
Resistance to Targeted Agents Used to Treat Paediatric ALK-Positive ALCL



1
Department of Pathology, University of Cambridge, Cambridge CB2 0QQ, UK
2
Department of Paediatric Oncology and Haematology, Addenbrooke’s Hospital, Cambridge CB2 0QQ, UK
3
CEITEC, Masaryk University, 62500 Brno, Czech Republic
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(23), 6003; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13236003
Submission received: 5 October 2021
/
Revised: 25 November 2021
/
Accepted: 25 November 2021
/
Published: 29 November 2021
(This article belongs to the Special Issue New Therapeutic Developments in Hematological Malignancies)
Abstract
:Simple Summary
In general, the non-Hodgkin lymphoma (NHL), anaplastic large cell lymphoma (ALCL) diagnosed in childhood has a good survival outcome when treated with multi-agent chemotherapy. However, side effects of treatment are common, and outcomes are poorer after relapse, which occurs in up to 30% of cases. New drugs are required that are more effective and have fewer side effects. Targeted therapies are potential solutions to these problems, however, the development of resistance may limit their impact. This review summarises the potential resistance mechanisms to these targeted therapies.
Abstract
Non-Hodgkin lymphoma (NHL) is the third most common malignancy diagnosed in children. The vast majority of paediatric NHL are either Burkitt lymphoma (BL), diffuse large B-cell lymphoma (DLBCL), anaplastic large cell lymphoma (ALCL), or lymphoblastic lymphoma (LL). Multi-agent chemotherapy is used to treat all of these types of NHL, and survival is over 90% but the chemotherapy regimens are intensive, and outcomes are generally poor if relapse occurs. Therefore, targeted therapies are of interest as potential solutions to these problems. However, the major problem with all targeted agents is the development of resistance. Mechanisms of resistance are not well understood, but increased knowledge will facilitate optimal management strategies