
Primary Mediastinal and Testicular Germ Cell Tumors in Adolescents and Adults: A Comparison of Genomic Alterations and Clinical Implications

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
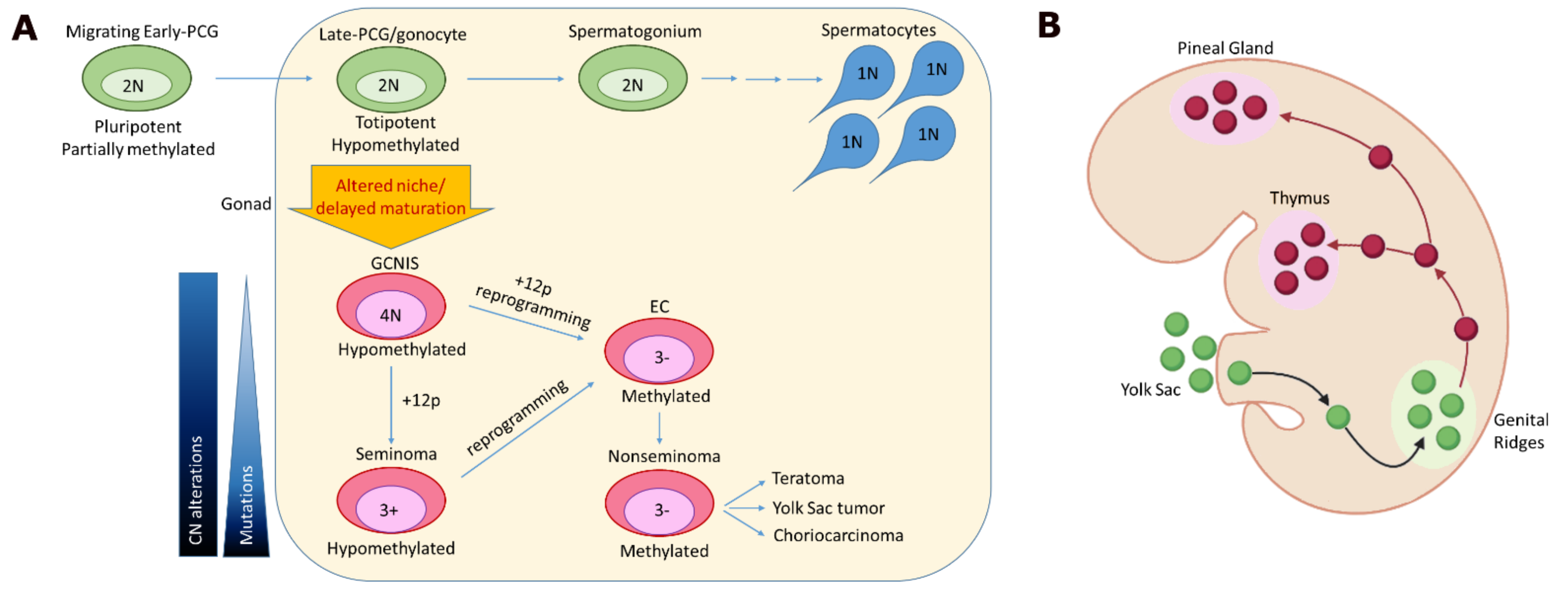
2. Origins
3. Comparison between MGCTs and TGCTs Molecular Alterations
4. MGCT: Clinico-Pathological Features and Treatment
5. MGCT and Concomitant Neoplasms
6. Final Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Rusner, C.; Trabert, B.; Katalinic, A.; Kieschke, J.; Emrich, K.; Stang, A. Network of German Cancer Registries (GEKID). In-cidence patterns and trends of malignant gonadal and extragonadal germ cell tumors in Germany, 1998–2008. Cancer Epidemiol. 2013, 37, 370–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, M.; Zehender, M.; Thalmann, G.N. Extragonadal retroperitoneal germ cell tumor: Evidence of origin in the testis. Ann. Oncol. 2002, 13, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Oldenburg, J.; Dunant, A.; Chen, I.; Salvioni, R.; Hartmann, J.T.; De Santis, M.; Daugaard, G.; Flechon, A.; de Giorgi, U.; et al. Assessing prognosis and optimizing treatment in patients with postchemotherapy viable nonseminomatous germ-cell tumors (NSGCT): Results of the sCR2 international study. Ann. Oncol. 2007, 19, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Mashaal, H.; Anjum, F. Mediastinal Germ Cell Tumors. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Mishra, S.; Das Majumdar, S.K.; Sable, M.; Parida, D.K. Primary malignant mediastinal germ cell tumors: A single institutional experience. S. Asian J. Cancer 2020, 09, 27–29. [Google Scholar] [CrossRef]
- Gillessen, S.; Sauvé, N.; Collette, L.; Daugaard, G.; de Wit, R.; Albany, C.; Tryakin, A.; Fizazi, K.; Stahl, O.; Gietema, J.A.; et al. Predicting Outcomes in Men With Metastatic Nonseminomatous Germ Cell Tumors (NSGCT): Results from the IGCCCG Update Consortium. J. Clin. Oncol. 2021, 39, 1563–1574. [Google Scholar] [CrossRef]
- Oosterhuis, J.W.; Looijenga, L.H.J. Human germ cell tumours from a developmental perspective. Nat. Rev. Cancer 2019, 19, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, J.; Terrasa, J.; Durán, I.; Germà-Lluch, J.R.; Gironés, R.; González-Billalabeitia, E.; Gumà, J.; Maroto, P.; Pinto, A.; García-Del-Muro, X. SEOM clinical guidelines for the management of germ cell testicular cancer (2016). Clin. Transl. Oncol. 2016, 18, 1187–1196. [Google Scholar] [CrossRef]
- Fizazi, K.; Culine, S.; Droz, J.P.; Kramar, A.; Théodore, C.; Ruffié, P.; Le Chevalier, T. Primary mediastinal nonseminomatous germ cell tumors: Results of modern therapy including cisplatin-based chemotherapy. J. Clin. Oncol. 1998, 16, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Skakkebæk, N.E.; Berthelsen, J.G.; Giwercman, A.; Müller, J. Carcinoma-in-situ of the testis: Possible origin from gonocytes and precursor of all types of germ cell tumours except spermatocytoma. Int. J. Androl. 1987, 10, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Rijlaarsdam, M.; Tax, D.M.J.; Gillis, A.J.M.; Dorssers, L.C.J.; Koestler, D..C.; de Ridder, J.; Looijenga, L.H.J. Genome Wide DNA Methylation Profiles Provide Clues to the Origin and Pathogenesis of Germ Cell Tumors. PLoS ONE 2015, 10, e0122146. [Google Scholar] [CrossRef] [Green Version]
- Looijenga, L.H.; de Munnik, H.; Oosterhuis, J.W. A molecular model for the development of germ cell cancer. Int. J. Cancer 1999, 83, 809–814. [Google Scholar] [CrossRef]
