
Locus-Specific DNA Methylation Editing in Melanoma Cell Lines Using a CRISPR-Based System

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
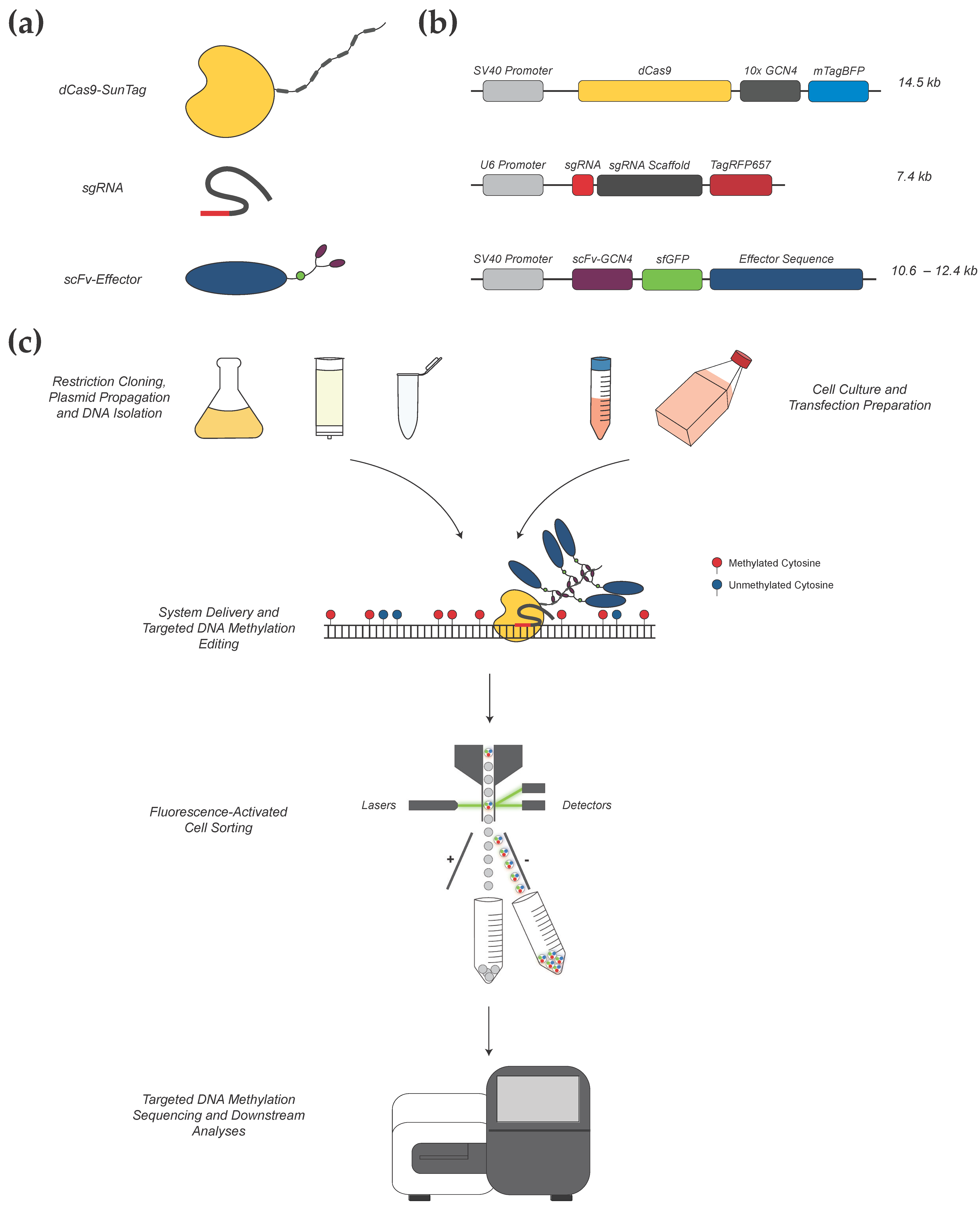
2.1. gRNA Design for CRISPR-Methylation Editing
2.2. CRISPR-Methylation Plasmid Preparation
2.2.1. Preparation of dCas9-SunTag and Effector Constructs
2.2.2. Preparation of Guide RNA (gRNA) Constructs
2.3. Transient Delivery of the CRISPR-Methylation Editing System
2.3.1. Cell Culture
2.3.2. DNA Preparation
2.3.3. Transfection
2.3.4. Cell Recovery and Maintenance
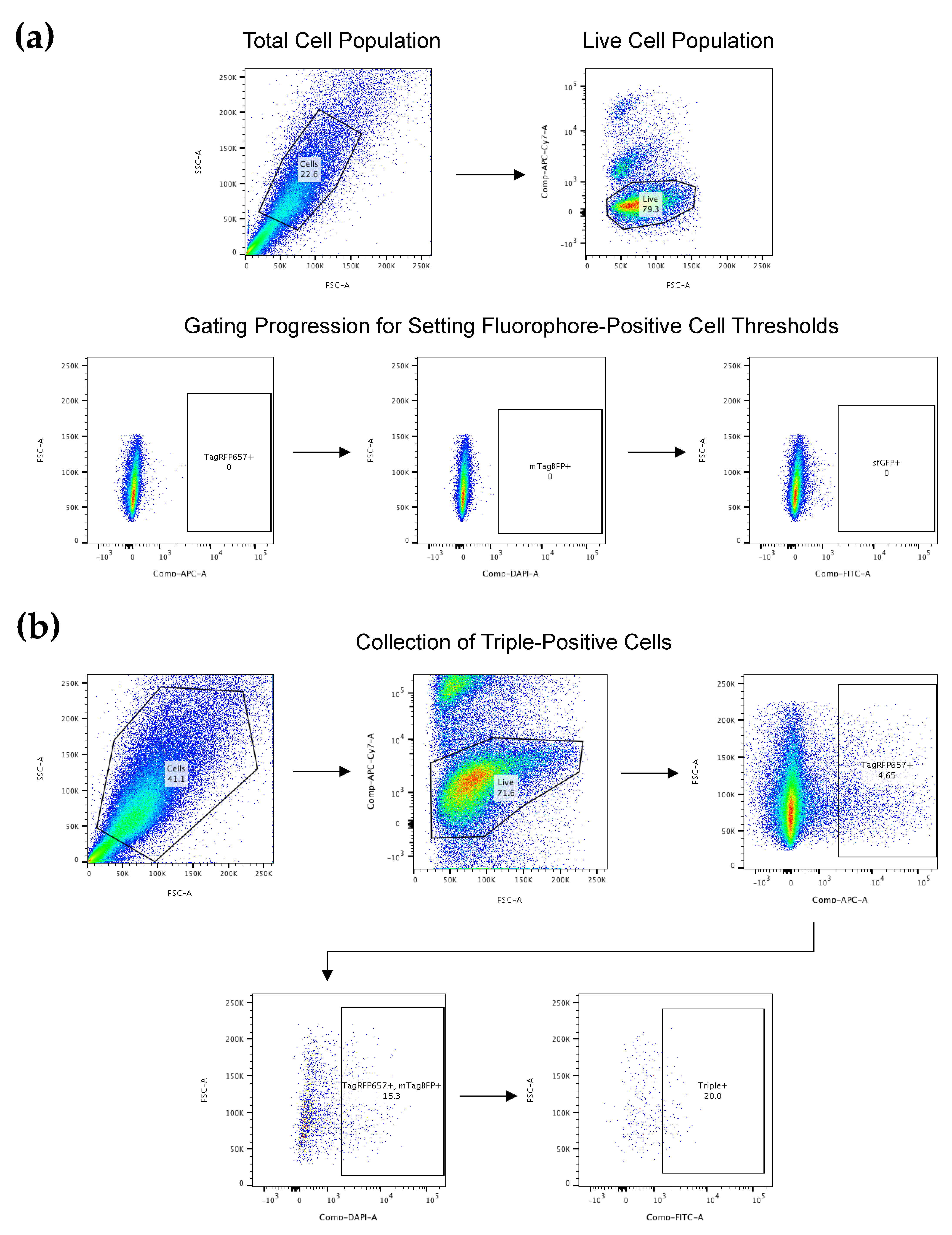
2.4. FACS for CRISPR-Edited Cells
2.4.1. Cell Preparation for FACS
2.4.2. FACS
2.5. Targeted DNA Methylation Analysis for Confirmation of CRISPR Methylation Editing
2.5.1. DNA Preparation and Sequencing
2.5.2. Analysis of Targeted DNA Methylation Sequencing Data
2.6. gRNA Evaluation for On-Target Specificity and Off-Target Activity
2.6.1. Selection of Predicted Off-Target Loci and Primer Design
2.6.2. gRNA and CRISPR-Cas9 Transfection
2.6.3. Illumina MiSeq Sequencing
2.6.4. Analysis of Sequencing Data
3. Results and Discussion
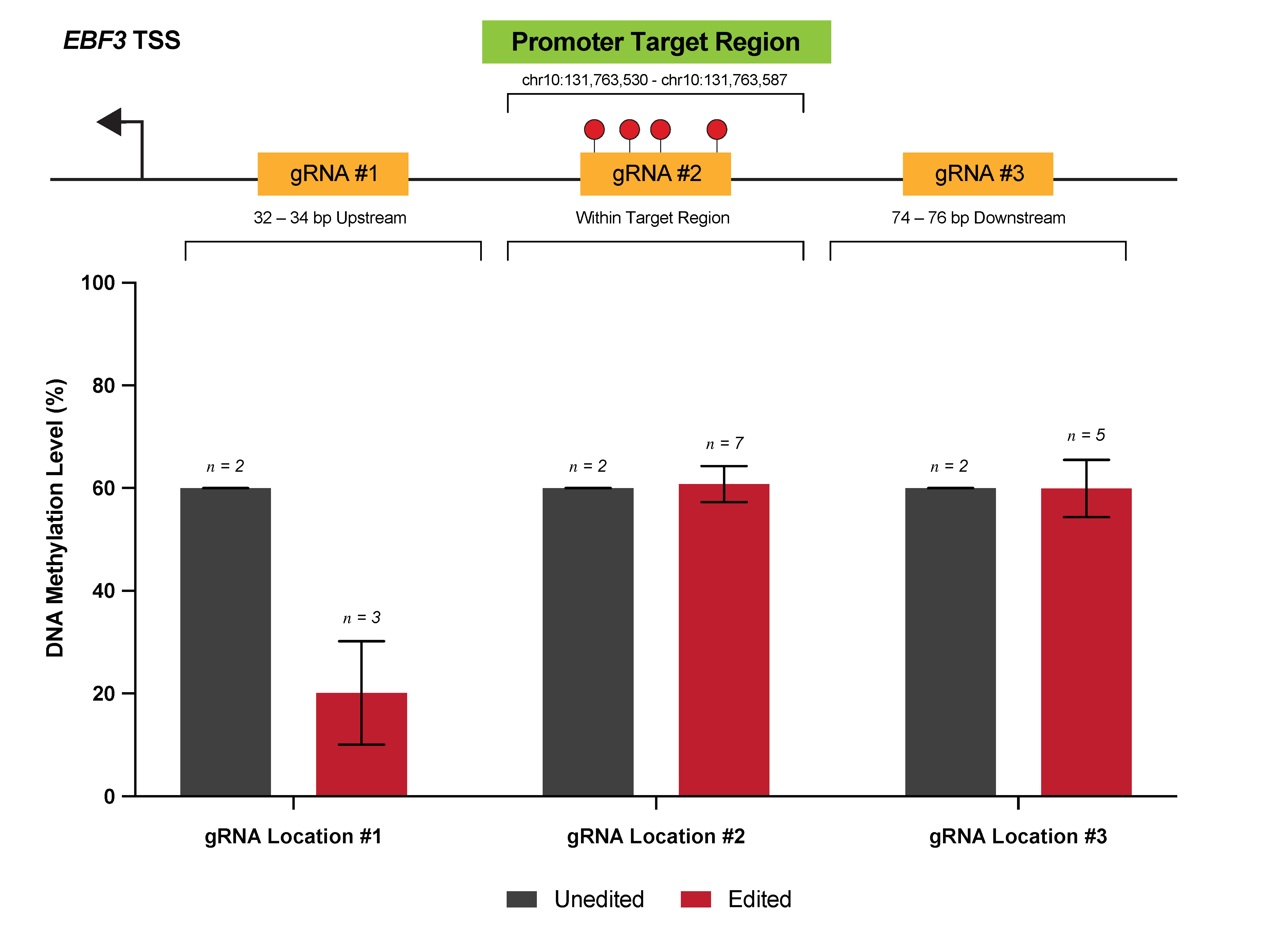
3.1. Locus-Specific gRNA Design for Targeted Methylation Editing
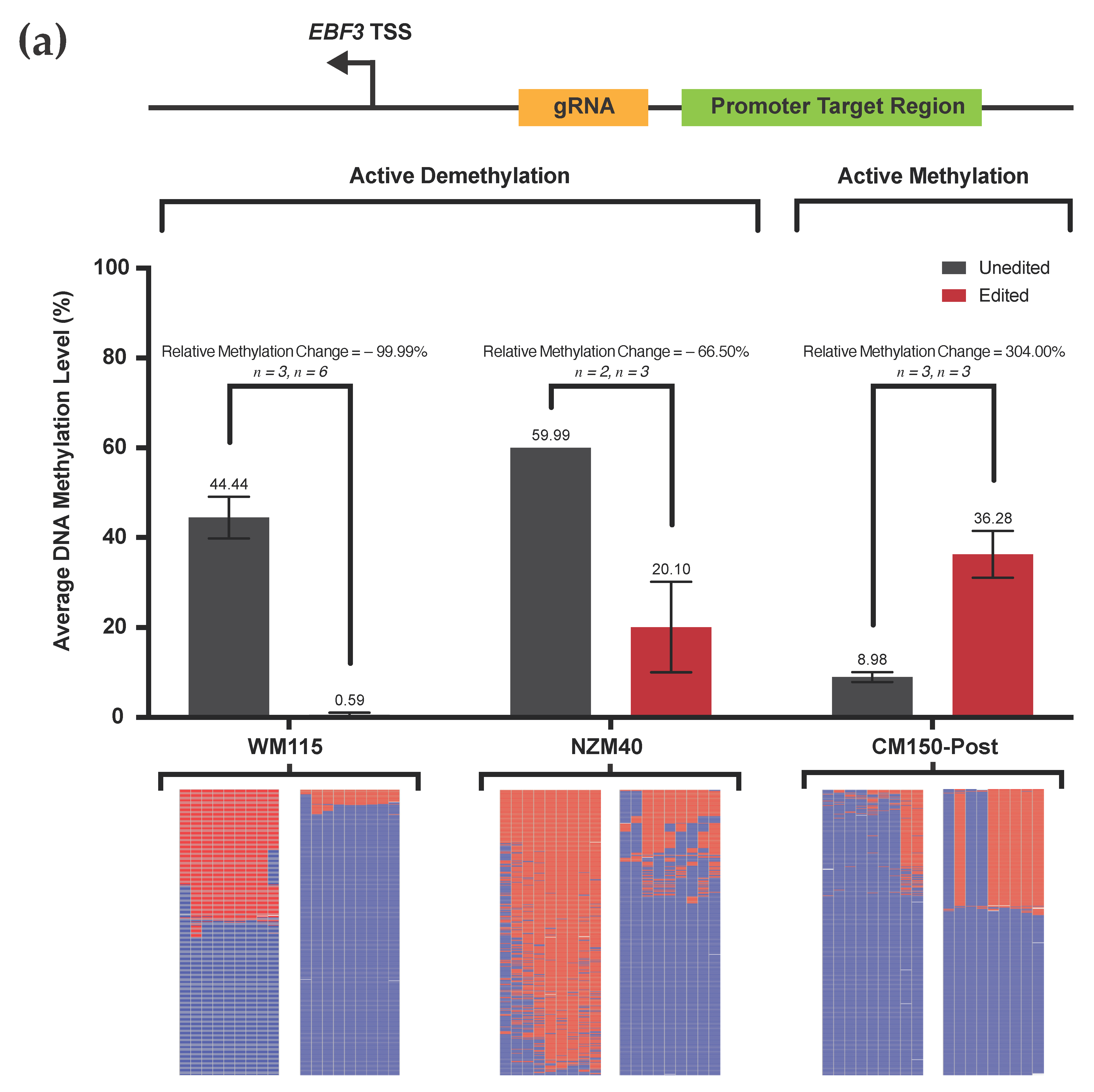
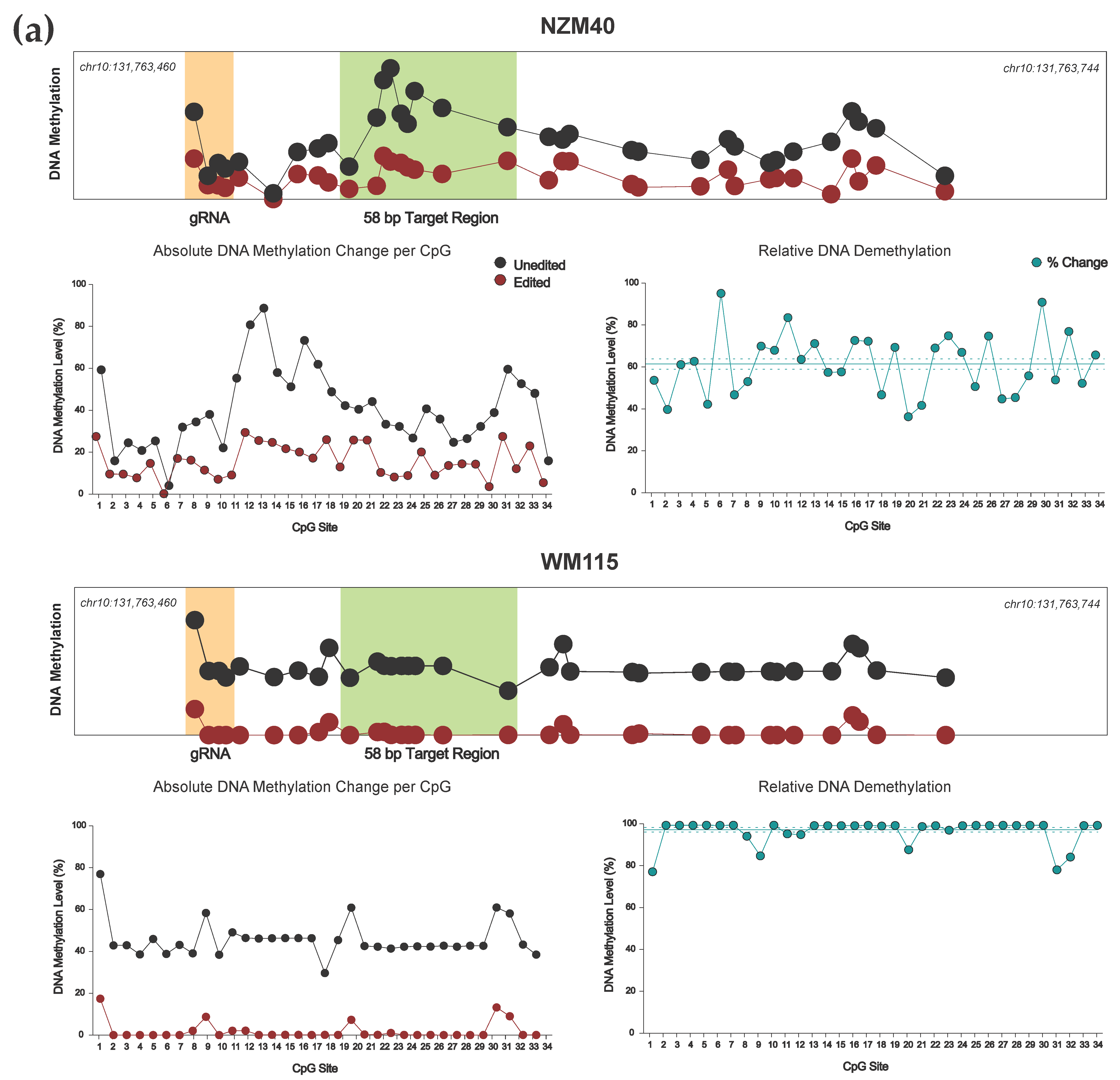
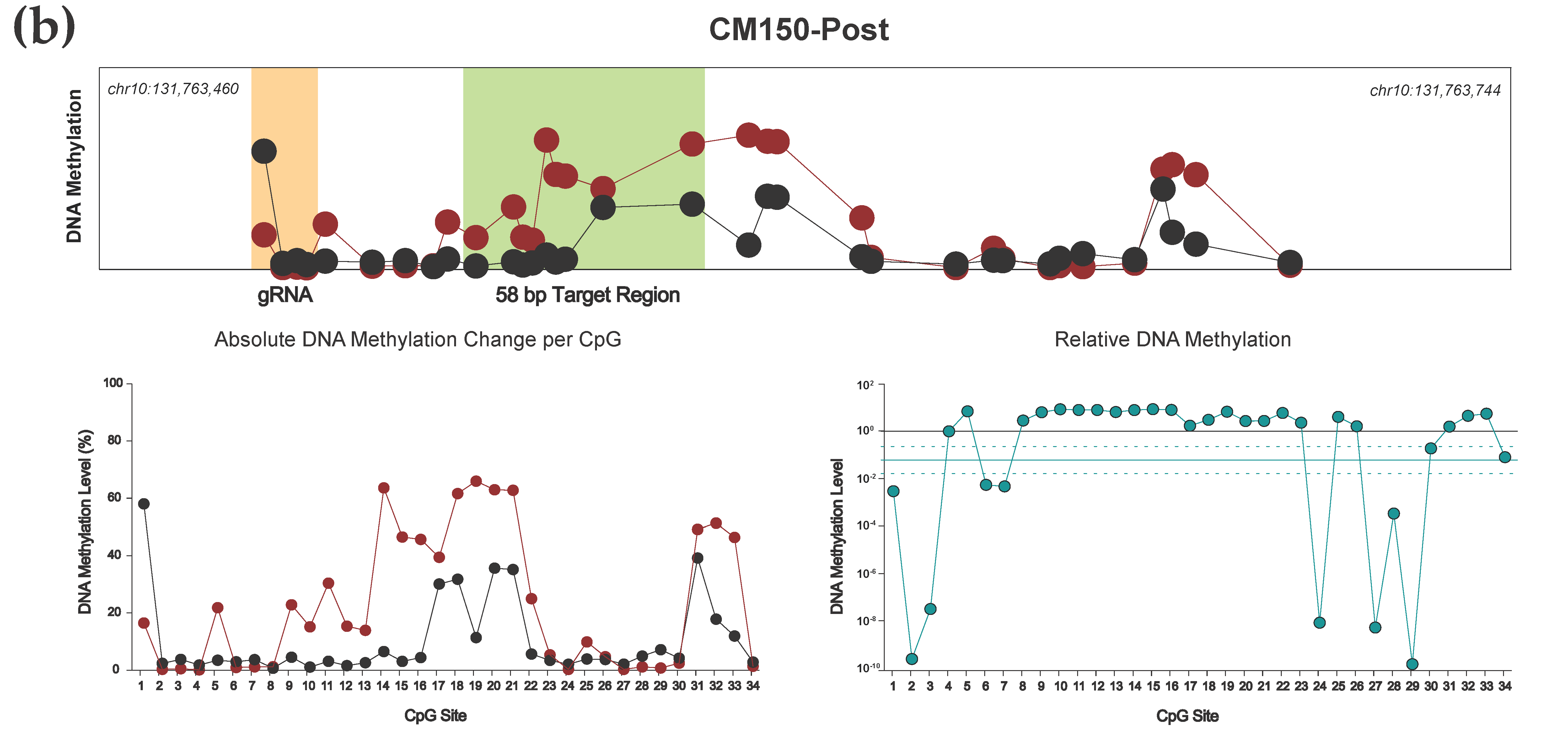
3.2. Efficient Locus-Specific Editing of the EBF3 Gene Promoter
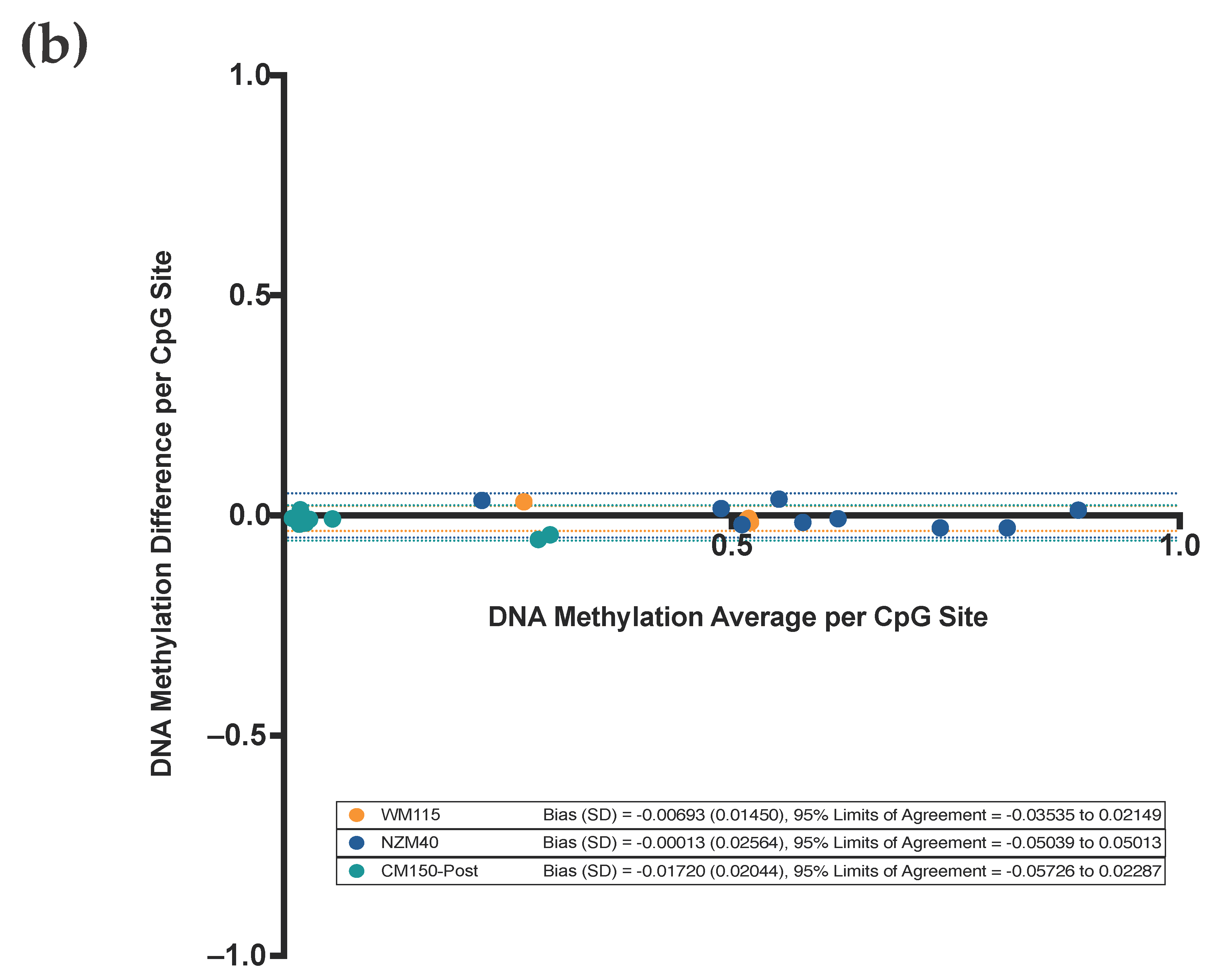
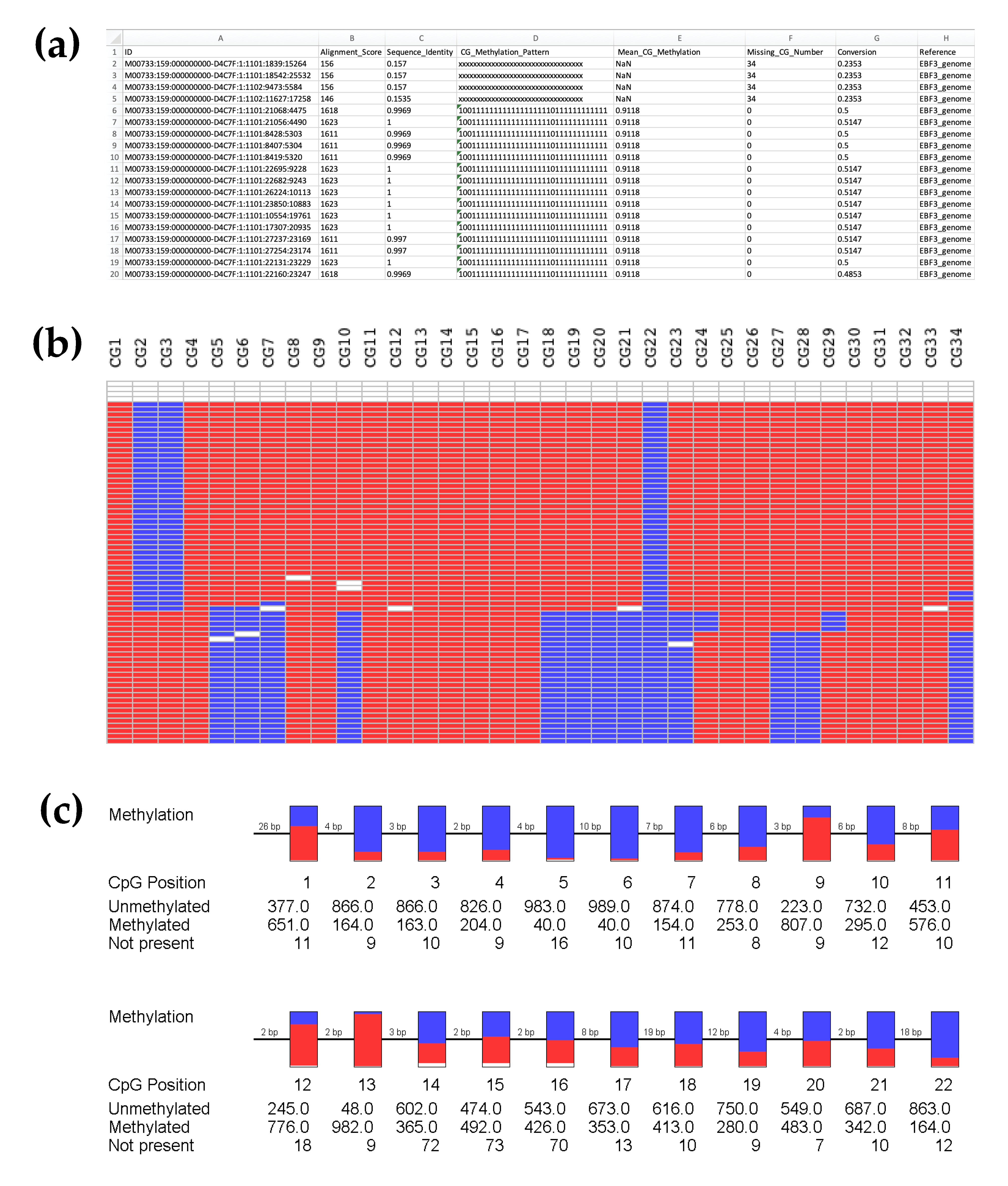
3.3. Highly Reproducible DNA Methylation Analysis Using Targeted Sequencing
3.4. Effective Editing Window of the dCas9-SunTag System
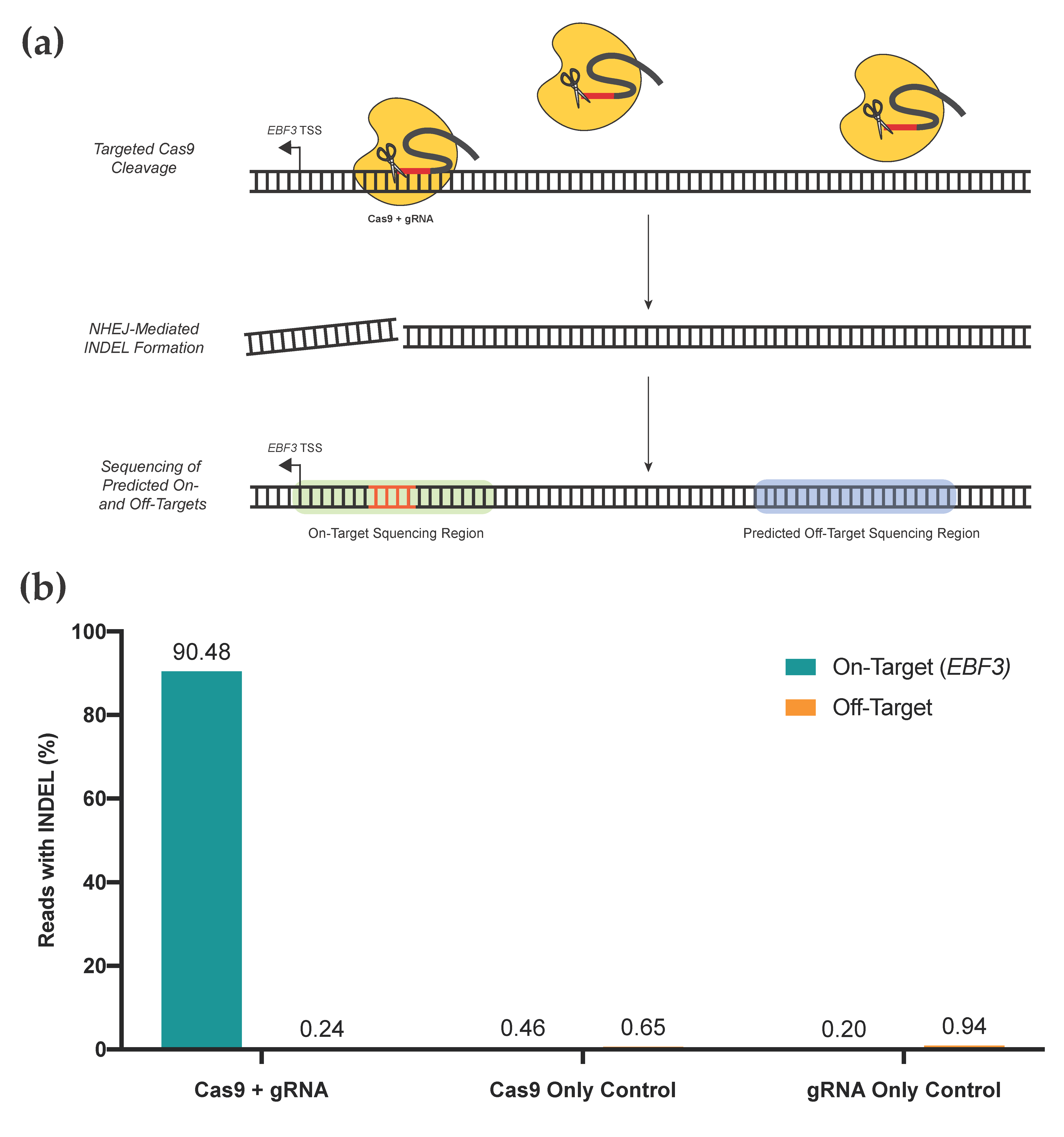
3.5. Assessment of On- and Off-Target Activity for Targeted Editing
3.6. Design and Delivery Considerations for Editing System Selection
3.7. Limitations and Troubleshooting
3.7.1. Timing of Transfection
3.7.2. DNA Quality and Total DNA Input
3.7.3. Exposure to Transfection Reagents
3.7.4. Spectral Overlap Considerations for FACS
3.7.5. Transfection Efficiency
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
- Human melanoma cell lines to be transfected (e.g., NZM40, WM115, CM150-Post)
- Appropriate cell culture medium for chosen cell line(s)
- 0.05% Trypsin-EDTA
- BsmBI restriction endonuclease and NEBuffer 3.1™ (New England Biolabs, Ipswich, MA, USA)
- Shrimp alkaline phosphatase (rSAP) (New England Biolabs, Ipswich, MA, USA)
- DNA Clean and Concentrator-5 Kit (Zymo Research, Irvine, CA, US), or equivalent
- T4 DNA Ligase and 10x T4 DNA Ligase Buffer (Invitrogen, Waltham, MA, USA)
