
OB-Folds and Genome Maintenance: Targeting Protein–DNA Interactions for Cancer Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
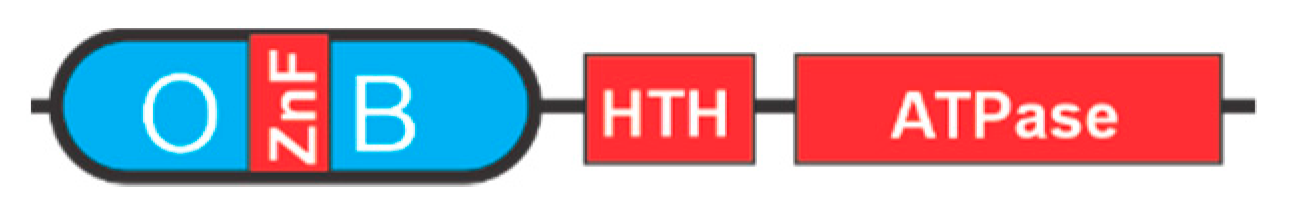{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
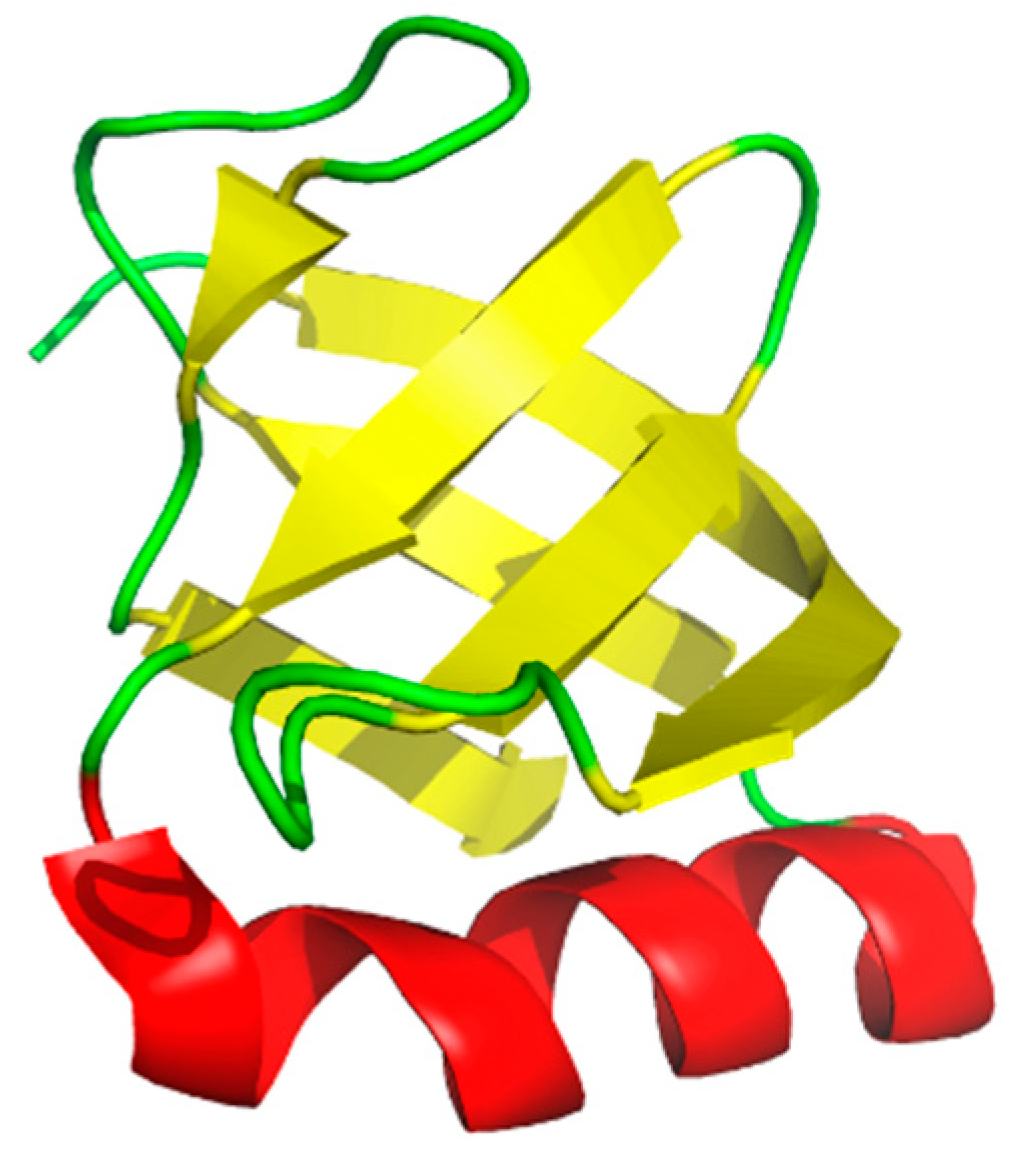
1. Oligonucleotide/Oligosaccharide Binding-Fold (OB-Folds)
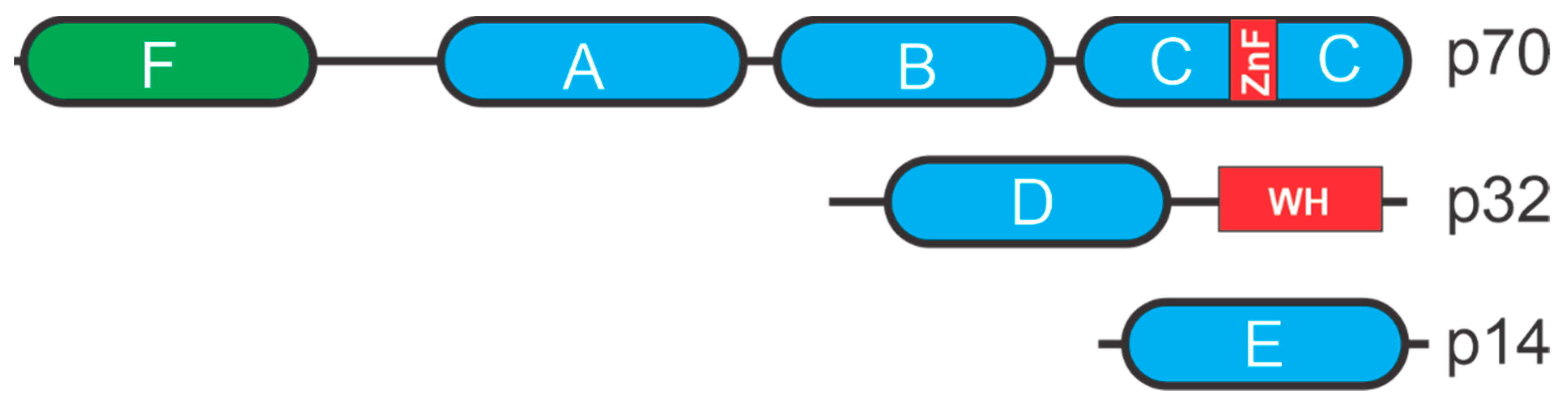
2. Replication Protein A (RPA)
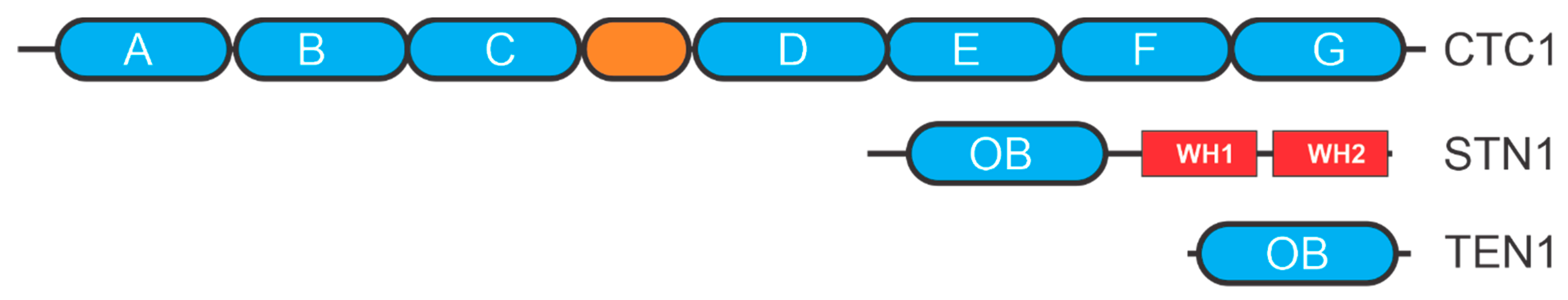
3. CST
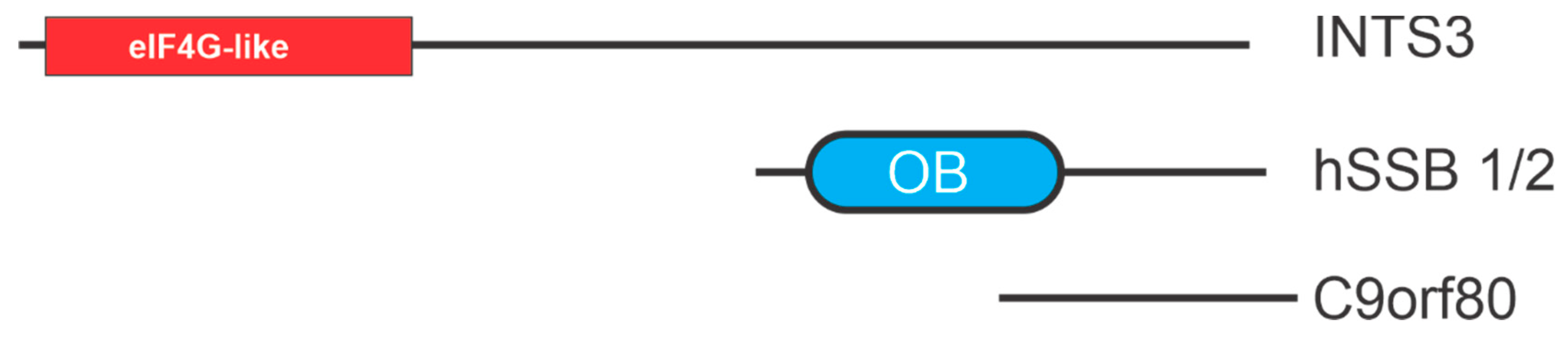
4. hSSB1/2
5. MCM (Minichromosome Maintenance Complex)
6. BRCA2
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murzin, A.G. OB (oligonucleotide/oligosaccharide binding)-fold: Common structural and functional solution for non-homologous sequences. EMBO J. 1993, 12, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Bochkarev, A.; Bochkareva, E. From RPA to BRCA2: Lessons from single-stranded DNA binding by the OB-fold. Curr. Opin. Struct. Biol. 2004, 14, 36–42. [Google Scholar] [CrossRef]
- Theobald, D.L.; Mitton-Fry, R.M.; Wuttke, D.S. Nucleic Acid Recognition by OB-Fold Proteins. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 115–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taib, N.; Gribaldo, S.; A MacNeill, S. Single-Stranded DNA-Binding Proteins in the Archaea. Methods Mol. Biol. 2021, 2281, 23–47. [Google Scholar]
- Amir, M.; Mohammad, T.; Dohare, R.; Islam, A.; Ahmad, F.; Imtaiyaz, H.M. Structure, function and therapeutic implications of OB-fold proteins: A lesson from past to present. Brief Funct Genom. 2020, 19, 377–389. [Google Scholar] [CrossRef]
- Ramilo, C.; Gu, L.; Guo, S.; Zhang, X.; Patrick, S.M.; Turchi, J.J.; Li, G.-M. Partial Reconstitution of Human DNA Mismatch Repair In Vitro: Characterization of the Role of Human Replication Protein A. Mol. Cell. Biol. 2002, 22, 2037–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wold, M.S. Replication Protein A: A Heterotrimeric, Single-Stranded DNA-Binding Protein Required for Eukaryotic DNA Metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef]
- Wu, Y.; Lu, J.; Kang, T. Human single-stranded DNA binding proteins: Guardians of genome stability. Acta Biochim. Biophys. Sin. 2016, 48, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Lindsey-Boltz, L.; Reardon, J.T.; Wold, M.; Sancar, A. In Vitro Analysis of the Role of Replication Protein A (RPA) and RPA Phosphorylation in ATR-mediated Checkpoint Signaling. J. Biol. Chem. 2012, 287, 36123–36131. [Google Scholar] [CrossRef] [Green Version]
- de Laat, W.L.; Appeldoorn, E.; Sugasawa, K.; Weterings, E.; Jaspers, N.G.J.; Hoeijmakers, J.H.J. DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev. 1998, 12, 2598–2609. [Google Scholar] [CrossRef] [Green Version]
- Binz, S.K.; Wold, M.S. Regulatory Functions of the N-terminal Domain of the 70-kDa Subunit of Replication Protein A (RPA). J. Biol. Chem. 2008, 283, 21559–21570. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Liu, Y.; Wu, X.; Shell, S.M. Functions of human replication protein A (RPA): From DNA replication to DNA damage and stress responses. J. Cell. Physiol. 2006, 208, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Arunkumar, A.I.; Stauffer, M.E.; Bochkareva, E.; Bochkarev, A.; Chazin, W.J. Independent and Coordinated Functions of Replication Protein A Tandem High Affinity Single-stranded DNA Binding Domains. J. Biol. Chem. 2003, 278, 41077–41082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glanzer, J.G.; Liu, S.; Oakley, G.G. Small molecule inhibitor of the RPA70 N-terminal protein interaction domain discovered using in silico and in vitro methods. Bioorg. Med. Chem. 2011, 19, 2589–2595. [Google Scholar] [CrossRef] [Green Version]