- Dorssers, L.C.J.; Gillis, A.J.M.; Stoop, H.; van Marion, R.; Nieboer, M.M.; van Riet, J.; van de Werken, H.J.G.; Oosterhuis, J.W.; de Ridder, J.; Looijenga, L.H.J. Molecular heterogeneity and early metastatic clone selection in testicular germ cell cancer development. Br. J. Cancer 2019, 120, 444–452. [Google Scholar] [CrossRef] [Green Version]
- Kier, M.G.G.; Lauritsen, J.; Almstrup, K.; Mortensen, M.S.; Toft, B.G.; Rajpert-De Meyts, E.; Skakkebaek, N.E.; Rørth, M.; von der Maase, H.; Agerbaek, M.; et al. Screening for carcinoma in situ in the contralateral testicle in patients with testicular cancer: A population-based study. Ann. Oncol. 2015, 26, 737–742. [Google Scholar] [CrossRef]
- Honecker, F.; Stoop, H.; Mayer, F.; Bokemeyer, C.; Castrillon, D.H.; Lau, Y.-F.C.; Looijenga, L.; Oosterhuis, J.W. Germ cell lineage differentiation in non-seminomatous germ cell tumours. J. Pathol. 2005, 208, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef]
- Cheng, M.L.; Donoghue, M.T.; Audenet, F.; Wong, N.C.; Pietzak, E.J.; Bielski, C.M.; Isharwal, S.; Iyer, G.; Funt, S.; Bagrodia, A.; et al. Germ Cell Tumor Molecular Heterogeneity Revealed Through Analysis of Primary and Metastasis Pairs. JCO Precis. Oncol. 2020, 4, 1307–1320. [Google Scholar] [CrossRef]
- Chaganti, R.; Rodriguez, E.; Mathew, S. Origin of adult male mediastinal germ-cell tumours. Lancet 1994, 343, 1130–1132. [Google Scholar] [CrossRef]
- Hasle, H.; Mellemgaard, A.; Nielsen, J.; Hansen, J. Cancer incidence in men with Klinefelter syndrome. Br. J. Cancer 1995, 71, 416–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, M.H.; Mai, P.L.; Loud, J.T.; Pathak, A.; Peters, J.A.; Mirabello, L.; McMaster, M.L.; Rosenberg, P.; Stewart, D.R. Familial testicular germ cell tumors (FTGCT)—Overview of a multidisciplinary etiologic study. Andrology 2014, 3, 47–58. [Google Scholar] [CrossRef]
- Looijenga, L.H.; Hersmus, R.; Oosterhuis, J.W.; Cools, M.; Drop, S.L.; Wolffenbuttel, K.P. Tumor risk in disorders of sex development (DSD). Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 480–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litchfield, K.; Levy, M.; Orlando, G.; Loveday, C.; Law, P.J.; Migliorini, G.; Holroyd, A.; Broderick, P.; Karlsson, R.; Haugen, T.B.; et al. Identification of 19 new risk loci and potential regulatory mechanisms influencing susceptibility to testicular germ cell tumor. Nat. Genet. 2017, 49, 1133–1140. [Google Scholar] [CrossRef]
- Wang, Z.; A McGlynn, K.; Meyts, E.R.-D.; Bishop, D.T.; Chung, C.C.; Dalgaard, M.D.; Greene, M.H.; Gupta, R.; Grotmol, T.; Haugen, T.B.; et al. Meta-analysis of five genome-wide association studies identifies multiple new loci associated with testicular germ cell tumor. Nat. Genet. 2017, 49, 1141–1147. [Google Scholar] [CrossRef]
- Aldubayan, S.H.; Pyle, L.C.; Gamulin, M.; Kulis, T.; Moore, N.D.; Taylor-Weiner, A.; Hamid, A.A.; Reardon, B.; Wubbenhorst, B.; Godse, R.; et al. Association of Inherited Pathogenic Variants in Checkpoint Kinase 2 (CHEK2) With Susceptibility to Testicular Germ Cell Tumors. JAMA Oncol. 2019, 5, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Main, K.M. Testicular dysgenesis syndrome: An increasingly common developmental disorder with environmental aspects. Hum. Reprod. 2001, 16, 972–978. [Google Scholar] [CrossRef]
- Rodriguez, E.; Mathew, S.; Reuter, V.; Ilson, D.H.; Bosl, G.J.; Chaganti, R.S. Cytogenetic analysis of 124 prospectively ascer-tained malegerm cell tumors. Cancer Res. 1992, 52, 2285–2291. [Google Scholar]
