
Biological Significance and Targeting of the FGFR Axis in Cancer


1
School of Life Sciences Pharmacy and Chemistry, Kingston University, Penrhyn Road, Kingston upon Thames KT1 2EE, UK
2
Centre for Tumour Biology, Barts Cancer Institute, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(22), 5681; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225681
Submission received: 27 September 2021
/
Revised: 8 November 2021
/
Accepted: 11 November 2021
/
Published: 13 November 2021
(This article belongs to the Collection The Development of Anti-cancer Agents)
Abstract
:Simple Summary
All cells within tissues and organ systems must communicate with each other to ensure they function in a coordinated manner. One form of communication is signalling mediated by small proteins (for example fibroblast growth factors; FGFs) that are secreted by one cell and bind to specialised receptors (for example FGF receptors) on nearby cells. These receptors propagate the signal to the nucleus of the receiving cell, which in turn dictates to the cell how it should react. FGFR signalling is versatile, tightly controlled and important for normal body homeostasis, facilitating growth, healing and replacing old cells. However, cancer cells can take command of this pathway and use it to their advantage. This review will first explain the biology of FGFR signalling and then describe how it can be corrupted, the implications in cancer, and how it can be targeted to improve cancer therapy.
Abstract
The pleiotropic effects of fibroblast growth factors (FGFs), the widespread expression of all seven signalling FGF receptors (FGFRs) throughout the body, and the dramatic phenotypes shown by many FGF/R knockout mice, highlight the diversity, complexity and functional importance of FGFR signalling. The FGF/R axis is critical during normal tissue development, homeostasis and repair. Therefore, it is not surprising that substantial evidence also pinpoints the involvement of aberrant FGFR signalling in disease, including tumourigenesis. FGFR aberrations in cancer include mutations, gene fusions, and amplifications as well as corrupted autocrine/paracrine loops. Indeed, many clinical trials on cancer are focusing on targeting the FGF/FGFR axis, using selective FGFR inhibitors, nonselective FGFR tyrosine kinase inhibitors, ligand traps, and monoclonal antibodies and some have already been approved for the treatment of cancer patients. The heterogeneous tumour microenvironment and complexity of FGFR signalling may be some of the factors responsible for the resistance or poor response to therapy with FGFR axis-directed therapeutic agents. In the present review we will focus on the structure and function of FGF(R)s, their common irregularities in cancer and the therapeutic value of targeting their function in cancer.
1. Introduction
Cancer is a disease of cells, starting with genetic alterations in one cell or a small group of cells. If the repair machinery of the cells fails, then accumulation of genetic alterations will lead to cancer and with time to metastasis. In order for cells to become cancerous, they need to adopt behavioural changes outlined as the “hallmarks of cancer” [1]. Of course, besides the classical hallmarks of cancers, many years of research from different angles has shed light onto novel emerging hallmarks of cancer, such as an altered microbiome, neuronal signalling, epigenetic dysregulation and transdifferentiation [2]. There are many targeted therapies that inhibit and block each of the developed competencies necessary for the growth and progression of tumour development. A number of these approaches target tyrosine kinase receptors, such as the human epidermal growth factor receptor 2 (HER2), epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor 2 (VEGFR2), platelet derived growth factor receptor (PDGFR) and FGFR, in various ways. Acknowledging that many other tyrosine kinase receptors merit therapeutic targeting, this review will focus and discuss further the importance and different ways of targeting the FGF/FGFR axis.
Fibroblast growth factor receptor (FGFR) signalling plays a pivotal role in a myriad of processes including embryonic development, cell differentiation, proliferation, wound healing, cell migration, angiogenesis and various endocrine signalling pathways [3]. Dysregulation of FGFR signalling can lead to an antiapoptotic, mutagenic and angiogenic response in cells, all of which are cancer hallmarks [4]. The oncogenic potential of FGFR signalling also lies in its potential to serve as an escape mechanism for acquired resistance to cancer therapy. To appreciate the therapeutic value of targeting FGFR signalling in cancer, we will first consider normal structure and function, then discuss how aberrant FGFR signalling can influence cancer progression and, finally, describe how it can be targeted.
2. FGF(R) Structure
In humans, the fibroblast growth factor (FGF) family comprises 22 members classified into seven subfamilies based on similarities in coding sequence, protein structure and biochemical function: FGF1 (FGF1 and FGF2), FGF4 (FGF4, -5, -6), FGF7 (FGF3, -7, -10, -22), FGF8 (FGF8, -17, -18), FGF9 (FGF9, -16, -20), FGF19 (FGF19, -21, -23) and FGF11 (FGF11, -12, -13, -14) [5,6,7]. Each FGF ligand comprises a conserved core region of 120 amino acids and shares between 35% and 50% sequence homology [6]. Despite being similar in structure, only eighteen FGFs are reported to signal via FGFR, namely FGF1 to FGF10 and FGF16 to FGF23 [8]. Other FGF ligands, such as FGF11 to FGF14, which also share a similar structure to other ligands, do not bind to these receptors but instead can function via voltage-gated sodium channels [9], although recent work casts doubt on this dogma [10]. Five FGF subfamilies (FGF1, -4, -7, -8, -9) are characterised as paracrine signalling molecules that signal by forming a three-way complex with FGFR and heparan sulphate proteoglycans (HSPGs). The other two subfamilies (FGF11 and FGF19) act differently; the FGF11 subfamily act intracellularly, while FGF19 have a reduced HSPG binding affinity and bind to αKlotho and βKlotho cofactors to function in an endocrine manner to have an impact on adult homeostasis and metabolism [8,11].
FGFRs have extracellular immunoglobulin-like (Ig) domains 1–3 (D1–D3), a transmembrane (TM) domain, tyrosine kinase I, and II domains, a carboxyl-terminal, and an acidic box [12] (Figure 1). The D2 and D3 regions form a ligand-binding pocket for two FGF ligands and two heparin molecules [13]. The acidic box is responsible for the auto-inhibition and regulation of optimal interactions from bivalent cations (Figure 1). The interaction between the acidic box and the heparan sulphate-binding site inhibits activation of the receptor when FGF is absent [8,14,15,16]. FGF binds in the Ig2 and Ig3 domains, where HSPGs protect FGFs from protease-mediated degradation, thus stabilising the FGF–FGFR complex (Figure 1 and Figure 2A) [17]. Hence, high-affinity FGFRs are activated upon FGF ligand binding. Paracrine FGFs bind strongly to HSPGs, which possess cofactor functions to prevent the FGFs from diffusing through the extracellular matrix (ECM) as well as regulating the FGFR specificity [11,18].
Aside from the four main FGFR family members (FGFR1–4), there is an additional receptor, fibroblast growth factor receptor like 1 (FGFRL1 or FGFR5), that can bind to FGFs and heparin, but lacks the tyrosine kinase domain and therefore cannot signal via transphosphorylation [19]. FGFR1L is believed to negatively regulate FGFR signalling by acting as a decoy receptor that neutralises FGFs by binding to them without activating any downstream signalling cascade [20]. FGFRL1 is expressed mainly in musculoskeletal tissues and the kidney and its main function is to control the growth of the metanephric kidney [21]. It is hypothesised that its function depends on Ig2 and Ig3 domains interacting together with an FGF ligand and another molecule from the surface of other cells from their microenvironment [21]. In fact binding of FGF8 to FGFRL1 plays an important role in developing kidneys by driving the formation of nephrons [22].
3. FGFR Splicing
Despite the high sequence homology between FGFR family members (55%–72%) and their similar structural characteristics, there are a variety of isoforms (Figure 2) [23]. FGFR diversity is not only attributed to the different genes that can encode FGFR1–4 and the multiple FGFs that can activate them, but also to the fact that FGFR genes can be alternatively spliced (Figure 2).
FGFR genes consists of 18 exons (Figure 2A). Each gene can be alternatively spliced and produce different mRNAs that consequently will result in FGFR protein diversity [25,26]. FGFRs 1–3 each can generate two splice variants of the immunoglobulin (Ig)-like domain 3b and 3c, which are fundamental to ligand-binding specificity during development and are often mis-spliced in cancer [5,8,11]. Hence, there are seven main signalling FGFRs, FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3b, FGFR3c and FGFR4, encoded by four genes. Each specific FGFR binds to specific FGFs and most FGF ligands can bind to several different variants of FGFRs. The FGF binding specificity to FGFRs is regulated by two distinct splice variants of exon 8 and exon 9 of domain 3 (D3) [6] (Figure 2A,B). The splicing variant of exon 7/8 and exon 7/9 encodes the carboxyl-terminal of the domain D3, resulting in the -b or -c isoform [27,28,29,30,31]. In human tissues, the -b isoforms are confined to epithelial cells, with the -c isoform predominating in mesenchymal lineages [6]. The specificity of the ligand binding to FGFRs differs amongst isoforms -b and -c (Figure 2B) [6]. For instance, FGF4 binds to the FGFR1-3c isoform, while FGF7 binds specifically to the FGFR1b and -2b isoforms (Figure 2B). In conclusion, the exon rearrangement at the Ig3 loop (D3) has a profound effect on the FGF spectrum for each receptor, with the FGFR1-3b isoforms having a more limited binding affinity with FGFs compared to the FGFR1-3c isoforms (Figure 2B) [6].
Interestingly, it has been reported that reversible switching of the FGFR2-3b isoform to the -c isoform was induced by exogenous and endogenous FGF1 and FGF2. This switch was confluence and cell cycle dependent [32]. Altered splicing has been associated with cancer progression [33,34,35], for example during EMT [33,36].
FGFR1 and -2 also have another isoform -a, in which exon 7 joins directly with exon 10, the TM domain. This truncated variant is a secreted protein that is incapable of signal transduction and has an autoinhibitory role [37]. In bladder cancer, the switch from the FGFR1a to the FGFR1b isoform increased FGF1-induced activation of the latter and was associated with the tumour grade and stage, likely due to it giving a proliferation advantage [38].
In addition, there are secreted FGFR isoforms that lack the TM domain and the entire cytoplasmic region [23,39,40] (Figure 2C). There are also reports of truncated FGFR isoforms lacking Ig1 [39,40,41,42] (Figure 2E). The truncated Ig1 isoform is known to be a high affinity oncogenic variant that can activate various downstream signalling pathways, due to the Ig1 region performing an autoinhibitory role [6,24,43,44]. Interestingly, there are also isoforms missing the acid box between the Ig1 and Ig2 loops (i.e., truncated Ig1 FGFR2b and FGFR3c) [45] and other isoforms missing the carboxyl terminal [13]. In fact, such a variant missing the inhibitory carboxyl terminal portion of FGFR2 was expressed in a breast cancer cell line (SUM-52PE), along with other splice variants, with the different splice variants having different transforming activities [43]. Variants expressing the C3 carboxyl terminus resulted in more autonomous signalling, cell growth, and invasion [43]. Recently, a novel FGFR3 splice variant was reported in African American prostate cancer (FGFR3-S) that was associated with a poor prognosis and increased cell proliferation and motility [44]. The FGFR3-S variant lacked exon 14, comprising 123 nucleotides that encode the activation loop in the split kinase domain [44]. FGFR4 is well defined as it is only produced in a single isoform homologous to the FGFR1-3c splice variant [46] (Figure 2D). Although their properties are not well understood at present, there are also reports of FGFRL1 isoforms with an absent Ig1 domain with and without the acid box [47].
4. FGFR Signalling
The multiple possibilities of FGFR activation, due to the wide range of FGFs and FGFR isoforms, drives several oncogenic signalling pathways. Typically, two FGF molecules are needed to bind to the Ig2 (D2) and Ig3 (D3) extracellular domains in the FGFR to drive dimerisation and activation (Figure 1). Studies on the structure of FGFR revealed that a 2:2 FGF–FGFR complex is formed between the FGF, D2 and D3 of the FGFR. Under physiological conditions FGF–FGFR interactions are not sufficient to stabilise FGFR dimers [14], with HSPG acting as a linker to stabilise the HSPG–FGF–FGFR complex [6,36,48,49]. The FGF–FGFR–HSPG complexes induce activation of downstream signalling cascades; mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), phospholipase Cγ (PLCγ) and signal tranducer and activator of transcription 1 (STAT 1) (Figure 1) [50].
As discussed, the specificity of FGF–FGFR binding is determined by alternative splicing, ligand specificity and tissue specific expression of both FGF ligands and receptors [5,6]. Further control of the FGF–FGFR coupling is provided by the interaction with secreted proteins and plasma membrane bound receptors, such as α and β Klotho proteins [5,51,52], and a single-pass transmembrane Klotho-related protein (KLPH). These act as cofactors for the endocrine FGFs by forming an FGF–FGFR–Klotho ternary complex [53,54].
Dimerisation of the FGFR causes a conformational shift in the receptor’s structure, leading to a 50- to 100-fold increase in the receptor kinase activity, resulting in the phosphorylation through mutual transphosphorylation of numerous tyrosine residues in the intracellular domain. Subsequently, various protein complexes are formed to initiate downstream signalling transduction [4,9,11,12,55,56]. One of the adaptor proteins that governs the downstream signalling cascade is the v-crk sarcoma virus CT10 oncogene homolog (Crk) (Figure 1) [48]. Upon FGFR phosphorylation, Crk gets transiently phosphorylated and can be associated with son of sevenless (SOS), that in turns activates small GTPases [49,51,52,57].
Furthermore, FGFR activation can trigger the phosphorylation of the docking protein FGFR substrate 2 (FRS2), accompanied by the recruitment of shp2 tyrosine phosphatase. Phosphorylation of shp2 facilities its link with growth factor receptor-bound 2 (GRB2) and SOS [55,56,58,59,60]. The recruitment of GRB2 associated binding protein 1 (GAB1) forming the FRS2 complex leads to the activation of the PI3K-Ak strain transforming (AKT) pathway, which regulates cell survival and fate [61,62,63] (Figure 1). Several other signalling molecules have also been reported to be activated by FGFRs, STATs, p90 ribosomal protein S6 kinase 2 (RSK2) and the nonreceptor tyrosine kinase Src [64,65,66,67]. The phospho-tyrosine residues in FGFR carboxyl terminal regions confer strong and selective binding to src homology two (SH2) domains accommodating proteins such as PLCγ. These interactions result in phosphatidylinositol 4, 5-bisphosphate (PIP2) hydrolysis to produce inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) [68,69,70]. IP3 accumulation stimulates Ca2+ release from internal stores, while activation of protein kinase C (PKC) and MAPK pathways are facilitated by DAG [71] (Figure 1).
With the inherent complexity of the modes of activation, transduction and biological output, it is not surprising that the orchestration of FGFR signalling is tightly regulated. We previously discussed the FGFR autoregulation via the acid box in Ig1 and its association with ligand binding Ig2 and Ig3 domains (Figure 1). However, there are several mediators associated with controlling the signalling output from activated FGFRs. Some of the known negative regulators are sprouty proteins that are induced by FGF signalling (Figure 1) [72,73]. Furthermore, FRS2 can be phosphorylated by MAPK on serine and threonine residues, inhibiting GRB2 recruitment and producing a negative feedback loop [74,75]. Other negative modulators of the FGF signalling pathway are the transmembrane proteins, similar expression to FGF genes (SEF), anosmin-1, fibronectin-leucine-rich transmembrane protein 3 (FLRT3), FGFRL1 and MAPK phosphatases (MKP) that can also interfere with the activation of downstream signalling pathways [76,77,78,79] (Figure 1). In addition to the above, the stimulated FGF–FGFR complex can be completely blocked by internalisation and subsequent lysosomal degradation. The E3 ubiquitin ligase Cbl binds to activated FRS2 and facilitates FGFR ubiquitination by acting as a signal for receptor degradation [80].
FGFR signalling clearly has profound direct effects on cancer cells. However, the FGFR axis also impacts on angiogenesis and this is an emerging field of translational medicine [81]. FGF2 has been heavily implicated as a proangiogenic factor, promoting endothelial proliferation and migration following FGFR1/2 signalling and VEGF/angiopoietin 2 secretion [82], and has been shown to mediate resistance to VEGFR targeted therapy in cancer [83]. In addition, other FGFs, such as FGF5 and FGF18, can promote angiogenesis through endothelial FGFR activation [84,85]. Interestingly, a recent study revealed an association of an FGFR1 mutation with spontaneous haemorrhage in paediatric and young adult low grade glioma, though the specific mechanism remains unclear [86]. In urothelial carcinomas, FGFR3 was able to induce a proangiogenic phenotype, suggesting that constitutive activation of FGFR3 may be able to potentiate growth factor signalling in the tumour microenvironment and implicating FGFR3 as a potential therapeutic target from an antiangiogenic perspective [87]. As with other behaviours, the effects of FGFR signalling can be context specific. In an embryoid body model, FGFR1 negatively regulated angiogenesis by altering the balance of cytokines, such as interleukin-4 and pleiotrophin [88].