- Yates, L.A.; Aramayo, R.J.; Pokhrel, N.; Caldwell, C.; Kaplan, J.A.; Perera, R.; Spies, M.; Antony, E.; Zhang, X. A structural and dynamic model for the assembly of Replication Protein A on single-stranded DNA. Nat. Commun. 2018, 9, 5447. [Google Scholar] [CrossRef]
- Wang, Q.-M.; Yang, Y.-T.; Wang, Y.-R.; Gao, B.; Xi, X.; Hou, X.-M. Human replication protein A induces dynamic changes in single-stranded DNA and RNA structures. J. Biol. Chem. 2019, 294, 13915–13927. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wold, M.S. Replication protein A: Single-stranded DNA’s first responder: Dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 2014, 36, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.; Sokoloski, J.; Galletto, R.; Elson, E.L.; Wold, M.; Lohman, T.M. Diffusion of Human Replication Protein A along Single-Stranded DNA. J. Mol. Biol. 2014, 426, 3246–3261. [Google Scholar] [CrossRef] [Green Version]
- Waterson, A.G.; Kennedy, J.P.; Patrone, J.; Pelz, N.F.; Feldkamp, M.D.; Frank, A.O.; Vangamudi, B.; Souza-Fagundes, E.M.; Rossanese, O.W.; Chazin, W.J.; et al. Diphenylpyrazoles as Replication Protein A Inhibitors. ACS Med. Chem. Lett. 2014, 6, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.O.; Vangamudi, B.; Feldkamp, M.D.; Souza-Fagundes, E.M.; Luzwick, J.W.; Cortez, D.; Olejniczak, E.T.; Waterson, A.G.; Rossanese, O.W.; Chazin, W.J.; et al. Discovery of a Potent Stapled Helix Peptide That Binds to the 70 N Domain of Replication Protein A. J. Med. Chem. 2014, 57, 2455–2461. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, J.G.; Carnes, K.A.; Soto, P.; Liu, S.; Parkhurst, L.J.; Oakley, G.G. A small molecule directly inhibits the p53 transactivation domain from binding to replication protein A. Nucleic Acids Res. 2012, 41, 2047–2059. [Google Scholar] [CrossRef] [Green Version]
- Glanzer, J.G.; Liu, S.; Wang, L.; Mosel, A.; Peng, A.; Oakley, G.G. RPA Inhibition Increases Replication Stress and Suppresses Tumor Growth. Cancer Res. 2014, 74, 5165–5172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anciano Granadillo, V.J.; Earley, J.N.; Shuck, S.C.; Georgiadis, M.M.; Fitch, R.W.; Turchi, J.J. Targeting the OB-Folds of Replication Protein A with Small Molecules. J. Nucleic. Acids 2010, 2010, 304035. [Google Scholar] [CrossRef] [Green Version]
- Turchi, J.J.; VanderVere-Carozza, P.S. Small-Molecule Inhibitor Screen for DNA Repair Proteins. Breast Cancer 2019, 1999, 217–221. [Google Scholar] [CrossRef]
- Shuck, S.; Turchi, J.J. Targeted Inhibition of Replication Protein A Reveals Cytotoxic Activity, Synergy with Chemotherapeutic DNA-Damaging Agents, and Insight into Cellular Function. Cancer Res. 2010, 70, 3189–3198. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.K.; Dormi, S.S.; Turchi, A.M.; Woods, D.S.; Turchi, J.J. Chemical inhibitor targeting the replication protein A–DNA interaction increases the efficacy of Pt-based chemotherapy in lung and ovarian cancer. Biochem. Pharm. 2015, 93, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Vernon, T.L.; Jordan, M.R.; Turchi, J.J. Structure-Guided Optimization of Replication Protein A (RPA)–DNA Interaction Inhibitors. ACS Med. Chem. Lett. 2020, 11, 1118–1124. [Google Scholar] [CrossRef]