through improving our understanding of when to select each targeted agent, and when a combinatorial approach may be helpful. This review summarises currently available knowledge regarding resistance to targeted therapies used in paediatric anaplastic lymphoma kinase (ALK)-positive ALCL. Specifically, we outline where gaps in knowledge exist, and further investigation is required in order to find a solution to the clinical problem of drug resistance in ALCL.
1. Introduction
1.1. Epidemiology and Pathologenesis of Paediatric ALCL
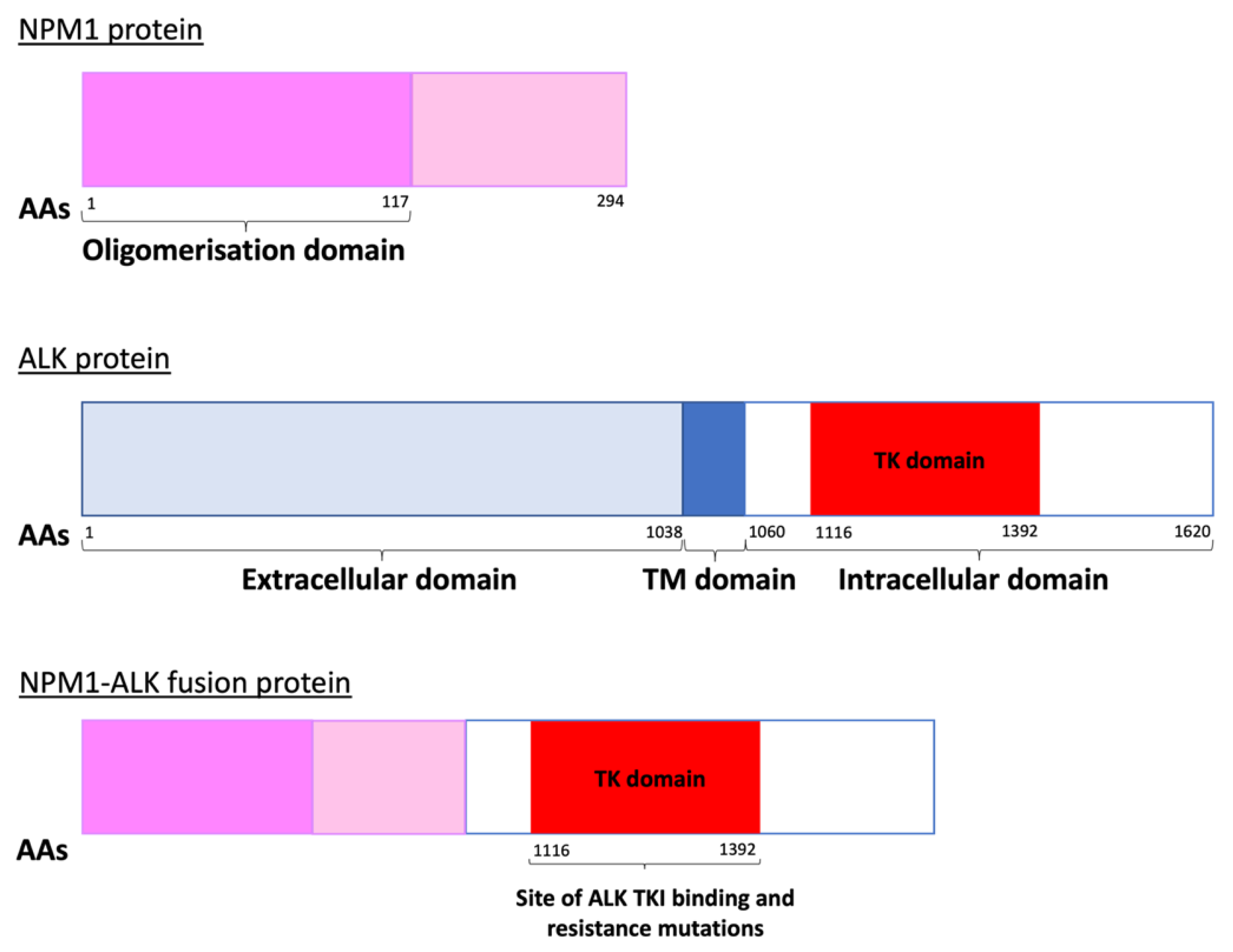
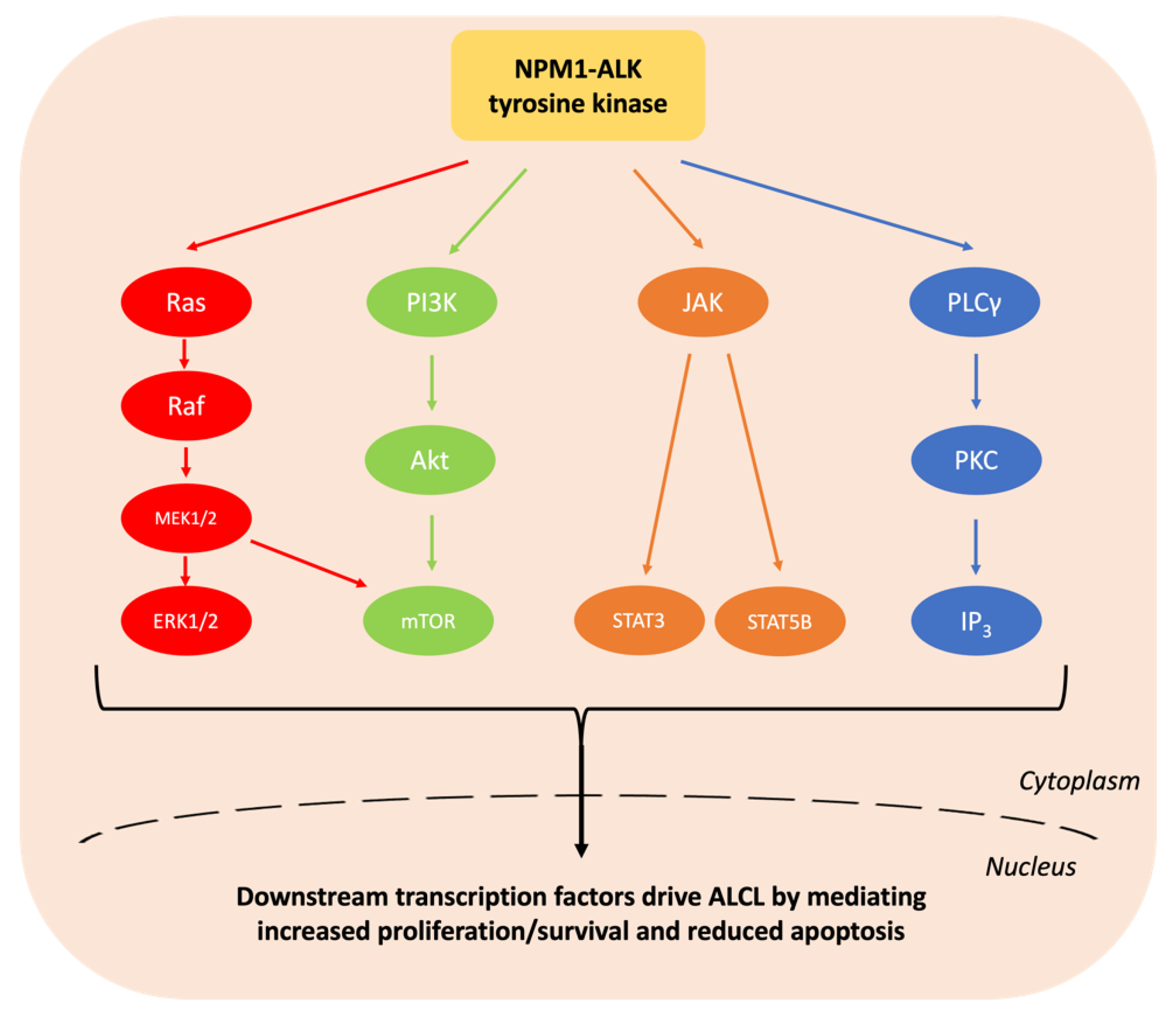
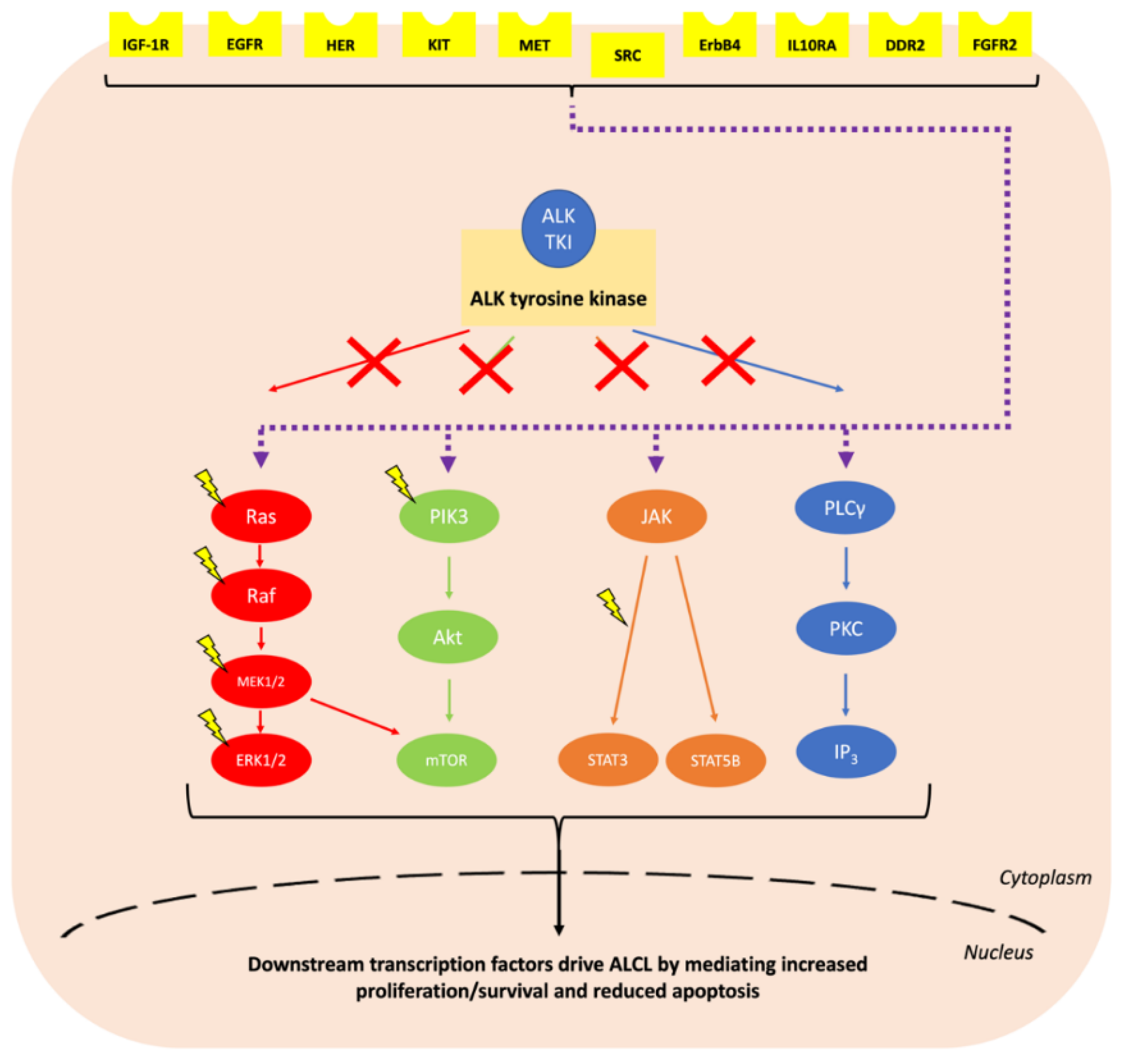
Anaplastic large cell lymphoma (ALCL) is a peripheral T cell non-Hodgkin lymphoma (NHL) with an annual incidence of 1.2 per million children aged under 15 [1]. The World Health Organisation (WHO) sub-classifies ALCL into anaplastic lymphoma kinase (ALK)-positive nodal/systemic, ALK-negative nodal/systemic, primary cutaneous and breast implant-associated ALCL [2]. The majority of paediatric ALCL is ALK-positive, usually due to a t(2;5)(p23;q35) chromosomal translocation causing the expression of the oncogenic breakpoint product NPM1-ALK [1,3]. This translocation results in unregulated ligand-independent activation of ALK, a receptor tyrosine kinase [2,4], due to homodimerization and auto-phosphorylation of its kinase domain [5] (Figure 1). The oncogenic activity of NPM1-ALK has been proven in a variety of genetically modified murine models and is therefore considered the driving event in these malignancies [6,7,8,9]. In essence, NPM1-ALK is able to activate a number of pathways that confer the hallmarks of cancer on incipient tumour cells [10] including pathways that facilitate increased survival and reduced apoptosis, such as the mitogen-activated protein kinase (MAPK) [11], phosphoinositide 3-kinase (PI3K)-Akt [12], Janus kinase (JAK)-signal transducer and activator of transcription (STAT) [13] and phospholipase C gamma (PLCγ) pathways [14] (Figure 2).