- Bussey, K.J.; Lawce, H.J.; Olson, S.B.; Arthur, D.C.; Kalousek, D.K.; Krailo, M.; Giller, R.; Heifetz, S.; Womer, R.; Magenis, R.E. Chromo-some abnormalities of eighty-one pediatric germ cell tumors: Sex-, age-, site-, and histopathology-related differences—A children’s cancer group study. Genes Chromosomes Cancer 1999, 25, 134–146. [Google Scholar] [CrossRef]
- Kao, C.-S.; Bangs, C.D.; Aldrete, G.; Cherry, A.M.; Ulbright, T.M. A Clinicopathologic and Molecular Analysis of 34 Mediastinal Germ Cell Tumors Suggesting Different Modes of Teratoma Development. Am. J. Surg. Pathol. 2018, 42, 1662–1673. [Google Scholar] [CrossRef]
- Schneider, D.T.; Schuster, A.E.; Fritsch, M.K.; Hu, J.; Olson, T.; Lauer, S.; Göbel, U.; Perlman, E.J. Multipoint imprinting analysis indicates a common precursor cell for gonadal and nongonadal pediatric germ cell tumors. Cancer Res. 2001, 61, 7268–7276. [Google Scholar]
- Schneider, D.T.; Schuster, A.E.; Fritsch, M.K.; Calaminus, G.; Göbel, U.; Harms, D.; Lauer, S.; Olson, T.; Perlman, E. Genetic analysis of mediastinal nonseminomatous germ cell tumors in children and adolescents. Genes Chromosom. Cancer 2002, 34, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Sperger, J.M.; Chen, X.; Draper, J.S.; Antosiewicz, J.E.; Chon, C.H.; Jones, S.B.; Brooks, J.D.; Andrews, P.W.; Brown, P.O.; Thomson, J.A. Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 13350–13355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, S.; Jafer, O.; Goker, H.; Summersgill, B.M.; Zafarana, G.; Gillis, A.J.M.; Van Gurp, R.J.H.L.M.; Oosterhuis, J.W.; Lu, Y.-J.; Huddart, R.; et al. Expression profile of genes from 12p in testicular germ cell tumors of adolescents and adults associated with i(12p) and amplification at 12p11.2–p12.1. Oncogene 2003, 22, 1880–1891. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Weiner, A.; Zack, A.T.-W.T.; O’Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.H.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 2016, 540, 114–118. [Google Scholar] [CrossRef]
- Loveday, C.; Litchfield, K.; Proszek, P.Z.; Cornish, A.J.; Santo, F.; Levy, M.; MacIntyre, G.; Holryod, A.; Broderick, P.; Dudakia, D.; et al. Genomic landscape of platinum resistant and sensitive testicular cancers. Nat. Commun. 2020, 11, 2189. [Google Scholar] [CrossRef]
- Litchfield, K.; Summersgill, B.; Yost, S.; Sultana, R.; LaBreche, K.; Dudakia, D.; Renwick, A.; Seal, S.; Al-Saadi, R.; Broderick, P.; et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat. Commun. 2015, 6, 5973. [Google Scholar] [CrossRef]
- McIntyre, A.; Summersgill, B.; Spendlove, H.E.; Huddart, R.; Houlston, R.; Shipley, J. Activating Mutations and/or Expression Levels of Tyrosine Kinase Receptors GRB7, RAS, and BRAF in Testicular Germ Cell Tumors. Neoplasia 2005, 7, 1047–1052. [Google Scholar] [CrossRef] [Green Version]
- Bagrodia, A.; Lee, B.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients With Advanced Germ Cell Tumors. J. Clin. Oncol. 2016, 34, 4000–4007. [Google Scholar] [CrossRef]
- Feldman, D.R.; Iyer, G.; Van Alstine, L.; Patil, S.; Al-Ahmadie, H.; Reuter, V.E.; Bosl, G.; Chaganti, R.S.; Solit, D.B. Presence of Somatic Mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in Cisplatin-Resistant Germ Cell Tumors. Clin. Cancer Res. 2014, 20, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Przygodzki, R.M.; Hubbs, A.E.; Zhao, F.-Q.; O’Leary, T.J. Primary mediastinal seminomas: Evidence of single and multiple KIT mutations. Lab. Investig. 2002, 82, 1369–1375. [Google Scholar] [CrossRef] [Green Version]
