
Large-Scale Transcriptomics-Driven Approach Revealed Overexpression of CRNDE as a Poor Survival Prognosis Biomarker in Glioblastoma


 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Biosamples
2.3. RNA Isolation and Sequencing
2.4. RNA Sequencing Data Collection and Processing
2.5. Quantization of Molecular Pathway Activation
2.6. Survival Analysis
2.7. Plots and Visualizations
2.8. Testing of Intersections Significance
2.9. Data and Code Availability
3. Results
3.1. RNA Sequencing of Experimental Glioblastoma Samples
3.2. Primary Comparison of RNAseq Profiles among the Experimental and Literature Datasets
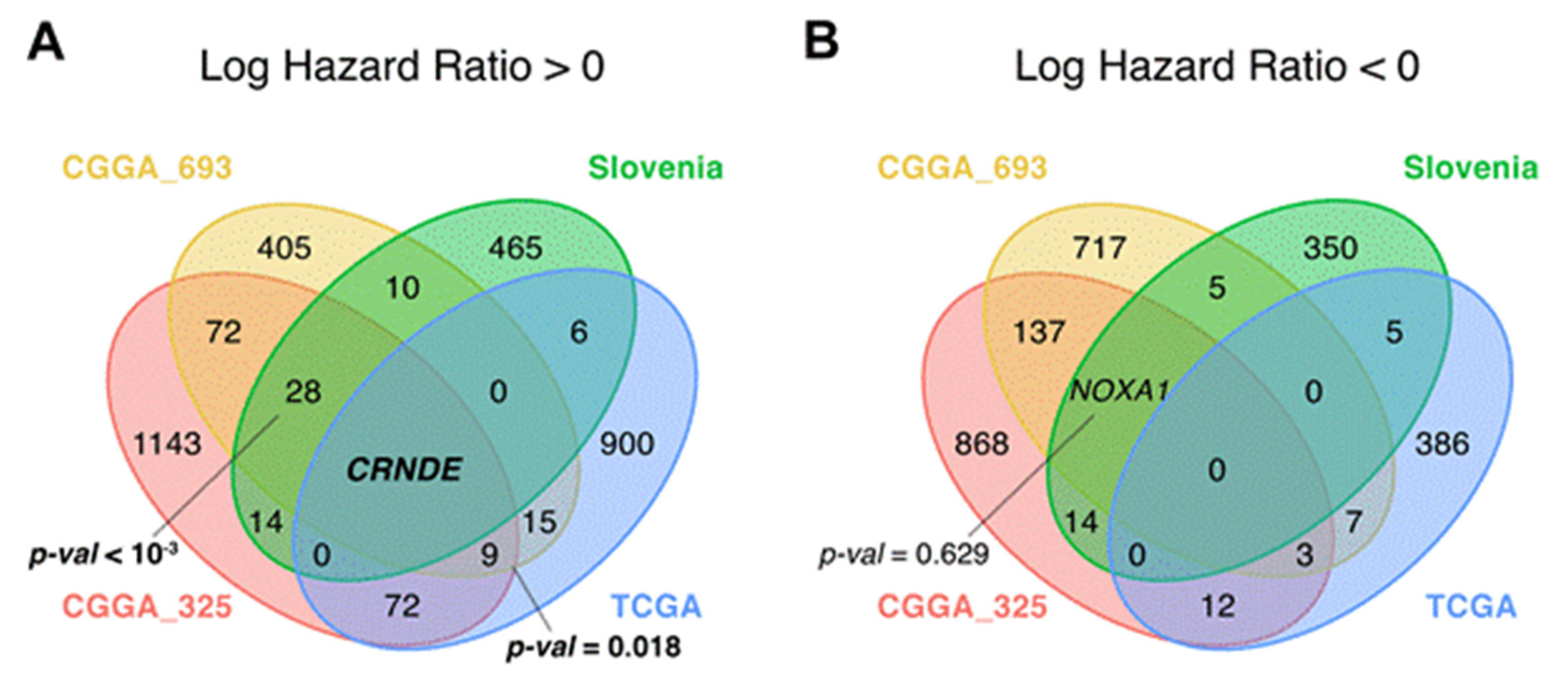
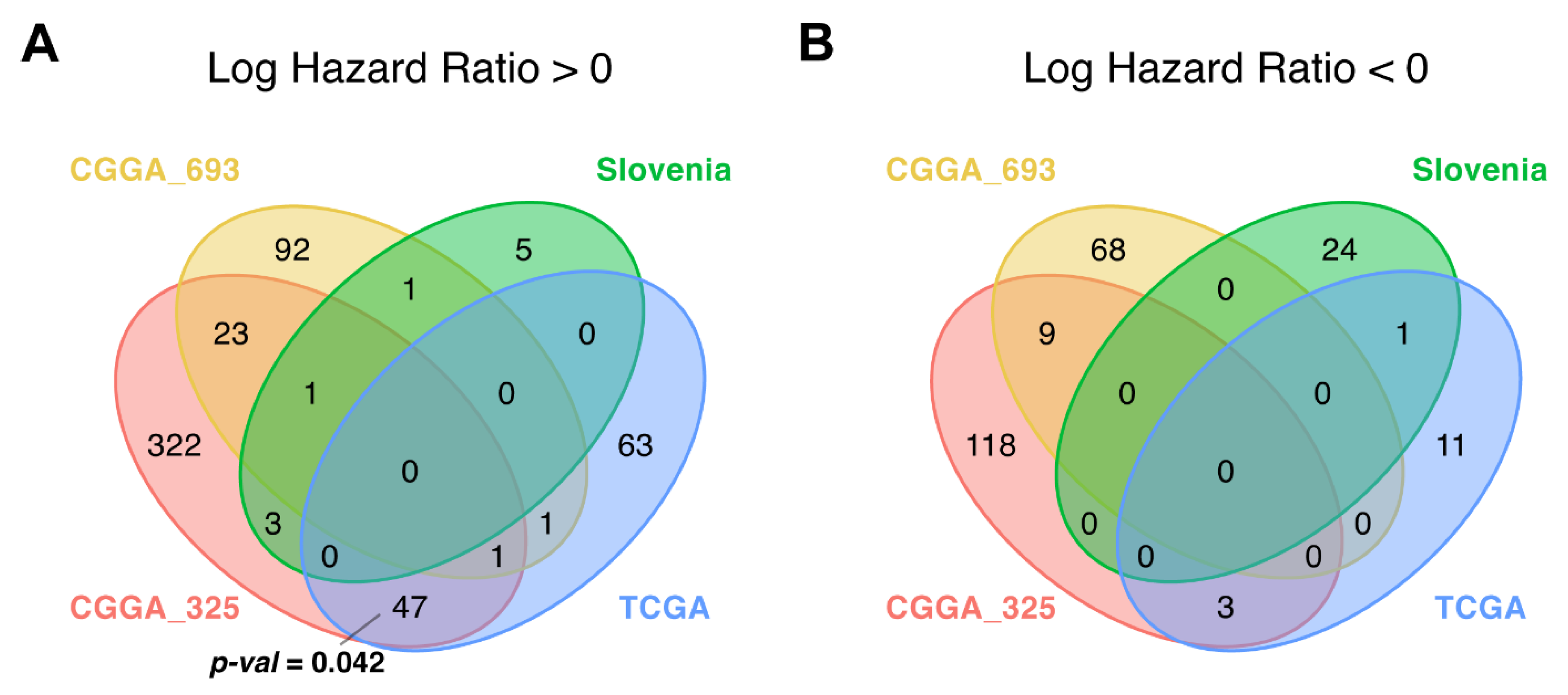
3.3. Survival-Linked Differential Gene Analysis
3.4. Differential Pathway Activation Analysis
3.5. CRNDE Overexpression Is Associated with Poor Patient Overall Survival
4. Discussion
CRNDE
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vecera, M.; Sana, J.; Lipina, R.; Smrcka, M.; Slaby, O. Long Non-Coding RNAs in Gliomas: From Molecular Pathology to Diagnostic Biomarkers and Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Paul, A.; Kellie, S.J.; O’Neill, G.M. Mesenchymal migration as a therapeutic target in glioblastoma. J. Oncol. 2010, 2010, 430142. [Google Scholar] [CrossRef]
- Ho, V.K.; Reijneveld, J.C.; Enting, R.H.; Bienfait, H.P.; Robe, P.; Baumert, B.G.; Visser, O. Dutch Society for, N.-O. Changing incidence and improved survival of gliomas. Eur. J. Cancer 2014, 50, 2309–2318. [Google Scholar] [CrossRef]