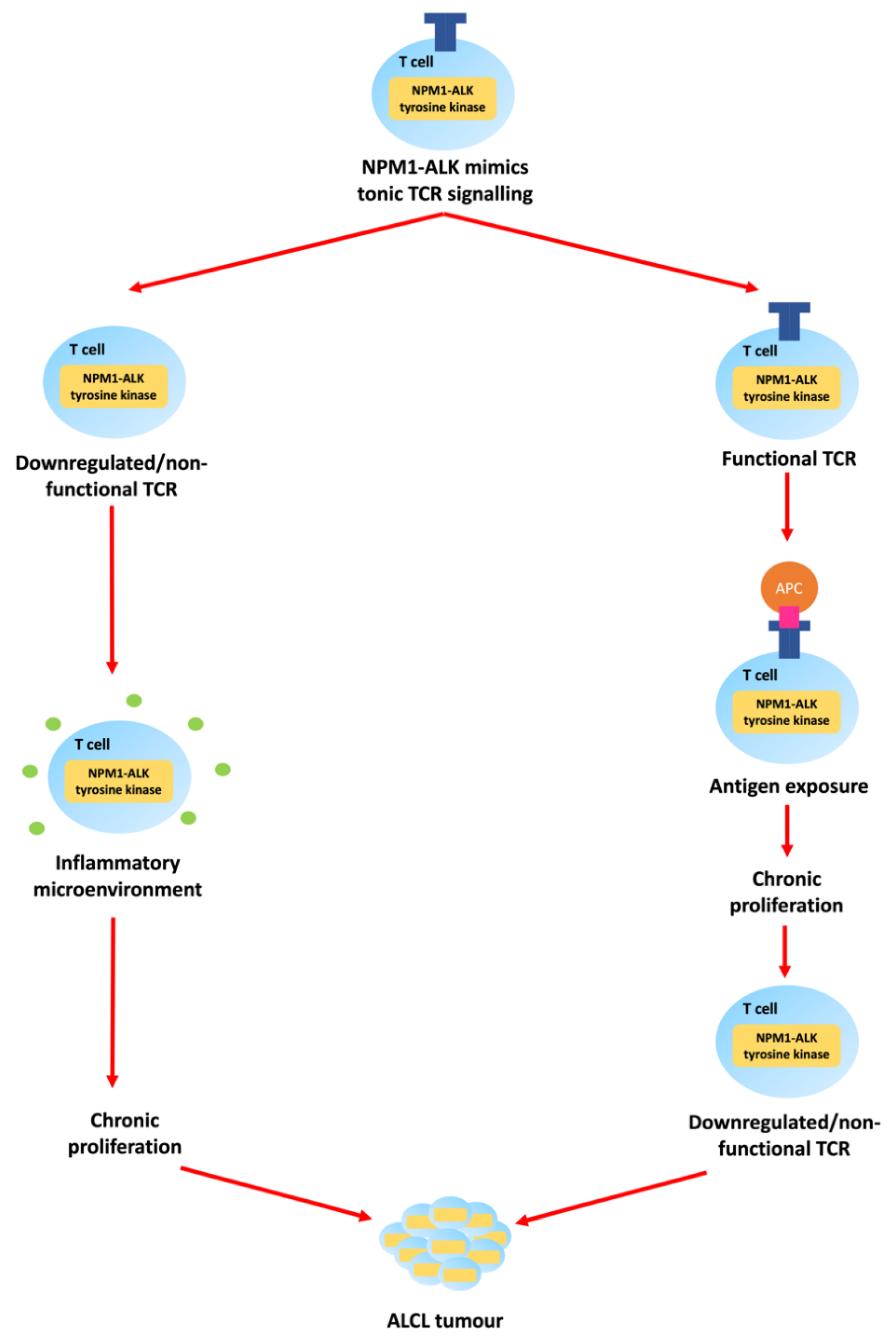
However, while NPM1-ALK is necessary for the development of ALK-positive ALCL, its presence alone is not sufficient for lymphomagenesis given that approximately 1% of newborn babies carry the t(2;5)(p23;q35) translocation (as evidenced by the presence of the translocation in cord blood stem cells) but, the incidence of ALCL is orders of magnitude lower [19]. In keeping with multi-step carcinogenesis, ‘second hits’ might include antigen-induced T-cell receptor (TCR) signalling, or antigen-independent microenvironmental factors [2,7,11,20,21] (Figure 3).
The uncertainty surrounding the precise pathogenesis of ALCL is made more complex by the illusive ‘cell of origin’. Most ALCL tumour cells express CD4 (indicative of helper T cells, although CD3 expression is seen in <20% tumours), granzyme B, TIA-1 and perforin (indicative of cytotoxic T cells), CD30 (indicative of activated lymphocytes) and sometimes CD25 (indicative of activated lymphocytes or regulatory T cells when co-expressed with CD4), with the common denominator being a T cell [2,20,22,23]. However, gene expression profiles of ALCL not only share features with T helper 17 (Th17) cells [24], but also with early thymic progenitors or haemopoietic stem cells [25]. Whether this latter genetic signature is a consequence of the cell of origin or NPM1-ALK-induced activity is debatable. In support of the former, NPM1-ALK enables thymocytes to skip beta-selection in mice, and it has been observed that two-thirds of human ALCL tumours have TCR rearrangements that would not normally be permissive of successful thymic development suggesting that NPM1-ALK is active in the aberrant thymocytes of children carrying this translocation [7,20]. However, NPM1-ALK is also able to transform primary human peripheral T cells to mimic ALCL and activate a gene signature associated with stemness [26,27,28,29]. Nevertheless, additional events, beyond the expression of NPM1-ALK, are required for the development of ALK-positive ALCL. In summary, ALCL is a rare paediatric NHL whose oncogenic driver is NPM1-ALK. Its exact cell of origin and pathogenesis remains the subject of continued research.
1.2. Clinical Presentation and Management of Paediatric ALCL
Children with ALCL usually present with high-grade advanced-stage nodal and extranodal disease [30]. Up to 61% have bone marrow involvement when minimal disseminated disease (MDD) is assessed using qualitative reverse-transcription polymerase chain reaction [31], and 5% have central nervous system (CNS) involvement [32]. Symptoms vary with disease location but typically include lymphadenopathy and B symptoms [30].
Paediatric ALCL is treated in many countries with the internationally recognised ALCL99 multi-agent chemotherapy regimen which has a 10-year overall survival (OS) of over 90% [20,33] but often leads to acute toxicities and late effects [34]. Additionally, progression-free survival (PFS) is only 70% and outcomes are poorer for relapsed cases [20,33]. The European Inter-Group for Childhood Non-Hodgkin Lymphoma (EICNHL) ALCL-RELAPSE trial suggested vinblastine monotherapy was effective for low-risk first relapse, and multi-agent chemotherapy followed by allogeneic stem cell transplant (SCT) was best for high-risk first relapse [2,35]. However, there is no absolute management consensus, particularly for subsequent relapses [20,33].
Current challenges in the treatment of paediatric ALCL are to find less toxic treatments, predict relapse, and treat it more effectively. Vinblastine monotherapy is minimally toxic and effective for a subgroup of patients with relapsed disease; it is the subject of an upcoming trial in newly diagnosed non-high-risk ALCL patients.
Vinblastine is a vinca alkaloid that binds to the mitotic spindle and stops cell division at metaphase. It is usually given at a weekly dose of 6 mg/m2 intravenously and is generally well tolerated, with the advantage that monotherapy can be delivered on an outpatient basis [36]. Vinblastine has shown efficacy in the treatment of relapsed/refractory ALCL whereby a study conducted by the French Society of Paediatric Oncology reported that vinblastine monotherapy (median duration 14 months) led to complete remission (CR) in 83% of relapsed patients, which was sustained for 7 years in 36% of cases [37]. Additionally, the EICNHL-ALCL-RELAPSE trial showed that 2 years of vinblastine monotherapy achieved a 3-year event-free survival (EFS) of 85% and OS of 90% for low-risk relapses [35]. This suggests that vinblastine-induced remissions are better sustained with longer treatment. However, in the first-line setting the efficacy of vinblastine is not as clear cut; EICNHL together with the Children’s Oncology Group (COG) investigated the addition of 1 year of vinblastine to standard first-line multi-agent chemotherapy for paediatric ALCL. Unfortunately, this approach did not reduce the relapse rate, and instead increased acute toxicity [38,39]. These data suggest that vinblastine is best utilised as a single agent if the goal is to reduce treatment-related toxicity. Therefore, EICNHL will investigate vinblastine monotherapy as a first-line treatment for low-risk patients, but with a longer treatment duration of 2 years in an upcoming trial [20]. Given the efficacy of the ALCL99 regimen, this is unlikely to further improve survival rates but is expected to reduce toxicity [33].
Resistance to vinblastine is a distinct possibility as has been reported for other malignancies. The major cause of vinblastine resistance in these cases is due to upregulation of expression of members of the ATP-binding cassette (ABC) transporter superfamily, such as ABCC1 and MDR-1 (P-glycoprotein). These transporters facilitate multi-drug resistance through their ability to efflux drugs, resulting in a reduced intracellular drug concentration. They have been shown to mediate vinblastine resistance specifically in renal cell carcinoma [40,41] and T cell lymphoblastic leukaemia [42] cell lines. Increased expression of these efflux pumps generally occurs through gene amplification or via downregulation of negative regulators of their expression, such as a decrease in MiR-210-3p which is a negative regulator of ABCC1 [40,43]. Many drugs have been investigated as potential therapies to overcome vinblastine resistance caused by ABC transporters. These include chloroquine, chlorpromazine, verapamil, cyclosporine A, quinine, valspodar, biricodar and zosuquidar [41,44,45,46,47]. However, these drugs did not perform well in clinical trials due to poor efficacy and/or toxicity problems and none are currently approved for the reversal of drug resistance caused by ABC transporters [46,47]. Indeed, through this mechanism, resistance to multiple standard toxic chemotherapy agents is induced and with the advent and increasing use of novel targeted agents, the search for inhibitors of multi-drug resistance in this form may become of decreasing necessity.