5. Examples of the Involvement of FGFR Signalling in Development
Before discussing how FGFR signalling can drive cancer, it is important to understand how it is involved in development and why such a pleiotropic and dynamic pathway can be key in disease development. FGFR signalling plays a fundamental role in cell proliferation and migration. However, during embryonic development, FGF signalling regulates differentiation and the cell cycle. FGF signalling is important as early as in the preimplantation of embryos in mammals. For example, FGF4 is expressed in the morula and then in epiblast cells of the inner cell mass [89] and facilitates cell proliferation and the formation of the ectoderm [90,91]. There are reports of FGFR1 and FGFR2 in the inner cell mass and FGFR2 also in the embryonic ectoderm [92]. Later in development it has a vital role in organogenesis, particularly regulating the reciprocal crosstalk between epithelial and mesenchymal cells [93,94]. For example, FGFR2 plays an important function in both the ectoderm and mesenchyme during limb development [7]. More broadly, mesenchymal cells express FGFs, such as FGF4, 7, and 10, leading to downstream signalling activation through the epithelial 3b splice variant of FGFR1 and -2 in the epithelium and as a result, facilitate lung, salivary gland, intestine and limb development [95,96,97]. In contrast, epithelial tissue can secret FGFs 8 and 9 that activate FGFR1 and FGFR2-3c isoforms in the mesenchymal tissue [98,99]. However, organogenesis is not always driven exclusively via paracrine loops [11]. During development of the central nervous system, FGF8 signals in the anterior neural primordium by acting as an autocrine/paracrine factor in the development of the inner ear [100]. The differentiation of the cochlear sensory epithelium is regulated by autocrine/paracrine FGF signalling [101]. More recently it was found that FGFR can interact with N-Cadherin and activated FGFR that in turn facilitates migration of neocortical projection neurons [102]. FGFRs could regulate multipolar neuronal orientation and change them into bipolar cells to enter the cortical plate [102].
Given the importance of FGF signalling to development, it is unsurprising that malfunction can lead to developmental defects. The absence of FGFR1 in genetically modified mice leads to early growth defects [103]. Activated FGFR germline mutations can lead to skeletal disorders, such as a mutation in FGFR3 which can lead to growth defects and human dwarfism achondroplasia (ACH) [104,105]. A variety of inherited syndromes are caused by germline irregularities in FGFR [106]. Furthermore, mutations, especially in FGFR2, can lead to craniofacial malformation syndromes [107].
6. Aberrant FGFR Signalling in Cancer
The pleiotropic function of FGFR and its involvement in crucial physiological processes makes the FGFR signalling pathway a good candidate for facilitating cancer progression. In this section we will highlight the different ways FGFR signalling can be involved in the pathogenesis of cancer (Figure 3) and briefly give examples of FGFRs’ genetic alterations in different cancers (Figure 4).
One way of facilitating malignant progression via FGFR signalling is via a corrupted autocrine/paracrine loop and exon switching. Dysregulation of FGF secretion and FGFR expression in stromal and cancer cells can be a driving force in cancer progression. Many FGFs and their elevated levels are associated with cancer progression, for example FGF1, -2, -6, -8, -10, -19 and -23 [108,109,110,111,112,113,114,115,116,117,118]. Interestingly, FGFs are implicated in EMT in cancer by attributing mesenchymal characteristics in epithelial cells [119,120,121]. In some cases, growth factors (e.g., FGFs) are produced and secreted by one type of cell (for example stromal cells) and stimulate via paracrine signalling another type of cell to signal cell functions, such as proliferation, differentiation and migration [1].
However, cancer cells can synthesise FGFs and create a positive feedback loop via autocrine signalling. For example, in breast and non-small cell lung carcinomas, FGF2 and FGF9 are expressed and activate their respective FGFRs in the same cells [122,123]. Furthermore, FGF10 has been implicated as a key paracrine signal in breast, pancreatic, stomach, skin, lung and prostate cancer [108,124].
The specificity of FGF ligands can be altered through isoform switching and alternative splicing of FGFRs, thereby increasing the range of FGFs that can stimulate cancer cells, depending on the FGFR isoforms they express [8,35]. For example, alternative splicing of FGFR1 is associated with a high tumour grade and stage in bladder cancer [38]. Similarly, FGFR1 alternative FGFR1α/FGFR1β splicing was found to play a key role in breast cancer [34] and FGFR3 splicing promoted aggressiveness in prostate cancer [125].
Deregulation of negative regulators of the FGFR axis can also contribute to aberrant FGFR signalling in cancer. For example, SEF and SPRY expression levels are associated with breast, prostate, ovarian and thyroid cancer progression, with high grade carcinomas expressing lower levels of these negative FGFR regulators [126,127,128]. In contrast, a recent study reported that loss of SPRY1 improved the response to targeted therapy in melanoma [129] and suppression of SPRY1 inhibited triple-negative breast cancer malignancy via enhancing the estrogen growth factor and its receptor (EGF/EGFR) mediated mesenchymal phenotype [130].
Genetic alterations of FGFR can also dysregulate signalling and contribute towards malignant progression. Next-Generation Sequencing (NGS) analysis of 4853 tumours revealed FGFR aberrations in 7.1% of cancers [131]. More specifically, 66% of the aberrations were gene amplification, 26% were gene mutations and 8% were rearrangements [131]. A recent study on advanced urothelial cancer using NGS to analyze cell-free DNA from the plasma of 997 patients, revealed that 20% had FGFR2 and FGFR3 genomic alterations, of which 14% were activating mutations [132].
6.1. Activating Mutations
The most common types of genetic variation are single nucleotide polymorphisms (SNPs). FGFR2 harbours one of the first SNPs to be identified as a breast cancer susceptibility locus by Genome-Wide association studies (GWAS) [133,134]. Risk alleles of various SNPs found in FGFR2, they are associated with ER-positive cancers [135], increased FGFR2 expression [136], lymph node metastasis in breast cancer [137] and radiation-induced breast cancer risk [138]. More recently another study identified an FGFR2 SNP that was linked with susceptibility to breast cancer in a Chinese population [139]. However, only a few SNP loci are confirmed in FGFR1 that correlate significantly with a breast cancer predisposition [140]. In contrast, a more recent study correlated three FGFR1 SNPs to reduced breast cancer risk [141]. SNPs in FGFR4 but not in FGFR3 were strongly correlated with breast cancer [142]. In breast cancer patients, FGFR4 and FGFR2 SNPs were previously suggested to be candidate pharmacogenomic factors to predict the response to chemotherapy [143]. Notably, SNPs in the FGF/FGFR axis (FGF1, FG18, FGF7, FGF23 and FGF5) were also associated with an increased risk of ovarian cancer [144].
A number of germ line activating point mutations in FGFR1, -2 and -3 are found in prostate, breast, bladder, endometrial, brain, lung, uterus, cervical, stomach, head and neck, colon and melanoma cancers (as reviewed by [145]). These mutations can alter the ligand binding, juxtamembrane and kinase domains and constitutively activate FGFR or impair FGFR degradation, leading to increased FGFR signalling (as reviewed by [111,145,146,147]. FGFR4 activating mutations are not detected very often, apart from in rhabdomyosarcoma [148] and gastric cancer [149]. Interestingly, some of the activating mutations can result in changes in the efficacy of several inhibitors that can target FGFR, such as AZD4547, BGJ-398, KTI258, AP24534 and JNJ42756493 [150].
6.2. FGFR Gene Amplification and Overexpression
Elevated FGFR levels can be achieved either via chromosomal amplification or aberrant transcription (Figure 3). In cancer, distinctive FGFR abnormalities are known such as the amplification of genes or post-transcriptional regulation, contributing to overexpression of the receptor. Mutations in FGFRs could generate receptors that are either consistently active or may demonstrate a reduced necessity of activation through ligand binding [11]. The most common abnormalities in malignancies are due to gene amplification of FGFR1, -2 and -3, as well as FGF ligands. Several studies have highlighted that FGFR is amplified in various cancers. For example, FGFR1 expression is amplified in bladder, oral, oesophageal squamous, NSCLC, prostate and ovarian cancers [151,152,153,154]. FGFR1 amplification and overexpression was observed in some patients with lymph node metastasis and advanced pathological stages of hypopharyngeal and laryngeal squamous cell carcinoma [155]. In addition, hormone receptor positive breast cancer patients with metastatic disease had FGFR1 amplification that was associated with a shorter time to progression on first line endocrine therapy [156]. Furthermore, it was suggested that FGFR1 amplification grants resistance to estrogen receptor (ER), PI3K, mammalian target of rapamycin (mTOR) and cyclin-dependent kinase (CDK)4/6 inhibitors [157]. FGFR2 amplification in gastric cancer is associated with a poor prognosis and response to chemotherapy [158].
6.3. Chromosomal Translocation
The exchange of chromosomal arms (or segments) between heterologous chromosomes, known as chromosomal translocation, is a type of structural chromosomal abnormality that results in fusion genes/proteins. The generated fusion proteins can have oncogenic properties. Chromosomal translocations in FGFRs have about an 8% incidence rate [131]. There are two types of FGFR gene fusions: (1) when only the FGFR tyrosine kinase domain is fused to the 5′ end of the fusion protein (the extracellular and transmembrane domain portion of the FGFR is missing from the fusion protein), therefore is constitutively dimerised and active; (2) when the whole FGFR remains intact and acts as the 5′ fusion gene that will bind to its partner at the 3′ end of the FGFR [147].
The first reports of FGFR fusion genes were in haematological malignancies. The FGFR kinase domain was fused with the N terminus of transcription factors such as ETV6, ZNF198 and BCR in lymphoma/leukaemia patients with myeloproliferative disorder stem cell syndrome [159,160,161,162]. A recent study reported EVT6-FGFR2 fusion protein in a mixed phenotype (T-myeloid/lymphoid) acute leukaemia, that resulted in aberrant FGFR2 tyrosine kinase expression and was correlated with aggressive clinical behaviour and a poor response to therapy [163]. FGFR1, FGFR2 and FGFR3 fusions are also identified in solid tumours, such as lung, colorectal, glioblastoma, breast, head and neck, bladder, cervical cancer and cholangiocarcinoma (as reviewed by [164]). A common fusion is FGFR3 with transforming acidic coiled-coil 3 (TACC3) that induces a constitutive phosphorylation of the tyrosine kinase domain and therefore activation of downstream MAPK and STAT1 pathways that further leads to increased metastatic cell behaviour (e.g., cell proliferation) [165,166,167]. There are several identified binding partners for FGFR2, some of them are TACC3 and CCDC6 in cholangiocarcinoma [166,168] and BICC1 in hepatocarcinoma and colorectal cancer [169]. Examples of FGFR1 fusion partners are HOOK3 in gastrointestinal stromal tumour, TACC1 in glioblastoma and ZNF703 in breast cancer [167,170,171,172]. A recent genomic profiling study identified ANO3 and NSD1 as fusion partners for FGFR4 in non-small cell lung cancer [173]. Although FGFR fusions are relatively rare in human cancers it might be of interest to identify how patients with FGFR fusions respond to therapy targeting the tyrosine kinase (TK) domain of FGFR.
7. Nuclear FGFR in Cancer
FGFRs have been shown to signal via the cell membrane and endosomal compartments via downstream signalling pathways. However, studies have suggested that other TK receptors as well as FGFRs and FGFs, can target the nucleus and carry out functions that might not be dependent on tyrosine kinase activity [174,175,176,177,178,179,180,181,182]. Examples of nuclear FGFs are FGF1, that stimulated DNA synthesis, and FGF2 that was associated with increased cell proliferation in glioma cells and invasion in pancreatic cancer [11,183,184]. Both FGFR1 and FGFR2 have been reported to function in the nucleus [183,185,186]. Nuclear FGFR2 was recently found to negatively regulate hypoxia-induced cell invasion in prostate cancer [187] and nuclear FGFR1 was positively corelated with pancreatic and breast cancer progression [178,179].
Although there are strong indications, it is still not fully understood how FGF(R)s travel to the nucleus and what their mode of action is once there. Several researchers have highlighted the mechanisms by which full length TK receptors translocate via the cell membrane to the nucleus. For example, upon binding of the ligands, the activated receptors get internalised to the early endosomal compartments either via the vesicular pathway or after retro-translocation from the endoplasmic reticulum (ER) to the cytosol [181,188,189,190]. The molecular mechanism by which the receptor escapes the endosomal pathway to travel to the nucleus remains elusive and conflicting data point to different trafficking possibilities. One of the possible mechanisms for nuclear translocation of full length FGFR involves retro-translocation of FGFR from the ER/Golgi apparatus [183]. Typically, after co-translational insertion into the ER membranes, FGFR1 traffics via the vesicular transport systems through the Golgi apparatus to reach the plasma membrane [185,191]. This process may be accompanied by retro-translocation of the pool of FGFR into the cytosol, with FGFR1 undergoing retrograde transport via the sec61 channel, similarly to ER-associated protein degradation [183]. Once in the cytosol, FGFRs interact with ribosomal S6-kinase 1 and FGF2 which facilitates receptor transport to the nucleus to directly regulate gene expression [185,191]. Full length FGFR is a molecule too large to pass through the nuclear membrane via diffusion, and another mechanism involves the full-length receptor in the cytoplasm activating the importin beta pathway to enter the nucleus [176]. The nuclear receptor can then interact with other nuclear proteins to control transcription [185,192,193].
An alternative is that the nuclear trafficking of the receptor is dependent on proteolytic cleavage of the intracellular domain allowing translocation to the nucleus of the unrestricted cytoplasmic portion [179,194]. There are several mechanisms utilised by tyrosine kinase receptors to reach the nucleus, but generation of nuclear RTK fragments via alternative splicing of the receptor or proteolytic cleavage of FGFRs/RTK with caspases, secretases, granzymes and other proteases (e.g., ADAM10/15/17) [179,184,186,188], are increasingly reported. The FGF receptor can be present in a cleaved form before trafficking to the nucleus, and there are indications suggesting this proteolytic pathway might be FGFR kinase activity-dependent [179]. Previous studies indicated that Notch1 and FGFR1 can be cleaved by Granzyme B (GrB) [189]. In breast cancer cells, FGFR activation-dependent cleavage of FGFR1 generates a C-terminal fragment that can translocate to the nucleus and control the expression of target genes [179]. Nuclear FGFR1 could control the oncogenic networks involved in organ development, tissue and cell pluripotency, cell cycle, cancer related TP53 pathway, neuroectodermal and mesodermal programming networks, axonal growth and synaptic plasticity pathways [190].
Therefore, there might be a novel mechanism by which FGFR signalling can control metastatic cancer cell behaviour. This further suggests a potentially novel therapeutic target for invasive cancer treatment.
8. Targeting FGFR Signalling in Cancer
One of the main obstacles in cancer therapy is chemoresistance and radioresistance. There is evidence highlighting the possible role of the FGFR axis in the development of drug resistance. For example, overexpression of FGF2 and FGF1 are linked with both in vivo and in vitro resistance to cancer drugs such as doxorubicin, 5-fluorouracil and paclitaxel [195]. Interestingly, a pan-FGFR inhibitor (BGJ398) was able to overcome paclitaxel resistance in FGFR1 expressing urothelial carcinoma [192]. Another study identified FGFR4 as a targetable element of drug resistance in colorectal cancer [193]. Increased FGFR1 and FGF3 expression was correlated with a poor response to anti-HER2 treatment in breast cancer patients, and this was overcome using a combination therapy of FGFR inhibitors together with lapatinib and trastuzumab [196]. Overexpression of FGFR3 was also linked with tamoxifen resistant breast cancer [197]. In afatinib-resistant non-small cell lung cancer cells, overexpression of FGFR1 and FGF2 played a role in overcoming cell survival by compensating the loss of the estrogen growth factor receptor (EGFR)-driven signalling pathway [198]. In addition, gefitinib sensitivity was also restored in non-small cell lung cancer cells when FGF2 and FGFR1 were inhibited via siRNA and treatment with a small molecule inhibitor, PD173074, suggesting FGFR activation as a potential mechanism of acquired resistance to EGFR-TKs [199]. In FGFR1 amplified lung cancer, a combination therapy approach overcame resistance to treatment with an FGFR inhibitor [200]. In EGFR-dependent cancers of multiple cell lineages, FGFR3-TACC3 fusion proteins are also characterised as “naturally occurring drivers of tumour resistance” by reactivating EGFR/ERK signalling [201]. Considering all the evidence together, this highlights the importance of targeting the FGFR axis in combination therapies tailored for different cohorts of patients.
Therapeutic targeting of FGFs and their receptors is a key area of drug development. Several drugs targeting FGF pathways are currently under clinical investigation (Table 1). However, abrogating FGFR signalling can be accomplished by targeting the diverse components present in the pathway, which include the ligands, receptors as well as the products of the downstream signalling pathway [61] (Figure 5). Nevertheless, converting knowledge into a treatment for patients has proven challenging as even specific inhibitors targeting FGFR have off-target effects [202,203,204]. Hence, further research is necessary to determine the mechanisms of effective targeting of FGFR signalling in cancer without obstructing its fundamental functions in healthy cells.
The FGFR targeting field has progressed significantly, as novel agents inhibiting FGF ligands or using monoclonal antibodies and FGF ligand traps have been developed as well as using FGFR non-selective and selective inhibitors (Figure 4). The ATP-competitive small molecules were the first FGFR inhibitors [205,206]. PDGFR and VEGFR share comparable structural homology to FGFRs, hence these inhibitors can act as multitarget tyrosine kinase inhibitors (TKIs) as they also bind and act on the conserved ATP-binding regions.
