
SARAF and EFHB Modulate Store-Operated Ca2+ Entry and Are Required for Cell Proliferation, Migration and Viability in Breast Cancer Cells

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
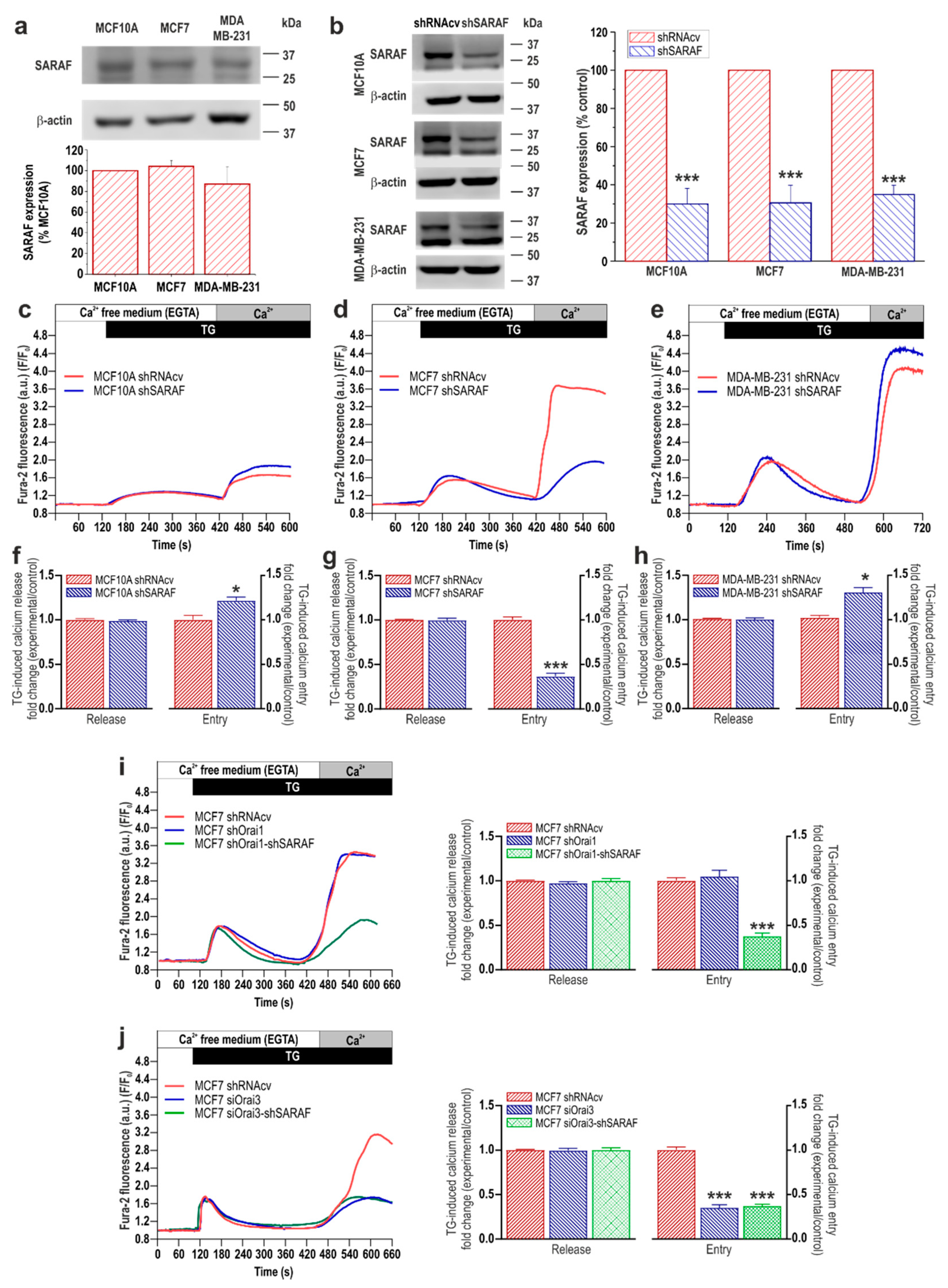
2.1. Expression of EFHB and SARAF, and Functional Role in SOCE, in Breast Cancer and Pre-Neoplastic Epithelial Cells
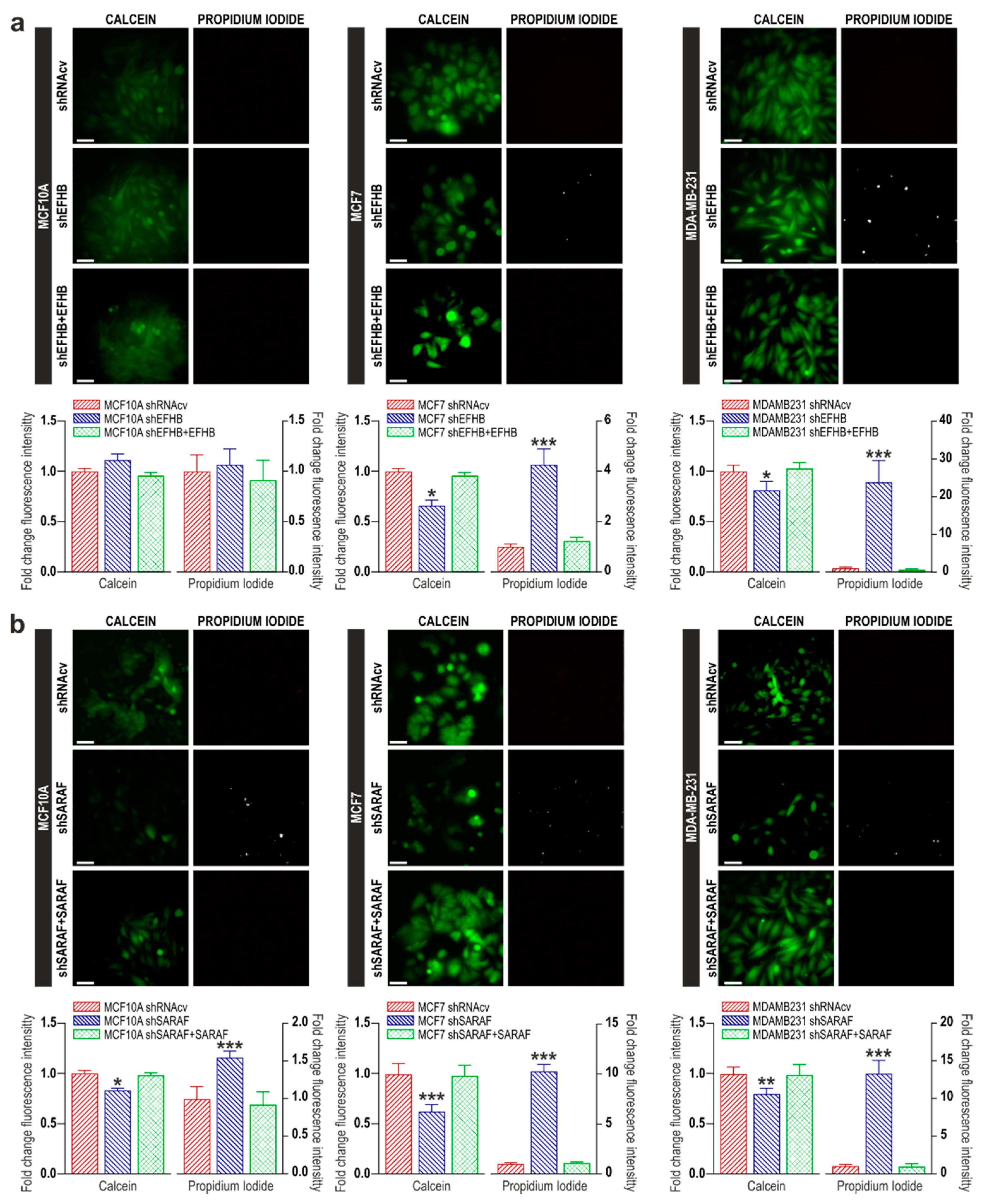
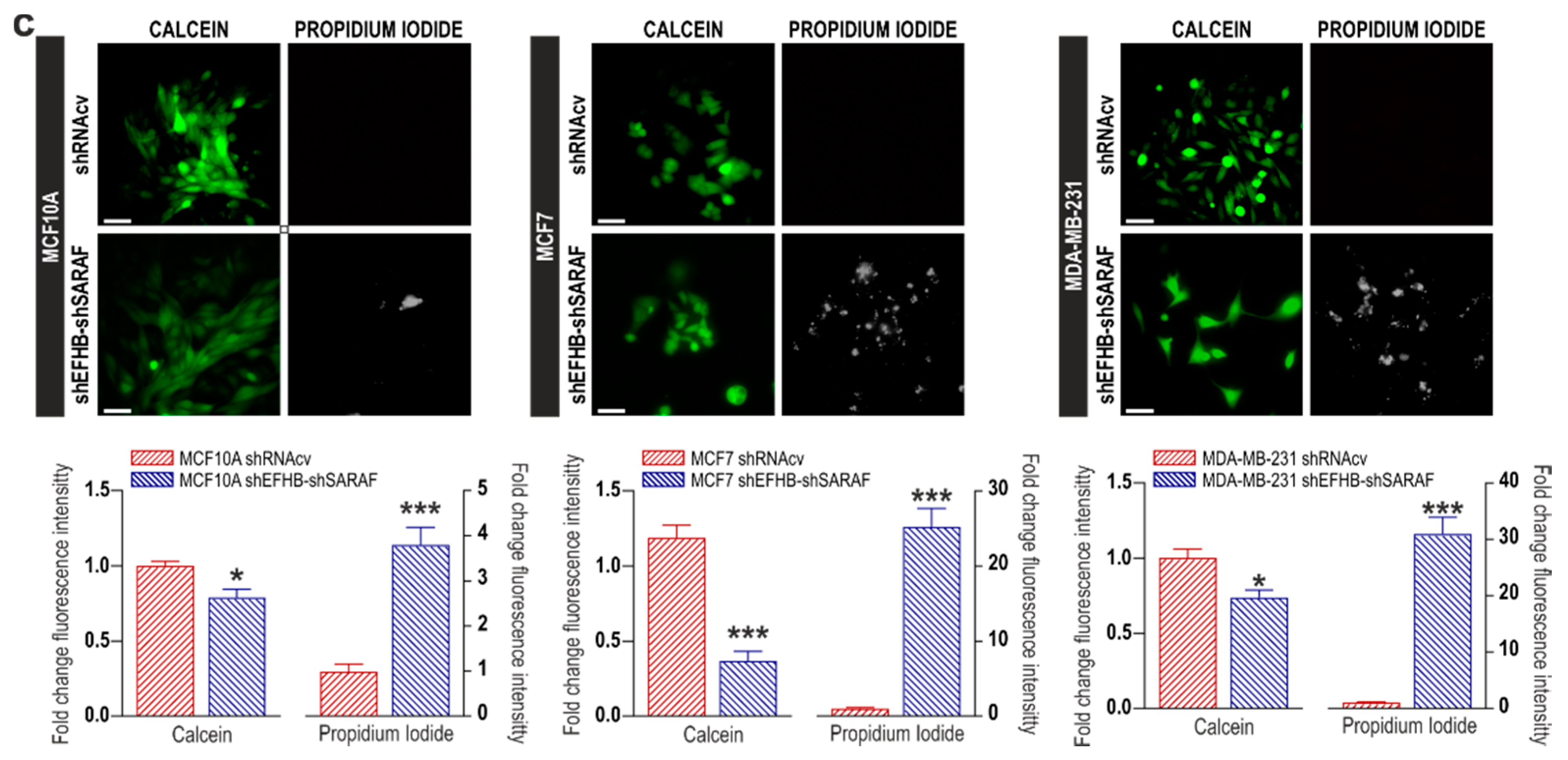
2.2. EFHB and SARAF Are Required for Cell Viability in Breast Cancer Cells
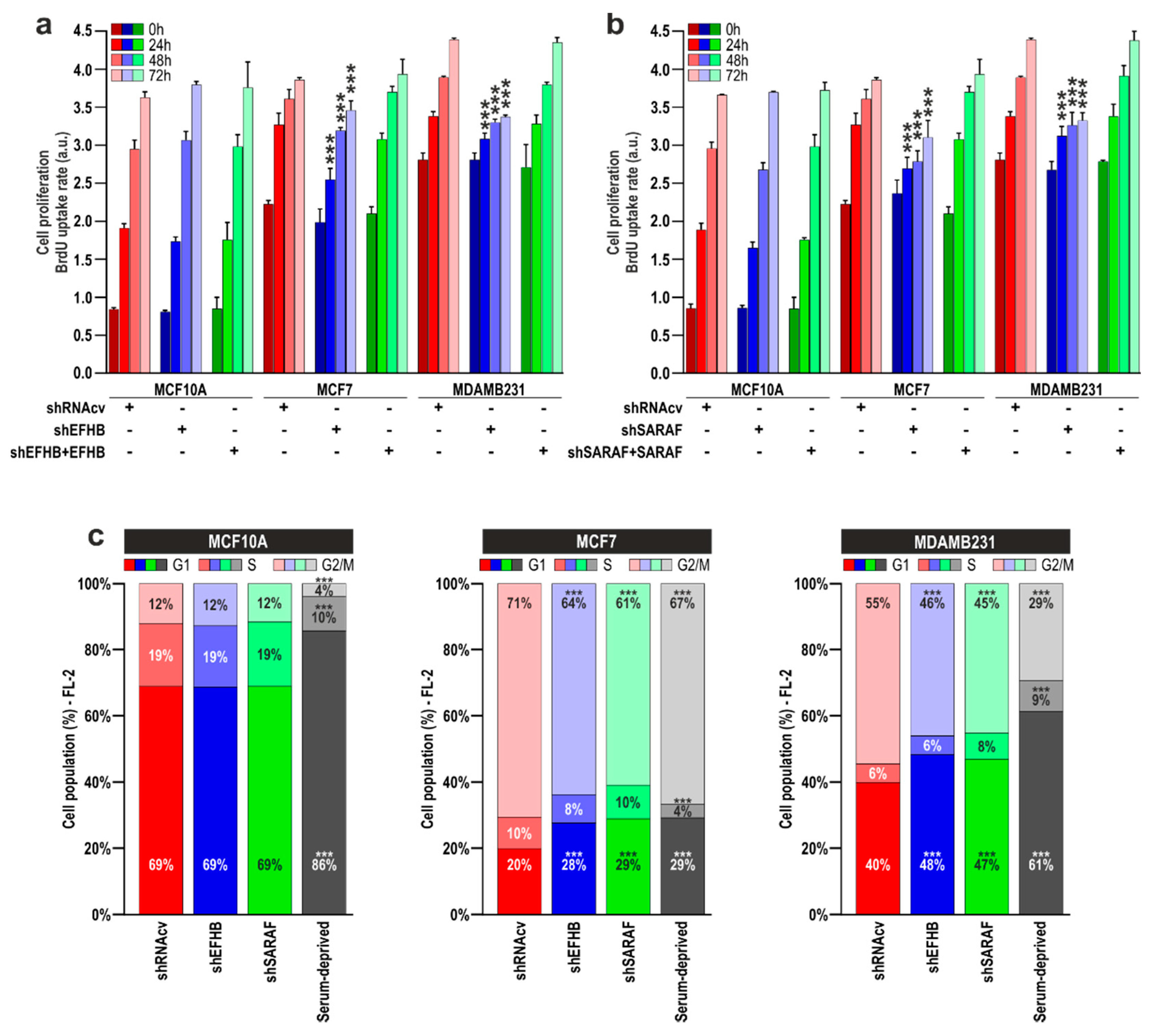
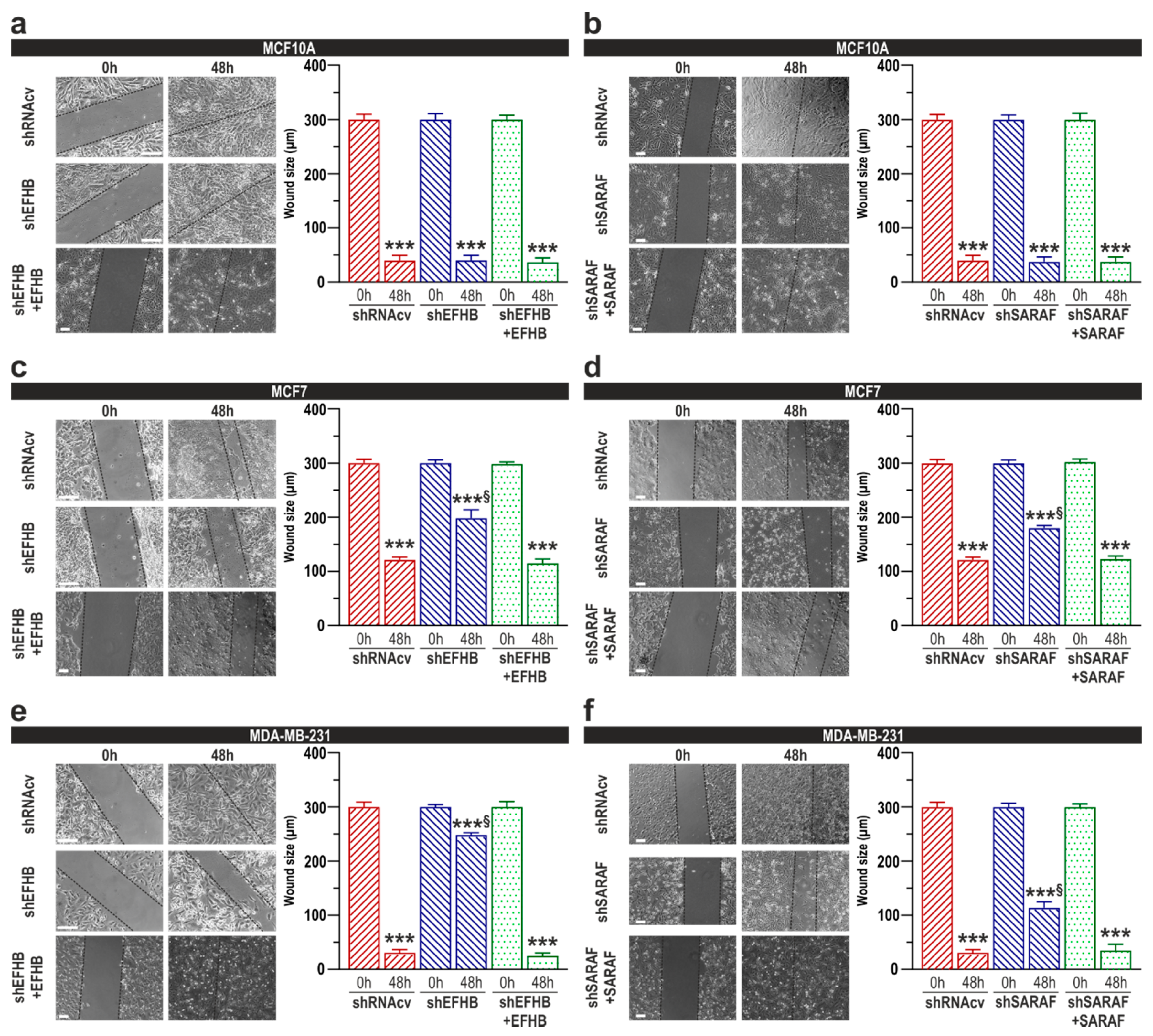
2.3. EFHB and SARAF Are Required for Proliferation and Migration in Breast Cancer Cells but Not in Pre-Neoplastic Epithelial Cells
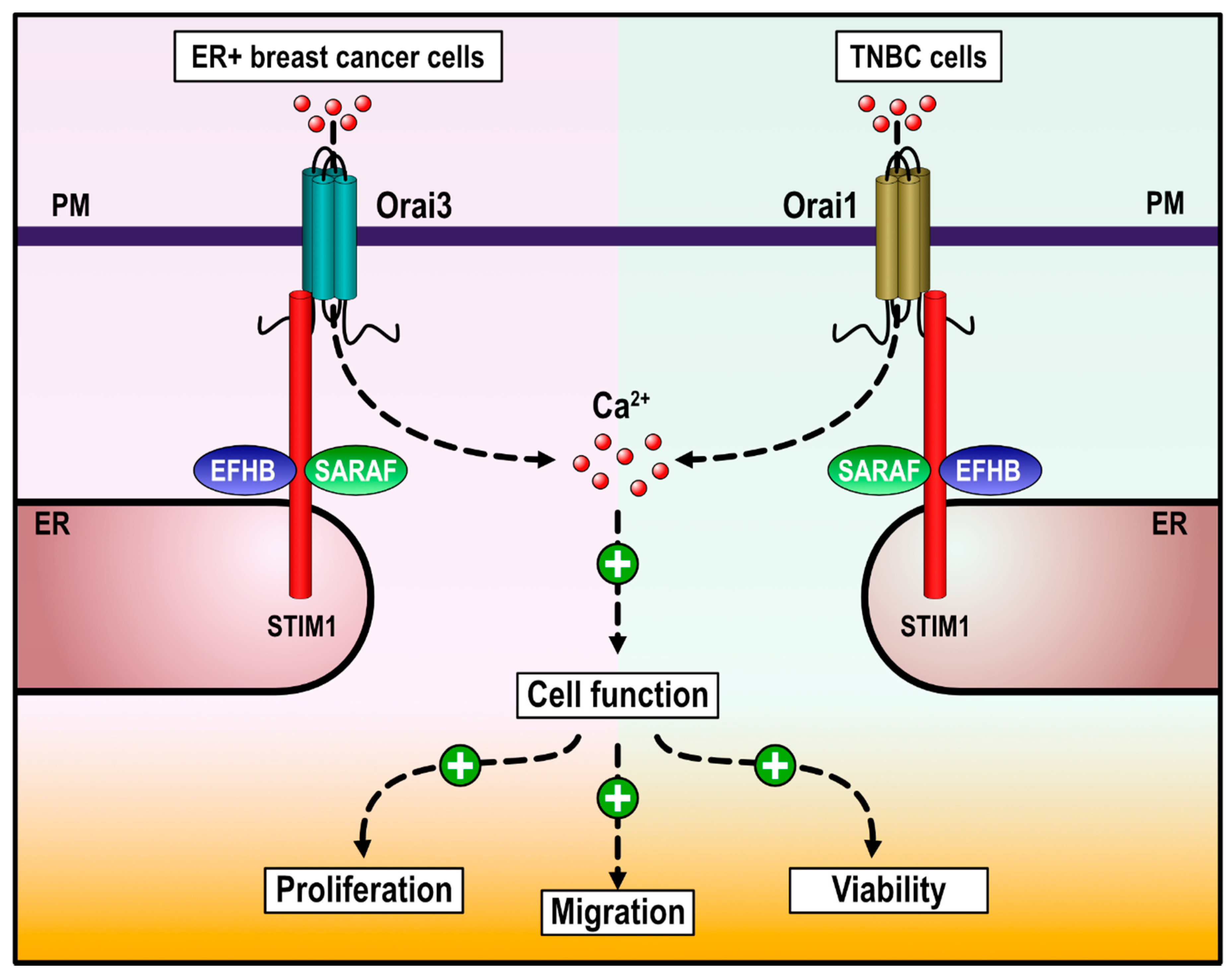
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Cell Culture and Transfections
4.3. Determination of Cytosolic Free-Ca2+ Concentration
4.4. Western Blotting
4.5. Cell Proliferation
4.6. Cell Viability Assay
4.7. Wound-Healing Assay