A second cause of vinblastine resistance is increased expression or activity of c-Jun, a member of the transcription factor, activator protein-1 (AP-1) family, which disrupts the ability of vinblastine to initiate apoptosis [48]. As AP-1 transcription factors are actively transcribed in ALCL, this mechanism of resistance is a distinct possibility [16]. A third cause of resistance is an alteration in tubulin content and polymerisation status, which interferes with the site of action of vinblastine [49]. However, despite these possible mechanisms, resistance to vinblastine monotherapy is not particularly common in ALK-positive ALCL, as it remains effective for subsequent relapses that occur after therapy cessation in the majority of patients [37]. Additionally, as previously mentioned, the increased use of novel targeted agents will likely negate any pressure to develop counter-active measures towards resistance to vinblastine.
Emerging agents targeting the pathways dysregulated by NPM1-ALK hold great promise for both newly diagnosed and relapsed patients. However, both primary (intrinsic) and acquired resistance to these drugs are a major problem and, at present clear solutions or guidelines to overcome this issue do not exist. A prerequisite for establishing solutions is a detailed understanding of the potential mechanisms of resistance and therefore this review will focus on the latter rather than providing solutions.
2. Targeted Agents for the Treatment of ALCL
Studying targeted agents for the treatment of paediatric ALK-positive ALCL is challenging due to the low disease incidence. However, coordinated international efforts to study the treatment of paediatric ALCL, in addition to knowledge gained from investigating adult ALCL and other ALK-positive diseases, has led to the suggestion that ALK tyrosine kinase inhibitors (ALK TKIs), armed antibodies to CD30 (such as brentuximab vedotin (BV)) and immune checkpoint inhibitors (such as nivolumab) may be useful drugs to overcome the current challenges in the treatment of paediatric ALCL.
2.1. ALK Tyrosine Kinase Inhibitors
ALK TKIs inhibit the kinase activity of aberrantly expressed ALK largely through binding to the ATP pocket. They are given orally and are generally well tolerated as monotherapy with few, largely manageable side effects [20,50]. As ALK is usually only expressed in neonatal neurons, off-target side effects were expected to be minimal [51], particularly as ALK knockout mice are viable without any obvious health defects [52,53]. The use of ALK TKIs to treat ALCL has benefitted from prior investigations of their use in ALK-positive non-small cell lung cancer (NSCLC) [1,54]. The first ALK TKI approved for NSCLC by the Food and Drug Administration (FDA) was crizotinib, but second (ceritinib, alectinib and brigatinib) and third (lorlatinib) generation inhibitors have subsequently also been authorised [1]. Approval for use in ALCL has been difficult, particularly in children due to disease rarity [20]. However, crizotinib recently gained FDA approval for relapsed/refractory ALK-positive ALCL in patients aged over one year [55]. This was based on evidence that crizotinib was well tolerated and led to a complete response in 67–83% of patients with relapsed ALCL [50,56,57]. Additionally, alectinib has recently gained approval in Japan for relapsed ALCL, due to a trial showing efficacy (PFS 58.3%, EFS and OS both 70%) [58].
How ALK TKIs should be combined with or replace existing frontline therapy remains to be resolved given the already high survival rate of multi-agent chemotherapy protocols such as ALCL99. A recently completed trial (NCT01606878) employed crizotinib in combination with ALCL99 therapy and showed unacceptable cytopenias and gastrointestinal toxicity in the paediatric ALCL relapse setting [59]. Another trial (ITCC053) closed the arm in which crizotinib and vinblastine were given in combination for relapsed paediatric ALCL due to toxicities [60]. The full results of an additional trial (NCT01979536) administering crizotinib with the ALCL99 chemotherapy regimen in newly diagnosed paediatric ALCL, are awaited, although a temporary pause in the trial had to be imposed due to a number of thromboses [1,61]. Whether other ALK inhibitors will result in different toxicity profiles remains to be seen. An industry-led trial (Takeda) combining brigatinib with ALCL99 multi-agent chemotherapy is planned. Brigatinib has shown efficacy in adults with NSCLC resistant to crizotinib and unlike crizotinib, brigatinib has shown good brain penetrance and durable responses in patients with NSCLC [62], and it is hoped that its use in the treatment of ALCL will be similarly effective.
2.2. Brentuximab Vedotin
All ALK-positive paediatric ALCL express the transmembrane receptor CD30, which facilitates cancer cell survival through the activation of, for example, the nuclear factor-kappa B (NF-kB), Interferon regulatory factor 4 (IRF4) and MYC proto-oncogene (MYC) pathways [63,64]. However, monoclonal antibodies (MABs) targeting CD30 alone were disappointing; only 17% of adult ALCL patients had any response to the anti-CD30 MAB SGN-30 in a Phase II trial [65]. Therefore, the antibody-dug conjugate BV was developed. This consists of an antibody targeting CD30 bound to the anti-tubulin agent monomethyl auristatin E (MMAE). When the antibody binds to CD30, the drug-receptor complex is internalised, and after lysosomal processing, MMAE is released inside the cell, driving apoptosis [63].
BV was approved by the FDA for adults with relapsed ALCL in 2011 [1]. This was based on early results from a phase II study (NCT00866047) which has since demonstrated durable remissions at five years [66,67]. BV was next recommended for use alongside multi-agent chemotherapy in newly diagnosed adult ALCL due to the results of the phase III ECHELON-2 trial [68]. Subsequently, BV has been studied for use in paediatric ALCL. A phase I/II study (NCT01492088) including paediatric patients with relapsed/refractory ALCL showed that the pharmacokinetic profiles were similar to those in adults; 53% of patients achieved an overall response, and side effects were manageable [69]. Another phase II study (ANHL12P1) showed that the addition of BV to the ALCL99 chemotherapy regimen for newly diagnosed ALK-positive paediatric ALCL led to an EFS of 79.1%, an improvement from the assumed EFS of 70% for ALCL99 therapy alone, without any additional toxicity. Moreover, this treatment approach almost completely eliminated progression/relapse during first-line treatment. This is particularly important because children who progress during first-line treatment have the poorest prognosis [63,70].
2.3. Checkpoint Inhibitors
ALK-positive ALCL cells constitutively express the immune checkpoint protein programmed cell-death ligand 1 (PD-L1) on their surface. This interacts with the programmed cell death 1 (PD-1) receptor to dampen the T cell immune response against the tumour, although the precise mechanism via which this occurs is currently under investigation [71,72]. MABs directed against PD-L1, nivolumab and pembrolizumab, may be particularly effective in ALK-positive ALCL because NPM1-ALK drives high levels of PD-L1 expression via STAT3 [1]. Indeed, initial case reports describe durable responses and limited toxicities in teenagers with relapsed/refractory ALK-positive ALCL [73,74]. Patients are currently being recruited to a phase II trial (NCT03703050, NIVO-ALCL) to study the use of nivolumab in adult and paediatric patients with relapsed/refractory ALK-positive ALCL previously treated with chemotherapy and either an ALK TKI or BV [1].
3. Mechanisms of Resistance to ALCL Therapy
It is anticipated that resistance to these novel agents may be a major problem. Understanding how this resistance develops, and how it can be overcome, will be essential to the successful integration of these novel approaches into the mainstream treatment of paediatric ALK-positive ALCL.
3.1. Mechanisms of Resistance to ALK Tyrosine Kinase Inhibitors
Despite the promise of ALK TKIs, it appears at least in some contexts, that therapy will either have to be for long periods of time or as a bridge to another treatment, as swift relapses have been observed on the discontinuation of crizotinib monotherapy [1,75]. In addition, resistance is expected as has been experienced for patients with ALK-positive NSCLC [17]. Indeed, reports of crizotinib resistance developing within months of treatment initiation in patients with ALCL have been published [56,57].