One of the non-selective FGFR TKIs is dovitinib (TKI 258, Novartis, Basel, Switzerland), which is in phase II/III clinical trials, and this has been shown to have a strong affinity to FGFR3 resulting in the inhibition of downstream signalling, blocking cell proliferation and promoting apoptosis [61,207]. Dovitinib likewise inhibits other members of the TK family due to a lack of drug specificity including FGFR1, PDGFR and VDGFR [208,209,210]. A pilot study evaluated the efficacy of an orally bioavailable multitargeted tyrosine kinase inhibitor, ponatinib, that inhibits all FGFRs as well as other kinases (such as KIT, RET, SRC, VEGFR and PDGFR) [211,212]. Their findings demonstrated a clinical benefit response in over 45% of the patients, suggesting a potential antitumour activity of ponatinib in biliary tract cancer patients with altered FGFR2 [212]. AZD4547 (AstraZeneca, Cambridge, UK) is a highly potent selective FGFR1-3 inhibitor. Phase I/II clinical trials have indicated that AZD4547 can target cancers, such as gastric/esophagogastric, bladder, gastric adenocarcinoma, lung and breast, with FGFR1 and -2 amplifications [213,214,215,216,217,218,219,220]. A recent detailed literature review using a wide range of databases and utilising a systematic review approach, demonstrated that clinical trials using selective FGFR inhibitors (i.e., erdafitinib JNJ 42756493, Infigratinib BGJ398, Rogaritinib BAY 1163877, PD173074, BLU9931, AZD4547, Pemigatinib INCB54828, LY2874455, DEBIO 1347, Futibatinib TAS-120) in advanced urothelial cancer had significant antitumour activity [221].
Infigratinib (a pan-FGFR kinase inhibitor) was evaluated in a phase 2 study for biliary tract carcinoma with FGFR alterations, with all responsive tumours containing FGFR2 fusions. The overall response rate for FGFR2 fusions was 18.8% and the disease control rate was 83.3% with an estimated median progression-free survival of 5.8 months [222]. Currently there are seven phase 1 and 2 clinical trials evaluating Infigratinib in gastric, adenocarcinoma, breast, advanced malignant solid neoplasm, bladder, renal pelvis and ureter urothelial carcinoma, advanced cholangiocarcinoma and glioblastoma (NCT05019794, NCT04504331, NCT04233567, NCT04972253, NCT04197986, NCT04228042, NCT02150967, NCT04424966). Most importantly, there are two phase 3 clinical trials investigating Infigratinib as a possible cancer treatment for upper tract urothelial carcinoma/urothelial bladder cancer (NCT04197986) and advanced cholangiocarcinoma (NCT03773302).
In trials using Erdafitinib (another a pan-FGFR kinase inhibitor), the rate of confirmed response in advanced metastatic urothelial carcinoma was 40%, with the median duration of progression-free survival at 5.5 months, and median duration of overall survival at 13.8 months [223]. In a phase 2 study on cholangiocarcinoma patients with FGFR alterations, it was reported that the disease control rate was 83.3% and median progression free survival was 5.59 months. In 10 evaluable FGFR2+ patients the disease control rate was 100% and the median progression-free survival was 12.35 months [224]. Currently, there are nineteen phase 1 and 2 clinical trials on Erdafitinib and cancers such as breast, bladder/urinary bladder, lung, advanced solid tumours, urothelial, prostate, and multiple myeloma (NCT03238196, NCT04917809, NCT04172675, NCT02699606, NCT04083976, NCT02365597, NCT03547037, NCT03999515, NCT04754425, NCT05052372, NCT03827850, NCT03210714, NCT03473743, NCT04963153, NCT03955913, NCT03088059 NCT02925234, NCT02465060, NCT03732703, NCT03155620). There is also a phase 3 clinical trial evaluating Erdafitinib in urothelial cancer (NCT03390504).
Another pan-FGFR inhibitor, Rogaratinib, showed excellent in vivo efficacy in FGFR overexpressing preclinical cancer models [225]. There are five phase 1 and 3 clinical trials studying Rogaratinib in breast, lung, gastrointestinal stromal, urothelial, and squamous cell head and neck cancers (NCT04483505, NCT03762122, NCT04595747, NCT03473756, NCT03088059).
Therapeutic monoclonal antibodies have been established with the rationale that they could target FGF ligands and FGFR isoforms with a high specificity, hence offering an alternative to inhibitors that might have side effects [226,227]. Antibodies can compromise the other benefit of employing the immune system to synergise with the antitumour activity via antibody-dependent cellular cytotoxicity or complement-dependent cytotoxicity. A number of anti-FGFR monoclonal antibodies have also been considered in preclinical studies [228,229]. Human anti-FGFR3 mAb, MFGR1877S (Genentech), is a monoclonal antibody against FGFR3 and has been used against multiple myeloma and MFGR1877S and has also shown antitumour activity for overexpressed FGFR3 in preclinical models of bladder cancer [221,229,230,231,232,233]. Phase I clinical trials of MFGR187S have been carried out in t(4;14) translocated multiple myeloma patients [233]. Furthermore, GP369 is a specific and potent anti-FGFR2b monoclonal antibody that suppresses phosphorylation and the downstream signalling induced by ligand binding. FGFR2 activated signalling in mice significantly inhibited the growth of human cancer xenografts in the presence of GP369 [234,235].
Antibodies against FGF2 and FGF8 have also shown promising results in inhibiting tumour progression and angiogenesis [236,237] A human single-chain variable fragment (ScFvs; 1A2) that binds to human FGF2 was identified via screening of a human scFv phage library [238]. This purified antibody inhibited various biological functions of FGF2, such as proliferation/growth, migration and tube formation of human umbilical vein endothelial cells and apoptosis in glioma cells in vitro [238].
An alternative method of inhibiting FGFR signalling is via a ligand trap to isolate FGF ligands preventing them from binding to and activating FGFRs [112,239,240]. FP-1039 (GlaxoSmithKline, GSK3052230) is a soluble fusion protein that consists of an extracellular FGFR1-IIIc domain fused to the Fc portion of IgG1 that inhibits the binding of FGF1, -2, and -4 to FGFR1 and has shown promising results in solid tumours [241,242,243]. Other FGF2 antagonists are small molecules such as sm27, PI-88, pentosan and pentraxin-3 [8]. Because of the ability to bind to heparin/heparan sulphate, chemical compounds mimicking heparin (i.e., suramin) could antagonise FGF2 binding and inhibit its action [244]. Peg-interferon alpha-2b was also able to suppress the plasma FGF2 level in melanoma patients with metastasis and gave a clinical response [245]. FGF2-induced angiogenesis was also inhibited by sulfonic acid polymers such as PAMPS, small molecules such as sirolimus, PI-88, thalidomide, suramin and platelet factor 4 protein (as reviewed by [246]).
Not much is known about the mechanism by which FGFR inhibitors induce cell death. Recent work on endometrial cancer showed that FGFR inhibitors (Infigratinib, AZD4547 and PD173074) caused mitochondrial depolarisation, cytochrome c release and impaired mitochondrial respiration in two FGFR2-mutant endometrial cancer cell lines (AN3CA and JHUEM2). However, they did not detect caspase activation following FGFR inhibition. When they were treated with the pan-caspase inhibitor (Z-VAD-FMK) they did not prevent cell death, suggesting that the cell death was caspase-independent [247]. Bcl-2 inhibitors enhanced FGFR inhibitor-induced mitochondrial-dependent endometrial cancer cell death [247]. Interestingly, in another study, Infigratinib induced cell death in non-small cell lung cancer cells (H1581) by activating the caspase-dependent mitochondrial and non-mitochondrial pathway [239]. In high-grade bladder cancer cells, a combination treatment with Infigratinib and a novel histone deacetylase inhibitor (OBP-801/YM753/spiruchostatin A), inhibited cell growth and markedly induced apoptosis, by activating caspase-3, -8 and -9. Interestingly, a pan-caspase inhibitor (Z-VAD-FMK) significantly reduced the apoptotic response to the combined treatment. The combination treatment was shown to be at least partially dependent on Bim [240].
9. Conclusions
Even though drugs targeting tyrosine kinase activity (e.g., HER2, FGFR, EGFR, VEGFR2), can prolong survival by inducing cancer regression, the lack of selectivity to a single target and/or development of drug resistance remains a problem. The heterogeneous nature of cancer, the involvement of the tumour microenvironment, together with the pleiotropic way FGFR signalling functions, highlights the need for a more personalised approach in cancer treatment and combination therapies. Experimental data and clinical trials focusing on targeting the FGFR axis have demonstrated positive outcomes. An awareness of FGFR genetic alterations or the FGFR mode of action in cancer patients (e.g., whether FGFR acts via a paracrine or autocrine mechanism in a specific tumour) is important for tailoring combinations of targeted therapies aiming at the FGFR axis. For example, using small molecule FGFR inhibitors, RNA based drugs, FGF traps and humanised/human anti-FGFR monoclonal antibodies in combination with targeting the immune system and/or other signalling pathways. A better understanding of FGFR biology could also help in identifying the mechanisms of drug resistance to FGFR inhibitors and facilitating their bypass. Developing diagnostic assays to screen patients for FGF and FGFR status for a targeted approach might help improve treatment efficacy.
Author Contributions
Conceptualisation, A.-M.C.; writing—original draft preparation, A.-M.C.; writing—review and editing, R.P.G.; visualisation, A.-M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Senga, S.S.; Grose, R.P. Hallmarks of cancer-the new testament. Open Biol. 2021, 11, 200358. [Google Scholar] [CrossRef]
- Ornitz, D.M. FGFs, heparan sulfate and FGFRs: Complex interactions essential for development. BioEssays 2000, 22, 108–112. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer. 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Xu, J.; Colvin, J.S.; McEwen, D.G.; MacArthur, C.A.; Coulier, F.; Gao, G.; Goldfarb, M. Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 1996, 271, 15292–15297. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Ibrahimi, O.A.; Olsen, S.K.; Umemori, H.; Mohammadi, M.; Ornitz, D.M. Receptor specificity of the fibroblast growth factor family—The complete mammalian FGF family. J. Biol. Chem. 2006, 281, 15694–15700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [Green Version]
- Olsen, S.K.; Garbi, M.; Zampieri, N.; Eliseenkova, A.V.; Ornitz, D.M.; Goldfarb, M.; Mohammadi, M. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J. Biol. Chem. 2003, 278, 34226–34236. [Google Scholar] [CrossRef] [Green Version]
- Sochacka, M.; Opalinski, L.; Szymczyk, J.; Zimoch, M.B.; Czyrek, A.; Krowarsch, D.; Otlewski, J.; Zakrzewska, M. FHF1 is a bona fide fibroblast growth factor that activates cellular signaling in FGFR-dependent manner. Cell Commun. Signal. 2020. 18, 69. [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Tiong, K.; Mah, L.; Leong, C.-O. Functional roles of fibroblast growth factor receptors (FGFRs) signaling in human cancers. Apoptosis 2013, 18, 1447–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Kan, M.; Yan, G.; Xu, J.; McKeehan, W. Alternately spliced NH2-terminal immunoglobulin-like loop I in the ectodomain of the fibroblast growth factor (FGF) receptor 1 lowers affinity for both heparin and FGF-1. J. Biol. Chem. 1995, 270, 10231–10235. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Olsen, S.K.; Ibrahimi, O.A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005, 16, 107–137. [Google Scholar] [CrossRef]
- Kalinina, J.; Dutta, K.; Ilghari, D.; Beenken, A.; Goetz, R.; Eliseenkova, A.V.; Cowburn, D.; Mohammadi, M. The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition. Structure 2012, 20, 77–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, I.; Kimura-Yoshida, C. Extracellular modulation of Fibroblast growth factor signaling through heparan sulfate proteoglycans in mammalian development. Curr. Opin. Genet. Dev. 2013, 23, 399–407. [Google Scholar] [CrossRef]
- Wiedemann, M.; Trueb, B. Characterization of a novel protein (FGFRL1) from human cartilage related to FGF receptors. Genomics 2000, 69, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, M.; Fraser, J.; McDonald, M.; Yuan, S.; White, D.; Grandison, P.; Kumble, K.; Watson, J.D.; Murison, J.G. Identification of a new fibroblast growth factor receptor, FGFR5. Gene 2001, 271, 171–182. [Google Scholar] [CrossRef]
- Gerber, S.D.; Beauchamp, P.; Zhuang, L.; Villiger, P.M.; Trueb, B. Functional domains of the FgfrL1 receptor. Dev. Biol. 2020, 461, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Vogel, M.; Villiger, P.M.; Trueb, B. Dissecting the interaction of FGF8 with receptor FGFRL1. Biomolecules 2020, 10, 1399. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Williams, L.T. Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res. 1993, 60, 1–41. [Google Scholar] [PubMed]
- Furdui, C.M.; Lew, E.D.; Schlessinger, J.; Anderson, K.S. Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol. Cell 2006, 21, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gorry, M.C.; Post, J.C.; Ehrlich, G.D. Genomic organization of the human fibroblast growth factor receptor 2 (FGFR2) gene and comparative analysis of the human FGFR gene family. Gene 1999, 230, 69–79. [Google Scholar] [CrossRef]
- Rebscher, N.; Deichmann, C.; Sudhop, S.; Fritzenwanker, J.H.; Green, S.; Hassel, M. Conserved intron positions in FGFR genes reflect the modular structure of FGFR and reveal stepwise addition of domains to an already complex ancestral FGFR. Dev. Genes Evol. 2009, 219, 455–468. [Google Scholar] [CrossRef]
- Johnson, D.E.; Lu, J.; Chen, H.; Werner, S.; Williams, L.T. The human fibroblast growth factor receptor genes: A common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol. Cell. Biol. 1991, 11, 4627–4634. [Google Scholar]
- Dell, K.R.; Williams, L.T. A novel form of fibroblast growth factor receptor 2. Alternative splicing of the third immunoglobulin-like domain confers ligand binding specificity. J. Biol. Chem. 1992, 267, 21225–21229. [Google Scholar]
- Werner, S.; Duan, D.S.; de Vries, C.; Peters, K.G.; Johnson, D.E.; Williams, L.T. Differential splicing in the extracellular region of fibroblast growth factor receptor 1 generates receptor variants with different ligand-binding specificities. Mol. Cell. Biol. 1992, 12, 82–88. [Google Scholar]
- Avivi, A.; Yayon, A.; Givol, D. A novel form of FGF receptor-3 using an alternative exon in the immunoglobulin domain III. FEBS Lett. 1993, 330, 249–252. [Google Scholar] [CrossRef] [Green Version]
- Chellaiah, A.T.; McEwen, D.G.; Werner, S.; Xu, J.; Ornitz, D.M. Fibroblast growth factor receptor (FGFR) 3. Alternative splicing in immunoglobulin-like domain III creates a receptor highly specific for acidic FGF/FGF-1. J. Biol. Chem. 1994, 269, 11620–11627. [Google Scholar] [CrossRef]
- Scotet, E.; Houssaint, E. Exon III splicing switch of fibroblast growth factor (FGF) receptor-2 and -3 can be induced by FGF-1 or FGF-2. Oncogene 1998, 17, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Ishiwata, T. Role of fibroblast growth factor receptor-2 splicing in normal and cancer cells. Front. Biosci. (Landmark Ed) 2018, 23, 626–639. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Zhuo, M.L.; Zheng, X.; Su, X.; Meric-Bernstam, F. FGFR1beta is a driver isoform of FGFR1 alternative splicing in breast cancer cells. Oncotarget 2019, 10, 30–44. [Google Scholar] [CrossRef]
- Holzmann, K.; Grunt, T.; Heinzle, C.; Sampl, S.; Steinhoff, H.; Reichmann, N.; Kleiter, M.; Hauck, M.; Marian, B. Alternative splicing of fibroblast growth factor receptor IgIII loops in cancer. J. Nucleic Acids 2012, 2012, 950508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy Burman, D.; Das, S.; Das, C.; Bhattacharya, R. Alternative splicing modulates cancer aggressiveness: Role in EMT/metastasis and chemoresistance. Mol. Biol. Rep. 2021, 48, 897–914. [Google Scholar] [CrossRef] [PubMed]
- Wheldon, L.M.; Khodabukus, N.; Patey, S.J.; Smith, T.G.; Heath, J.K.; Hajihosseini, M.K. Identification and characterization of an inhibitory Fibroblast growth factor receptor 2 (FGFR2) molecule, upregulated in an Apert Syndrome mouse model. Biochem. J. 2011, 436, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, D.C.; Knowles, M.A. Altered splicing of FGFR1 is associated with high tumor grade and stage and leads to increased sensitivity to FGF1 in bladder cancer. Am. J. Pathol. 2010, 177, 2379–2386. [Google Scholar] [CrossRef]
- Johnson, D.E.; Lee, P.L.; Lu, J.; Williams, L.T. Diverse forms of a receptor for acidic and basic fibroblast growth factors. Mol. Cell. Biol. 1990, 10, 4728–4736. [Google Scholar]
- Reid, H.H.; Wilks, A.F.; Bernard, O. Two forms of the basic fibroblast growth factor receptor-like mRNA are expressed in the developing mouse brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1596–1600. [Google Scholar] [CrossRef] [Green Version]