- VanderVere-Carozza, P.S.; Pawelczak, K.S.; Gavande, N.S.; Jalal, S.I.; Pollok, K.E.; Ekinci, E.; Heyza, J.; Patrick, S.M.; Turchi, J.J. Chemical exhaustion of RPA in cancer treatment. bioRxiv 2020. [Google Scholar] [CrossRef]
- Toledo, L.; Neelsen, K.J.; Lukas, J. Replication Catastrophe: When a Checkpoint Fails because of Exhaustion. Mol. Cell 2017, 66, 735–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surovtseva, Y.V.; Churikov, D.; Boltz, K.A.; Song, X.; Lamb, J.C.; Warrington, R.; Leehy, K.; Heacock, M.; Price, C.M.; Shippen, D.E. Conserved Telomere Maintenance Component 1 Interacts with STN1 and Maintains Chromosome Ends in Higher Eukaryotes. Mol. Cell 2009, 36, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, Y.; Nakamura, M.; Nabetani, A.; Shimamura, S.; Tamura, M.; Yonehara, S.; Saito, M.; Ishikawa, F. RPA-like Mammalian Ctc1-Stn1-Ten1 Complex Binds to Single-Stranded DNA and Protects Telomeres Independently of the Pot1 Pathway. Mol. Cell 2009, 36, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Nugent, C.I.; Hughes, T.R.; Lue, N.F.; Lundblad, V. Cdc13p: A Single-Strand Telomeric DNA-Binding Protein with a Dual Role in Yeast Telomere Maintenance. Science 1996, 274, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Grandin, N.; Reed, S.I.; Charbonneau, M. Stn1, a new Saccharomyces cerevisiae protein, is implicated in telomere size regulation in association with Cdc13. Genes Dev. 1997, 11, 512–527. [Google Scholar] [CrossRef] [Green Version]
- Grandin, N.; Damon, C.; Charbonneau, M. Ten1 functions in telomere end protection and length regulation in association with Stn1 and Cdc13. EMBO J. 2001, 20, 1173–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.J.; Barbour, A.T.; Zaug, A.J.; Goodrich, K.J.; McKay, A.E.; Wuttke, D.S.; Cech, T.R. The structure of human CST reveals a decameric assembly bound to telomeric DNA. Science 2020, 368, 1081–1085. [Google Scholar] [CrossRef]
- Sun, J.; Yu, E.Y.; Yang, Y.; Confer, L.A.; Sun, S.H.; Wan, K.; Lue, N.F.; Lei, M. Stn1-Ten1 is an Rpa2-Rpa3-like complex at telomeres. Genes Dev. 2009, 23, 2900–2914. [Google Scholar] [CrossRef] [Green Version]
- Fanning, E.; Klimovich, V.; Nager, A.R. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006, 34, 4126–4137. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, A.; Wang, Y.; Diao, J.; Price, C.M. Dynamic DNA binding, junction recognition and G4 melting activity underlie the telomeric and genome-wide roles of human CST. Nucleic Acids Res. 2017, 45, 12311–12324. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, A.; Stewart, J.; Chaiken, M.; Price, C.M. STN1 OB Fold Mutation Alters DNA Binding and Affects Selective Aspects of CST Function. PLoS Genet. 2016, 12, e1006342. [Google Scholar] [CrossRef] [Green Version]