- Tykocki, T.; Eltayeb, M. Ten-year survival in glioblastoma. A systematic review. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2018, 54, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.L.; Burkhard, C.; Schuler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [Green Version]
- Olar, A.; Aldape, K.D. Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 2014, 232, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Wrensch, M.; Fisher, J.L.; Schwartzbaum, J.A.; Bondy, M.; Berger, M.; Aldape, K.D. The molecular epidemiology of gliomas in adults. Neurosurg. Focus 2005, 19, E5. [Google Scholar] [CrossRef]
- Nicolaidis, S. Biomarkers of glioblastoma multiforme. Metab. Clin. Exp. 2015, 64, S22–S27. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2009, 16, 748–754. [Google Scholar] [CrossRef]
- Gourlay, J.; Morokoff, A.P.; Luwor, R.B.; Zhu, H.J.; Kaye, A.H.; Stylli, S.S. The emergent role of exosomes in glioma. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2017, 35, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Aoyagi, M.; Wakimoto, H.; Ando, N.; Nariai, T.; Yamamoto, M.; Ohno, K. Accumulation of CD133-positive glioma cells after high-dose irradiation by Gamma Knife surgery plus external beam radiation. J. Neurosurg. 2010, 113, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chakraborty, S.; Yang, C.R.; Hatanpaa, K.J.; Cipher, D.J.; Puliyappadamba, V.T.; Rehman, A.; Jiwani, A.J.; Mickey, B.; Madden, C.; et al. An EGFR wild type-EGFRvIII-HB-EGF feed-forward loop regulates the activation of EGFRvIII. Oncogene 2014, 33, 4253–4264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre-Gregoire, G.; Gavard, J. Spitting