- Dionne, C.A.; Crumley, G.; Bellot, F.; Kaplow, J.M.; Searfoss, G.; Ruta, M.; Burgess, W.H.; Jaye, M.; Schlessinger, J. Cloning and expression of two distinct high-affinity receptors cross-reacting with acidic and basic fibroblast growth factors. EMBO J. 1990, 9, 2685–2692. [Google Scholar] [CrossRef]
- Crumley, G.; Bellot, F.; Kaplow, J.M.; Schlessinger, J.; Jaye, M.; Dionne, C.A. High-affinity binding and activation of a truncated FGF receptor by both aFGF and bFGF. Oncogene 1991, 6, 2255–2262. [Google Scholar] [PubMed]
- Moffa, A.B.; Tannheimer, S.L.; Ethier, S.P. Transforming potential of alternatively spliced variants of fibroblast growth factor receptor 2 in human mammary epithelial cells. Mol. Cancer Res. 2004, 2, 643–652. [Google Scholar] [PubMed]
- Olender, J.; Wang, B.D.; Ching, T.; Garmire, L.X.; Garofano, K.; Ji, Y.; Knox, T.; Latham, P.; Nguyen, K.; Rhim, J.; et al. A novel FGFR3 splice variant preferentially expressed in african american prostate cancer drives aggressive phenotypes and docetaxel resistance. Mol. Cancer Res. 2019, 17, 2115–2125. [Google Scholar] [CrossRef]
- Shimizu, A.; Tada, K.; Shukunami, C.; Hiraki, Y.; Kurokawa, T.; Magane, N.; Kurokawa-Seo, M. A novel alternatively spliced fibroblast growth factor receptor 3 isoform lacking the acid box domain is expressed during chondrogenic differentiation of ATDC5 cells. J. Biol. Chem. 2001, 276, 11031–11040. [Google Scholar] [CrossRef] [Green Version]
- Vainikka, S.; Partanen, J.; Bellosta, P.; Coulier, F.; Birnbaum, D.; Basilico, C.; Jaye, M.; Alitalo, K. Fibroblast growth factor receptor-4 shows novel features in genomic structure, ligand binding and signal transduction. EMBO J. 1992, 11, 4273–4280. [Google Scholar] [CrossRef]
- Trueb, B. Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cell. Mol. Life Sci. 2011, 68, 951–964. [Google Scholar] [CrossRef] [Green Version]
- Larsson, H.; Klint, P.; Landgren, E.; Claesson-Welsh, L. Fibroblast growth factor receptor-1-mediated endothelial cell proliferation is dependent on the Src homology (SH) 2/SH3 domain-containing adaptor protein Crk. J. Biol. Chem. 1999, 274, 25726–25734. [Google Scholar] [CrossRef] [Green Version]
- Minden, A.; Lin, A.; Claret, F.X.; Abo, A.; Karin, M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell 1995, 81, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, A.N.; Schlessinger, J.; Hubbard, S.R.; Mohammadi, M. Structural basis for fgf receptor dimerization and activation. Cell 1999, 98, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, M.; Hashimoto, Y.; Muroya, K.; Hasegawa, H.; Kurata, T.; Tanaka, S.; Nakamura, S.; Hattori, S. CRK protein binds to two guanine nucleotide-releasing proteins for the Ras family and modulates nerve growth factor-induced activation of Ras in PC12 cells. Mol. Cell. Biol. 1994, 14, 5495–5500. [Google Scholar]
- Mochizuki, N.; Ohba, Y.; Kobayashi, S.; Otsuka, N.; Graybiel, A.M.; Tanaka, S.; Matsuda, M. Crk activation of JNK via C3G and R-Ras. J. Biol. Chem. 2000, 275, 12667–12671. [Google Scholar] [CrossRef] [Green Version]
- Kuro, O.M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef]
- Hu, M.C.; Shiizaki, K.; Kuro-o, M.; Moe, O.W. Fibroblast growth factor 23 and Klotho: Physiology and pathophysiology of an endocrine network of mineral metabolism. Annu. Rev. Physiol. 2013, 75, 503–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, Z.; Timsah, Z.; Suen, K.M.; Cook, N.P.; Lee, G.R.; Lin, C.-C.; Gagea, M.; Marti, A.A.; Ladbury, J.E. Grb2 monomer-dimer equilibrium determines normal versus oncogenic function. Nat. Commun. 2015, 6, 7354. [Google Scholar] [CrossRef] [PubMed]
- Kouhara, H.; Hadari, Y.R.; Spivak-Kroizman, T.; Schilling, J.; Bar-Sagi, D.; Lax, I.; Schlessinger, J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 1997, 89, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Dolfi, F.; Garcia-Guzman, M.; Ojaniemi, M.; Nakamura, H.; Matsuda, M.; Vuori, K. The adaptor protein Crk connects multiple cellular stimuli to the JNK signaling pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 15394–15399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoh, N.; Laks, S.; Nakashima, M.; Lax, I.; Schlessinger, J. FRS2 family docking proteins with overlapping roles in activation of MAP kinase have distinct spatial-temporal patterns of expression of their transcripts. FEBS Lett. 2004, 564, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Hadari, Y.R.; Kouhara, H.; Lax, I.; Schlessinger, J. Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol. Cell. Biol. 1998, 18, 3966–3973. [Google Scholar] [CrossRef] [Green Version]
- Harada, A.; Katoh, H.; Negishi, M. Direct interaction of Rnd1 with FRS2 beta regulates Rnd1-induced down-regulation of RhoA activity and is involved in fibroblast growth factor-induced neurite outgrowth in PC12 cells. J. Biol. Chem. 2005, 280, 18418–18424. [Google Scholar] [CrossRef] [Green Version]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.-C. Targeting FGFR signaling in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.H.; Hadari, Y.R.; Gotoh, N.; Guy, G.R.; Schlessinger, J.; Lax, I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 6074–6079. [Google Scholar] [CrossRef] [Green Version]
- Suryawanshi, A.; Schaefer, K.; Holz, O.; Apel, D.; Lange, E.; Hayward, D.C.; Miller, D.J.; Hassel, M. What lies beneath: Hydra provides cnidarian perspectives into the evolution of FGFR docking proteins. Dev. Genes Evol. 2020, 230, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Su, W.C.; Kitagawa, M.; Xue, N.; Xie, B.; Garofalo, S.; Cho, J.; Deng, C.; Horton, W.A.; Fu, X.Y. Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature 1997, 386, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, L.; Iwata, T.; Kitagawa, M.; Fu, X.Y.; Deng, C.X. A Lys644Glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Hum. Mol. Genet. 1999, 8, 35–44. [Google Scholar] [CrossRef]
- Hart, K.C.; Robertson, S.C.; Kanemitsu, M.Y.; Meyer, A.N.; Tynan, J.A.; Donoghue, D.J. Transformation and stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene 2000, 19, 3309–3320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudka, A.A.; Sweet, S.M.; Heath, J.K. Signal transducers and activators of transcription-3 binding to the fibroblast growth factor receptor is activated by receptor amplification. Cancer Res. 2010, 70, 3391–3401. [Google Scholar] [CrossRef] [Green Version]
- Bunney, T.D.; Wan, S.; Thiyagarajan, N.; Sutto, L.; Williams, S.V.; Ashford, P.; Koss, H.; Knowles, M.A.; Gervasio, F.L.; Coveney, P.V.; et al. The effect of mutations on drug sensitivity and kinase activity of fibroblast growth factor receptors: A combined experimental and theoretical study. EBioMedicine 2015, 2, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, M.; Honegger, A.M.; Rotin, D.; Fischer, R.; Bellot, F.; Li, W.; Dionne, C.A.; Jaye, M.; Rubinstein, M.; Schlessinger, J. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-gamma 1. Mol. Cell. Biol. 1991, 11, 5068–5078. [Google Scholar] [PubMed] [Green Version]
- Berridge, M.J. Inositol trisphosphate and calcium signalling. Nature 1993, 361, 315–325. [Google Scholar] [CrossRef]
- Cross, M.J.; Lu, L.; Magnusson, P.; Nyqvist, D.; Holmqvist, K.; Welsh, M.; Claesson-Welsh, L. The Shb adaptor protein binds to tyrosine 766 in the FGFR-1 and regulates the Ras/MEK/MAPK pathway via FRS2 phosphorylation in endothelial cells. Mol. Biol. Cell 2002, 13, 2881–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felfly, H.; Klein, O.D. Sprouty genes regulate proliferation and survival of human embryonic stem cells. Sci. Rep. 2013, 3, 2277. [Google Scholar] [CrossRef] [Green Version]
- Akbulut, S.; Reddi, A.L.; Aggarwal, P.; Ambardekar, C.; Canciani, B.; Kim, M.K.H.; Hix, L.; Vilimas, T.; Mason, J.; Basson, M.A.; et al. Sprouty proteins inhibit receptor-mediated activation of phosphatidylinositol-specific phospholipase C. Mol. Biol. Cell 2010, 21, 3487–3496. [Google Scholar] [CrossRef] [Green Version]
- Lake, D.; Corrêa, S.; Müller, J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef] [Green Version]
- Peters, K.G.; Marie, J.; Wilson, E.; Ives, H.E.; Escobedo, J.; Rosario, M.D.; Mirda, D.; Williams, L.T. Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature 1992, 358, 678–681. [Google Scholar] [CrossRef]
- Korsensky, L.; Ron, D. Regulation of FGF signaling: Recent insights from studying positive and negative modulators. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2016; Volume 53, pp. 101–114. [Google Scholar]
- Grothe, C.; Claus, P.; Haastert, K.; Lutwak, E.; Ron, D. Expression and regulation of Sef, a novel signaling inhibitor of receptor tyrosine kinases-mediated signaling in the nervous system. Acta Histochem. 2008, 110, 155–162. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, X.; Yan, L.; Li, M. Similar expression to FGF (Sef) reduces endometrial adenocarcinoma cells proliferation via inhibiting fibroblast growth factor 2-mediated MAPK/ERK signaling pathway. Gynecol. Oncol. 2011, 122, 669–674. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Z.Y. The mechanism of dephosphorylation of extracellular signal-regulated kinase 2 by mitogen-activated protein kinase phosphatase 3. J. Biol. Chem. 2001, 276, 32382–32391. [Google Scholar] [CrossRef] [Green Version]
- Haugsten, E.M.; Malecki, J.; Bjorklund, S.M.S.; Olsnes, S.; Wesche, J. Ubiquitination of fibroblast growth factor receptor 1 is required for its intracellular sorting but not for its endocytosis. Mol. Biol. Cell 2008, 19, 3390–3403. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. Therapeutics targeting FGF signaling network in human diseases. Trends Pharm. Sci. 2016, 37, 1081–1096. [Google Scholar] [CrossRef]
- House, S.L.; Castro, A.M.; Lupu, T.S.; Weinheimer, C.; Smith, C.; Kovacs, A.; Ornitz, D.M. Endothelial fibroblast growth factor receptor signaling is required for vascular remodeling following cardiac ischemia-reperfusion injury. Am. J. Physiol.-Heart Circ. Physiol. 2016, 310, H559–H571. [Google Scholar] [CrossRef]
- Welti, J.C.; Gourlaouen, M.; Powles, T.; Kudahetti, S.C.; Wilson, P.; Berney, D.M.; Reynolds, A.R. Fibroblast growth factor 2 regulates endothelial cell sensitivity to sunitinib. Oncogene 2011, 30, 1183–1193. [Google Scholar] [CrossRef] [Green Version]
- Sonvilla, G.; Allerstorfer, S.; Stattner, S.; Karner, J.; Klimpfinger, M.; Fischer, H.; Grasl-Kraupp, B.; Holzmann, K.; Berger, W.; Wrba, F.; et al. FGF18 in colorectal tumour cells: Autocrine and paracrine effects. Carcinogenesis 2008, 29, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Allerstorfer, S.; Sonvilla, G.; Fischer, H.; Spiegl-Kreinecker, S.; Gauglhofer, C.; Setinek, U.; Czech, T.; Marosi, C.; Buchroithner, J.; Pichler, J.; et al. FGF5 as an oncogenic factor in human glioblastoma multiforme: Autocrine and paracrine activities. Oncogene 2008, 27, 4180–4190. [Google Scholar] [CrossRef] [Green Version]
- Ishi, Y.; Yamaguchi, S.; Hatanaka, K.C.; Okamoto, M.; Motegi, H.; Kobayashi, H.; Terasaka, S.; Houkin, K. Association of the FGFR1 mutation with spontaneous hemorrhage in low-grade gliomas in pediatric and young adult patients. J. Neurosurg. 2020, 134, 733–741. [Google Scholar] [CrossRef]
- Bertz, S.; Abee, C.; Schwarz-Furlan, S.; Alfer, J.; Hofstadter, F.; Stoehr, R.; Hartmann, A.; Gaumann, A.K. Increased angiogenesis and FGFR protein expression indicate a favourable prognosis in bladder cancer. Virchows Arch. 2014, 465, 687–695. [Google Scholar] [CrossRef]
- Magnusson, P.U.; Dimberg, A.; Mellberg, S.; Lukinius, A.; Claesson-Welsh, L. FGFR-1 regulates angiogenesis through cytokines interleukin-4 and pleiotrophin. Blood 2007, 110, 4214–4222. [Google Scholar] [CrossRef]
- Yuan, H.; Corbi, N.; Basilico, C.; Dailey, L. Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes Dev. 1995, 9, 2635–2645. [Google Scholar] [CrossRef] [Green Version]
- Feldman, B.; Poueymirou, W.; Papaioannou, V.E.; DeChiara, T.M.; Goldfarb, M. Requirement of FGF-4 for postimplantation mouse development. Science 1995, 267, 246–249. [Google Scholar] [CrossRef]
- Krawchuk, D.; Honma-Yamanaka, N.; Anani, S.; Yamanaka, Y. FGF4 is a limiting factor controlling the proportions of primitive endoderm and epiblast in the ICM of the mouse blastocyst. Dev. Biol. 2013, 384, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Orr-Urtreger, A.; Givol, D.; Yayon, A.; Yarden, Y.; Lonai, P. Developmental expression of two murine fibroblast growth factor receptors, flg and bek. Development 1991, 113, 1419–1434. [Google Scholar] [CrossRef]
- Perantoni, A.O.; Timofeeva, O.; Naillat, F.; Richman, C.; Pajni-Underwood, S.; Wilson, C.; Vainio, S.; Dove, L.F.; Lewandoski, M. Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development 2005, 132, 3859–3871. [Google Scholar] [CrossRef] [Green Version]
- Pownall, M.; Isaacs, H. FGF signalling in vertebrate development. Morgan Claypool Life Sci. 2010, 1, 1–75. [Google Scholar] [CrossRef]
- Yu, K.; Ornitz, D.M. FGF signaling regulates mesenchymal differentiation and skeletal patterning along the limb bud proximodistal axis. Development 2008, 135, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Yu, Y.; Perdue, Y.; Werb, Z. The apical ectodermal ridge is a timer for generating distal limb progenitors. Development 2008, 135, 1395–1405. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Rhee, S.; Palaria, A.; Tremblay, K.D. FGF signaling is required for anterior but not posterior specification of the murine liver bud. Dev. Dyn. 2015, 244, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Yin, Y. Signaling networks regulating development of the lower respiratory tract. Cold Spring Harb. Perspect. Biol. 2012, 4, a008318. [Google Scholar] [CrossRef]
- Benazet, J.D.; Zeller, R. Vertebrate limb development: Moving from classical morphogen gradients to an integrated 4-dimensional patterning system. Cold Spring Harb. Perspect. Biol. 2009, 1, a001339. [Google Scholar] [CrossRef]
- Ladher, R.K.; Wright, T.J.; Moon, A.M.; Mansour, S.L.; Schoenwolf, G.C. FGF8 initiates inner ear induction in chick and mouse. Genes Dev. 2005, 19, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Ebeid, M.; Huh, S.-H. FGF signaling: Diverse roles during cochlear development. BMB Rep. 2017, 50, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Kon, E.; Calvo-Jimenez, E.; Cossard, A.; Na, Y.; Cooper, J.A.; Jossin, Y. N-cadherin-regulated FGFR ubiquitination and degradation control mammalian neocortical projection neuron migration. Elife 2019, 8, e47673. [Google Scholar] [CrossRef]
- Deng, C.; Wynshaw-Boris, A.; Zhou, F.; Kuo, A.; Leder, P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 1996, 84, 911–921. [Google Scholar] [CrossRef] [Green Version]
- Iwata, T.; Li, C.L.; Deng, C.X.; Francomano, C.A. Highly activated Fgfr3 with the K644M mutation causes prolonged survival in severe dwarf mice. Hum. Mol. Genet. 2001, 10, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Gudernova, I.; Vesela, I.; Balek, L.; Buchtova, M.; Dosedelova, H.; Kunova, M.; Pivnicka, J.; Jelinkova, I.; Roubalova, L.; Kozubik, A.; et al. Multikinase activity of fibroblast growth factor receptor (FGFR) inhibitors SU5402, PD173074, AZD1480, AZD4547 and BGJ398 compromises the use of small chemicals targeting FGFR catalytic activity for therapy of short-stature syndromes. Hum. Mol. Genet. 2016, 25, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: Biologic and clinical implications. Cancer Metastasis Rev. 2015, 34, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.M. Genetic syndromes associated with craniosynostosis. J. Korean Neurosurg. Soc. 2016, 59, 187–191. [Google Scholar] [CrossRef]
- Clayton, N.S.; Grose, R.P. Emerging roles of fibroblast growth factor 10 in cancer. Front. Genet. 2018, 9, 499. [Google Scholar] [CrossRef] [Green Version]
- Giacomini, A.; Matarazzo, S.; Pagano, K.; Ragona, L.; Rezzola, S.; Corsini, M.; di Salle, E.; Presta, M.; Ronca, R. A long pentraxin-3-derived pentapeptide for the therapy of FGF8b-driven steroid hormone-regulated cancers. Oncotarget 2015, 6, 13790–13802. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Corn, P.G.; Yang, J.; Palanisamy, N.; Starbuck, M.W.; Efstathiou, E.; Tapia, E.M.L.N.; Zurita, A.J.; Aparicio, A.; Ravoori, M.K.; et al. Prostate cancer cell-stromal cell crosstalk via FGFR1 mediates antitumor activity of dovitinib in bone metastases. Sci. Transl. Med. 2014, 6, 252ra122. [Google Scholar] [CrossRef] [Green Version]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Tanner, Y.; Grose, R.P. Dysregulated FGF signalling in neoplastic disorders. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2016; Volume 53, pp. 126–135. [Google Scholar]
- Tuomela, J.; Harkonen, P. Tumor models for prostate cancer exemplified by fibroblast growth factor 8-induced tumorigenesis and tumor progression. Reprod Biol. 2014, 14, 16–24. [Google Scholar] [CrossRef]
- Feng, S.; Dakhova, O.; Creighton, C.J.; Ittmann, M. Endocrine fibroblast growth factor FGF19 promotes prostate cancer progression. Cancer Res. 2013, 73, 2551–2562. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Wang, J.; Zhang, Y.; Creighton, C.J.; Ittmann, M. FGF23 promotes prostate cancer progression. Oncotarget 2015, 6, 17291–17301. [Google Scholar] [CrossRef] [Green Version]