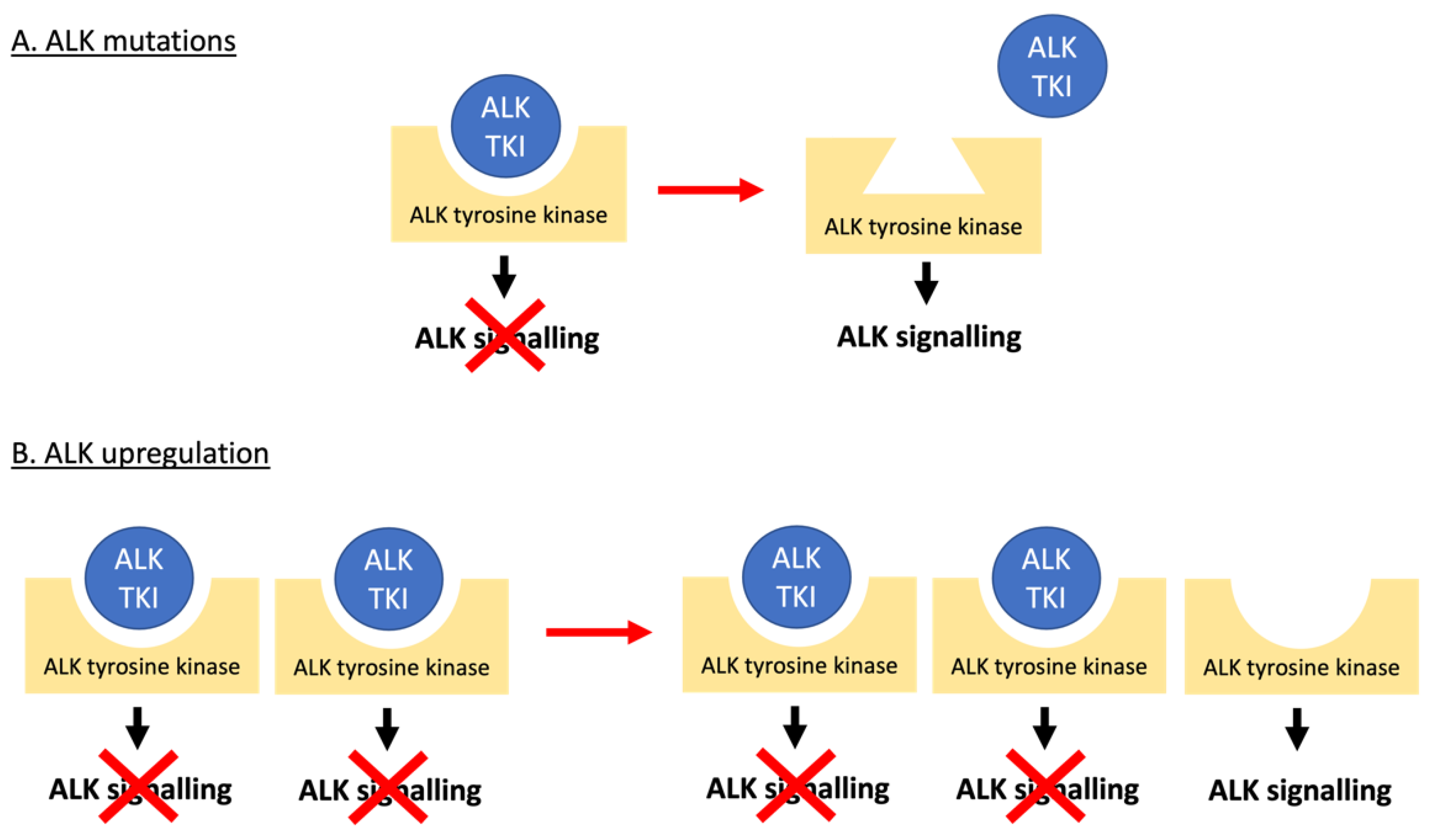
ALK TKI resistance can either be ALK-dependent or -independent with the former largely occurring due to mutations in ALK (Figure 1 and Figure 4). These have been reported as either mutations in residues at the TKI binding site, or those that result in conformational changes that increase aberrant ALK activity [76,77]. Extensive studies of ALK-dependent resistance mechanisms in NSCLC [17,78] have identified numerous mutations which in some cases are specific to a certain ALK TKI and in others are ubiquitous amongst the ALK TKIs (Table 1). In the treatment of NSCLC, clinicians usually start by treating with crizotinib, and then swap to second- or third-generation inhibitors depending on the ALK mutation that has developed. Ultimately this increases the risk of compound mutations developing, which confer resistance to second and even third-generation inhibitors [17,79]. However, some of these compound mutations can actually re-sensitise patients to crizotinib, even if it has been administered to the patient previously [17]. Cycling of TKIs may therefore be an approach to preventing or responding to the development of resistance.
Another form of ALK-dependent resistance is caused by the amplification of the ALK gene (Figure 4). This results in resistance due to a target excess [17,78] as has been reported for NSCLC cell lines [80,81] and patients [78,82,83], as well as ALCL cell lines [84,85] but not yet for ALCL patients. Interestingly, ALK upregulation in response to ALK TKIs results in overwhelming ALK signalling such that if the ALK TKI treatment is stopped, the excess in ALK signalling can counterintuitively drive apoptosis. Therefore, a non-continuous dosing regimen may be beneficial [85,86,87].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of reported ALK mutants conferring resistance to ALK TKIs. Sites of the identified mutations are reported according to their position in the full-length ALK protein. Resistance mutations refer to those reported in the context of conferring resistance to ALK TKIs, some of which have been proven to confer sensitivity to ALK TKIs and others with conflicting evidence as to their ALK TKI response. Ins = insertion, del = deletion.
Table 1.
Summary of reported ALK mutants conferring resistance to ALK TKIs. Sites of the identified mutations are reported according to their position in the full-length ALK protein. Resistance mutations refer to those reported in the context of conferring resistance to ALK TKIs, some of which have been proven to confer sensitivity to ALK TKIs and others with conflicting evidence as to their ALK TKI response. Ins = insertion, del = deletion.
| ALK TKI | ALK Mutation | ||
|---|---|---|---|
| Resistance | Sensitivity | Conflicting | |
| Crizotinib | C1156T [88] C1156Y [17,79,89,90,91,92,93,94] | L1198F [79,89,95,96] C1156Y/L1198F [95] | |
| D1203N [79,96,97,98] | G1202R/L1198F [91] | ||
| E1210K [79,90,99] | I1171N/L1265F [91] | ||
| F1174C [76,79,89,91,99] F1174I [91] F1174L [100,101] F1174V [91] F1245V [92] | |||
| G1128A [102] | |||
| G1202R [78,79,89,90,94] G1202del [79,89] | |||
| G1269A [79,82,89,90,94,96,99] G1269S [103] | |||
| I1171N [79,89,90,103,104,105] I1171S [79,89,90,103] I1171T [76,79,89,90,91,92,98] I1171X [106] | |||
| I1268L [91] | |||
| L1152P [103,107] | |||
| L1152R [81,94] | |||
| L1196M [17,78,79,80,82,89,90,91,92,93,94,96,98,99] L1196Q [91,96,104] | |||
| L1198P [97] | |||
| R1192P [108] | |||
| S1206C [90,103] S1206Y [78,79,90,94] | |||
| T1151K [109,110] T1151M [108] | |||
| V1180L [96] | |||
| Q1188_L1190del [111] | |||
| 1151Tins [78,90] | |||
| D1203N/E1210K [79,89] | |||
| D1203N/F1174C [79,89] | |||
| F1174L/G1269A [98] | |||
| Ceritinib | F1174L [79,90,103,107,108] | E1210K [79,89] | C1156Y [79,89,90,103,107,112] |
| F1174S [90] | F1245C [113] | D1203N [79,89,90,96] | |
| F1174V [103] | I1171T [79,89,107] | F1174C [79,89,107,112] | |
| G1123S [114] | I1268L [91] | G1202R [79,89,90,92,98,103,112] | |
| G1128A [98] | L1196Q [91] | G1269A [79,89,96,98,107,108] | |
| G1202del [79,89,103] | S1206Y [107,115] | I1171N [79,89,90,107] | |
| L1122V [96] | V1185L [91] | I1171S [79,89,90] | |
| L1152P [103] | G1269A/I1171S [116] | L1196M [79,89,90,91,96,107,115] | |
| L1152R [103,107] | G1269A/I1171N [91] | ||
| L1198F [79,89,96] | |||
| R1192P [108] | |||
| R1275Q [17] | |||
| T1151K [109] | |||
| T1151M [108] | |||
| T1151Sins [117] | |||
| Q1188_L1190del [111] | |||
| 1151Tins [103,107] | |||
| C1156Y/I1171N [79] | |||
| C1156Y/G1202del/V1180L [79] | |||
| D1203N/E1210K [79,89] | |||
| D1203N/F1174C [79,89] | |||
| E1210K/I1171T [98] | |||
| G1202R/F1174L [79] | |||
| G1202R/F1174V [92] | |||
| G1202R/L1196M [91] | |||
| Alectinib | F1174I [91] | C1156Y [79,89,118] | F1174C [76,79,89] |
| F1174L [79,90,91,103,107,108] | D1203N [79,89,96] | I1171T [76,79,89,90,91,119] | |
| F1174S [90] | E1210K [79,89] | L1196M [79,89,90,91,96,118] | |
| F1174V [91,103] | F1174L [118] | 1151Tins [118] | |
| G1202R [79,89,90,92,99,118] | G1269A [79,89,96,118] | ||
| G1202S [99] | I1268L [91] | ||
| G1202del [79,89] | L1152R [118] | ||
| G1210K [120] | L1198F [79,89] | ||
| G1269A [108] | L1256F [91] | ||
| I1171N [79,89,90,91,92] | S1206Y [118] | ||
| I1171S [79,89,90,91,108] | T1151K [110] | ||
| I1171 X [106] | V1185L [91] | ||
| L1122V [96] | I1171N/L1256F [91] | ||
| L1196Q [91] | |||
| L1198F [96] | |||
| R1192P [108] | |||
| T1151M [108] | |||
| V1180L [119] | |||
| W1295C [98] | |||
| D1203N/E1210K [79,89] | |||
| D1203N/F1174C [79,89] | |||
| F1174L/G1269A [98] | |||
| G1202R/L1196M [98] | |||
| L1196M/V1185L [91] | |||
| Brigatinib | G1202L [121] | C1156Y [79,89,107] | D1203N [79,89,90,96,107] |
| G1202del [79,89] | F1174C [79,89,107] | E1210K [79,89,90] | |
| L1122V [84,96] | F1174L [107] | G1202R [62,79,89,90,107] | |
| S1206C [90] | G1269A [96,107] | I1171N [79,89,104,107] | |
| D1203N/F1174C [89] | I1171S [79,89,106] | L1198F [79,89,96,107] | |