- Lue, N.F.; Zhou, R.; Chico, L.; Mao, N.; Steinberg-Neifach, O.; Ha, T. The Telomere Capping Complex CST Has an Unusual Stoichiometry, Makes Multipartite Interaction with G-Tails, and Unfolds Higher-Order G-Tail Structures. PLoS Genet. 2013, 9, e1003145. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-C.; Shih, J.-W.; Hsu, C.-L.; Lin, J.-J. Binding and Partial Denaturing of G-quartet DNA by Cdc13p ofSaccharomyces cerevisiae. J. Biol. Chem. 2001, 276, 47671–47674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.J.; Cech, T.R. Shaping human telomeres: From shelterin and CST complexes to telomeric chromatin organization. Nat. Rev. Mol. Cell Biol. 2021, 22, 283–298. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Ackerson, S.M.; Gable, C.I.; Stewart, J.A. Human CTC1 promotes TopBP1 stability and CHK1 phosphorylation in response to telomere dysfunction and global replication stress. Cell Cycle 2020, 19, 3491–3507. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Hsu, S.-J.; Bhattacharjee, A.; Wang, Y.; Diao, J.; Price, C.M. CTC1-STN1 terminates telomerase while STN1-TEN1 enables C-strand synthesis during telomere replication in colon cancer cells. Nat. Commun. 2018, 9, 2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.-Y.; Redon, S.; Lingner, J. The human CST complex is a terminator of telomerase activity. Nature 2012, 488, 540–544. [Google Scholar] [CrossRef]
- Wan, M.; Qin, J.; Songyang, Z.; Liu, D. OB Fold-containing Protein 1 (OBFC1), a Human Homolog of Yeast Stn1, Associates with TPP1 and Is Implicated in Telomere Length Regulation. J. Biol. Chem. 2009, 284, 26725–26731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Wang, B.; Li, T.; Liu, R.; Xiao, Y.; Geng, X.; Li, G.; Liu, Q.; Price, C.; Liu, Y.; et al. Mammalian CST averts replication failure by preventing G-quadruplex accumulation. Nucleic Acids Res. 2019, 47, 5243–5259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.A.; Wang, F.; Chaiken, M.F.; Kasbek, C.; Ii, P.D.C.; Wright, W.E.; Price, C.M. Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J. 2012, 31, 3537–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.A.; Wang, Y.; Ackerson, S.M.; Schuck, P.L. Emerging roles of CST in maintaining genome stability and human disease. Front. Biosci. 2018, 23, 1564–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, X.; Lei, K.; Sang, P.B.; Shiva, O.; Chastain, M.; Chi, P.; Chai, W. Human CST complex protects stalled replication forks by directly blocking MRE11 degradation of nascent-strand DNA. EMBO J. 2021, 40, e103654. [Google Scholar] [CrossRef]
- Chastain, M.; Zhou, Q.; Shiva, O.; Fadri-Moskwik, M.; Whitmore, L.; Jia, P.; Dai, X.; Huang, C.; Ye, P.; Chai, W. Human CST Facilitates Genome-wide RAD51 Recruitment to GC-Rich Repetitive Sequences in Response to Replication Stress. Cell Rep. 2016, 16, 1300–1314. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Brady, K.S.; Caiello, B.P.; Ackerson, S.M.; A Stewart, J. Human CST suppresses origin licensing and promotes AND-1/Ctf4 chromatin association. Life Sci. Alliance 2019, 2, e201800270. [Google Scholar] [CrossRef] [Green Version]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef]
- Setiaputra, D.; Durocher, D. Shieldin—The protector of DNA ends. EMBO Rep. 2019, 20, e47560. [Google Scholar] [CrossRef]