- Nagamatsu, H.; Teishima, J.; Goto, K.; Shikuma, H.; Kitano, H.; Shoji, K.; Inoue, S.; Matsubara, A. FGF19 promotes progression of prostate cancer. Prostate 2015, 75, 1092–1101. [Google Scholar] [CrossRef]
- Lee, E.K.; Martinez, M.C.; Blakely, K.; Santos, K.D.; Hoang, V.C.; Chow, A.; Emmenegger, U. FGF23: Mediator of poor prognosis in a sizeable subgroup of patients with castration-resistant prostate cancer presenting with severe hypophosphatemia? Med. Hypotheses 2014, 83, 482–487. [Google Scholar] [CrossRef]
- Cheng, Y.-M.; Chou, C.-Y.; Hsu, Y.-C.; Chen, M.-J. Influence of HPV16 E6/7 on the expression of FGF2 and FGFR type B in cervical carcinogenesis. Reprod. Sci. 2012, 19, 580–586. [Google Scholar] [CrossRef]
- Abolhassani, A.; Riazi, G.H.; Azizi, E.; Amanpour, S.; Muhammadnejad, S.; Haddadi, M.; Zekri, A.; Shirkoohi, R. FGF10: Type III epithelial mesenchymal transition and invasion in breast cancer cell lines. J. Cancer 2014, 5, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Song, W.; Li, L.; Cao, L.; Yu, Y.; Song, C.; Wang, Y.; Zhang, F.; Li, Y.; Zhang, B.; et al. FGF4 induces epithelial-mesenchymal transition by inducing store-operated calcium entry in lung adenocarcinoma. Oncotarget 2016, 7, 74015–74030. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Xiao, Y.; Liao, X.; Tang, S.; Xie, X.; Liu, R.; Chen, Q. FGF8 induces epithelial-mesenchymal transition and promotes metastasis in oral squamous cell carcinoma. Int. J. Oral. Sci. 2021, 13, 6. [Google Scholar] [CrossRef]
- Marek, L.; Ware, K.E.; Fritzsche, A.; Hercule, P.; Helton, W.R.; Smith, J.E.; McDermott, L.A.; Coldren, C.D.; Nemenoff, R.A.; Merrick, D.T.; et al. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in Non-Small-Cell lung cancer cells. Mol. Pharmacol. 2009, 75, 196. [Google Scholar] [CrossRef]
- Sharpe, R.; Pearson, A.; Herrera-Abreu, M.; Johnson, D.; Mackay, A.; Welti, J.C.; Natrajan, R.; Reynolds, A.R.; Reis-Filho, J.; Ashworth, A.; et al. FGFR signaling promotes the growth of triple-negative and basal-like breast cancer cell lines both in vitro and in vivo. Clin. Cancer Res. 2011, 17, 5275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memarzadeh, S.; Xin, L.; Mulholland, D.J.; Mansukhani, A.; Wu, H.; Teitell, M.A.; Witte, O.N. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell 2007, 12, 572–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.D.; Ceniccola, K.; Hwang, S.; Andrawis, R.; Horvath, A.; Freedman, J.A.; Olender, J.; Knapp, S.; Ching, T.; Garmire, L.; et al. Alternative splicing promotes tumour aggressiveness and drug resistance in African American prostate cancer. Nat. Commun. 2017, 8, 15921. [Google Scholar] [CrossRef]
- Zisman-Rozen, S.; Fink, D.; Ben-Izhak, O.; Fuchs, Y.; Brodski, A.; Kraus, M.H.; Bejar, J.; Ron, D. Downregulation of Sef, an inhibitor of receptor tyrosine kinase signaling, is common to a variety of human carcinomas. Oncogene 2007, 26, 6093–6098. [Google Scholar] [CrossRef] [Green Version]
- Darby, S.; Murphy, T.; Thomas, H.; Robson, C.N.; Leung, H.Y.; Mathers, M.E.; Gnanapragasam, V.J. Similar expression to FGF (Sef) inhibits fibroblast growth factor-induced tumourigenic behaviour in prostate cancer cells and is downregulated in aggressive clinical disease. Br. J. Cancer 2009, 101, 1891–1899. [Google Scholar] [CrossRef]
- Fritzsche, S.; Kenzelmann, M.; Hoffmann, M.J.; Muller, M.; Engers, R.; Grone, H.J.; Schulz, W.A. Concomitant down-regulation of SPRY1 and SPRY2 in prostate carcinoma. Endocr.-Relat. Cancer 2006, 13, 839–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montico, B.; Colizzi, F.; Giurato, G.; Rizzo, A.; Salvati, A.; Baboci, L.; Benedetti, D.; Pivetta, E.; Covre, A.; Bo, M.D.; et al. Loss of Spry1 reduces growth of BRAF(V600)-mutant cutaneous melanoma and improves response to targeted therapy. Cell Death Dis. 2020, 11, 392. [Google Scholar] [CrossRef]
- He, Q.; Jing, H.; Liaw, L.; Gower, L.; Vary, C.; Hua, S.; Yang, X. Suppression of Spry1 inhibits triple-negative breast cancer malignancy by decreasing EGF/EGFR mediated mesenchymal phenotype. Sci. Rep. 2016, 6, 23216. [Google Scholar] [CrossRef] [Green Version]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR landscape in cancer: Analysis of 4,853 tumors by next-generation sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Grivas, P.; Kiedrowski, L.A.; Sonpavde, G.P.; Gupta, S.V.; Thomas, R.A.; Gourdin, T.S.; Hardin, A.I.; Hamann, K.M.; Faltas, B.M.; Vogelzang, N.J. Spectrum of FGFR2/3 alterations in cell-free DNA of patients with advanced urothelial carcinoma. Bladder Cancer 2021, 7, 143–148. [Google Scholar] [CrossRef]
- Easton, D.F.; Pooley, K.A.; Dunning, A.M.; Pharoah, P.D.; Thompson, D.; Ballinger, D.G.; Struewing, J.P.; Morrison, J.; Field, H.; Luben, R.; et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007, 447, 1087–1093. [Google Scholar] [CrossRef] [Green Version]
- Hunter, D.J.; Kraft, P.; Jacobs, K.B.; Cox, D.G.; Yeager, M.; Hankinson, S.E.; Wacholder, S.; Wang, Z.; Welch, R.; Hutchinson, A.; et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 2007, 39, 870–874. [Google Scholar] [CrossRef]
- Garcia-Closas, M.; Chanock, S. Genetic susceptibility loci for breast cancer by estrogen receptor status. Clin. Cancer Res. 2008, 14, 8000–8009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huijts, P.E.; van Dongen, M.; de Goeij, M.C.; van Moolenbroek, A.J.; Blanken, F.; Vreeswijk, M.P.; de Kruijf, E.M.; Mesker, W.E.; van Zwet, E.W.; Tollenaar, R.A.; et al. Allele-specific regulation of FGFR2 expression is cell type-dependent and may increase breast cancer risk through a paracrine stimulus involving FGF10. Breast Cancer Res. 2011, 13, R72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.H.; Li, X.Q.; Chen, Y.; Feng, Y.M. Risk of aggressive breast cancer in women of Han nationality carrying TGFB1 rs1982073 C allele and FGFR2 rs1219648 G allele in North China. Breast Cancer Res. Treat. 2011, 125, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.P.; van Leeuwen, F.E.; Cooke, R.; Broeks, A.; Enciso-Mora, V.; Olver, B.; Lloyd, A.; Broderick, P.; Russell, N.S.; Janus, C.; et al. FGFR2 genotype and risk of radiation-associated breast cancer in Hodgkin lymphoma. Blood 2012, 119, 1029–1031. [Google Scholar] [CrossRef] [Green Version]
- Shu, J.; Hui, X.; Zheng, X.; Zhao, J.; Xu, Z.; Chen, Y.; Lu, C.; Li, J. Correlation of FGFR2 rs2981582 polymorphisms with susceptibility to breast cancer: A case-control study in a Chinese population. J. Int. Med. Res. 2019, 47, 4753–4763. [Google Scholar] [CrossRef]
- Agarwal, D.; Pineda, S.; Michailidou, K.; Herranz, J.; Pita, G.; Moreno, L.T.; Alonso, M.R.; Dennis, J.; Wang, Q.; Bolla, M.K.; et al. FGF receptor genes and breast cancer susceptibility: Results from the breast cancer association consortium. Br. J. Cancer 2014, 110, 1088–1100. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, Y.; Liu, J.; Chen, Q.; Pang, D.; Jiang, Y. Effects of FGFR1 gene polymorphisms on the risk of breast cancer and FGFR1 protein expression. Cell. Physiol. Biochem. 2018, 47, 2569–2578. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, S.; Wei, W.; Ren, Y.; Liu, J.; Pang, D. Association of FGFR3 and FGFR4 gene polymorphisms with breast cancer in Chinese women of Heilongjiang province. Oncotarget 2015, 6, 34023–34029. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Qi, H.; Zhang, L.; Li, H.; Shao, J.; Chen, H.; Zhong, M.; Shi, X.; Ye, T.; Li, Q. Effects of FGFR gene polymorphisms on response and toxicity of cyclophosphamide-epirubicin-docetaxel-based chemotherapy in breast cancer patients. BMC Cancer 2018, 18, 1038. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.H.; Xu, E.; Hildebrandt, M.A.; Liang, D.; Lu, K.; Ye, Y.; Wagar, E.A.; Wu, X. Genetic variants in the fibroblast growth factor pathway as potential markers of ovarian cancer risk, therapeutic response, and clinical outcome. Clin. Chem. 2014, 60, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, I.; Iwata, T.; Leung, H.Y. Mechanisms of FGFR-mediated carcinogenesis. BBA-Mol. Cell Res. 2012, 1823, 850–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haugsten, E.M.; Wiedlocha, A.; Olsnes, S.; Wesche, J. Roles of fibroblast growth factor receptors in carcinogenesis. Mol. Cancer Res. MCR 2010, 8, 1439–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, L.H.; Nelson, K.N.; Meyer, A.N.; Donoghue, D.J. Functions of Fibroblast Growth Factor Receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev. 2015, 26, 425–449. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.G.T.; Cheuk, A.T.; Tsang, P.S.; Chung, J.Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 2009, 119, 3395–3407. [Google Scholar]
- Futami, T.; Kawase, T.; Mori, K.; Asaumi, M.; Kihara, R.; Shindoh, N.; Kuromitsu, S. Identification of a novel oncogenic mutation of FGFR4 in gastric cancer. Sci. Rep. 2019, 9, 14627. [Google Scholar] [CrossRef]
- Patani, H.; Bunney, T.D.; Thiyagarajan, N.; Norman, R.A.; Ogg, D.; Breed, J.; Ashford, P.; Potterton, A.; Edwards, M.; Williams, S.V.; et al. Landscape of activating cancer mutations in FGFR kinases and their differential responses to inhibitors in clinical use. Oncotarget 2016, 7, 24252–24268. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.; Krishna, N.S.; Witton, C.J.; Bartlett, J.M.S. Gene amplifications associated with the development of hormone-resistant prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 5271–5281. [Google Scholar]
- Freier, K.; Schwaenen, C.; Sticht, C.; Flechtenmacher, C.; Muhling, J.; Hofele, C.; Radlwimmer, B.; Lichter, P.; Joos, S. Recurrent FCFR1 amplification and high FGFR1 protein expression in oral squamous cell carcinoma (OSCC). Oral Oncol. 2007, 43, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci. Transl. Med. 2010, 2, 62ra93. [Google Scholar] [CrossRef] [Green Version]
- Dutt, A.; Alex, H.R.; Peter, S.H.; Mermel, C.; Cho, J.; Sharifnia, T.; Chande, A.; Kumiko, E.T.; Stransky, N.; Greulich, H.; et al. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PLoS ONE 2011, 6, e20351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.K.; Cho, Y.A.; Koh, Y.W.; Shin, H.A.; Cho, B.C.; Yoon, S.O. Prognostic implications of Fibroblast growth factor receptor 1 (FGFR1) gene amplification and protein overexpression in hypopharyngeal and laryngeal squamous cell carcinoma. BMC Cancer 2020, 20, 348. [Google Scholar] [CrossRef] [Green Version]
- Drago, J.Z.; Niemierko, A.; Spring, L.; Moy, B.; Juric, D.; Isakoff, S.J.; Iafrate, A.J.; Ellisen, L.W.; Bardia, A. FGFR gene amplification and response to endocrine therapy in metastatic hormone receptor positive (HR+) breast cancer. J. Clin. Oncol. 2017, 35 (Suppl. 15), 1013. [Google Scholar] [CrossRef]
- Drago, J.Z.; Formisano, L.; Juric, D.; Niemierko, A.; Servetto, A.; Wander, S.A.; Spring, L.M.; Vidula, N.; Younger, J.; Peppercorn, J.; et al. Amplification mediates endocrine resistance but retains TORC sensitivity in metastatic hormone receptor–positive (HR+) breast cancer. Transl. Cancer Mech. Ther. 2019, 25, 6443–6451. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.Y.; Chao, J.; Kim, K.; Kim, S.T.; Kim, K.M.; Klempner, S.J.; Lee, J. High-level FGFR2 amplification is associated with poor prognosis and Lower response to chemotherapy in gastric cancers. Pathol. Res. Pract. 2020, 216, 152878. [Google Scholar] [CrossRef]
- Roumiantsev, S.; Krause, D.S.; Neumann, C.A.; Dimitri, C.A.; Asiedu, F.; Cross, N.C.; van Etten, R.A. Distinct stem cell myeloproliferative/T lymphoma syndromes induced by ZNF198-FGFR1 and BCR-FGFR1 fusion genes from 8p11 translocations. Cancer Cell 2004, 5, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Yagasaki, F.; Wakao, D.; Yokoyama, Y.; Uchida, Y.; Murohashi, I.; Kayano, H.; Taniwaki, M.; Matsuda, A.; Bessho, M. Fusion of ETV6 to fibroblast growth factor receptor 3 in peripheral T-cell lymphoma with a t(4;12)(p16;p13) chromosomal translocation. Cancer Res. 2001, 61, 8371–8374. [Google Scholar]
- Xiao, S.; Nalabolu, S.R.; Aster, J.C.; Ma, J.; Abruzzo, L.; Jaffe, E.S.; Stone, R.; Weissman, S.M.; Hudson, T.J.; Fletcher, J.A. FGFR1 is fused with a novel zinc-finger gene, ZNF198, in the t(8;13) leukaemia/lymphoma syndrome. Nat. Genet. 1998, 18, 84–87. [Google Scholar] [CrossRef]
- Jackson, C.C.; Medeiros, L.J.; Miranda, R.N. 8p11 myeloproliferative syndrome: A review. Hum. Pathol. 2010, 41, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Carll, T.; Patel, A.; Derman, B.; Hyjek, E.; Lager, A.; Wanjari, P.; Segal, J.; Odenike, O.; Fidai, S.; Arber, D. Diagnosis and treatment of mixed phenotype (T-myeloid/lymphoid) acute leukemia with novel ETV6-FGFR2 rearrangement. Blood Adv. 2020, 4, 4924–4928. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Abate, R.E.; Rachiglio, A.M.; Maiello, M.R.; Esposito, C.; Schettino, C.; Izzo, F.; Nasti, G.; Normanno, N. FGFR Fusions in cancer: From diagnostic approaches to therapeutic intervention. Int. J. Mol. Sci. 2020, 21, 6856. [Google Scholar] [CrossRef]
- Williams, S.V.; Hurst, C.D.; Knowles, M.A. Oncogenic FGFR3 gene fusions in bladder cancer. Hum. Mol. Genet. 2013, 22, 795–803. [Google Scholar] [CrossRef]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ding, X.; Wang, S.; Moser, C.D.; Shaleh, H.M.; Mohamed, E.A.; Chaiteerakij, R.; Allotey, L.K.; Chen, G.; Miyabe, K.; et al. Antitumor effect of FGFR inhibitors on a novel cholangiocarcinoma patient derived xenograft mouse model endogenously expressing an FGFR2-CCDC6 fusion protein. Cancer Lett. 2016, 380, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Shi, E.; Chmielecki, J.; Tang, C.M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J. Transl. Med. 2016, 14, 339. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; McBride, D.J.; Lin, M.L.; Varela, I.; Pleasance, E.D.; Simpson, J.T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Mudie, L.J.; et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009, 462, 1005–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stefano, A.L.; Fucci, A.; Frattini, V.; Labussiere, M.; Mokhtari, K.; Zoppoli, P.; Marie, Y.; Bruno, A.; Boisselier, B.; Giry, M.; et al. Detection, characterization, and inhibition of FGFR-TACC Fusions in IDH wild-type glioma. Clin. Cancer Res. 2015, 21, 3307–3317. [Google Scholar] [CrossRef] [Green Version]
- Qin, A.; Johnson, A.; Ross, J.S.; Miller, V.A.; Ali, S.M.; Schrock, A.B.; Gadgeel, S.M. Detection of known and novel FGFR fusions in non-small cell lung cancer by comprehensive genomic profiling. J. Thorac. Oncol. 2019, 14, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Yang, L.; Shi, L.; Li, Q.; Zhang, G.; Wu, J.; Zheng, J.; Jiao, B. Nuclear translocation of fibroblast growth factor-2 (FGF2) is regulated by Karyopherin-β2 and Ran GTPase in human glioblastoma cells. Oncotarget 2015, 6, 21468–21478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.N.; Yamaguchi, H.; Hsu, J.M.; Hung, M.C. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene 2010, 29, 3997–4006. [Google Scholar] [CrossRef] [Green Version]
- Reilly, J.F.; Maher, P.A. Importin β–mediated nuclear import of fibroblast growth factor receptor. J. Cell Biol. 2001, 152, 1307–1312. [Google Scholar] [CrossRef] [Green Version]
- Reilly, J.F.; Mizukoshi, E.; Maher, P.A. Ligand dependent and independent internalization and nuclear translocation of fibroblast growth factor (FGF) receptor 1. DNA Cell Biol. 2004, 23, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.J.; Chioni, A.M.; Ghallab, M.; Anderson, R.K.; Lemoine, N.R.; Kocher, H.M.; Grose, R.P. Nuclear translocation of FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med. 2014, 6, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Chioni, A.M.; Grose, R. FGFR1 cleavage and nuclear translocation regulates breast cancer cell behavior. J. Cell Biol. 2012, 197, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Popovici, C.; Fallet, M.; Marguet, D.; Birnbaum, D.; Roubin, R. Intracellular trafficking of LET-756, a fibroblast growth factor of C. elegans, is controlled by a balance of export and nuclear signals. Exp. Cell Res. 2006, 312, 1484–1495. [Google Scholar] [CrossRef]
- Coleman, S.J.; Bruce, C.; Chioni, A.M.; Kocher, H.M.; Grose, R.P. The ins and outs of fibroblast growth factor receptor signalling. Clin. Sci. 2014, 127, 217–231. [Google Scholar] [CrossRef]
- Bryant, D.M.; Stow, J.L. Nuclear translocation of cell-surface receptors: Lessons from fibroblast growth factor. Traffic 2005, 6, 947–954. [Google Scholar] [CrossRef]