| D1203N/E1210K [89] | I1171T [79,89] | S1206Y [17,90,107] | |
| E1210K/S1206C [89,99,103] | L1152P [107] | ||
| F1174V/L1198F [84] | L1152R [107] | ||
| F1174L/L1198V [99] | L1196M [79,80,89,96,107] | ||
| G1202R/L1196M [122] | L1196Q [104] | ||
| V1180L [96,107] | |||
| 1151Tins [107] | |||
| G1269A/I1171S [116] | |||
| G1269A/I1171N [91] | |||
| I1171N/L1196M [91] | |||
| I1171N/L1198F [91] | |||
| I1171N/L1256F [91] | |||
| Lorlatinib | C1156F [123] | C1156Y [79,89] | E1201K [79,89,123] |
| G1128S [123] | D1203N [79,89] | G1269A [79,89,96,124] | |
| L1256F [91] | F1174C [79,89] | I1171N [79,89,123] | |
| C1156F/D1203N [123] | F1174I [123] | I1171T [79,89,123] | |
| C1156F/L1198F [124,125] | F1174L [126] | ||
| C1156Y/D1203N [125] | F1245C [126] | ||
| C1156Y/F1174C [125] | G1202del [79,89] | ||
| C1156Y/F1174I [125] | G1202K [120] | ||
| C1156Y/F1174V [125] | G1202L [121] | ||
| C1156Y/G1269A [125,127] | G1202R [79,89,128] | ||
| C1156Y/I1171T [125] | I1171S [79,89] | ||
| C1156Y/L1196M [125] | L1196M [79,89,96] | ||
| C1156Y/L1198F [125] | R1275Q [126] | ||
| C1156Y/S1256F [125] | V1180L [96] | ||
| D1203N/F1174C [79,89] | |||
| D1203N/L1196M [127] | |||
| F1174C/G1202R [91] | |||
| F1174C/G1269A [125] | |||
| F1174C/L1196M [125] | |||
| F1174L/G1202R [91,127] | |||
| G1202R/G1269A [91,98,124] | |||
| G1202R/I1171N [91] | |||
| G1202R/L1196M [92,122,125] | |||
| G1202R/L1198F [91,125] | |||
| G1269A/I1171S [116] | |||
| G1269A/I1171N [125] | |||
| G1269A/I1171T [125] | |||
| G1269A/L1196M [91,125] | |||
| G1269A/N1178H [124] | |||
| I1171N/C1156Y [91] | |||
| I1171N/L1198F [91] | |||
| I1171N/L1256F [91] | |||
| L1196M/F1174C [125] | |||
| L1196M/F1174L [125] | |||
| L1196M/F1174V [125] | |||
| L1196M/I1171S [125] | |||
| L1196M/I1179V [125] | |||
| L1196M/L1198F [125] | |||
| L1196M/L1198H [125] | |||
| L1196M/L1256F [125] | |||
ALK-independent mechanisms of resistance occur when the need for NPM1-ALK is bypassed through the activation of its downstream targets via alternative signalling cascades, so-called bypass tracks (Figure 5 and Table 2) [17,78]. For example, increased signalling through the insulin-like growth factor receptor (IGF-1R) activates the JAK/STAT, MAPK and PI3K/Akt pathways normally stimulated by EML4-ALK or NPM1-ALK, causing crizotinib resistance in NSCLC and ALCL respectively [129,130].
Alternatively, ALK-independent resistance can be induced due to activation of the pathways downstream of aberrant ALK activity via mutation of genes encoding proteins involved in these pathways (Figure 4). For example, mutations in the MAPK pathway components KRAS [82,99,131,144,145], NRAS [79,98], BRAF [79,98,132,140] and MEK [132,146], as well as reduced DUSP6 (a MAPK phosphatase) [144] and mutations in the RAS negative regulators neurofibromin 1 and 2 (NF1/2) [99,127], cause ALK TKI resistance in NSCLC. Furthermore, lorlatinib and ceritinib resistance has been associated with upregulation of the MAPK pathway in ALCL xenografts [124] and neuroblastoma cell lines [147], respectively. Additionally, PI3KCA mutations cause ALK TKI resistance in NSCLC [79,98,132,146], and lorlatinib resistance of ALCL xenografts has been associated with PI3K/Akt pathway upregulation [124]. Finally, NOTCH1 mutations in NSCLC [99] and ALCL [148], and PIM1 overexpression in neuroblastoma [149], also cause ALK TKI resistance through their effects on the JAK-STAT pathway. In a similar manner, we have shown that activation of signalling via the IL10R bypasses NPM-ALK to activate STAT3 in ALCL mediating resistance to crizotinib, alectinib, brigatinib and lorlatinib [142].
Besides ALK-dependent and independent mechanisms of resistance, there are some additional mechanisms through which ALK TKIs cease to be effective. Some are well studied, such as the CNS relapses that occur if crizotinib and ceritinib are effluxed from the CNS by P-glycoprotein in the blood-brain barrier. These effects can be overcome by using alectinib, brigatinib or lorlatinib as they are not substrates for P-glycoprotein [107]. Other resistance mechanisms are very specific to individual tumour types. For example, NSCLC acquires resistance through epithelial–mesenchymal transition, whereby cells lose their polarity and become more fibroblastic and invasive [79,127,150,151]. NSCLC also acquire resistance through transforming to a small cell lung cancer (SCLC) phenotype, although given that these cells retain their ALK rearrangements, further investigation is required to determine why these transformations mediate resistance [152,153,154,155]. Additionally, neuroblastoma cells driven by MYCN and ALK amplifications can acquire resistance via a multi-step process in which they downregulate MYCN, and instead upregulate and become dependent on BORIS, a DNA binding protein that increases the proliferation and survival of cancer cells, and here causes increased expression of transcription factors that promote the transformation to an ALK TKI-resistant phenotype [156,157]. Further resistance mechanisms are less well studied, such as the generation or loss of different ALK fusions. Indeed, in the case of NSCLC with 3 co-existing rare ALK fusions at diagnosis (COX7A2L-ALK, LINC01210-ALK and ATP13A4-ALK), the generation of an additional SLCO2A1-ALK fusion mediated crizotinib resistance, and the subsequent loss of the ATP13A4-ALK and SLCO2A1-ALK fusions mediated ceritinib resistance [158]. Additionally, increased autophagy in which cells break down obsolete constituents of their own cytoplasm as a way of generating additional energy for tumour growth and proliferation leads to crizotinib resistance in ALCL [159] and NSCLC [160] by allowing the cell to overcome the metabolic stress of the ALK TKI. Indeed, treatment of ALCL cells (both in vitro and in vivo) with crizotinib and chloroquine, which inhibits autophagy, resulted in a greater inhibitory effect than treatment with crizotinib alone, suggesting that there are potential ways of overcoming resistance caused by this mechanism [159].