- Pre-prepared CRISPR-methylation plasmids
- LB agar and broth for propagation of pre-prepared CRISPR-methylation plasmids
- Appropriate antibiotic for plasmid selection (e.g., 100 µg/mL ampicillin)
- Plasmid isolation system (e.g., GenCatch™ Plasmid Plus DNA Maxiprep Kit (Epoch Life Science Inc, Missouri, TX, USA))
- Opti-MEM serum-free medium (Invitrogen, Waltham, MA, USA)
- Lipofectamine 3000 reagent and P3000 reagent (Invitrogen, Waltham, MA, USA)
- 1 × Dulbecco’s phosphate buffered saline (DPBS) (Invitrogen, Waltham, MA, USA)
- Sterile autoMACS buffer (1 x DPBS + 1% FCS + 2 mM EDTA)
- Ice for keeping cells pre- and post-fluorescence-activated cell sorting (FACS)
- DNA isolation kit for transfected FACS-selected cells
- Bisulfite conversion kit (e.g., EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA, USA)
- For low cell numbers, the combined DNA isolation and bisulfite conversion kit (e.g., EZ DNA Methylation-Direct Kit (Zymo Research, Irvine, CA, USA))
- Reagents for targeted DNA methylation analysis (see Section 2.5)
- Equipment
- Access to the Benchling online platform for gRNA Design (http://benchling.com, accessed on 1 May 2019)
- Cell culture facilities and incubator with appropriate conditions for chosen cell line(s)
- Cell culture flasks
- Tube(s) for cell resuspension (e.g., 15 mL or 50 mL Falcon conical centrifuge tubes)
- Centrifuge suitable for 15 mL or 50 mL Falcon tubes
- Cell counting apparatus (e.g., hemocytometer)
- Shaking incubator suitable for mid-scale (200–500 mL) bacterial cultures
- 1.5 mL microcentrifuge tubes
- 6-well cell culture plates
- 15 mL Falcon tubes for FACS cell preparation and collection
- FACS system with capacity for sorting cells that are positive for at least three fluorophores simultaneously (e.g., BD FACSAria Fusion (BD Biosciences))
- Equipment for targeted DNA methylation analysis (see Section 2.5)
- Access to the NCBI Primer-BLAST online tool for optional gRNA assessment experiments (https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/tools/primer-blast/index.cgi, accessed on 1 May 2019)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Target Strand | Forward/Reverse | Sequence a |
|---|---|---|---|
| gRNA Location #1 | Sense | Forward | CACCGAAAAACCAAGCGGACGCCGC |
| Reverse | AAACGCGGCGTCCGCTTGGTTTTTC | ||
| Antisense | Forward | CACCGCGCGGCGTCCGCTTGGTTTT | |
| Reverse | AAACAAAACCAAGCGGACGCCGCGC | ||
| gRNA Location #2 | Sense | Forward | CACCGCGGCGCGCGGCTTCCCGACC |
| Reverse | AAACGGTCGGGAAGCCGCGCGCCGC | ||
| Antisense | Forward | CACCGGCGCGCTCACCCGGGTCCGG | |