- Porębska, N.; Latko, M.; Kucińska, M.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Targeting cellular trafficking of fibroblast growth factor receptors as a strategy for selective cancer treatment. J. Clin. Med. 2018, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, G.; Schanz, W.; Pappas, B.A.; Maretzky, T. Members of the Fibroblast growth factor receptor superfamily are proteolytically cleaved by two differently activated metalloproteases. Int. J. Mol. Sci. 2021, 22, 3165. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G.; Liao, H.-J. Receptor tyrosine kinases in the nucleus. Cold Spring Harb. Perspect. Biol. 2013, 5, a008979. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; Evers, A.; Zhou, W.; Swendeman, S.L.; Wong, P.M.; Rafii, S.; Reiss, K.; Blobel, C.P. Migration of growth factor-stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat. Commun. 2011, 2, 229. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Shin, S.H.; Shin, H.W.; Chun, Y.S.; Park, J.W. Nuclear FGFR2 negatively regulates hypoxia-induced cell invasion in prostate cancer by interacting with HIF-1 and HIF-2. Sci. Rep. 2019, 9, 3480. [Google Scholar] [CrossRef] [Green Version]
- Merilahti, J.A.M.; Elenius, K. Gamma-secretase-dependent signaling of receptor tyrosine kinases. Oncogene 2019, 38, 151–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeb, C.R.; Harris, J.L.; Craik, C.S. Granzyme B proteolyzes receptors important to proliferation and survival, tipping the balance toward apoptosis. J. Biol. Chem. 2006, 281, 28326–28335. [Google Scholar] [CrossRef] [Green Version]
- Terranova, C.; Narla, S.T.; Lee, Y.W.; Bard, J.; Parikh, A.; Stachowiak, E.K.; Tzanakakis, E.S.; Buck, M.J.; Birkaya, B.; Stachowiak, M.K. Global Developmental Gene Programing Involves a Nuclear Form of Fibroblast Growth Factor Receptor-1 (FGFR1). PLoS ONE 2015, 10, e0123380. [Google Scholar]
- Chen, M.-K.; Hung, M.-C. Proteolytic cleavage, trafficking, and functions of nuclear receptor tyrosine kinases. FEBS J. 2015, 282, 3693–3721. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Ryu, H.; Ock, C.Y.; Suh, K.J.; Lee, J.Y.; Kim, J.W.; Lee, J.O.; Kim, J.W.; Kim, Y.J.; Lee, K.W.; et al. BGJ398, A Pan-FGFR inhibitor, overcomes paclitaxel resistance in urothelial carcinoma with FGFR1 overexpression. Int. J. Mol. Sci. 2018, 19, 3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turkington, R.C.; Longley, D.B.; Allen, W.L.; Stevenson, L.; McLaughlin, K.; Dunne, P.D.; Blayney, J.K.; Salto-Tellez, M.; van Schaeybroeck, S.; Johnston, P.G. Fibroblast growth factor receptor 4 (FGFR4): A targetable regulator of drug resistance in colorectal cancer. Cell Death Dis. 2014, 5, e1046. [Google Scholar] [CrossRef] [Green Version]
- Bray, S. Notch signalling: A simple pathway becomes complex. Nat. Reviews. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Wientjes, M.G.; Gan, Y.; Au, J.L. Fibroblast growth factors: An epigenetic mechanism of broad spectrum resistance to anticancer drugs. Proc. Natl. Acad. Sci. USA 2000, 97, 8658–8663. [Google Scholar] [CrossRef] [Green Version]
- Hanker, A.B.; Garrett, J.T.; Estrada, M.V.; Moore, P.D.; Ericsson, P.G.; Koch, J.P.; Langley, E.; Singh, S.; Kim, P.S.; Frampton, G.M.; et al. HER2-Overexpressing Breast Cancers Amplify FGFR Signaling upon Acquisition of Resistance to Dual Therapeutic Blockade of HER2. Clin. Cancer Res. 2017, 23, 4323–4334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, D.C.; Knowles, M.A.; Speirs, V. Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int. J. Cancer 2012, 130, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Kawahara, A.; Sonoda, K.; Nakashima, K.; Tashiro, K.; Watari, K.; Izumi, H.; Kage, M.; Kuwano, M.; Ono, M.; et al. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget 2014, 5, 5908–5919. [Google Scholar] [CrossRef] [Green Version]
- Terai, H.; Soejima, K.; Yasuda, H.; Nakayama, S.; Hamamoto, J.; Arai, D.; Ishioka, K.; Ohgino, K.; Ikemura, S.; Sato, T.; et al. Activation of the FGF2-FGFR1 autocrine pathway: A novel mechanism of acquired resistance to gefitinib in NSCLC. Mol. Cancer Res. MCR 2013, 11, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Malchers, F.; Ercanoglu, M.; Schutte, D.; Castiglione, R.; Tischler, V.; Michels, S.; Dahmen, I.; Bragelmann, J.; Menon, R.; Heuckmann, J.M.; et al. Mechanisms of primary drug resistance in FGFR1-amplified lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 5527–5536. [Google Scholar] [CrossRef] [Green Version]
- Daly, C.; Castanaro, C.; Zhang, W.; Zhang, Q.; Wei, Y.; Ni, M.; Young, T.M.; Zhang, L.; Burova, E.; Thurston, G. FGFR3-TACC3 fusion proteins act as naturally occurring drivers of tumor resistance by functionally substituting for EGFR/ERK signaling. Oncogene 2017, 36, 471–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Ahmed, M.; Legrand, C.; Relimpio, A.Y.; Beretta, C.A.; Muschko, A.; Wegehingel, S.; Müller, H.M.; Sehr, P.; Will, D.W.; Lewis, J.D.; et al. A time-resolved live cell imaging assay to identify small molecule inhibitors of FGF2 signaling. FEBS Lett. 2019, 593, 2162–2176. [Google Scholar] [CrossRef]
- Dai, S.; Zhou, Z.; Chen, Z.; Xu, G.; Chen, Y. Fibroblast growth factor receptors (FGFRs): Structures and small molecule inhibitors. Cells 2019, 8, 614. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Liu, Z.; Wu, J.; Cai, Y.; Li, X. Anticancer molecules targeting fibroblast growth factor receptors. Trends Pharmacol. Sci. 2012, 33, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Chen, H.; Wortmann, A.; Loch, D.; Gartside, M.G.; Dehkhoda, F.; Blais, S.P.; Neubert, T.A.; Mohammadi, M.; Pollock, P.M. The N550K/H mutations in FGFR2 confer differential resistance to PD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neoplasia 2013, 15, 975–988. [Google Scholar] [CrossRef] [Green Version]
- Musolino, A.; Campone, M.; Neven, P.; Denduluri, N.; Barrios, C.H.; Cortes, J.; Blackwell, K.; Soliman, H.; Kahan, Z.; Bonnefoi, H.; et al. Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR(+), HER2(-) breast cancer that had progressed during or after prior endocrine therapy. Breast Cancer Res. 2017, 19, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knights, V.; Cook, S.J. De-regulated FGF receptors as therapeutic targets in cancer. Pharmacol. Ther. 2010, 125, 105–117. [Google Scholar] [CrossRef]
- Angevin, E.; Lopez-Martin, J.A.; Lin, C.C.; Gschwend, J.E.; Harzstark, A.; Castellano, D.; Soria, J.C.; Sen, P.; Chang, J.; Shi, M.; et al. Phase I study of dovitinib (TKI258), an oral FGFR, VEGFR, and PDGFR inhibitor, in advanced or metastatic renal cell carcinoma. Clin. Cancer Res. 2013, 19, 1257–1268. [Google Scholar] [CrossRef] [Green Version]
- André, F.; Cortés, J. Rationale for targeting fibroblast growth factor receptor signaling in breast cancer. Breast Cancer Res. Treat. 2015, 150, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Gozgit, J.M.; Wong, M.J.; Moran, L.; Wardwell, S.; Mohemmad, Q.K.; Narasimhan, N.I.; Shakespeare, W.C.; Wang, F.; Clackson, T.; Rivera, V.M. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol. Cancer Ther. 2012, 11, 690–699. [Google Scholar] [CrossRef] [Green Version]
- Ahn, D.H.; Junior, P.L.S.U.; Masci, P.; Kosiorek, H.; Halfdanarson, T.R.; Mody, K.; Babiker, H.; DeLeon, T.; Sonbol, M.B.; Gores, G.; et al. A pilot study of Pan-FGFR inhibitor ponatinib in patients with FGFR-altered advanced cholangiocarcinoma. Investig. New Drugs 2021. [Google Scholar] [CrossRef]
- Smyth, E.C.; Turner, N.C.; Peckitt, C.; Pearson, A.; Brown, G.; Chua, S.; Gillbanks, A.; Johnston, S.R.D.; Tarazona, N.; Cutts, R.; et al. Phase II multicenter proof of concept study of AZD4547 in FGFR amplified tumours. J. Clin. Oncol. 2015, 33 (Suppl. 15), 2508. [Google Scholar] [CrossRef]
- Jimenez-Pascual, A.; Siebzehnrubl, F.A. Fibroblast growth factor receptor functions in glioblastoma. Cells 2019, 8, 715. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Choi, Y.J.; Seo, B.Y.; Jo, U.; Park, S.I.; Kim, Y.H.; Park, K.H. A Selective FGFR inhibitor AZD4547 suppresses RANKL/M-CSF/OPG-dependent ostoclastogenesis and breast cancer growth in the metastatic bone microenvironment. Sci. Rep. 2019, 9, 8726. [Google Scholar] [CrossRef] [Green Version]
- Paik, P.K.; Shen, R.; Berger, M.F.; Ferry, D.; Soria, J.C.; Mathewson, A.; Rooney, C.; Smith, N.R.; Cullberg, M.; Kilgour, E.; et al. A phase Ib open-label multicenter study of AZD4547 in patients with advanced squamous cell lung cancers. Clin. Cancer Res. 2017, 23, 5366–5373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seckl, M.; Badman, P.D.; Liu, X.; MacPherson, I.R.; Zubairi, I.H.; Baird, R.D.; Garcia-Corbacho, J.; Cresti, N.; Plummer, E.R.; Armstrong, A.C.; et al. RADICAL trial: A phase Ib/IIa study to assess the safety and efficacy of AZD4547 in combination with either anastrozole or letrozole in ER positive breast cancer patients progressing on these aromatase inhibitors (AIs). J. Clin. Oncol. 2017, 35 (Suppl. 15), 1059. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Bang, Y.J.; Mansoor, W.; Petty, R.D.; Chao, Y.; Cunningham, D.; Ferry, D.R.; Smith, N.R.; Frewer, P.; Ratnayake, J.; et al. A randomized, open-label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann. Oncol. 2017, 28, 1316–1324. [Google Scholar] [CrossRef]
- Perez-Garcia, J.; Munoz-Couselo, E.; Soberino, J.; Racca, F.; Cortes, J. Targeting FGFR pathway in breast cancer. Breast 2018, 37, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Casadei, C.; Dizman, N.; Schepisi, G.; Cursano, M.C.; Basso, U.; Santini, D.; Pal, S.K.; de Giorgi, U. Targeted therapies for advanced bladder cancer: New strategies with FGFR inhibitors. Ther. Adv. Med. Oncol. 2019, 11, 1758835919890285. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.; Gizzi, M.; Bahleda, R.; Loriot, Y. Clinical development of FGFR3 inhibitors for the treatment of urothelial cancer. Bladder Cancer 2019, 5, 87–102. [Google Scholar] [CrossRef] [Green Version]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-O.; Feng, Y.-H.; Chen, Y.-Y.; Su, W.C.; Oh, D.Y.; Shen, L.; Kim, K.p.; Liu, X.; Bai, Y.; Liao, H.; et al. Updated results of a phase IIa study to evaluate the clinical efficacy and safety of erdafitinib in Asian advanced cholangiocarcinoma (CCA) patients with FGFR alterations. J. Clin. Oncol. 2019, 37 (Suppl. 15), 4117. [Google Scholar] [CrossRef]
- Grunewald, S.; Politz, O.; Bender, S.; Heroult, M.; Lustig, K.; Thuss, U.; Kneip, C.; Kopitz, C.; Zopf, D.; Collin, M.P.; et al. Rogaratinib: A potent and selective pan-FGFR inhibitor with broad antitumor activity in FGFR-overexpressing preclinical cancer models. Int. J. Cancer 2019, 145, 1346–1357. [Google Scholar] [CrossRef] [Green Version]
- Daniele, G.; Corral, J.; Molife, L.; de Bono, J. FGF receptor inhibitors: Role in cancer therapy. Curr. Oncol. Rep. 2012, 14, 111–119. [Google Scholar] [CrossRef]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araujo, A.; Reclusa, P.; Franchina, T.; van der Steen, N.; van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Adams, G.; Weiner, L. Monoclonal antibody therapy of cancer. Nat. Biotechnol. 2005, 23, 1147–1157. [Google Scholar] [CrossRef]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052–16074. [Google Scholar] [CrossRef] [Green Version]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A.; Eliseenkova, A.V.; Yeh, B.K.; Yayon, A.; Linhardt, R.J.; Mohammadi, M. Crystal structure of a ternary FGF-FGFR-Heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Hadari, Y.; Schlessinger, J. FGFR3-targeted mAb therapy for bladder cancer and multiple myeloma. J. Clin. Investig. 2009, 119, 1077–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, J.; Du, X.; Chen, Y.; Chan, P.; Li, H.; Wu, P.; Marsters, S.; Stawicki, S.; Tien, J.; Totpal, K.; et al. Antibody-based targeting of FGFR3 in bladder carcinoma and t(4;14)-positive multiple myeloma in mice. J. Clin. Investig. 2009, 119, 1216–1229. [Google Scholar] [CrossRef] [Green Version]
- Ocio, E.M.; Richardson, P.G.; Rajkumar, S.V.; Palumbo, A.; Mateos, M.V.; Orlowski, R.; Kumar, S.; Usmani, S.; Roodman, D.; Niesvizky, R.; et al. New drugs and novel mechanisms of action in multiple myeloma in 2013: A report from the International Myeloma Working Group (IMWG). Leukemia 2014, 28, 525–542. [Google Scholar] [CrossRef] [Green Version]
- Bai, A.; Meetze, K.; Vo, N.Y.; Kollipara, S.; Mazsa, E.K.; Winston, W.M.; Weiler, S.; Poling, L.L.; Chen, T.; Ismail, N.S.; et al. GP369, an FGFR2-IIIb-specific antibody, exhibits potent antitumor activity against human cancers driven by activated FGFR2 signaling. Cancer Res. 2010, 70, 7630–7639. [Google Scholar] [CrossRef] [Green Version]
- Borek, A.; Sokolowska-Wedzina, A.; Chodaczek, G.; Otlewski, J. Generation of high-affinity, internalizing anti-FGFR2 single-chain variable antibody fragment fused with Fc for targeting gastrointestinal cancers. PLoS ONE 2018, 13, e0192194. [Google Scholar]
- Wang, L.; Park, H.; Chhim, S.; Ding, Y.; Jiang, W.; Queen, C.; Kim, K.J. A novel monoclonal antibody to fibroblast growth factor 2 effectively inhibits growth of hepatocellular carcinoma xenografts. Mol. Cancer Ther. 2012, 11, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Furuya, A.; Yamasaki, M.; Hanai, N.; Kuriki, K.; Kamiakito, T.; Kobayashi, Y.; Yoshida, H.; Koike, M.; Fukayama, M. High frequency of fibroblast growth factor (FGF) 8 expression in clinical prostate cancers and breast tissues, immunohistochemically demonstrated by a newly established neutralizing monoclonal antibody against FGF 8. Cancer Res. 1998, 58, 2053–2056. [Google Scholar]
- Tao, J.; Xiang, J.J.; Li, D.; Deng, N.; Wang, H.; Gong, Y.P. Selection and characterization of a human neutralizing antibody to human fibroblast growth factor-2. Biochem. Biophys. Res. Commun. 2010, 394, 767–773. [Google Scholar] [CrossRef]
- Goke, A.; Goke, R.; Ofner, A.; Herbst, A.; Lankat-Buttgereit, B. The FGFR inhibitor NVP-BGJ398 induces NSCLC cell death by activating caspase-dependent pathways as well as caspase-independent apoptosis. Anticancer Res. 2015, 35, 5873–5879. [Google Scholar]
- Takamura, T.; Horinaka, M.; Yasuda, S.; Toriyama, S.; Aono, Y.; Sowa, Y.; Miki, T.; Ukimura, O.; Sakai, T. FGFR inhibitor BGJ398 and HDAC inhibitor OBP-801 synergistically inhibit cell growth and induce apoptosis in bladder cancer cells. Oncol. Rep. 2018, 39, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Harding, T.C.; Long, L.; Palencia, S.; Zhang, H.; Sadra, A.; Hestir, K.; Patil, N.; Levin, A.; Hsu, A.W.; Charych, D.; et al. Blockade of nonhormonal fibroblast growth factors by FP-1039 inhibits growth of multiple types of cancer. Sci. Transl. Med. 2013, 5, 178ra39. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Papadopoulos, K.P.; Patnaik, A.; Wilson, K.; Thayer, S.; Zanghi, J.; Gemo, A.T.; Kavanaugh, W.M.; Keer, H.N.; LoRusso, P.M. A phase I, first in human study of FP-1039 (GSK3052230), a novel FGF ligand trap, in patients with advanced solid tumors. Ann. Oncol. 2016, 27, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, C.; Sherk, C.; Fricko, M.; Ganji, G.; Barnette, M.; Hoang, B.; Tunstead, J.; Skedzielewski, T.; Alsaid, H.; Jucker, B.M.; et al. Inhibition of FGF/FGFR autocrine signaling in mesothelioma with the FGF ligand trap, FP-1039/GSK3052230. Oncotarget 2016, 7, 39861–39871. [Google Scholar] [CrossRef] [Green Version]
- Tayel, A.; el Galil, K.H.A.; Ebrahim, M.A.; Ibrahim, A.S.; El-Gayar, A.M.; Al-Gayyar, M.M. Suramin inhibits hepatic tissue damage in hepatocellular carcinoma through deactivation of heparanase enzyme. Eur. J. Pharmacol. 2014, 728, 151–160. [Google Scholar] [CrossRef]
- Go, R.S.; Lee, S.J.; Shin, D.; Callister, S.M.; Jobe, D.A.; Conry, R.M.; Tarhini, A.A.; Kirkwood, J.M. ECOG phase II trial of graded-dose peginterferon alpha-2b in patients with metastatic melanoma overexpressing basic fibroblast growth factor (E2602). Clin. Cancer Res. 2013, 19, 6597–6604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akl, M.; Nagpal, P.; Ayoub, N.; Tai, B.; Prabhu, S.; Capac, C.; Gliksman, M.; Goy, A. Molecular and clinical significance of fibroblast growth factor 2 (FGF2 /bFGF) in malignancies of solid and hematological cancers for personalized therapies. Oncotarget 2016, 7, 44735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, L.M.; Stehbens, S.J.; Bonazzi, V.F.; Gunter, J.H.; Ju, R.J.; Ward, M.; Gartside, M.G.; Byron, S.A.; Pollock, P.M. Bcl-2 inhibitors enhance FGFR inhibitor-induced mitochondrial-dependent cell death in FGFR2-mutant endometrial cancer. Mol. Oncol. 2019, 13, 738–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Fibroblast growth factor receptor (FGFR) structure and function. FGFR possesses three immunoglobulin-like domains (Ig1–3), an acid box (AB), a transmembrane domain (TM), and an intracellular tyrosine kinase domain that is split into two (TK1 and TK2). The FGF–FGFR complex is composed by two FGFs, two FGFRs and one heparin sulphate proteoglycan (HSPG). The TK domains are transphosphorylated upon ligand binding between Ig2-Ig3 and receptor dimerisation. This initiates the interaction between a network of downstream signalling molecules that can activate key pathways, such as MAPK, AKT, PLCγ, STAT1 and in turn regulate target genes involved in cell proliferation, migration, differentiation, survival, resistance to anticancer agents and neoangiogenesis. Signalling can be negatively regulated by SEF, FGFR-like 1 (FGFRL1), sprouty (SPRY) and MAPK phosphatase 1 and 3 (MKP1 and MKP3) at different levels. Created with BioRender.com (accessed on 26 September 2021).