out the demons: Extracellular vesicles in glioblastoma. Cell Adhes. Migr. 2017, 11, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Kessler, T.; Sahm, F.; Sadik, A.; Stichel, D.; Hertenstein, A.; Reifenberger, G.; Zacher, A.; Sabel, M.; Tabatabai, G.; Steinbach, J.; et al. Molecular differences in IDH wildtype glioblastoma according to MGMT promoter methylation. Neuro-Oncology 2018, 20, 367–379. [Google Scholar] [CrossRef]
- Westphal, M.; Lamszus, K. Circulating biomarkers for gliomas. Nat. Rev. Neurol. 2015, 11, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Cairncross, G.; Berkey, B.; Shaw, E.; Jenkins, R.; Scheithauer, B.; Brachman, D.; Buckner, J.; Fink, K.; Souhami, L.; Laperierre, N.; et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2006, 24, 2707–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Bent, M.J.; Carpentier, A.F.; Brandes, A.A.; Sanson, M.; Taphoorn, M.J.; Bernsen, H.J.; Frenay, M.; Tijssen, C.C.; Grisold, W.; Sipos, L.; et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: A randomized European Organisation for Research and Treatment of Cancer phase III trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 2715–2722. [Google Scholar] [CrossRef] [Green Version]
- Suntsova, M.; Gaifullin, N.; Allina, D.; Reshetun, A.; Li, X.; Mendeleeva, L.; Surin, V.; Sergeeva, A.; Spirin, P.; Prassolov, V.; et al. Atlas of RNA sequencing profiles for normal human tissues. Sci. Data 2019, 6, 36. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas. Available online: https://portal.gdc.cancer.gov/repository (accessed on 2 December 2020).