- Necchi, A.; Bratslavsky, G.; Chung, J.; Millis, S.; Gay, L.M.; Ali, S.M.; Ross, J.S. Genomic Features for Therapeutic Insights of Chemotherapy-Resistant, Primary Mediastinal Nonseminomatous Germ Cell Tumors and Comparison with Gonadal Counterpart. Oncologist 2018, 24, e142–e145. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 world health organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [Green Version]
- Rosti, G.; Secondino, S.; Necchi, A.; Fornarini, G.; Pedrazzoli, P. Primary mediastinal germ cell tumors. Semin. Oncol. 2019, 46, 107–111. [Google Scholar] [CrossRef]
- De Giorgi, U.; Richard, S.; Badoglio, M.; Kanfer, E.; Bourrhis, J.H.; Nicolas-Virelizier, E.; Vettenranta, K.; Lioure, B.; Martin, S.; Dreger, P.; et al. Salvage high-dose chemotherapy in female patients with relapsed/refractory germ-cell tumors: A retrospective analysis of the European Group for Blood and Marrow Transplantation (EBMT). Ann. Oncol. 2017, 28, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Domínguez Malagón, H.; Pérez Montiel, D. Mediastinal germ cell tumors. Semin. Diagn. Pathol. 2005, 22, 230–240. [Google Scholar] [CrossRef]
- Drevelegas, A.; Palladas, P.; Scordalaki, A. Mediastinal germ cell tumors: A radiologic–pathologic review. Eur. Radiol. 2001, 11, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Beyer, J.; Collette, L.; Sauvé, N.; Daugaard, G.; Feldman, D.R.; Tandstad, T.; Tryakin, A.; Stahl, O.; Gonzalez-Billalabeitia, E.; De Giorgi, U.; et al. International germ cell cancer classification update consortium. survival and new prognosticators in metastatic seminoma: Results from the IGCCCG-update consortium. J. Clin. Oncol. 2021, 39, 1553–1562. [Google Scholar] [CrossRef]
- van Dijk, M.R.; Steyerberg, E.W.; Habbema, J.D. Survival of non-seminomatous germ cell cancer patients according to the IGCC classification: An update based on meta-analysis. Eur. J. Cancer 2006, 42, 820–826. [Google Scholar] [CrossRef]
- De Giorgi, U.; Rosti, G.; Salvioni, R.; Papiani, G.; Ballardini, M.; Pizzocaro, G.; Marangolo, M. Long-term outcome of salvage high-dose chemotherapy in patients with germ cell tumor with poor prognostic features. Urol. Oncol. Semin. Orig. Investig. 2011, 29, 284–290. [Google Scholar] [CrossRef]
- Einhorn, L.H.; Williams, S.D.; Chamness, A.; Brames, M.J.; Perkins, S.M.; Abonour, R. High-dose chemotherapy and stem-cell rescue for metastatic germ-cell tumors. N. Engl. J. Med. 2007, 357, 340–348. [Google Scholar] [CrossRef]
- Kopf, B.; De Giorgi, U.; Vertogen, B.; Kopf, B.; De Giorgi, U.; Vertogen, B.; Monti, G.; Molinari, A.; Turci, D.; Dazzi, C.; et al. A randomized study comparing filgrastim versus lenograstim versus molgramostim plus chemotherapy for peripheral blood progenitor cell mobilization. Bone Marrow Transplant. 2006, 38, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Prognostic Factors Study Group; Lorch, A.; Beyer, J.; Bascoul-Mollevi, C.; Kramar, A.; Einhorn, L.H.; Necchi, A.; Massard, C.; De Giorgi, U.; Fléchon, A.; et al. Prognostic factors in patients with metastatic germ cell tumors who experienced treatment failure with cisplatin-based first-line chemotherapy. J. Clin. Oncol. 2010, 28, 4906–4911. [Google Scholar] [PubMed]
- Bokemeyer, C.; Schleucher, N.; Metzner, B.; Thomas, M.S.C.; Rick, O.; Schmoll, H.-J.; Kollmannsberger, C.; Boehlke, I.; Kanz, L.; Hartmann, J.T. First-line sequential high-dose VIP chemotherapy with autologous transplantation for patients with primary mediastinal nonseminomatous germ cell tumours: A prospective trial. Br. J. Cancer 2003, 89, 29–35. [Google Scholar] [CrossRef]