| Reverse | AAACCCGGACCCGGGTGAGCGCGCC | ||
| gRNA Location #3 | Sense | Forward | CACCGCAAAGGACGTCTGCGCGACA |
| Reverse | AAACTGTCGCGCAGACGTCCTTTGC | ||
| Antisense | Forward | CACCGCGTGTCGCGCAGACGTCCTT | |
| Reverse | AAACAAGGACGTCTGCGCGACACGC |


References
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484. [Google Scholar] [CrossRef]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005, 2, S4–S11. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Ahn, A.; Stockwell, P.A.; Parry, M.; Motwani, J.; Gallagher, S.J.; Shklovskaya, E.; Tiffen, J.; Eccles, M.R.; et al. Marked Global DNA Hypomethylation Is Associated with Constitutive PD-L1 Expression in Melanoma. iScience 2018, 4, 312–325. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 2018, 51, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Witte, T.; Plass, C.; Gerhauser, C. Pan-cancer patterns of DNA methylation. Genome Med. 2014, 6, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Sen, S.; Weeks, R.J.; Eccles, M.R.; Chatterjee, A. Promoter DNA Hypermethylation and Paradoxical Gene Activation. Trends Cancer 2020, 6, 392–406. [Google Scholar] [CrossRef]
- Guilleret, I.; Yan, P.; Grange, F.; Braunschweig, R.; Bosman, F.T.; Benhattar, J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int. J. Cancer 2002, 101, 335–341. [Google Scholar] [CrossRef]
- Castelo-Branco, P.; Choufani, S.; Mack, S.; Gallagher, D.; Zhang, C.; Lipman, T.; Zhukova, N.; Walker, E.J.; Martin, D.; Merino, D. Methylation of the TERT promoter and risk stratification of childhood brain tumours: An integrative genomic and molecular study. Lancet Oncol. 2013, 14, 534–542. [Google Scholar] [CrossRef]
- Bert Saul, A.; Robinson Mark, D.; Strbenac, D.; Statham Aaron, L.; Song Jenny, Z.; Hulf, T.; Sutherland Robert, L.; Coolen Marcel, W.; Stirzaker, C.; Clark Susan, J. Regional Activation of the Cancer Genome by Long-Range Epigenetic Remodeling. Cancer Cell 2013, 23, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Takasawa, K.; Arai, Y.; Yamazaki-Inoue, M.; Toyoda, M.; Akutsu, H.; Umezawa, A.; Nishino, K. DNA hypermethylation enhanced telomerase reverse transcriptase expression in human-induced pluripotent stem cells. Hum. Cell 2018, 31, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spainhour, J.C.G.; Lim, H.S.; Yi, S.V.; Qiu, P. Correlation patterns between DNA methylation and gene expression in The Cancer Genome Atlas. Cancer Inform. 2019, 18, 1176935119828776. [Google Scholar] [CrossRef]