- Chinese Glioma Genome Atlas. Available online: http://cgga.org.cn (accessed on 2 December 2020).
- Zhao, Y.; Wong, L.; Goh, W.W.B. How to do quantile normalization correctly for gene expression data analyses. Sci. Rep. 2020, 10, 15534. [Google Scholar] [CrossRef]
- Borisov, N.; Sorokin, M.; Garazha, A.; Buzdin, A. Quantitation of Molecular Pathway Activation Using RNA Sequencing Data. Methods Mol. Biol. 2020, 2063, 189–206. [Google Scholar] [CrossRef]
- Sorokin, M.; Borisov, N.; Kuzmin, D.; Gudkov, A.; Zolotovskaia, M.; Garazha, A.; Buzdin, A. Algorithmic Annotation of Functional Roles for Components of 3,044 Human Molecular Pathways. Front. Genet. 2021, 12, 617059. [Google Scholar] [CrossRef]
- Therneau, T.M. Survival Analysis [R Package Survival Version 3.2-11]; 2021. [Google Scholar]
- Drawing Survival Curves using “ggplot2” [R Package Survminer Version 0.4.9]; 2021.
- Dusa, A. Draw Venn Diagrams [R Package Venn Version 1.10]; 2021. [Google Scholar]
- Sorokin, M.; Ignatev, K.; Barbara, V.; Vladimirova, U.; Muraveva, A.; Suntsova, M.; Gaifullin, N.; Vorotnikov, I.; Kamashev, D.; Bondarenko, A.; et al. Molecular Pathway Activation Markers Are Associated with Efficacy of Trastuzumab Therapy in Metastatic HER2-Positive Breast Cancer Better than Individual Gene Expression Levels. Biochemistry 2020, 85, 758–772. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. Available online: https://bioconductor.org/packages/release/bioc/vignettes/EnhancedVol-cano/inst/doc/EnhancedVolcano.html#references (accessed on 19 March 2021).
- GitHub Repository. Available online: https://github.com/raevskymichail/CRNDE_glioblastoma/tree/v0.1 (accessed on 1 July 2021).
- Buzdin, A.; Sorokin, M.; Garazha, A.; Sekacheva, M.; Kim, E.; Zhukov, N.; Wang, Y.; Li, X.; Kar, S.; Hartmann, C.; et al. Molecular pathway activation—New type of biomarkers for tumor morphology and personalized selection of target drugs. Semin. Cancer Biol. 2018, 53, 110–124. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Seals, S.; Aban, I. Survival analysis and regression models. J. Nucl. Cardiol. 2014, 21, 686–694. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Mukhopadhyay, K. Survival analysis in clinical trials: Basics and must know areas. Perspect. Clin. Res. 2011, 2, 145–148. [Google Scholar] [CrossRef]
- Spruance, S.L.; Reid, J.E.; Grace, M.; Samore, M. Hazard ratio in clinical trials. Antimicrob. Agents Chemother. 2004, 48, 2787–2792. [Google Scholar] [CrossRef] [Green Version]
- Guan, Q.; Chen, R.; Yan, H.; Cai, H.; Guo, Y.; Li, M.; Li, X.; Tong, M.; Ao, L.; Li, H.; et al. Differential expression analysis for individual cancer samples based on robust within-sample relative gene expression orderings across multiple profiling platforms. Oncotarget 2016, 7, 68909–68920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Sha, H.; Sun, X.; Zhang, Y.; Wu, Y.; Zhang, J.; Zhang, H.; Wu, J.; Feng, J. CRNDE: An oncogenic long non-coding RNA in cancers. Cancer Cell Int. 2020, 20, 162. [Google Scholar] [CrossRef]