4.8. Determination of Cell Cycle by Flow Cytometry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kappel, S.; Borgstrom, A.; Stoklosa, P.; Dorr, K.; Peinelt, C. Store-operated calcium entry in disease: Beyond STIM/Orai expression levels. Semin. Cell. Dev. Biol. 2019, 94, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Smani, T.; Shapovalov, G.; Skryma, R.; Prevarskaya, N.; Rosado, J.A. Functional and physiopathological implications of TRP channels. Biochim. Biophys. Acta 2015, 1853, 1772–1782. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoast, R.E.; Emrich, S.M.; Zhang, X.; Xin, P.; Johnson, M.T.; Fike, A.J.; Walter, V.; Hempel, N.; Yule, D.I.; Sneyd, J.; et al. The native ORAI channel trio underlies the diversity of Ca2+ signaling events. Nat. Commun. 2020, 11, 2444. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell. Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berna-Erro, A.; Jardin, I.; Salido, G.M.; Rosado, J.A. Role of STIM2 in cell function and physiopathology. J. Physiol. 2017, 595, 3111–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [Green Version]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers 2018, 10, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 2012, 149, 425–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, A.; Ahuja, M.; Maleth, J.; Moreno, C.M.; Yuan, J.P.; Kim, M.S.; Muallem, S. The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J. Cell. Biol. 2013, 202, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albarran, L.; Lopez, J.J.; Woodard, G.E.; Salido, G.M.; Rosado, J.A. Store-operated Ca2+ entry-associated regulatory factor (SARAF) plays an important role in the regulation of arachidonate-regulated Ca2+ (ARC) channels. J. Biol. Chem. 2016, 291, 6982–6988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albarran, L.; Lopez, J.J.; Ben Amor, N.; Martín-Cano, F.E.; Berna-Erro, A.; Smani, T.; Salido, G.M.; Rosado, J.A. Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store-operated calcium entry. Sci. Rep. 2016, 6, 24452. [Google Scholar] [CrossRef] [PubMed]