Another potential cause of resistance to ALK TKIs is p53 disruption. It is already known that TP53 mutation can cause resistance to traditional genotoxic chemotherapeutic agents by preventing apoptosis despite chemotherapy-induced DNA damage [161,162]. However, the resulting genetic instability caused by p53 disruption enables mutations to accumulate over time, and it is these later changes that may drive resistance to targeted agents. This has been shown to be the case in chronic lymphocytic leukaemia [161,162]. Additionally, in neuroblastoma, it has been shown that combination treatment with an ALK TKI and a p53 activator may prevent the resistance seen with ALK TKI monotherapy because the combination stimulates apoptosis rather than reversible growth arrest seen in cells treated with monotherapy [163]. However, p53-mediated resistance to targeted agents has not yet been demonstrated in ALCL. This should be investigated further because p53 is inactivated in some cases of ALCL, occasionally due to TP53 gene mutations [164] but more usually via NPM1-ALK stimulated induction of JNK and MDM2 activity [165]. Additionally, it has been shown that the p53 activator nutlin-3a can induce apoptosis of ALCL and thereby enhance the efficacy of chemotherapy [166].
In summary, due to a large number of possible ALK TKI resistance mechanisms, it is essential that tumours are assessed at the time of resistance to identify the underlying cause, to inform on the next best therapeutic approach towards a cure. For example, the identification of an ALK mutation could dictate which ALK TKI to use next, and the emergence of ALK-independent bypass mechanisms may identify other druggable targets to overcome resistance. Indeed IGF-1R [129], HER [99], HGF [139], SRC [146], MEK [146] and mTOR [167] inhibitors have been shown to overcome ALK TKI resistance caused by activation of these bypass pathways. Therefore, these could be considered as future treatments in these cases. It is also imperative that further potential resistance mechanisms continue to be identified in the laboratory as for many patients, the mechanisms cannot be explained by known pathways. In evidence, a large study of NSCLC patients resistant to ALK TKIs found that only 33–44% of the resistance phenotype could be explained by currently known resistance mechanisms [98]. This is even more important for ALCL, as well as neuroblastoma, because the majority of known ALK TKI resistance mechanisms have been studied in NSCLC and may differ substantially between diseases due to differing underlying biology and activities specific to the type of aberrant ALK expression observed, including overexpression of full-length ALK, compared to a fusion protein.
Additionally, the schedule of ALK TKI delivery and the combination of drugs with which it is administered may also impact resistance mechanisms. For example, resistance may be delayed or prevented when combination therapies are given upfront rather than as sequential monotherapies; when therapy is given in a metronomic manner; or when treatments are cycled before resistance has developed rather than changing treatment once resistance has already developed. In particular, alternating ALK TKIs based on the additional proteins that they target other than ALK may be useful in reducing the selective pressure leading to specific ALK mutations and bypass resistance tracks. For example, crizotinib (also an MET and ROS1 inhibitor [90]) could be cycled with alectinib (also an RET, LTK and GAK inhibitor [90]). Furthermore, it has been demonstrated in NSCLC that MET-driven ALK TKI bypass resistance is present at lower levels in patients treated with a less selective ALK TKI [168]. This requires investigation specific to ALCL but suggests that the development of drugs with potent ALK inhibition, but not necessarily more ALK specificity, is required.
3.2. Mechanisms of Resistance to Brentuximab Vedotin
Resistance to BV develops relatively frequently, with around half of patients with relapsed/refractory ALCL treated with BV either progressing on therapy or requiring additional treatments [169]. The most obvious mechanism of resistance is a reduction in CD30 protein expression, the target of BV activity. This has been observed in at least one case of adult ALK-negative ALCL [170] and in epithelioid inflammatory myofibroblastic sarcoma patient-derived xenografts (PDXs) [171]. It is unclear whether these reductions in CD30 were due to downregulation of the CD30 target, increased turnover of CD30, CD30 internalisation, or the selective outgrowth of sub-clones with lower levels of CD30 expression [171]. However, CD30 target reduction does not account for all BV resistance because CD30 expression is maintained in some BV-resistant Hodgkin lymphoma and ALCL [172]. A second potential resistance mechanism is upregulation of ABC transporters, which has been demonstrated in BV-resistant Hodgkin lymphoma cell lines and patient samples [172,173,174], and epithelioid inflammatory myofibroblastic sarcoma PDXs [171]. However, this also cannot account for all BV resistance because Hodgkin lymphoma cells treated with BV and an MDR-1 inhibitor eventually stop responding to this combination [174].
In summary, further work is required to fully elucidate how to use BV in paediatric ALK-positive ALCL. In order to explore whether BV might allow traditional chemotherapy to be reduced, a better understanding of BV resistance mechanisms is required. This will help to establish whether it can be used as a monotherapy, with factors put in place to prevent the acquisition of resistance, or whether it should only ever be used alongside other agents.
3.3. Mechanisms of Resistance to Immune Checkpoint Inhibitors
Resistance to anti-PD-L1 immune checkpoint inhibitors will eventually occur in the majority of patients, although there are differences in rates between tumour types. This can be a consequence of PD-L1 upregulation [175,176,177], which can occur due to increased JAK-STAT signalling, caused by: loss of the tumour suppressor FBP1 [176,178], mutations in JAK1/2 [179], or stimulation of tumour cells’ interferon-gamma receptor 2 by interferon-gamma released from activated CD8+ T cells in the tumour microenvironment [180,181]. PD-L1 upregulation can also occur when loss of the tumour suppressor PTEN results in increased PI3K/Akt signalling [176,177].
Alternatively, resistance to anti-PD-L1 immune checkpoint inhibitors can occur as a result of a skew in the distribution of immune cells in the tumour microenvironment towards an immunosuppressive one, compensating for the effects of loss of PD-L1 signalling. This skew can be caused by a low overall T cell abundance [181], an increased ratio of regulatory T cells to effector T cells [182], dysfunctional CD8+ T lymphocytes [183,184], increased myeloid-derived suppressor cells (MDSCs) [185] and/or increased PD-L1 positive M2 macrophages [185,186]. These changes are mediated in a variety of ways (Table 3), which might be therapeutically targetable. For example, a clinical trial (NCT03048500) is investigating whether metformin used alongside nivolumab in NSCLC can overcome the nivolumab resistance caused by the detrimental effects of hypoxia on CD8+ lymphocytes [184,187].
Another cause of resistance to anti-PD-L1 immune checkpoint inhibitors is a reduction in neoantigen expression on the tumour cell surface. A lack of neoantigens, either due to a low tumour mutational burden, or altered antigen processing and presentation, results in a reduced immune response to the tumour, which compensates for the immunosuppressive effects of PD-L1 signalling that have been lost [180,185,191].
It is important to note that although there is some understanding of mechanisms of resistance to immune checkpoint inhibitors, these may not all be relevant to ALCL. For example, there is an endogenous immune response to ALCL via the production of anti-ALK autoantibodies amongst other mechanisms, therefore, an immunosuppressive microenvironment and a lack of neoantigens may not be as relevant in ALCL as it is in other diseases [1]. Further research is required to determine whether these potential resistance mechanisms apply in ALCL, to find other resistance mechanisms, and to determine how to overcome these.
4. Conclusions
In summary, there is a trade-off between reducing side effects and the potential for resistance to develop when choosing targeted therapies for ALK-positive paediatric ALCL. Knowledge of potential resistance mechanisms is starting to develop but large gaps remain. Further investigation into how resistance develops is an essential prerequisite for finding a solution to this resistance problem so that targeted agents can be successfully integrated into the mainstream treatment of ALK-positive paediatric ALCL. This will include the investigation into potential differences between primary (intrinsic) resistance, where there is a lack of response to treatments from the start of therapy, and acquired resistance, where the selective pressure of exposure to a treatment results in the outgrowth of resistant sub-clones and eventual resistance after an initial response [17]. Overall, the ability to evaluate patient tumour samples for known resistance mechanisms prior to treatment will enable the prediction of treatment response, and after the development of resistance will enable identification of the causes. This will inform on the best therapeutic approach towards a cure on an individual patient basis.
Author Contributions
L.H., S.D.T. and G.A.A.B. all contributed to the writing and editing of this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
L.H. is supported with funding from a Cancer Research UK Cambridge Centre clinical research fellowship (grant number: C9685/A25117).
Conflicts of Interest
S.D.T. and L.H. declare no conflict of interest. G.A.A.B. has received institutional consultancy fees from Roche, Takeda, Novartis, and Janssen.