- Chatterjee, A.; Stockwell, P.A.; Ahn, A.; Rodger, E.J.; Leichter, A.L.; Eccles, M.R. Genome-wide methylation sequencing of paired primary and metastatic cell lines identifies common DNA methylation changes and a role for EBF3 as a candidate epigenetic driver of melanoma metastasis. Oncotarget 2017, 8, 6085. [Google Scholar] [CrossRef] [Green Version]
- Rodger, E.J.; Chatterjee, A.; Stockwell, P.A.; Eccles, M.R. Characterisation of DNA methylation changes in EBF3 and TBC1D16 associated with tumour progression and metastasis in multiple cancer types. Clin. Epigenet. 2019, 11, 114. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Urbano, A.; Smith, J.; Weeks, R.J.; Chatterjee, A. Gene-Specific Targeting of DNA Methylation in the Mammalian Genome. Cancers 2019, 11, 1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K. Targeted DNA demethylation in vivo using dCas9–peptide repeat and scFv–TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.C.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017, 18, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Shao, X.; Gao, L.; Zhang, S. Systematic DNA methylation analysis of multiple cell lines reveals common and specific patterns within and across tissues of origin. Hum. Mol. Genet. 2015, 24, 4374–4384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodger, E.J.; Almomani, S.N.; Ludgate, J.L.; Stockwell, P.A.; Baguley, B.C.; Eccles, M.R.; Chatterjee, A. Comparison of Global DNA Methylation Patterns in Human Melanoma Tissues and Their Derivative Cell Lines. Cancers 2021, 13, 2123. [Google Scholar] [CrossRef]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569. [Google Scholar] [CrossRef]
- Lo, C.-L.; Choudhury, S.R.; Irudayaraj, J.; Zhou, F.C. Epigenetic Editing of Ascl1 Gene in Neural Stem Cells by Optogenetics. Sci. Rep. 2017, 7, 42047. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, R. How to Synthesize your gRNAs for CRISPR. Available online: https://www.benchling.com/2016/02/23/how-to-synthesize-your-grnas-for-crispr/ (accessed on 16 May 2020).
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient targeted DNA methylation with chimeric dCas9–Dnmt3a–Dnmt3L methyltransferase. Nucleic Acids Res. 2016, 45, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA methylation in the mammalian genome. Cell 2016, 167, 233–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281. [Google Scholar] [CrossRef] [Green Version]
- Franco, A.V.; Zhang, X.D.; Van Berkel, E.; Sanders, J.E.; Zhang, X.Y.; Thomas, W.D.; Nguyen, T.; Hersey, P. The role of NF-κB in TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis of melanoma cells. J. Immunol. 2001, 166, 5337–5345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vojta, A.; Dobrinić, P.; Tadić, V.; Bočkor, L.; Korać, P.; Julg, B.; Klasić, M.; Zoldoš, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [Green Version]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 2021, 184, 2503–2519. [Google Scholar] [CrossRef] [PubMed]