- Barazas, M.; Annunziato, S.; Pettitt, S.J.; de Krijger, I.; Ghezraoui, H.; Roobol, S.J.; Lutz, C.; Frankum, J.; Song, F.F.; Brough, R.; et al. The CST Complex Mediates End Protection at Double-Strand Breaks and Promotes PARP Inhibitor Sensitivity in BRCA1-Deficient Cells. Cell Rep. 2018, 23, 2107–2118. [Google Scholar] [CrossRef]
- Gu, P.; Min, J.-N.; Wang, Y.; Huang, C.; Peng, T.; Chai, W.; Chang, S. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J. 2012, 31, 2309–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casteel, D.E.; Zhuang, S.; Zeng, Y.; Perrino, F.W.; Boss, G.R.; Goulian, M.; Pilz, R.B. A DNA polymerase-{alpha}{middle dot}primase cofactor with homology to replication protein A-32 regulates DNA replication in mammalian cells. J. Biol. Chem. 2009, 284, 5807–5818. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Stewart, J.; Price, C.M. Human CST abundance determines recovery from diverse forms of DNA damage and replication stress. Cell Cycle 2014, 13, 3488–3498. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, Q.; Sack, L.; Kang, C.; Elledge, S.J. A gain-of-function senescence bypass screen identifies the homeobox transcription factor DLX2 as a regulator of ATM–p53 signaling. Genes Dev. 2016, 30, 293–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, S.A. Beginning at the ends: Telomeres and human disease. F1000Res 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, M.; Khan, P.; Queen, A.; Dohare, R.; Alajmi, M.F.; Hussain, A.; Islam, A.; Ahmad, F.; Hassan, I. Structural Features of Nucleoprotein CST/Shelterin Complex Involved in the Telomere Maintenance and Its Association with Disease Mutations. Cells 2020, 9, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, K.M.; Codd, V.; Rice, T.; Nelson, C.P.; Smirnov, I.V.; McCoy, L.S.; Hansen, H.M.; Elhauge, E.; Ojha, J.; Francis, S.S.; et al. Longer genotypically-estimated leukocyte telomere length is associated with increased adult glioma risk. Oncotarget 2015, 6, 42468–42477. [Google Scholar] [CrossRef] [Green Version]
- Gudmundsson, J.; Thorleifsson, G.; Sigurdsson, J.K.; Stefansdottir, L.; Jonasson, J.G.; Gudjonsson, S.A.; Gudbjartsson, D.F.; Masson, G.; Johannsdottir, H.; Halldorsson, G.H.; et al. A genome-wide association study yields five novel thyroid cancer risk loci. Nat. Commun. 2017, 8, 14517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojha, J.; Codd, V.; Nelson, C.P.; Samani, N.J.; Smirnov, I.V.; Madsen, N.R.; Hansen, H.M.; De Smith, A.J.; Bracci, P.M.; Wiencke, J.K.; et al. Genetic Variation Associated with Longer Telomere Length Increases Risk of Chronic Lymphocytic Leukemia. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Phelan, C.M.; AOCS Study Group; Kuchenbaecker, K.B.; Tyrer, J.P.; Kar, S.P.; Lawrenson, K.; Winham, S.; Dennis, J.; Pirie, A.; Riggan, M.J.; et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 2017, 49, 680–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, K.M.; Whitehead, T.P.; de Smith, A.J.; Smirnov, I.V.; Park, M.; Endicott, A.A.; Francis, S.S.; Codd, V.; ENGAGE Consortium Telomere Group; Samani, N.J.; et al. Common genetic variants associated with telomere length confer risk for neuroblastoma and other childhood cancers. Carcinogenesis 2016, 37, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, M.H.; Bishop, D.T.; Lee, J.E.; Brossard, M.; Martin, N.G.; Moses, E.; Song, F.; Barrett, J.H.; Kumar, R.; Easton, D.F.; et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat. Genet. 2015, 47, 987–995. [Google Scholar] [CrossRef] [Green Version]
- Győrffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online Survival Analysis Software to Assess the Prognostic Value of Biomarkers Using Transcriptomic Data in Non-Small-Cell Lung Cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.M.; Xia, N.X.; Yang, L.; Li, Z.; Yang, H.; Yu, H.J.; Liu, Y.; Lei, H.; Zhou, F.X.; Xie, C.H.; et al. CTC1 increases the radioresistance of human melanoma cells by inhibiting telomere shortening and apoptosis. Int. J. Mol. Med. 2014, 33, 1484–1490. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Hsu, S.-J.; Kasbek, C.; Chaiken, M.; Price, C.M. CTC1-mediated C-strand fill-in is an essential step in telomere length maintenance. Nucleic Acids Res. 2017, 45, 4281–4293. [Google Scholar] [CrossRef] [Green Version]
- Richard, D.; Bolderson, E.; Cubeddu, L.; Wadsworth, R.I.M.; Savage, K.; Sharma, G.G.; Nicolette, M.L.; Tsvetanov, S.; Mcilwraith, M.; Pandita, R.K.; et al. Single-stranded DNA-binding protein hSSB1 is critical for genomic stability. Nature 2008, 453, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Lawson, T.; El-Kamand, S.; Boucher, D.; Duong, D.C.; Kariawasam, R.; Bonvin, A.M.J.J.; Richard, D.; Gamsjaeger, R.; Cubeddu, L. The structural details of the interaction of single-stranded DNA binding protein hSSB2 (NABP1/OBFC2A) with UV-damaged DNA. Proteins Struct. Funct. Bioinform. 2020, 88, 319–326. [Google Scholar] [CrossRef]
- Croft, L.V.; Bolderson, E.; Adams, M.N.; El-Kamand, S.; Kariawasam, R.; Cubeddu, L.; Gamsjaeger, R.; Richard, D.J. Human single-stranded DNA binding protein 1 (hSSB1, OBFC2B), a critical component of the DNA damage response. Semin. Cell Dev. Biol. 2019, 86, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Bolderson, E.; Kumar, R.; Muniandy, P.A.; Xue, Y.; Richard, D.; Seidman, M.; Pandita, T.K.; Khanna, K.K.; Wang, W. hSSB1 and hSSB2 Form Similar Multiprotein Complexes That Participate in DNA Damage Response. J. Biol. Chem. 2009, 284, 23525–23531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, E.A.; Cortez, D. SOSS1/2: Sensors of single-stranded DNA at a break. Mol. Cell 2009, 35, 258–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, N.W.; Loo, D.; Paquet, N.; O’Byrne, K.J.; Richard, D.J. Novel insight into the composition of human single-stranded DNA-binding protein 1 (hSSB1)-containing protein complexes. BMC Mol. Biol. 2016, 17, 24. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zheng, L.; Hu, K.; Wang, X.; Zhang, R.; Zou, Y.; Zhong, L.; Wang, S.; Wu, Y.; Kang, T. SUMOylation stabilizes hSSB1 and enhances the recruitment of NBS1 to DNA damage sites. Signal. Transduct. Target. 2020, 5, 80. [Google Scholar] [CrossRef]
- Huang, J.; Gong, Z.; Ghosal, G.; Chen, J. SOSS Complexes Participate in the Maintenance of Genomic Stability. Mol. Cell 2009, 35, 384–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tye, B.K. MCM proteins in DNA replication. Annu. Rev. Biochem. 1999, 68, 649–686. [Google Scholar] [CrossRef] [PubMed]