- Vuky, J.; Bains, M.; Bacik, J.; Higgins, G.; Bajorin, D.F.; Mazumdar, M.; Bosl, G.J.; Motzer, R.J. Role of postchemotherapy adjunctive surgery in the management of patients with nonseminoma arising from the mediastinum. J. Clin. Oncol. 2001, 19, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Einhorn, L.; Nichols, C.R.; Droz, J.P.; Horwich, A.; Gerl, A.; Fossa, S.D.; Beyer, J.; Pont, J.; Schmoll, H.J.; et al. Second-line chemotherapy in patients with relapsed extragonadal nonseminomatous germ cell tumors: Results of an international multicenter analysis. J. Clin. Oncol. 2001, 19, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.H.; Feldman, D.R.; Bosl, G.J.; Motzer, R.J. A review of second-line chemotherapy and prognostic models for disseminated germ cell tumors. Hematol. Oncol. Clin. N. Am. 2011, 25, 557–576. [Google Scholar] [CrossRef] [Green Version]
- Lorch, A.; Bascoul-Mollevi, C.; Kramar, A. Conventional-dose versus high-dose chemotherapy as first salvage treatment in male patients with metastatic germ cell tumors: Evidence from a large international database. J. Clin. Oncol. 2011, 29, 2178–2184. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, U.; Demirer, T.; Wandt, H.; Taverna, C.; Siegert, W.; Bornhauser, M.; Kozak, T.; Papiani, G.; Ballardini, M.; Rosti, G. Second-line high-dose chemotherapy in patients with mediastinal and retroperitoneal primary non-seminomatous germ cell tumors: The EBMT experience. Ann. Oncol. 2005, 16, 146–151. [Google Scholar] [CrossRef]
- De Giorgi, U.; Rosti, G.; Aieta, M.; Testore, F.; Burattini, L.; Fornarini, G.; Naglieri, E.; Re, G.L.; Zumaglini, F.; Marangolo, M. Phase II Study of Oxaliplatin and Gemcitabine Salvage Chemotherapy in Patients with Cisplatin-Refractory Nonseminomatous Germ Cell Tumor. Eur. Urol. 2006, 50, 1032–1039. [Google Scholar] [CrossRef]
- Adra, N.; Abonour, R.; Althouse, S.K.; Albany, C.; Hanna, N.H.; Einhorn, L.H. High-dose chemotherapy and autologous peripheral-blood stem-cell transplantation for relapsed metastatic germ cell tumors: The indiana university experience. J. Clin. Oncol. 2017, 35, 1096–1102. [Google Scholar] [CrossRef]
- Cursano, M.C.; Kopf, B.; Scarpi, E.; Menna, C.; Casadei, C.; Schepisi, G.; Lolli, C.; Altavilla, A.; Gallà, V.; Santini, D.; et al. Prognostic Role of Systemic Inflammatory Indexes in Germ Cell Tumors Treated With High-Dose Chemotherapy. Front. Oncol. 2020, 10, 1325. [Google Scholar] [CrossRef] [PubMed]
- Chovanec, M.; Cierna, Z.; Miskovska, V.; Machalekova, K.; Kalavska, K.; Rejlekova, K.; Svetlovska, D.; Macak, D.; Spanik, S.; Kajo, K.; et al. Systemic immune-inflammation index in germ-cell tumours. Br. J. Cancer 2018, 118, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fankhauser, C.D.; Sander, S.; Roth, L.; Gross, O.; Eberli, D.; Sulser, T.; Seifert, B.; Beyer, J.; Hermanns, T. Systemic inflammatory markers have independent prognostic value in patients with metastatic testicular germ cell tumours undergoing first-line chemotherapy. Br. J. Cancer 2018, 118, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Bolat, D.; Aydoğdu, Ö.; Polat, S.; Yarımoğlu, S.; Bozkurt, İ.H.; Yonguç, T.; Şen, V. Predictive value of preoperative neutrophil-to-lymphocyte ratio on the prognosis of germ cell testicular tumors. Turk. J. Urol. 2017, 43, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Jankovich, M.; Jankovichova, T.; Ondrus, D.; Breza, J. Neutrophil-to-lymphocyte ratio as a predictor of preoperative tumor staging in testicular germ cell tumors. Bratisl. Med. J. 2017, 118, 510–512. [Google Scholar] [CrossRef]