Figure 1.
Fibroblast growth factor receptor (FGFR) structure and function. FGFR possesses three immunoglobulin-like domains (Ig1–3), an acid box (AB), a transmembrane domain (TM), and an intracellular tyrosine kinase domain that is split into two (TK1 and TK2). The FGF–FGFR complex is composed by two FGFs, two FGFRs and one heparin sulphate proteoglycan (HSPG). The TK domains are transphosphorylated upon ligand binding between Ig2-Ig3 and receptor dimerisation. This initiates the interaction between a network of downstream signalling molecules that can activate key pathways, such as MAPK, AKT, PLCγ, STAT1 and in turn regulate target genes involved in cell proliferation, migration, differentiation, survival, resistance to anticancer agents and neoangiogenesis. Signalling can be negatively regulated by SEF, FGFR-like 1 (FGFRL1), sprouty (SPRY) and MAPK phosphatase 1 and 3 (MKP1 and MKP3) at different levels. Created with BioRender.com (accessed on 26 September 2021).

Figure 2.
Key FGFR splice variants and ligand specificity. (A) A generalised diagram representing the key FGFR features. (B) Splice variant at the Ig3 loop occurs in FGFR1–3. This splice variant is responsible for ligand binding specificity and is generated by alternative splicing at the Ig3. The first portion of the Ig3 is the exon “a” (exon 7) that is spliced to either exon “b” (exon 8) or exon “c” (exon 9) and then to the exon that encodes the TM domain. The “b” isoform is mainly expressed by epithelial tissues/cells, whereas the “c” isoform is expressed by mesenchymal tissues. FGFs have differential specificity to different isoforms. (C) Splice variants can generate soluble variants without TK activity, truncated to one or more Ig domains and missing the TM domain. Variants lacking the TK domain can heterodimerise with full length FGFRs to generate nonfunctional dimers and therefore act as down regulators [24]. (D) FGFR4 can generate a single isoform containing the “c” exon (exon 9) in the Ig3 domain. (E) FGFR2 can generate a splice variant missing Ig1 and Ig3 containing the “b” exon (exon 8). (F) FGFR1 and FGFR2 can also generate a splice variant with truncated Ig1 and Ig3 containing the “c” exon (exon 9). SP: signal peptide, Ig: Immunoglobulin, AB: acid box; TM: transmembrane domain, UTR: untranslated region. Created with BioRender.com (accessed on 26 September 2021).
Figure 2.
Key FGFR splice variants and ligand specificity. (A) A generalised diagram representing the key FGFR features. (B) Splice variant at the Ig3 loop occurs in FGFR1–3. This splice variant is responsible for ligand binding specificity and is generated by alternative splicing at the Ig3. The first portion of the Ig3 is the exon “a” (exon 7) that is spliced to either exon “b” (exon 8) or exon “c” (exon 9) and then to the exon that encodes the TM domain. The “b” isoform is mainly expressed by epithelial tissues/cells, whereas the “c” isoform is expressed by mesenchymal tissues. FGFs have differential specificity to different isoforms. (C) Splice variants can generate soluble variants without TK activity, truncated to one or more Ig domains and missing the TM domain. Variants lacking the TK domain can heterodimerise with full length FGFRs to generate nonfunctional dimers and therefore act as down regulators [24]. (D) FGFR4 can generate a single isoform containing the “c” exon (exon 9) in the Ig3 domain. (E) FGFR2 can generate a splice variant missing Ig1 and Ig3 containing the “b” exon (exon 8). (F) FGFR1 and FGFR2 can also generate a splice variant with truncated Ig1 and Ig3 containing the “c” exon (exon 9). SP: signal peptide, Ig: Immunoglobulin, AB: acid box; TM: transmembrane domain, UTR: untranslated region. Created with BioRender.com (accessed on 26 September 2021).

Figure 3.
FGFR dysregulations. There are several mechanisms of oncogenic FGFR function. (A) Gene amplification results in accumulation of FGFRs that usually translate into protein overexpression and activation of the FGFR axis. (B) Gain of function mutations can lead to constitutive FGFR activation with or without FGF binding. (C) Corrupted autocrine and paracrine loops, either via alternative splicing affecting ligand binding specificity or FGFRs getting overstimulated by FGFs produced in an autocrine fashion, by the cancer cells themselves, or by the tumour microenvironment, in a paracrine fashion. (D) Chromosomal rearrangements can lead to the creation of hybrid oncogenic FGFRs by fusing with binding partners at the carboxyl or amino termini. Created with BioRender.com (accessed on 7 November 2021).
Figure 3.
FGFR dysregulations. There are several mechanisms of oncogenic FGFR function. (A) Gene amplification results in accumulation of FGFRs that usually translate into protein overexpression and activation of the FGFR axis. (B) Gain of function mutations can lead to constitutive FGFR activation with or without FGF binding. (C) Corrupted autocrine and paracrine loops, either via alternative splicing affecting ligand binding specificity or FGFRs getting overstimulated by FGFs produced in an autocrine fashion, by the cancer cells themselves, or by the tumour microenvironment, in a paracrine fashion. (D) Chromosomal rearrangements can lead to the creation of hybrid oncogenic FGFRs by fusing with binding partners at the carboxyl or amino termini. Created with BioRender.com (accessed on 7 November 2021).

Figure 4.
FGFR1–4 genetic alteration in cancer. Genetic alterations by cancer type and mutations of FGFR1 (A), FGFR2 (B), FGFR3 (C) and FGFR4 (D) were found in 662 (~6%), 360 (~3%), 351 (~3%) and 285 (~3%) patients, respectively out of 10,953 in total using cBioPortal. Mutation diagram circles and histograms are coloured with respect to the corresponding mutation types: Green = gene mutations; purple = structural variants; blue = deep deletions; grey = multiple alterations; red = gene amplifications. The lollipop diagrams of each FGF receptor (FGFR1–4), below each histogram, represent the mutation types in relation to the gene location (i.e., missense, truncating, in-frame, splice, SV/fusion). In the case of different mutation types at a single position, the colour of the circle is determined with respect to the most frequent mutation type.
Figure 4.
FGFR1–4 genetic alteration in cancer. Genetic alterations by cancer type and mutations of FGFR1 (A), FGFR2 (B), FGFR3 (C) and FGFR4 (D) were found in 662 (~6%), 360 (~3%), 351 (~3%) and 285 (~3%) patients, respectively out of 10,953 in total using cBioPortal. Mutation diagram circles and histograms are coloured with respect to the corresponding mutation types: Green = gene mutations; purple = structural variants; blue = deep deletions; grey = multiple alterations; red = gene amplifications. The lollipop diagrams of each FGF receptor (FGFR1–4), below each histogram, represent the mutation types in relation to the gene location (i.e., missense, truncating, in-frame, splice, SV/fusion). In the case of different mutation types at a single position, the colour of the circle is determined with respect to the most frequent mutation type.

Figure 5.
Targeting the FGFR axis. Aberrant FGFR signalling can contribute to cancer progression and therefore targeting FP-1039. can also block the FGF–FGFR interaction and therefore prohibit FGFR activation. In addition, ligand binding inhibitors that act as antagonists (e.g., PI-88 and sm27) can prevent FGFR activation. Monoclonal antibodies targeting specific FGFR isoforms (e.g., MFGR1877s by Genentech) can also have anti-tumour activity. Created with BioRender.com (accessed on 12 November 2021).
Figure 5.
Targeting the FGFR axis. Aberrant FGFR signalling can contribute to cancer progression and therefore targeting FP-1039. can also block the FGF–FGFR interaction and therefore prohibit FGFR activation. In addition, ligand binding inhibitors that act as antagonists (e.g., PI-88 and sm27) can prevent FGFR activation. Monoclonal antibodies targeting specific FGFR isoforms (e.g., MFGR1877s by Genentech) can also have anti-tumour activity. Created with BioRender.com (accessed on 12 November 2021).