- Riera, A.; Barbon, M.; Noguchi, Y.; Reuter, L.M.; Schneider, S.; Speck, C. From structure to mechanism—Understanding initiation of DNA replication. Genes Dev. 2017, 31, 1073–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froelich, C.A.; Kang, S.; Epling, L.B.; Bell, S.P.; Enemark, E.J. A conserved MCM single-stranded DNA binding element is essential for replication initiation. eLife 2014, 3, e01993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, M. The MCM Complex: Its Role in DNA Replication and Implications for Cancer Therapy. Curr. Cancer Drug Targets 2005, 5, 365–380. [Google Scholar] [CrossRef]
- Bochman, M.L.; Schwacha, A. The Mcm Complex: Unwinding the Mechanism of a Replicative Helicase. Microbiol. Mol. Biol. Rev. 2009, 73, 652–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, J.; Marians, K.J.; Kowalczykowski, S.C. Independent and Stochastic Action of DNA Polymerases in the Replisome. Cell 2017, 169, 1201–1213.e17. [Google Scholar] [CrossRef] [Green Version]
- Todorov, I.T.; A Werness, B.; Wang, H.Q.; Buddharaju, L.N.; Todorova, P.D.; Slocum, H.K.; Brooks, J.S.; A Huberman, J. HsMCM2/BM28: A novel proliferation marker for human tumors and normal tissues. Lab. Investig. 1998, 78, 73–78. [Google Scholar]
- Misono, S.; Mizuno, K.; Suetsugu, T.; Tanigawa, K.; Nohata, N.; Uchida, A.; Sanada, H.; Okada, R.; Moriya, S.; Inoue, H.; et al. Molecular Signature of Small Cell Lung Cancer after Treatment Failure: The MCM Complex as Therapeutic Target. Cancers 2021, 13, 1187. [Google Scholar] [CrossRef]
- Gonzalez, M.A.; Pinder, S.E.; Callagy, G.; Vowler, S.L.; Morris, L.S.; Bird, K.; Bell, J.A.; Laskey, R.A.; Coleman, N. Minichromosome Maintenance Protein 2 Is a Strong Independent Prognostic Marker in Breast Cancer. J. Clin. Oncol. 2003, 21, 4306–4313. [Google Scholar] [CrossRef]
- Cao, T.; Yi, S.-J.; Wang, L.-X.; Zhao, J.-X.; Xiao, J.; Xie, N.; Zeng, Z.; Han, Q.; Tang, H.-O.; Li, Y.-K.; et al. Identification of the DNA Replication Regulator MCM Complex Expression and Prognostic Significance in Hepatic Carcinoma. Biomed. Res. Int. 2020, 2020, 1–18. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, S.; Liu, J.; Yan, W.; Han, P.; Tian, D. Identification of MCM family as potential therapeutic and prognostic targets for hepatocellular carcinoma based on bioinformatics and experiments. Life Sci. 2021, 272, 119227. [Google Scholar] [CrossRef]
- Wan, W.; Shen, Y.; Li, Q. MCM10 Acts as a Potential Prognostic Biomarker and Promotes Cell Proliferation in Hepatocellular Carcinoma: Integrated Bioinformatics Analysis and Experimental Validation. Cancer Manag. Res. 2020, 12, 9609–9619. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Lai, G.; Li, R.; Hao, Y.; Cai, L.; Jia, J. Increased expression of MCM4 is associated with poor prognosis in patients with hepatocellular carcinoma. J. Gastrointest. Oncol. 2021, 12, 153–173. [Google Scholar] [CrossRef]
- Li, H.-T.; Wei, B.; Li, Z.-Q.; Wang, X.; Jia, W.-X.; Xu, Y.-Z.; Liu, J.-Y.; Shao, M.-N.; Chen, S.-X.; Mo, N.-F.; et al. Diagnostic and prognostic value of MCM3 and its interacting proteins in hepatocellular carcinoma. Oncol. Lett. 2020, 20, 308. [Google Scholar] [CrossRef]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited Mutations in 17 Breast Cancer Susceptibility Genes among a Large Triple-Negative Breast Cancer Cohort Unselected for Family History of Breast Cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, H.P.; Ma, X.; Vaquero, J.; Brinkmeyer, M.; Guo, F.; Heyer, W.-D.; Liu, J. DSS1 and ssDNA regulate oligomerization of BRCA2. Nucleic Acids Res. 2020, 48, 7818–7833. [Google Scholar] [CrossRef] [PubMed]