- Hartmann, J.T.; Nichols, C.R.; Droz, J.; Horwich, A.; Gerl, A.; Fossa, S.D.; Beyer, J.; Pont, J.; Einhorn, L.; Kanz, L.; et al. The relative risk of second nongerminal malignancies in patients with extragonadal germ cell tumors. Cancer 2000, 88, 2629–2635. [Google Scholar] [CrossRef]
- Nichols, C.R.; Hoffman, R.; Einhorn, L.H.; Williams, S.D.; Wheeler, L.A.; Garnick, M.B. Hematologic malignancies associated with primary mediastinal germ cell tumors. Ann. Intern. Med. 1985, 102, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Ibrahimi, S.; John, S.; Adnan, M.M.; Scordino, T.; Khalil, M.O.; Cherry, M. Non-seminomatous mediastinal germ cell tumor and acute megakaryoblastic leukemia. Ann. Hematol. 2017, 96, 1435–1439. [Google Scholar] [CrossRef]
- Orazi, A.; Neiman, R.S.; Ulbright, T.M.; Heerema, N.A.; John, K.; Nichols, C.R. Hematopoietic precursor cells within the yolk sac tumor component are the source of secondary hematopoietic malignancies in patients with mediastinal germ cell tumors. Cancer 1993, 71, 3873–3881. [Google Scholar] [CrossRef]
- Taylor, J.; Donoghue, M.T.; Ho, C.; Petrova-Drus, K.; Al-Ahmadie, H.A.; Funt, S.A.; Zhang, Y.; Aypar, U.; Rao, P.N.; Chavan, S.S.; et al. Germ cell tumors and associated hematologic malignancies evolve from a common shared precursor. J. Clin. Investig. 2020, 130, 6668–6676. [Google Scholar] [CrossRef]
- Chaganti, R.S.K.; Ladanyi, M.; Samaniego, F.; Offit, K.; Reuter, V.E.; Jhanwar, S.C.; Bosl, G.J. Leukemic differentiation of a mediastinal germ cell tumor. Genes Chromosom. Cancer 1989, 1, 83–87. [Google Scholar] [CrossRef]
- Ladanyi, M.; Samaniego, F.; Reuter, V.E.; Motzer, R.J.; Jhanwar, S.C.; Bosl, G.J.; Chaganti, R.S.K. Cytogenetic and Immunohistochemical Evidence for the Germ Cell Origin of a Subset of Acute Leukemias Associated With Mediastinal Germ Cell Tumors. J. Natl. Cancer Inst. 1990, 82, 221–227. [Google Scholar] [CrossRef]
- Vlasveld, L.; Splinter, T.A.W.; Hagemeijer, A.; Van Lom, K.; Löwenberg, B. Acute myeloid leukaemia with +i(12p) shortly after treatment of mediastinal germ cell tumour. Br. J. Haematol. 1994, 88, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Sowithayasakul, P.; Sinlapamongkolkul, P.; Treetipsatit, J.; Vathana, N.; Narkbunnam, N.; Sanpakit, K.; Buaboonnam, J. Hematologic Malignancies Associated With Mediastinal Germ Cell Tumors: 10 Years’ Experience at Thailand’s National Pediatric Tertiary Referral Center. J. Pediatr. Hematol. 2018, 40, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Riedell, P.; Miller, C.; Hagemann, I.S.; Westervelt, P.; Ozenberger, B.A.; O’Laughlin, M.; Magrini, V.; Demeter, R.T.; Duncavage, E.J.; et al. A common founding clone withTP53andPTENmutations gives rise to a concurrent germ cell tumor and acute megakaryoblastic leukemia. Mol. Case Stud. 2016, 2, a000687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, J.T.; Raess, P.W.; Dunlap, J.; Hayes-Lattin, B.; Tyner, J.W.; Traer, E. Functional and genetic screening of acute myeloid leukemia associated with mediastinal germ cell tumor identifies MEK inhibitor as an active clinical agent. J. Hematol. Oncol. 2016, 9, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Oshrine, B.R.; Olsen, M.N.; Heneghan, M.; Wertheim, G.; Daber, R.; Wilmoth, D.M.; Biegel, J.A.; Pawel, B.; Aplenc, R.; King, R.L. Acquired isochromosome 12p, somatic TP53 and PTEN mutations, and a germline ATM variant in an adolescent male with concurrent acute megakaryoblastic leukemia and mediastinal germ cell tumor. Cancer Genet. 2014, 207, 153–159. [Google Scholar] [CrossRef]