- Masser, D.R.; Stanford, D.R.; Freeman, W.M. Targeted DNA methylation analysis by next-generation sequencing. J. Vis. Exp. JoVE 2015, 96, 52488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheishvili, D.; Petropoulos, S.; Christiansen, S.; Szyf, M. Targeted DNA methylation analysis methods. In Epigenetics and Gene Expression in Cancer, Inflammatory and Immune Diseases; Springer: New York City, NY, USA, 2017; pp. 33–50. [Google Scholar]
- Li, L.-C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, A.; Rodger, E.J.; Morison, I.M.; Eccles, M.R.; Stockwell, P.A. Tools and strategies for analysis of genome-wide and gene-specific DNA methylation patterns. In Oral Biology; Springer: New York City, NY, USA, 2017; pp. 249–277. [Google Scholar]
- Almomani, S.N.; Alsaleh, A.A.; Weeks, R.J.; Chatterjee, A.; Day, R.C.; Honda, I.; Homma, H.; Fukuzawa, R.; Slatter, T.L.; Hung, N.A. Identification and validation of DNA methylation changes in pre-eclampsia. Placenta 2021, 110, 16–23. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2013, 30, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Lutsik, P.; Feuerbach, L.; Arand, J.; Lengauer, T.; Walter, J.; Bock, C. BiQ Analyzer HT: Locus-specific analysis of DNA methylation by high-throughput bisulfite sequencing. Nucleic Acids Res. 2011, 39 (Suppl. 2), W551–W556. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Devesa-Guerra, I.; Morales-Ruiz, T.; Pérez-Roldán, J.; Parrilla-Doblas, J.T.; Dorado-León, M.; García-Ortiz, M.V.; Ariza, R.R.; Roldán-Arjona, T. DNA Methylation Editing by CRISPR-guided Excision of 5-Methylcytosine. J. Mol. Biol 2020, 432, 2204–2216. [Google Scholar] [CrossRef]
- Xiong, T.; Meister, G.E.; Workman, R.E.; Kato, N.C.; Spellberg, M.J.; Turker, F.; Timp, W.; Ostermeier, M.; Novina, C.D. Targeted DNA methylation in human cells using engineered dCas9-methyltransferases. Sci. Rep. 2017, 7, 6732. [Google Scholar] [CrossRef]