- Sunada, S.; Nakanishi, A.; Miki, Y. Crosstalk of DNA double-strand break repair pathways in poly(ADP-ribose) polymerase inhibitor treatment of breast cancer susceptibility gene 1/2-mutated cancer. Cancer Sci. 2018, 109, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-Y.; Singh, T.R.; Nassar, N.; Zhang, F.; Freund, M.; Hanenberg, H.; Meetei, A.R.; Andreassen, P.R. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene 2014, 33, 4803–4812. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Zhang, F.; Chen, Z.J. BRCA2 in Ovarian Development and Function. N. Engl. J. Med. 2019, 380, 1086. [Google Scholar]
- Zhang, F.; Shi, J.; Bian, C.; Yu, X. Poly(ADP-Ribose) Mediates the BRCA2-Dependent Early DNA Damage Response. Cell Rep. 2015, 13, 678–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target. Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef]
- De Dosso, S.; Siebenhüner, A.R.; Winder, T.; Meisel, A.; Fritsch, R.; Astaras, C.; Szturz, P.; Borner, M. Treatment landscape of metastatic pancreatic cancer. Cancer Treat. Rev. 2021, 96, 102180. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Posadas, E.M.; Bhowmick, N.; Kim, H.L.; Daskivich, T.J.; Gupta, A.; Sandler, H.M.; Kamrava, M.; Zumsteg, Z.S.; Freedland, S.J.; et al. Integrating PARP Inhibitors Into Advanced Prostate Cancer Therapeutics. Oncology 2021, 35, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Mahtani, R.; Kittaneh, M.; Kalinsky, K.; Mamounas, E.; Badve, S.; Vogel, C.; Lower, E.; Schwartzberg, L.; Pegram, M. Advances in Therapeutic Approaches for Triple-Negative Breast Cancer. Clin. Breast Cancer 2020. [Google Scholar] [CrossRef]
- Schettini, F.; Giudici, F.; Bernocchi, O.; Sirico, M.; Corona, S.P.; Giuliano, M.; Locci, M.; Paris, I.; Scambia, G.; De Placido, S.; et al. Poly (ADP-ribose) polymerase inhibitors in solid tumours: Systematic review and meta-analysis. Eur. J. Cancer 2021, 149, 134–152. [Google Scholar] [CrossRef] [PubMed]
- Shao, F.; Duan, Y.; Zhao, Y.; Li, Y.; Liu, J.; Zhang, C.; He, S. PARP inhibitors in breast and ovarian cancer with BRCA mutations: A meta-analysis of survival. Aging 2021, 13, 8975–8988. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Par, S.; Vaides, S.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Stewart, J.; Turchi, J.J. OB-Folds and Genome Maintenance: Targeting Protein–DNA Interactions for Cancer Therapy. Cancers 2021, 13, 3346. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133346
Par S, Vaides S, VanderVere-Carozza PS, Pawelczak KS, Stewart J, Turchi JJ. OB-Folds and Genome Maintenance: Targeting Protein–DNA Interactions for Cancer Therapy. Cancers. 2021; 13(13):3346. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133346
Chicago/Turabian StylePar, Sui, Sofia Vaides, Pamela S. VanderVere-Carozza, Katherine S. Pawelczak, Jason Stewart, and John J. Turchi. 2021. "OB-Folds and Genome Maintenance: Targeting Protein–DNA Interactions for Cancer Therapy" Cancers 13, no. 13: 3346. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133346