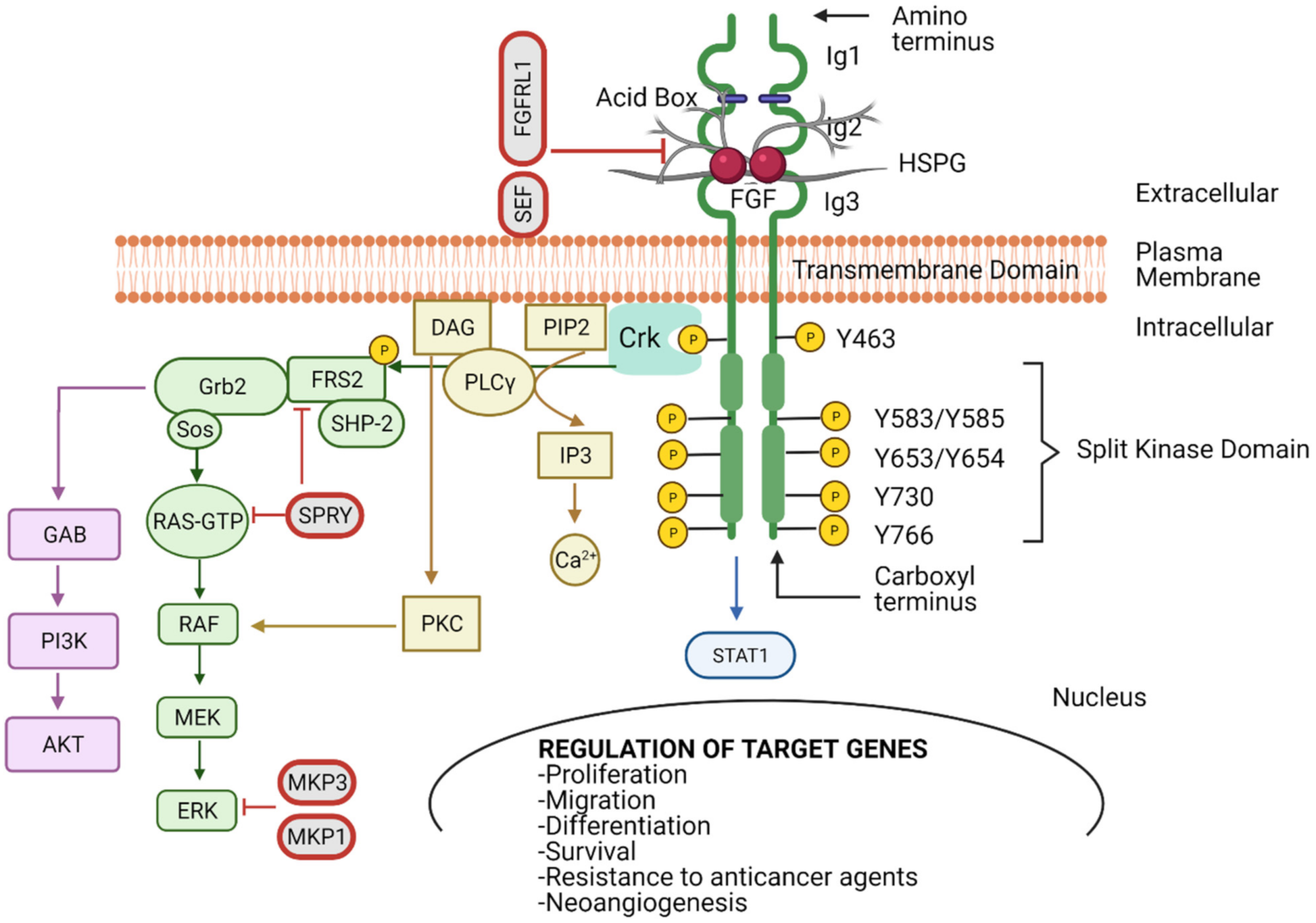{kind=link}
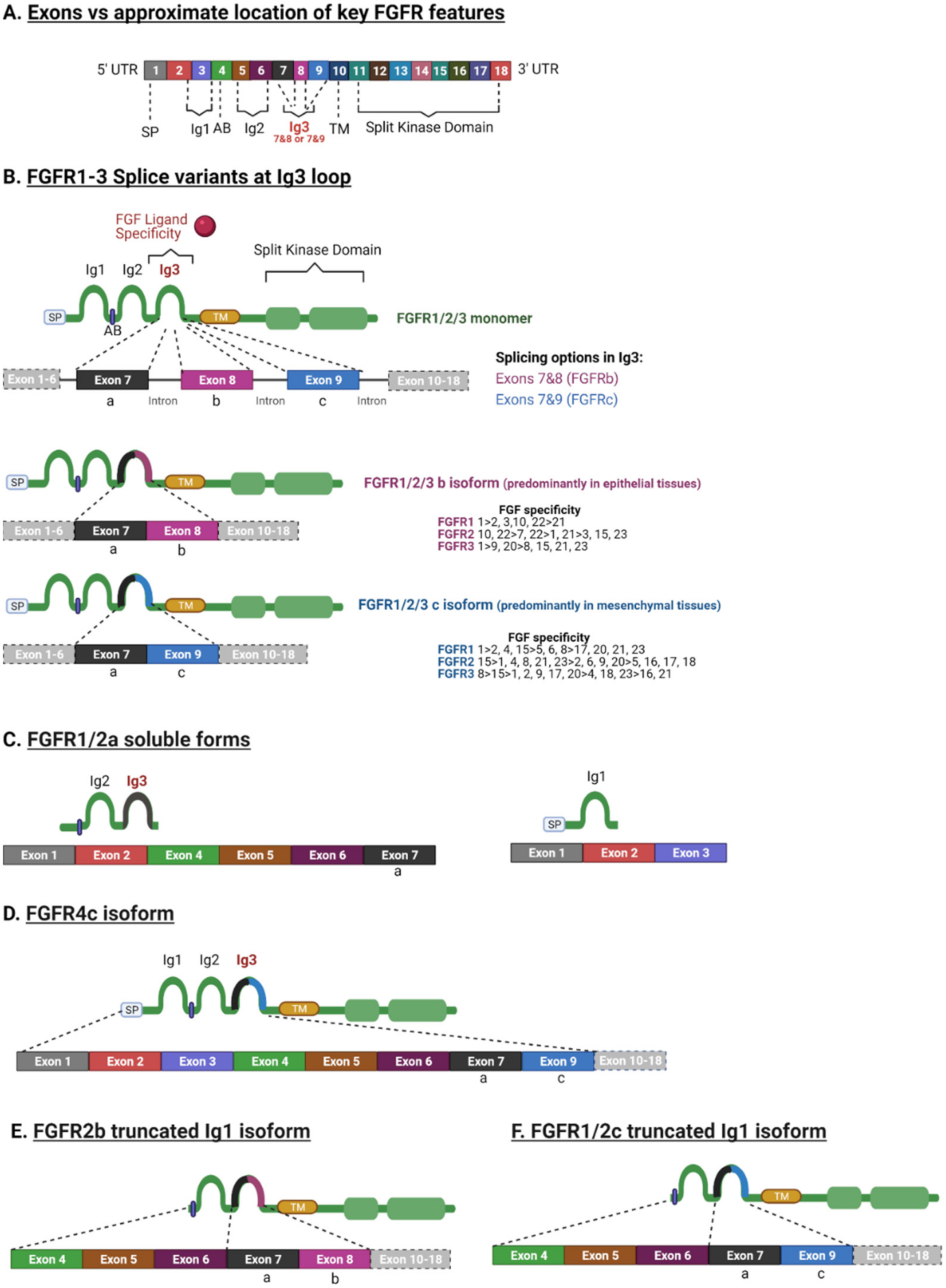{kind=link}
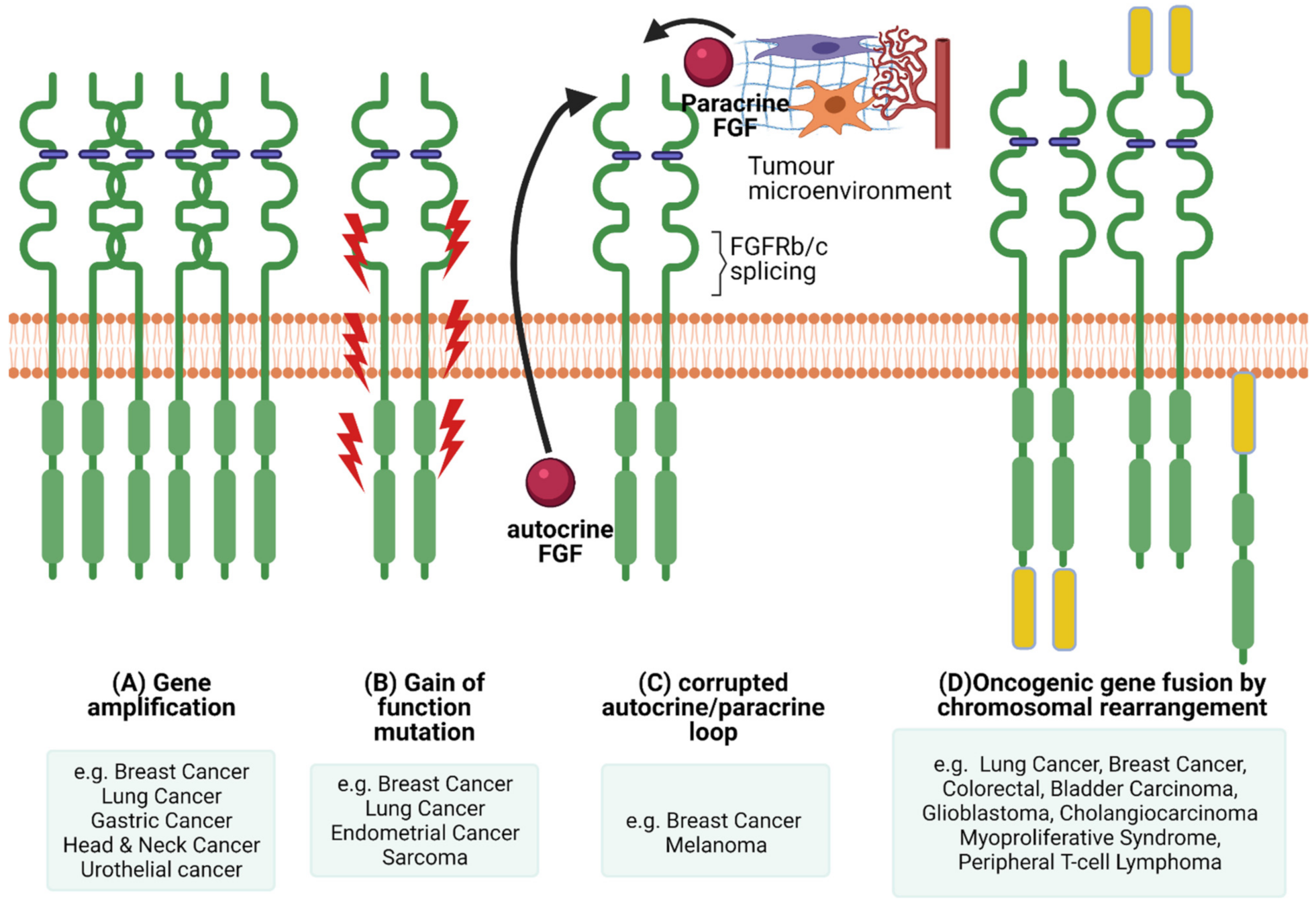{kind=link}
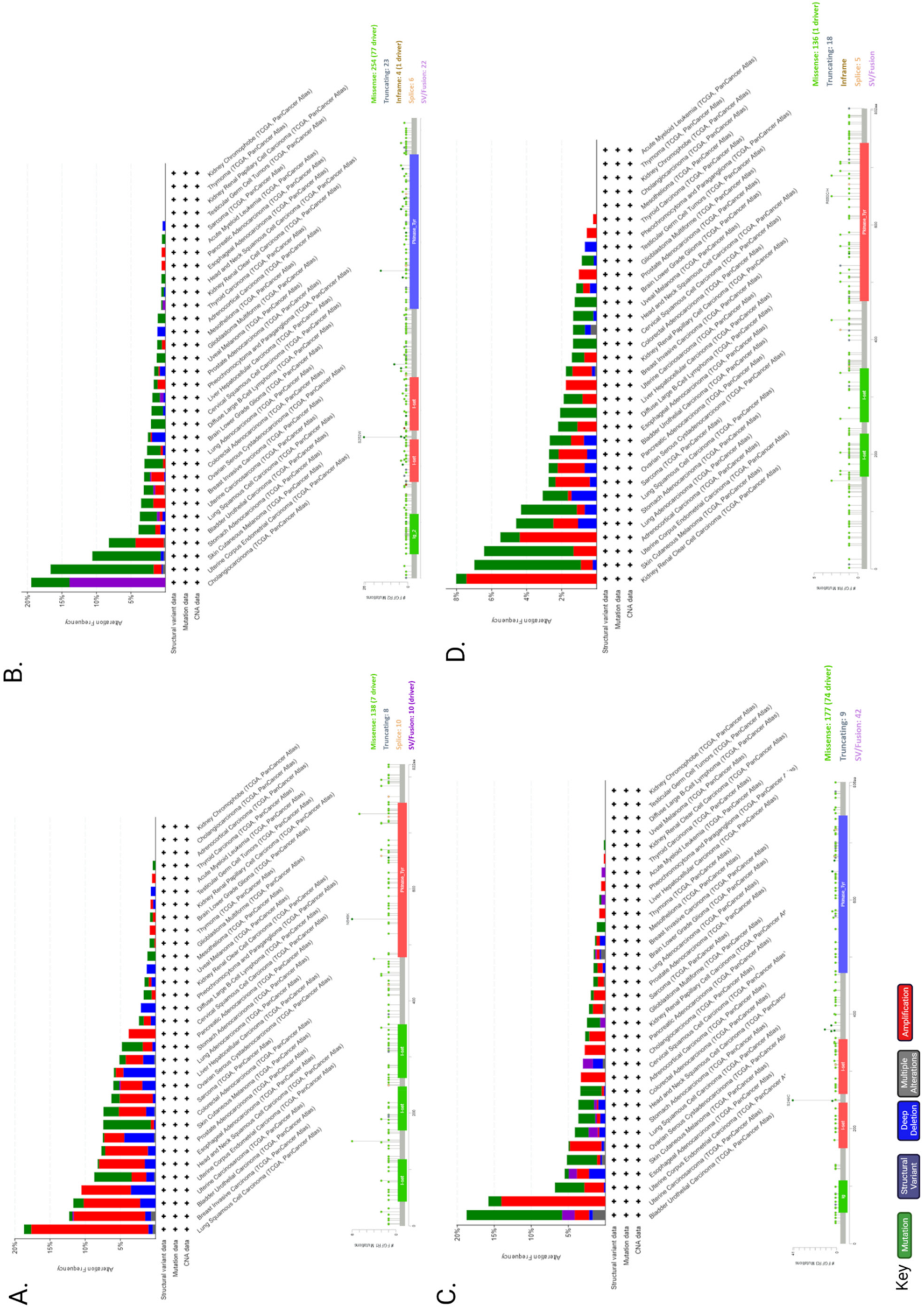{kind=link}
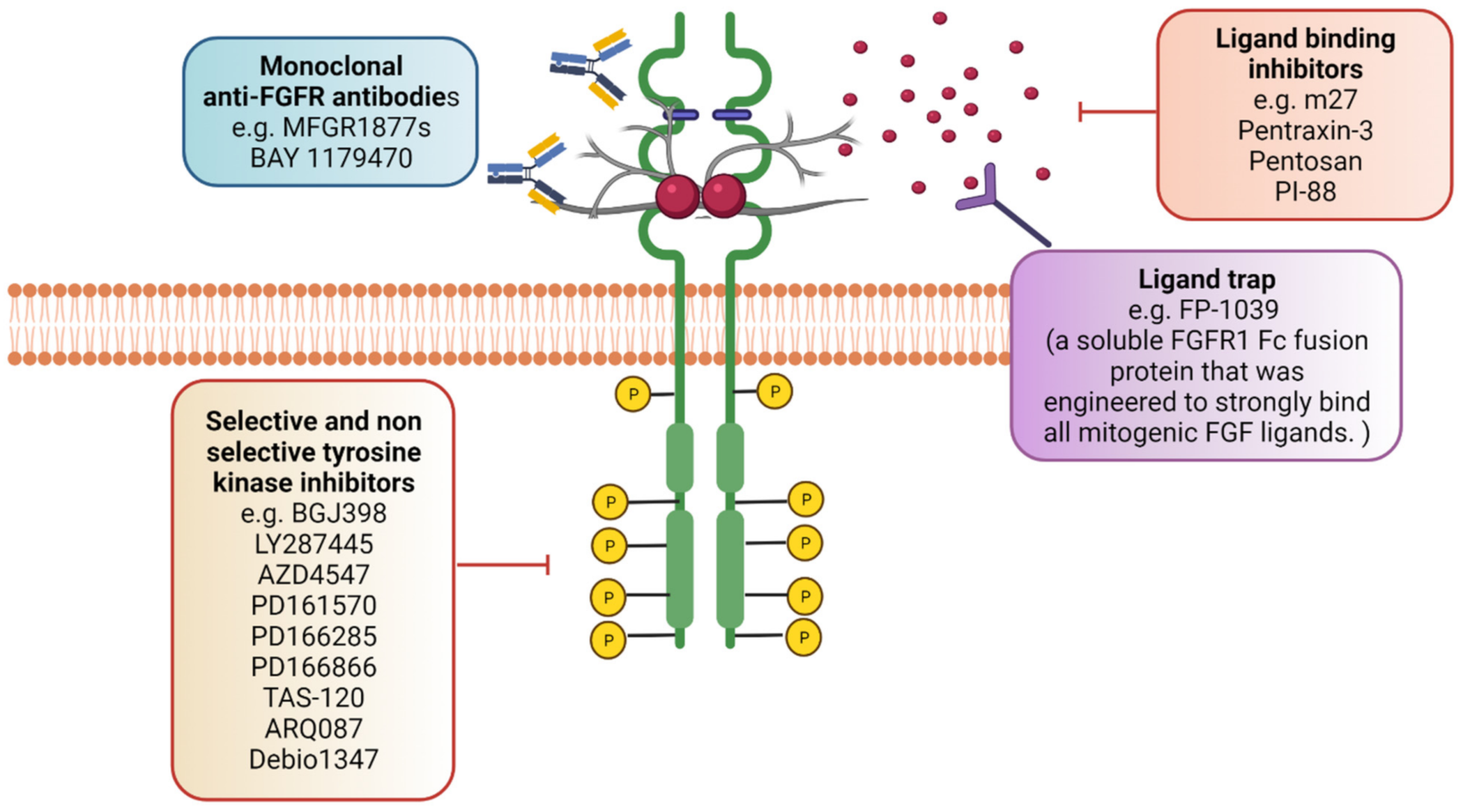{kind=link}
Table 1.
Phase 3 Interventional clinical studies targeting FGF receptors in cancer. Currently there are no phase 4 clinical trials, however, there are over 90 phase 1 and 2 clinical trials targeting FGFR in different types of cancers, and a number of phase 3 trials not yet recruiting. FGFR inhibitors are indicated in bold.
Table 1.
Phase 3 Interventional clinical studies targeting FGF receptors in cancer. Currently there are no phase 4 clinical trials, however, there are over 90 phase 1 and 2 clinical trials targeting FGFR in different types of cancers, and a number of phase 3 trials not yet recruiting. FGFR inhibitors are indicated in bold.
| NCT Number | Title | Conditions | Interventions | Enrolment |
|---|---|---|---|---|
| NCT03390504 | A Study of Erdafitinib compared with Vinflunine or Docetaxel or Pembrolizumab in participants with advanced urothelial cancer and selected Fibroblast Growth Factor Receptor (FGFR) gene aberrations | Urothelial Cancer | Erdafitinib, Vinflunine, Docetaxel, Pembrolizumab | 631 |
| NCT04197986 | Study of oral Infigratinib for the adjuvant Treatment of subjects with invasive urothelial carcinoma with susceptible FGFR3 genetic alterations | Upper Tract Urothelial Carcinomas, Urothelial Bladder Cancer | Infigratinib | 218 |
| NCT04093362 | Futibatinib versus Gemcitabine-Cisplatin chemotherapy as first-line treatment of patients with advanced cholangiocarcinoma harboring FGFR2 gene rearrangements | Advanced Cholangiocarcinoma; FGFR2 Gene Rearrangements | Futibatinib, Cisplatin/Gemcitabine | 216 |
| NCT03773302 | Phase 3 study of BGJ398 (Oral Infigratinib) in first line cholangiocarcinoma with FGFR2 gene fusions/translocations | Advanced Cholangiocarcinoma, FGFR2 Gene Mutation | Infigratinib, Gemcitabine, Cisplatin | 300 |
| NCT03656536 | A study to evaluate the efficacy and safety of Pemigatinib versus chemotherapy in unresectable or metastatic cholangiocarcinoma | Unresectable Cholangiocarcinoma, Metastatic Cholangiocarcinoma | Pemigatinib, Gemcitabine, Cisplatin | 432 |
| NCT03784014 | Molecular profiling of advanced soft-tissue sarcomas | Soft Tissue Sarcoma | Nilotinib, Ceritinib, Capmatinib, Lapatinib, Trametinib, Combination of Trametinib and Dabrafenib, Combination of Olaparib and Durvalumab, Palbociclib, Futibatinib | 960 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chioni, A.-M.; Grose, R.P. Biological Significance and Targeting of the FGFR Axis in Cancer. Cancers 2021, 13, 5681. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225681
AMA Style
Chioni A-M, Grose RP. Biological Significance and Targeting of the FGFR Axis in Cancer. Cancers. 2021; 13(22):5681. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225681
Chicago/Turabian StyleChioni, Athina-Myrto, and Richard P. Grose. 2021. "Biological Significance and Targeting of the FGFR Axis in Cancer" Cancers 13, no. 22: 5681. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225681
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.