- Li, J.; Mahata, B.; Escobar, M.; Goell, J.; Wang, K.; Khemka, P.; Hilton, I.B. Programmable human histone phosphorylation and gene activation using a CRISPR/Cas9-based chromatin kinase. Nat. Commun. 2021, 12, 896. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 2016, 7, 46545. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Pastor, W.A.; Shen, Y.; Tahiliani, M.; Liu, D.R.; Rao, A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE 2010, 5, e8888. [Google Scholar] [CrossRef] [Green Version]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.-L.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Zhang, X.; Su, J.; Jeong, M.; Gundry, M.C.; Huang, Y.-H.; Zhou, Y.; Li, W.; Goodell, M.A. Targeted DNA methylation in vivo using an engineered dCas9-MQ1 fusion protein. Nat. Commun. 2017, 8, 16026. [Google Scholar] [CrossRef]
- Lu, A.; Wang, J.; Sun, W.; Huang, W.; Cai, Z.; Zhao, G.; Wang, J. Reprogrammable CRISPR/dCas9-based recruitment of DNMT1 for site-specific DNA demethylation and gene regulation. Cell Discov. 2019, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Pflueger, C.; Tan, D.; Swain, T.; Nguyen, T.V.; Pflueger, J.; Nefzger, C.; Polo, J.M.; Ford, E.; Lister, R. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 2018, 28, 1193–1206. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.R.; Pan, Y.; Lee, C.M.; Davis, T.H.; Bao, G. Tools for experimental and computational analyses of off-target editing by programmable nucleases. Nat. Protoc. 2021, 16, 10–26. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Morita, S.; Horii, T.; Kimura, M.; Hatada, I. Synergistic Upregulation of Target Genes by TET1 and VP64 in the dCas9–SunTag Platform. Int. J. Mol. Sci. 2020, 21, 1574. [Google Scholar] [CrossRef] [Green Version]
- Marx, N.; Dhiman, H.; Schmieder, V.; Freire, C.M.; Nguyen, L.N.; Klanert, G.; Borth, N. Enhanced targeted DNA methylation of the CMV and endogenous promoters with dCas9-DNMT3A3L entails distinct subsequent histone modification changes in CHO cells. Metab. Eng. 2021, 66, 268–282. [Google Scholar] [CrossRef]
- Taghbalout, A.; Du, M.; Jillette, N.; Rosikiewicz, W.; Rath, A.; Heinen, C.D.; Li, S.; Cheng, A.W. Enhanced CRISPR-based DNA demethylation by Casilio-ME-mediated RNA-guided coupling of methylcytosine oxidation and DNA repair pathways. Nat. Commun. 2019, 10, 4296. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.; Findlay, G.M.; Kircher, M.; Shendure, J. Concurrent genome and epigenome editing by CRISPR-mediated sequence replacement. BMC Biol. 2019, 17, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, A.; Lagisz, M.; Rodger, E.J.; Zhen, L.; Stockwell, P.A.; Duncan, E.J.; Horsfield, J.A.; Jeyakani, J.; Mathavan, S.; Ozaki, Y. Sex differences in DNA methylation and expression in zebrafish brain: A test of an extended ‘male sex drive’hypothesis. Gene 2016, 590, 307–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, A.; Stockwell, P.A.; Horsfield, J.A.; Morison, I.M.; Nakagawa, S. Base-resolution DNA methylation landscape of zebrafish brain and liver. Genom. Data 2014, 2, 342–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]