- Zhang, J.; Yin, M.; Peng, G.; Zhao, Y. CRNDE: An important oncogenic long non-coding RNA in human cancers. Cell Prolif. 2018, 51, e12440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Li, X.D.; Wang, P.; Liu, X.B.; Xue, Y.X.; Hu, Y.; Li, Z.; Li, Z.Q.; Wang, Z.H.; Liu, Y.H. CRNDE affects the malignant biological characteristics of human glioma stem cells by negatively regulating miR-186. Oncotarget 2015, 6, 25339–25355. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, Q.; Zhao, Y.; Hou, R. lncRNA CRNDE is Upregulated in Glioblastoma Multiforme and Facilitates Cancer Progression Through Targeting miR-337-3p and ELMOD2 Axis. OncoTargets Ther. 2020, 13, 9225–9234. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Graham, L.D.; Pedersen, S.K.; Brown, G.S.; Ho, T.; Kassir, Z.; Moynihan, A.T.; Vizgoft, E.K.; Dunne, R.; Pimlott, L.; Young, G.P.; et al. Colorectal Neoplasia Differentially Expressed (CRNDE), a Novel Gene with Elevated Expression in Colorectal Adenomas and Adenocarcinomas. Genes Cancer 2011, 2, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Ellis, B.C.; Molloy, P.L.; Graham, L.D. CRNDE: A Long Non-Coding RNA Involved in CanceR, Neurobiology, and DEvelopment. Front. Genet. 2012, 3, 270. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Guan, G.; Li, X.; Wei, C.; Wu, J.; Cheng, P.; Wu, A.; Cheng, W. Profiling pro-neural to mesenchymal transition identifies a lncRNA signature in glioma. J. Transl. Med. 2020, 18, 378. [Google Scholar] [CrossRef]
- Katsushima, K.; Jallo, G.; Eberhart, C.G.; Perera, R.J. Long non-coding RNAs in brain tumors. NAR Cancer 2021, 3. [Google Scholar] [CrossRef]
- Moorcraft, S.Y.; Gonzalez, D.; Walker, B.A. Understanding next generation sequencing in oncology: A guide for oncologists. Crit. Rev. Oncol. Hematol. 2015, 96, 463–474. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.F.; Wang, Z.B.; Zhu, T.; Mao, C.X.; Mao, X.Y.; Li, L.; Yin, J.Y.; Zhou, H.H.; Liu, Z.Q. A critical overview of long non-coding RNA in glioma etiology 2016: An update. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 14403–14413. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, S.A.; Bassett, S.D.; Kaczorowski, D.; Blackburn, J.; Barton, K.; Bartonicek, N.; Carswell, S.L.; Tilgner, H.U.; Loy, C.; Halliday, G.; et al. Targeted, High-Resolution RNA Sequencing of Non-coding Genomic Regions Associated with Neuropsychiatric Functions. Front. Genet. 2019, 10, 309. [Google Scholar] [CrossRef] [Green Version]
- Rusconi, F.; Battaglioli, E.; Venturin, M. Psychiatric Disorders and lncRNAs: A Synaptic Match. Int. J. Mol. Sci. 2020, 21, 3030. [Google Scholar] [CrossRef]