- Akizuki, K.; Sekine, M.; Kogure, Y.; Kameda, T.; Shide, K.; Koya, J.; Kamiunten, A.; Kubuki, Y.; Tahira, Y.; Hidaka, T.; et al. TP53 and PTEN mutations were shared in concurrent germ cell tumor and acute megakaryoblastic leukemia. BMC Cancer 2020, 20, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Amra, N.; Zarate, L.V.; Punia, J.N.; Mahajan, P.; Stevens, A.M.; Roy, A.; Curry, C.V.; Cortes-Santiago, N.; Fisher, K.E. Mediastinal Germ Cell Tumor and Acute Megakaryoblastic Leukemia With Co-occurring KRAS Mutation and Complex Cytogenetics. Pediatr. Dev. Pathol. 2020, 23, 461–466. [Google Scholar] [CrossRef]
- Oosterhuis, J.W.; Looijenga, L.H. Mediastinal germ cell tumors: Many questions and perhaps an answer. J. Clin. Investig. 2020, 130, 6238–6241. [Google Scholar] [CrossRef]
- Nichols, C.R.; Heerema, N.A.; Palmer, C.; Loehrer, P.J.; Williams, S.D.; Einhorn, L.H. Klinefelter’s syndrome associated with mediastinal germ cell neoplasms. J. Clin. Oncol. 1987, 5, 1290–1294. [Google Scholar] [CrossRef]
- Levy, D.R.; Agaram, N.P.; Kao, C.-S.; Franks, S.E.; Kesler, K.A.; Stram, A.R.; Einhorn, L.H.; Bangs, C.D.; Ulbright, T.M. Vasculogenic Mesenchymal Tumor: A Clinicopathologic and Molecular Study of 55 Cases of a Distinctive Neoplasm Originating From Mediastinal Yolk Sac Tumor and an Occasional Precursor to Angiosarcoma. Am. J. Surg. Pathol. 2020, 45, 463–476. [Google Scholar] [CrossRef]
- Oing, C.; Kollmannsberger, C.; Oechsle, K.; Bokemeyer, C. Investigational targeted therapies for the treatment of testicular germ cell tumors. Expert Opin. Investig. Drugs 2016, 25, 1033–1043. [Google Scholar] [CrossRef]
- Mego, M.; Svetlovska, D.; Chovanec, M.; Rečkova, M.; Rejlekova, K.; Obertova, J.; Palacka, P.; Sycova-Mila, Z.; De Giorgi, U.; Mardiak, J. Phase II study of avelumab in multiple relapsed/refractory germ cell cancer. Investig. New Drugs 2019, 37, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Adra, N.; Einhorn, L.H.; Althouse, S.K.; Ammakkanavar, N.R.; Musapatika, D.; Albany, C.; Vaughn, D.; Hanna, N.H. Phase II trial of pembrolizumab in patients with platinum refractory germ-cell tumors: A hoosier cancer research network study GU14-206. Ann. Oncol. 2018, 29, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Ward, J.E.; Bao, R.; Hall, C.R.; Brockstein, B.E.; Luke, J.J. Clinical response of a patient to anti-PD-1 immunotherapy and the immune landscape of testicular germ cell tumors. Cancer Immunol. Res. 2016, 4, 903–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bono, J.S.; Scher, H.I.; Montgomery, R.B.; Parker, C.; Miller, M.C.; Tissing, H.; Doyle, G.V.; Terstappen, L.W.; Pienta, K.J.; Raghavan, D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate. Clin. Cancer Res. 2008, 14, 6302–6309. [Google Scholar]
- Nicolazzo, C.; Busetto, G.M.; Gradilone, A.; Sperduti, I.; Del Giudice, F.; Loreni, F.; Cortesi, E.; De Berardinis, E.; Gazzaniga, P.; Raimondi, C. Circulating Tumor Cells Identify Patients with Super-High-Risk Non-Muscle-Invasive Bladder Cancer: Updated Outcome Analysis of a Prospective Single-Center Trial. Oncologist 2019, 24, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Nastaly, P.; Ruf, C.; Becker, P.; Bednarz-Knoll, N.; Stoupiec, M.; Kavsur, R.; Isbarn, H.; Matthies, C.; Wagner, W.; Höppner, D.; et al. Circulating Tumor Cells in Patients with Testicular Germ Cell Tumors. Clin. Cancer Res. 2014, 20, 3830–3841. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Gene | GCT | TGCT | MGCT | ||
|---|---|---|---|---|---|
| Sensitive | Resistant | Seminomas | Nonseminomas | All | |