| Group | System | Description | Features | Delivery | Stability |
|---|---|---|---|---|---|
| Vojta et al. (2016) [29] | dCas9-Effector | Described the first dCas9 fusion with DNMT3A for targeted methylation editing in a ≈35 bp wide region for the BACH2 and IL6ST loci, with associated gene expression change | Simple design Higher off-target activity Moderate editing window (≈150 bp) | Lipofection | Peak increase in methylation at day 6–7 post-transfection; persistent increases observed to at least 42 days |
| Xu et al. (2016) [45] | dCas9, Tet1-MS2 | Designed a Effector-MS2 and dCas9 with modified gRNAs for effector multimerization | Allows multimerization of effector constructs Lower editing efficacy Moderate editing window (>100 but <300 bp; no editing within 100 bp of gRNA) | Polyethylenimine; Lipofection | mRNA expression change peaks at 4 days post-transfection only |
| Morita et al. (2016) [18] | dCas9-SunTag | Adapted the SunTag protein scaffold for multimerized DNA methylation editing, increasing the amino acid linker length to 22 nt to achieve demethylation efficacies of >90% both in vitro and in vivo | Allows multimerization of effector constructs Higher efficacy than dCas9-Effector, -MS2, -MQ1, -sMTase systems Low off-target activity Broader editing window (≤1 kb) | Lipofection | N/A |
| Lei et al. (2017) [46] | dCas9-MQ1 | Fused dCas9 with an engineered prokaryotic CpG DNA methyltransferase “MQ1” derived from Mollicutes spiroplasma (M.SssI), strain MQ1 | Quicker onset of effective methylation change (24 h) Narrow editing window (≈30–50 bp) | Lipofection | ≥3 weeks |
| Xiong et al. (2017) [40] | dCas9-sMTase | Developed a targeted, “split methyltransferase” derived from M.SssI where the two split components bind at the target CpG site | Low off-target activity Narrow editing window (8–25 bp) | Lipofection | N/A |
| Taghbalout et al. (2019) [53] | Casilio-ME | Adapted the Casilio platform for methylation editing, providing co-delivery of TET1 and BER-associated proteins GADD45A or NEIL2 | Allows multimerization of effector constructs Comparable or higher efficacy than SunTag systems Low off-target activity | Lipofection | N/A |
| Lu et al. (2019) [47] | dCas9-R2 | Developed a dCas9-R2 module that specifically binds and inhibits the action of endogenous DNMT1 to prevent local DNA methylation at a target locus | Narrow editing window (<100 bp) No requirement for exogenous effectors Similar efficacy to dCas9, Tet1-MS2 | Lipofection | Peaked at 7 days post-transfection |
