + Pembrolizumab (2nd Line) | 24 | Safety and Immunogenicity of a personalized neoantigen DNA vaccine. | Recruiting |
| NCT03941626 | Unresectable HCC/no effective treatment | CAR-T/TCR-T cells immunotherapy | 50 | Safety | Recruiting |
| NCT03829501 | Advanced | KY1044 monotherapy vs. KY1044 + Atezolizumab | 412 | Safety | Recruiting |
| No. Clinical Trial | Population | Phase | Arms | N | Aim | Status |
|---|---|---|---|---|---|---|
| NCT01246986 (Parts A and B) | Advanced | II | Galunisertib | 149 | Association of circulating AFP and TGF-β1 levels with OS | Completed |
| NCT01246986 (Part C) | Advanced | Galunisertib + Sorafenib | 47 | Safety, TTP, OS, PFF and ORR | Completed | |
| NCT02240433 | Unresectable | I | Galunisertib + Sorafenib | 14 | Safety, tolerability, PK, TTP and PFS | Completed |
| NCT02906397 | Advanced | I | Galunisertib + SBRT | 15 | Safety, PFS, OS | Active, no Recruiting |
| NCT02423343 | Advanced | I/II | Galunisertib + Nivolumab | 75 (10 HCC) | Safety, tolerability, PFS, ORR | Completed |
| NCT02699515 | Advanced | I | Bintrafusp alfa MSB0011359C (M7824) | 114 | Safety, tolerability and ORR | Active, no Recruiting |
| NCT03192345 | Advanced | I (Basket trial) | SAR439459 monotherapy vs. SAR439459 + Cemiplimab | ~350 | Safety, tolerability, TTP and PFS | Recruiting |
| NCT02947165 | Advanced | I (Basket trial) | NI5793 monotherapy vs. NI5793 + PDR001 | 120 | ORR and PFS | Active, no Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Sanchez, E.; Vaquero, J.; Férnandez-Barrena, M.G.; Lasarte, J.J.; Avila, M.A.; Sarobe, P.; Reig, M.; Calvo, M.; Fabregat, I. The TGF-β Pathway: A Pharmacological Target in Hepatocellular Carcinoma? Cancers 2021, 13, 3248. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133248
Gonzalez-Sanchez E, Vaquero J, Férnandez-Barrena MG, Lasarte JJ, Avila MA, Sarobe P, Reig M, Calvo M, Fabregat I. The TGF-β Pathway: A Pharmacological Target in Hepatocellular Carcinoma? Cancers. 2021; 13(13):3248. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133248
Chicago/Turabian StyleGonzalez-Sanchez, Ester, Javier Vaquero, Maite G. Férnandez-Barrena, Juan José Lasarte, Matías A. Avila, Pablo Sarobe, María Reig, Mariona Calvo, and Isabel Fabregat. 2021. "The TGF-β Pathway: A Pharmacological Target in Hepatocellular Carcinoma?" Cancers 13, no. 13: 3248. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133248