| KIT | 17% | 4% | 18–25% | 2% | na |
| RAS | 14–18% | 12% | 18% | 9% | 37% |
| TP53 | 0% | 16% | 4% | 20% | 61–82% |
| Refs. | Histology 1 | Molecular Analysis 2 | Molecular Alterations 3 | |||
|---|---|---|---|---|---|---|
| MGCT | HM | MGCT | HM | MGCT | HM | |
| [66] | na | AML M7 | na | CG | na | - |
| na | AML M7 | na | CG | na | - | |
| na | AML M7 | na | CG | na | - | |
| na | AML M7 | na | CG | na | - | |
| na | AML M5 | na | CG | na | - | |
| na | AUL | na | CG | na | i12p | |
| na | ET | na | CG | na | - | |
| na | ET | na | CG | na | - | |
| na | MM | na | CG | na | - | |
| na | MM | na | CG | na | - | |
| [68] | MT, Y | AML M6 + M7 | na | CG | na | - |
| MT, Y | AML M4 | na | CG | na | i12p | |
| MT, Y | AML M6 | na | CG | na | i12p | |
| MT, Y, SARC | AML M7 | na | CG | na | - | |
| MT, IT, Y | AML M7 | na | CG | na | - | |
| [70] | Y, IT, SARC | AML M2 | CG | CG | i12p | i12p |
| [71] | Y, IT | AML M4 | na | CG | na | - |
| IT, S, Y | AML M4 | na | CG | na | - | |
| IT | AML M5 | na | CG | na | i12p | |
| [72] | MT+YS | MDS; AML M2+M6 | na | CG | na | i12p |
| [73] | mixed | AML M1 | na | CG | na | i12p |
| MT, IT | AML M0 | na | CG | na | i12p | |
| mixed | MS | CG | CG | i12p | i12p | |
| [74] | na | AML M7 | WES | WES | +12p,TP53, PTEN | +12p,TP53, PTEN |
| [75] | MT+SARC | AML M6 | sanger | CG+TS | TP53, NRAS | TP53, NRAS |
| [76] | mixed | AML M7 | array + TS | CG + array +TS | i12p,TP53, PTEN | i12p,TP53, PTEN |
| [77] | IT, Y | AML M7 | CG+WES | CG+WES | +12p,TP53, PTEN | +12p,TP53, PTEN |
| [78] | MT, SARC | AML M7 | CG+WES | CG+WES | +12p,KRAS,TP53, PTEN | +12p,KRAS,TP53, PTEN |
| [69] | T | Mastocytosis | TS | CG | TP53, PTEN | i12p |
| Y, T | MDS + HS | WES | WES +CG (all) | i12p,RRAS2,BCOR,TP53 | MDS: i12p,RRAS2,BCOR,TP53; HS:RRAS2,BCOR,TP53 4,NRAS. | |
| T, Y | HS + CMML + AML M7 | WES + CG | WES + CG (all) | TP53,PIK3CD, +1q, +21q | AML:TP53,PIK3CD, +1q, +21q, −7q; HS and CMML: TP53,PIK3CD,NRAS | |
| T, Y | AML (non-M7) + MDS | na | TS | na | TP53 | |
| T | AML (non-M7) | TS | na | TP53,NRAS,RRAS2 | na | |
| Y, T | MDS + AML M7 | WES + CG | WES +CG (MDS) | i12p,KRAS,TP53 4,ARID1A | i12p,KRAS,TP53 4 | |
| CC | MDS | WES | WES+CG | TP53 | TP53 | |
| T | HS | na | TS | na | TP53,KRAS | |
| na | HLH | na | TS | na | TP53,PTEN,NRAS | |
| S, E | TCL + HLH | TS | TS (TCL) | TP53,PTEN | TP53 | |
| T, Y | CMML+ HLH | WES | WES +CG (CMML) | i12p,KRAS,AKT1 | i12p,KRAS,MED12 | |
| Y | AML M7 | CG | CG | i12p | i12p | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbini, M.; Schepisi, G.; Bleve, S.; Virga, A.; Gianni, C.; Gurioli, G.; Ulivi, P.; De Giorgi, U. Primary Mediastinal and Testicular Germ Cell Tumors in Adolescents and Adults: A Comparison of Genomic Alterations and Clinical Implications. Cancers 2021, 13, 5223. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205223
Urbini M, Schepisi G, Bleve S, Virga A, Gianni C, Gurioli G, Ulivi P, De Giorgi U. Primary Mediastinal and Testicular Germ Cell Tumors in Adolescents and Adults: A Comparison of Genomic Alterations and Clinical Implications. Cancers. 2021; 13(20):5223. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205223
Chicago/Turabian StyleUrbini, Milena, Giuseppe Schepisi, Sara Bleve, Alessandra Virga, Caterina Gianni, Giorgia Gurioli, Paola Ulivi, and Ugo De Giorgi. 2021. "Primary Mediastinal and Testicular Germ Cell Tumors in Adolescents and Adults: A Comparison of Genomic Alterations and Clinical Implications" Cancers 13, no. 20: 5223. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205223