- Zeng, T.; Li, L.; Zhou, Y.; Gao, L. Exploring Long Noncoding RNAs in Glioblastoma: Regulatory Mechanisms and Clinical Potentials. Int. J. Genom. 2018, 2018, 2895958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiang, K.M.; Zhang, X.Q.; Leung, G.K. Long Non-Coding RNAs: The Key Players in Glioma Pathogenesis. Cancers 2015, 7, 1406–1424. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Qian, J.; Wang, Y.Y.; Zhang, J.X.; You, Y.P. Long noncoding RNA profiles reveal three molecular subtypes in glioma. CNS Neurosci. Ther. 2014, 20, 339–343. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Sun, S.; Lam, K.F.; Kiang, K.M.; Pu, J.K.; Ho, A.S.; Lui, W.M.; Fung, C.F.; Wong, T.S.; Leung, G.K. A long non-coding RNA signature in glioblastoma multiforme predicts survival. Neurobiol. Dis. 2013, 58, 123–131. [Google Scholar] [CrossRef]
- Han, L.; Zhang, K.; Shi, Z.; Zhang, J.; Zhu, J.; Zhu, S.; Zhang, A.; Jia, Z.; Wang, G.; Yu, S.; et al. LncRNA pro fi le of glioblastoma reveals the potential role of lncRNAs in contributing to glioblastoma pathogenesis. Int. J. Oncol. 2012, 40, 2004–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, T.F.; Greiner, A.; Guibourt, V.; Lisec, K.; Kretzschmar, H.A. Identification of Stably Expressed lncRNAs as Valid Endogenous Controls for Profiling of Human Glioma. J. Cancer 2015, 6, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wu, J.J.; Lin, X.B.; Bao, Y.; Chen, Z.H.; Zhang, C.R.; Cai, Z.; Zhou, J.Y.; Ding, M.H.; Wu, X.J.; et al. Differential lncRNA expression profiles in recurrent gliomas compared with primary gliomas identified by microarray analysis. Int. J. Clin. Exp. Med. 2015, 8, 5033–5043. [Google Scholar] [PubMed]
- Chen, W.; Xu, X.K.; Li, J.L.; Kong, K.K.; Li, H.; Chen, C.; He, J.; Wang, F.; Li, P.; Ge, X.S.; et al. MALAT1 is a prognostic factor in glioblastoma multiforme and induces chemoresistance to temozolomide through suppressing miR-203 and promoting thymidylate synthase expression. Oncotarget 2017, 8, 22783–22799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Yuan, X.; Yan, D.; Li, D.; Guan, F.; Dong, Y.; Wang, H.; Liu, X.; Yang, B. Long Non-Coding RNA MALAT1 Decreases the Sensitivity of Resistant Glioblastoma Cell Lines to Temozolomide. Cell Physiol. Biochem. 2017, 42, 1192–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, T.; Liu, Y.; Xiao, J. Long noncoding RNA MALAT1 knockdown reverses chemoresistance to temozolomide via promoting microRNA-101 in glioblastoma. Cancer Med. 2018, 7, 1404–1415. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Tian, Y.; Chen, Y.; Zhang, G. The silencing of LncRNA-H19 decreases chemoresistance of human glioma cells to temozolomide by suppressing epithelial-mesenchymal transition via the Wnt/beta-Catenin pathway. OncoTargets Ther. 2018, 11, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Wang, P.; Sun, X.; Yuan, Z.; Zhan, R.; Ma, X.; Li, W. Knockdown of long noncoding RNA H19 sensitizes human glioma cells to temozolomide therapy. OncoTargets Ther. 2016, 9, 3501–3509. [Google Scholar] [CrossRef] [Green Version]
- Barry, G. Integrating the roles of long and small non-coding RNA in brain function and disease. Mol. Psychiatry 2014, 19, 410–416. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, S.; Pu, J.K.; Tsang, A.C.; Lee, D.; Man, V.O.; Lui, W.M.; Wong, S.T.; Leung, G.K. Long non-coding RNA expression profiles predict clinical phenotypes in glioma. Neurobiol. Dis. 2012, 48, 1–8. [Google Scholar] [CrossRef]