| Alexander et al. (2019) [54] | Dual Cas9, Template | Excised a 1120 bp CGI from the HPRT1 promoter using dual gRNA-guided Cas9 modules and replaced with either completely methylated or unmethylated fragments via NHEJ | Ensures complete de/methylation NHEJ-mediated insertion has low efficiency (<1%) Prone to repair-induced INDEL formation | Lipofection | N/A |
| Devesa-Guerra et al. (2020) [39] | dCas9-ROS1 | Described a dCas9-Effector using the plant-specific DNA glycosylase ROS1, which directly excises 5mC | Higher gene reactivation efficacy than dCas9-Effector fusions alone; ineffective at fully methylated loci Modest impact on measured methylation levels | Lipofection | N/A |
| Nuñez et al. (2021) [30] | CRISPRoff | Developed a novel dCas9-Dnmt3A-3L-ZNF10 KRAB fusion (CRISPRoff) to induce heritable, persistent gene silencing | High efficacy of methylation editing Applicable to genome-wide screening Stable editing effect despite transient delivery Non-isolated effect of multiple enzymes (e.g., DNMTs plus KRAB) | Lipofection; Nucleofection; Lentiviral | Construct expression lost at 10 days; methylation change peaks at 9 days, stable for at least 30–50 days post-transfection |
| Smith et al. (2021) (Current Manuscript) | dCas9-SunTag | Applied dCas9-SunTag with scFv-TET1CD and scFv-DNMT3A effectors to manipulate EBF3 promoter methylation in melanoma | Achieved near-complete demethylation and high methylation Comprehensive workflow for methylation editing and downstream analyses | Lipofection | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, J.; Banerjee, R.; Waly, R.; Urbano, A.; Gimenez, G.; Day, R.; Eccles, M.R.; Weeks, R.J.; Chatterjee, A. Locus-Specific DNA Methylation Editing in Melanoma Cell Lines Using a CRISPR-Based System. Cancers 2021, 13, 5433. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215433
Smith J, Banerjee R, Waly R, Urbano A, Gimenez G, Day R, Eccles MR, Weeks RJ, Chatterjee A. Locus-Specific DNA Methylation Editing in Melanoma Cell Lines Using a CRISPR-Based System. Cancers. 2021; 13(21):5433. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215433
Chicago/Turabian StyleSmith, Jim, Rakesh Banerjee, Reema Waly, Arthur Urbano, Gregory Gimenez, Robert Day, Michael R. Eccles, Robert J. Weeks, and Aniruddha Chatterjee. 2021. "Locus-Specific DNA Methylation Editing in Melanoma Cell Lines Using a CRISPR-Based System" Cancers 13, no. 21: 5433. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215433