- Albarran, L.; Lopez, J.J.; Gomez, L.J.; Salido, G.M.; Rosado, J.A. SARAF modulates TRPC1, but not TRPC6, channel function in a STIM1-independent manner. Biochem. J. 2016, 473, 3581–3595. [Google Scholar] [CrossRef] [PubMed]
- Galeano-Otero, I.; Del Toro, R.; Khatib, A.M.; Rosado, J.A.; Ordonez-Fernandez, A.; Smani, T. SARAF and Orai1 Contribute to Endothelial Cell Activation and Angiogenesis. Front. Cell. Dev. Biol. 2021, 9, 639952. [Google Scholar] [CrossRef] [PubMed]
- Albarran, L.; Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Berna-Erro, A.; Smani, T.; Camello, P.J.; Salido, G.M.; Rosado, J.A. EFHB is a Novel Cytosolic Ca2+ Sensor That Modulates STIM1-SARAF Interaction. Cell. Physiol. Biochem. 2018, 51, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Bong, A.H.; Poo, G.X.H.; Armitage, K.; Lok, D.; Roberts-Thomson, S.J.; Monteith, G.R. Pharmacological inhibition of store-operated calcium entry in MDA-MB-468 basal A breast cancer cells: Consequences on calcium signalling, cell migration and proliferation. Cell. Mol. Life Sci. 2018, 75, 4525–4537. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Diez-Bello, R.; Falcon, D.; Alvarado, S.; Regodon, S.; Salido, G.M.; Smani, T.; Rosado, J.A. Melatonin downregulates TRPC6, impairing store-operated calcium entry in triple negative breast cancer cells. J. Biol. Chem. 2020, 296, 100254. [Google Scholar] [CrossRef]
- Gueder, N.; Allan, G.; Telliez, M.S.; Hague, F.; Fernandez, J.M.; Sanchez-Fernandez, E.M.; Ortiz-Mellet, C.; Ahidouch, A.; Ouadid-Ahidouch, H. sp(2) -Iminosugar alpha-glucosidase inhibitor 1-C-octyl-2-oxa-3-oxocastanospermine specifically affected breast cancer cell migration through Stim1, beta1-integrin, and FAK signaling pathways. J. Cell. Physiol. 2017, 232, 3631–3640. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Collado, J.; Lopez, J.J.; Jardin, I.; Camello, P.J.; Falcon, D.; Regodon, S.; Salido, G.M.; Smani, T.; Rosado, J.A. Adenylyl Cyclase Type 8 Overexpression Impairs Phosphorylation-Dependent Orai1 Inactivation and Promotes Migration in MDA-MB-231 Breast Cancer Cells. Cancers 2019, 11, 