- Kiang, K.M.Y.; Leung, G.K.K. Clinical significance of CRNDE transcript variants in glioblastoma multiforme. Noncoding RNA Res. 2017, 2, 119–121. [Google Scholar] [CrossRef] [Green Version]
- Kiang, K.M.; Zhang, X.Q.; Zhang, G.P.; Li, N.; Cheng, S.Y.; Poon, M.W.; Pu, J.K.; Lui, W.M.; Leung, G.K. CRNDE Expression Positively Correlates with EGFR Activation and Modulates Glioma Cell Growth. Target. Oncol. 2017, 12, 353–363. [Google Scholar] [CrossRef]
- Jing, S.Y.; Lu, Y.Y.; Yang, J.K.; Deng, W.Y.; Zhou, Q.; Jiao, B.H. Expression of long non-coding RNA CRNDE in glioma and its correlation with tumor progression and patient survival. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3992–3996. [Google Scholar]
- Zheng, J.; Liu, X.; Wang, P.; Xue, Y.; Ma, J.; Qu, C.; Liu, Y. CRNDE Promotes Malignant Progression of Glioma by Attenuating miR-384/PIWIL4/STAT3 Axis. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1199–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.X.; Fei, X.R.; Dong, Y.F.; Cheng, C.D.; Yang, Y.; Deng, X.F.; Huang, H.L.; Niu, W.X.; Zhou, C.X.; Xia, C.Y.; et al. The long non-coding RNA CRNDE acts as a ceRNA and promotes glioma malignancy by preventing miR-136-5p-mediated downregulation of Bcl-2 and Wnt2. Oncotarget 2017, 8, 88163–88178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, Y.; Li, J.; Zhang, Y.; Yin, H.; Han, B. CRNDE, a long-noncoding RNA, promotes glioma cell growth and invasion through mTOR signaling. Cancer Lett. 2015, 367, 122–128. [Google Scholar] [CrossRef]
- Li, H.; Li, Q.; Guo, T.; He, W.; Dong, C.; Wang, Y. LncRNA CRNDE triggers inflammation through the TLR3-NF-kappaB-Cytokine signaling pathway. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317703821. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Gender | Age at Diagnosis | KPS | WHO Grade | Recurrent Tumor | OS (Months) | IDH1 R132H Mutation | ATRX Loss | p53 | MGMT Methylation | GTR | Therapy | Adjuvant Therapy | TTP (Months) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NB-00131/12 | M | 42 | 90 | IV | No | 13 | Negative | Not performed | Negative | No | No | 60 Gy radiotherapy + TMZ | Adjuvant TMZ | / |
| NB-00094/13 | M | 47 | 90 | IV | No | 16 | Negative | Not performed | Positive | Yes | Yes | 60 Gy radiotherapy + TMZ | Adjuvant TMZ | 10 |
| 17-H-21518 | M | 64 | 100 | IV | No | 16 | Negative | No | Negative | No | Yes | 60 Gy radiotherapy + TMZ | Adjuvant TMZ | 8 |
| NB-00233/12 | F | 42 | 100 | IV | No | 18 | Negative | Not performed | Negative | No | Yes | 60 Gy radiotherapy + TMZ | No (TMZ side effects) | 6 |
| NB-00464/12 | M | 62 | 90 | IV | No | 19 | Not performed | Not performed | Not performed | No | No | 30 Gy radiotherapy + TMZ | Adjuvant TMZ | 14 |
| NB-00079/14 | M | 43 | 90 | IV | No | 20 | Positive | Not performed | Positive | No | Yes | 60 Gy radiotherapy + TMZ | Adjuvant TMZ | 15 |
| 17-H-05191 | M | 68 | 100 | IV | No | 21 | Negative | No | Positive | No | Yes | 60 Gy radiotherapy + TMZ | Adjuvant TMZ | 12 |