1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.J.; Siegfried, G.; Cantonero, C.; Soulet, F.; Descarpentrie, J.; Smani, T.; Badiola, I.; Pernot, S.; Evrard, S.; Rosado, J.A.; et al. Furin Prodomain ppFurin Enhances Ca2+ Entry Through Orai and TRPC6 Channels’ Activation in Breast Cancer Cells. Cancers 2021, 13, 1670. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.P.; Elmi, A.; Alcantara-Adap, E.; Hubrack, S.; Nader, N.; Yu, F.; Dib, M.; Ramachandran, V.; Najafi Shoushtari, H.; Machaca, K. miRNA-dependent regulation of STIM1 expression in breast cancer. Sci. Rep. 2019, 9, 13076. [Google Scholar] [CrossRef]
- Jardin, I.; Albarran, L.; Salido, G.M.; Lopez, J.J.; Sage, S.O.; Rosado, J.A. Fine-tuning of store-operated calcium entry by fast and slow Ca2+-dependent inactivation: Involvement of SARAF. Biochim. Biophys. Acta. Mol. Cell. Res. 2018, 1865, 463–469. [Google Scholar] [CrossRef]
- Lopez, E.; Frischauf, I.; Jardin, I.; Derler, I.; Muik, M.; Cantonero, C.; Salido, G.M.; Smani, T.; Rosado, J.A.; Redondo, P.C. STIM1 phosphorylation at Y(316) modulates its interaction with SARAF and the activation of SOCE and I CRAC. J. Cell. Sci. 2019, 132, jcs226019. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Collado, J.; Lopez, J.J.; Gonzalez-Gutierrez, L.; Cantonero, C.; Jardin, I.; Salido, G.M.; Rosado, J.A. Functional role of TRPC6 and STIM2 in cytosolic and endoplasmic reticulum Ca2+ content in resting estrogen receptor-positive breast cancer cells. Biochem. J. 2020, 477, 3183–3197. [Google Scholar] [CrossRef] [PubMed]
- Albarran, L.; Regodon, S.; Salido, G.M.; Lopez, J.J.; Rosado, J.A. Role of STIM1 in the surface expression of SARAF. Channels 2016, 13, 1670. [Google Scholar] [CrossRef]
- Diez-Bello, R.; Jardin, I.; Lopez, J.J.; El Haouari, M.; Ortega-Vidal, J.; Altarejos, J.; Salido, G.M.; Salido, S.; Rosado, J.A. (–)-Oleocanthal inhibits proliferation and migration by modulating Ca2+ entry through TRPC6 in breast cancer cells. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 474–485. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jardin, I.; Nieto-Felipe, J.; Alvarado, S.; Diez-Bello, R.; Lopez, J.J.; Salido, G.M.; Smani, T.; Rosado, J.A. SARAF and EFHB Modulate Store-Operated Ca2+ Entry and Are Required for Cell Proliferation, Migration and Viability in Breast Cancer Cells. Cancers 2021, 13, 4160. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13164160
Jardin I, Nieto-Felipe J, Alvarado S, Diez-Bello R, Lopez JJ, Salido GM, Smani T, Rosado JA. SARAF and EFHB Modulate Store-Operated Ca2+ Entry and Are Required for Cell Proliferation, Migration and Viability in Breast Cancer Cells. Cancers. 2021; 13(16):4160. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13164160
Chicago/Turabian StyleJardin, Isaac, Joel Nieto-Felipe, Sandra Alvarado, Raquel Diez-Bello, Jose J. Lopez, Ginés M. Salido, Tarik Smani, and Juan A. Rosado. 2021. "SARAF and EFHB Modulate Store-Operated Ca2+ Entry and Are Required for Cell Proliferation, Migration and Viability in Breast Cancer Cells" Cancers 13, no. 16: 4160. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13164160