| 16-H-17976 | M | 66 | 70 | IV | No | 23 | Negative | No | Negative | Yes | Yes | 60 Gy radiotherapy + TMZ | Adjuvant TMZ | 16 |
| NB-00173/14 | M | 55 | 90 | IV | No | 26 | Negative | Not performed | Negative | Yes | Yes | 60 Gy radiotherapy + TMZ | Adjuvant TMZ | 21 |
| NB-00369/13 | F | 62 | 90 | IV | No | 31 | Negative | Not performed | Positive | No | Yes | 60 Gy radiotherapy + TMZ | Adjuvant TMZ | 26 |
| NB-00450/12 | F | 58 | 80 | IV | No | 50 | Negative | Not performed | Negative | Yes | No | 60 Gy radiotherapy + TMZ | Adjuvant TMZ | 37 |
| NB-00038/13 | F | 61 | 80 | IV | No | 58 | Negative | Not performed | Positive | No | Yes | 60 Gy radiotherapy + TMZ | Adjuvant TMZ | 14 |
| NB-00339/13 | M | 22 | 100 | IV | No | 91 | Negative | Not performed | Negative | No | Yes | 60 Gy radiotherapy + TMZ | Adjuvant TMZ | 90 * |
| NB-00046/12 | M | 62 | 80 | IV | No | 2 | Negative | Not performed | Negative | Yes | No | 34 Gy radiotherapy + TMZ | No | 2 |
| NB-00003/15 | F | 68 | 70 | IV | No | 2 | Negative | Yes | Negative | No | No | 30 Gy radiotherapy | / | 2 |
| 17-H-31914 | M | 70 | 50 | IV | No | 2 | Negative | No | Negative | Yes | No | None | None | 2 |
| Sample ID | Library ID | Uniquely Mapped Reads | Total Reads |
|---|---|---|---|
| NB-00369/13 | GB_1 | 6.57 | 38.16 |
| 17-H-21518 | GB_2 | 6.56 | 32.52 |
| NB-00464/12 | GB_3 | 11.09 | 36.74 |
| NB-00079/14 | GB_4 | 5.85 | 28.14 |
| 17-H-05191 | GB_5 | 7.18 | 38.79 |
| 16-H-17976 | GB_6 | 6.19 | 34.93 |
| NB-00173/14 | GB_7 | 5.80 | 38.31 |
| NB-00131/12 | GB_8 | 10.55 | 38.99 |
| NB-00450/12 | GB_10 | 8.36 | 28.62 |
| NB-00450/12 | GB_11 | 5.84 | 26.36 |
| NB-00233/12 | GB_12 | 6.97 | 30.42 |
| NB-00003/15 | GB_16 | 5.53 | 28.93 |
| 17-H-31914 | GB_17 | 7.10 | 28.03 |
| NB-00094/13 | GB_18 | 8.52 | 28.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorokin, M.; Raevskiy, M.; Zottel, A.; Šamec, N.; Skoblar Vidmar, M.; Matjašič, A.; Zupan, A.; Mlakar, J.; Suntsova, M.; Kuzmin, D.V.; et al. Large-Scale Transcriptomics-Driven Approach Revealed Overexpression of CRNDE as a Poor Survival Prognosis Biomarker in Glioblastoma. Cancers 2021, 13, 3419. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143419
Sorokin M, Raevskiy M, Zottel A, Šamec N, Skoblar Vidmar M, Matjašič A, Zupan A, Mlakar J, Suntsova M, Kuzmin DV, et al. Large-Scale Transcriptomics-Driven Approach Revealed Overexpression of CRNDE as a Poor Survival Prognosis Biomarker in Glioblastoma. Cancers. 2021; 13(14):3419. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143419
Chicago/Turabian StyleSorokin, Maxim, Mikhail Raevskiy, Alja Zottel, Neja Šamec, Marija Skoblar Vidmar, Alenka Matjašič, Andrej Zupan, Jernej Mlakar, Maria Suntsova, Denis V. Kuzmin, and et al. 2021. "Large-Scale Transcriptomics-Driven Approach Revealed Overexpression of CRNDE as a Poor Survival Prognosis Biomarker in Glioblastoma" Cancers 13, no. 14: 3419. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143419