References
- Prokoph, N.; Larose, H.; Lim, M.S.; Burke, G.A.A.; Turner, S.D. Treatment options for paediatric anaplastic large cell lymphoma (ALCL): Current standard and beyond. Cancers 2018, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Abla, O.; Alexander, S.; Attarbaschi, A.; Batchelor, T.T.; Beishuizen, A.; Bond, J.; Borkhardt, A.; Brugieres, L.; Burke, A.; Burkhardt, B.; et al. Non-Hodgkin’s Lymphoma in Childhood and Adolescence, 1st ed.; Springer International Publishing: Cham, Switzerland, 2019. [Google Scholar]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Bischof, D.; Pulford, K.; Mason, D.Y.; Morris, S.W. Role of the nucleophosmin (NPM) portion of the non-Hodgkin’s lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol. Cell. Biol. 1997, 17, 2312–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, S.D.; Tooze, R.; Maclennan, K.; Alexander, D.R. Vav-promoter regulated oncogenic fusion protein NPM-ALK in transgenic mice causes B-cell lymphomas with hyperactive Jun kinase. Oncogene 2003, 22, 7750–7761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malcolm, T.I.; Villarese, P.; Fairbairn, C.J.; Lamant, L.; Trinquand, A.; Hook, C.E.; Burke, G.A.A.; Brugières, L.; Hughes, K.; Payet, D.; et al. Anaplastic large cell lymphoma arises in thymocytes and requires transient TCR expression for thymic egress. Nat. Commun. 2016, 7, 10087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarle, R.; Gong, J.Z.; Guasparri, I.; Pesci, A.; Cai, J.; Liu, J.; Simmons, W.J.; Dhall, G.; Howes, J.; Piva, R.; et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood 2003, 101, 1919–1927. [Google Scholar] [CrossRef] [Green Version]
- Turner, S.D.; Merz, H.; Yeung, D.; Alexander, D.R. CD2 promoter regulated nucleophosmin-anaplastic lymphoma kinase in transgenic mice causes B lymphoid malignancy. Anticancer Res. 2006, 26, 3275–3279. [Google Scholar]
- Werner, M.T.; Zhao, C.; Zhang, Q.; Wasik, M.A. Nucleophosmin-anaplastic lymphoma kinase: The ultimate oncogene and therapeutic target. Blood 2017, 129, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Turner, S.D.; Yeung, D.; Hadfield, K.; Cook, S.J.; Alexander, D.R. The NPM-ALK tyrosine kinase mimics TCR signalling pathways, inducing NFAT and AP-1 by RAS-dependent mechanisms. Cell. Signal. 2007, 19, 740–747. [Google Scholar] [CrossRef]
- Slupianek, A.; Nieborowska-Skorska, M.; Hoser, G.; Morrione, A.; Majewski, M.; Xue, L.; Morris, S.W.; Wasik, M.A.; Skorski, T. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001, 61, 2194–2199. [Google Scholar]
- Zhang, Q.; Raghunath, P.N.; Xue, L.; Majewski, M.; Carpentieri, D.F.; Odum, N.; Morris, S.; Skorski, T.; Wasik, M.A. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J. Immunol. 2002, 168, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, R.Y.; Dieter, P.; Peschel, C.; Morris, S.W.; Duyster, J. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol. Cell. Biol. 1998, 18, 6951–6961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. Anaplastic lymphoma kinase (ALK) inhibitors in the treatment of ALK-driven lung cancers. Pharmacol. Res. 2017, 117, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The Transcriptional Roles of ALK Fusion Proteins in Tumorigenesis. Cancers 2019, 11, 1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.G.; Mota, I.; Mologni, L.; Patrucco, E.; Gambacorti-Passerini, C.; Chiarle, R. Tumor resistance against ALK targeted therapy—Where it comes from and where it goes. Cancers 2018, 10, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trigg, R.M.; Turner, S.D. ALK in neuroblastoma: Biological and therapeutic implications. Cancers 2018, 10, 113. [Google Scholar] [CrossRef] [Green Version]
- Laurent, C.; Lopez, C.; Desjobert, C.; Berrebi, A.; Damm-Welk, C.; Delsol, G.; Brousset, P.; Lamant, L. Circulating t(2;5)-positive cells can be detected in cord blood of healthy newborns. Leukemia 2012, 26, 188–190. [Google Scholar] [CrossRef]
- Turner, S.D.; Lamant, L.; Kenner, L.; Brugières, L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br. J. Haematol. 2016, 173, 560–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knörr, F.; Damm-Welk, C.; Ruf, S.; Singh, V.K.; Zimmermann, M.; Reiter, A.; Woessmann, W. Blood cytokine concentrations in pediatric patients with anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Haematologica 2018, 103, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonzheim, I.; Geissinger, E.; Roth, S.; Zettl, A.; Marx, A.; Rosenwald, A.; Müller-Hermelink, H.K.; Rüdiger, T. Anaplastic large cell lymphomas lack the expression of T-cell receptor molecules or molecules of proximal T-cell receptor signaling. Blood 2004, 104, 3358–3360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malcolm, T.I.M.; Hodson, D.J.; Macintyre, E.A.; Turner, S.D. Challenging perspectives on the cellular origins of lymphoma. Open Biol. 2016, 6, 160232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleussner, N.; Merkel, O.; Costanza, M.; Liang, H.C.; Hummel, F.; Romagnani, C.; Durek, P.; Anagnostopoulos, I.; Hummel, M.; Jöhrens, K.; et al. The AP-1-BATF and -BATF3 module is essential for growth, survival and TH17/ILC3 skewing of anaplastic large cell lymphoma. Leukemia 2018, 32, 1994–2007. [Google Scholar] [CrossRef] [PubMed]
- Moti, N.; Malcolm, T.; Hamoudi, R.; Mian, S.; Garland, G.; Hook, C.E.; Burke, G.A.A.; Wasik, M.A.; Merkel, O.; Kenner, L.; et al. Anaplastic large cell lymphoma-propagating cells are detectable by side population analysis and possess an expression profile reflective of a primitive origin. Oncogene 2015, 34, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Congras, A.; Hoareau-Aveilla, C.; Caillet, N.; Tosolini, M.; Villarese, P.; Cieslak, A.; Rodriguez, L.; Asnafi, V.; Macintyre, E.; Egger, G.; et al. ALK-transformed mature T lymphocytes restore early thymus progenitor features. J. Clin. Investig. 2020, 130, 6395–6408. [Google Scholar] [CrossRef]
- Kasprzycka, M.; Marzec, M.; Liu, X.; Zhang, Q.; Wasik, M.A. Nucleophosmin/anaplastic lymphoma kinase (NPM/ALK) oncoprotein induces the T regulatory cell phenotype by activating STAT3. Proc. Natl. Acad. Sci. USA 2006, 103, 9964–9969. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wei, F.; Wang, H.Y.; Liu, X.; Roy, D.; Xiong, Q.B.; Jiang, S.; Medvec, A.; Danet-Desnoyers, G.; Watt, C.; et al. The potent oncogene NPM-ALK mediates malignant transformation of normal human CD4(+) T lymphocytes. Am. J. Pathol. 2013, 183, 1971–1980. [Google Scholar] [CrossRef] [Green Version]
- Marzec, M.; Halasa, K.; Liu, X.; Wang, H.Y.; Cheng, M.; Baldwin, D.; Tobias, J.W.; Schuster, S.J.; Woetmann, A.; Zhang, Q.; et al. Malignant transformation of CD4+ T lymphocytes mediated by oncogenic kinase NPM/ALK recapitulates IL-2–induced cell signaling and gene expression reprogramming. J. Immunol. 2013, 191, 6200. [Google Scholar] [CrossRef] [Green Version]
- Brugières, L.; Le Deley, M.-C.; Rosolen, A.; Williams, D.; Horibe, K.; Wrobel, G.; Mann, G.; Zsiros, J.; Uyttebroeck, A.; Marky, I.; et al. Impact of the methotrexate administration dose on the need for intrathecal treatment in children and adolescents with anaplastic large-cell lymphoma: Results of a randomized trial of the EICNHL group. J. Clin. Oncol. 2009, 27, 897–903. [Google Scholar] [CrossRef]
- Mussolin, L.; Pillon, M.; d’Amore, E.S.; Santoro, N.; Lombardi, A.; Fagioli, F.; Zanesco, L.; Rosolen, A. Prevalence and clinical implications of bone marrow involvement in pediatric anaplastic large cell lymphoma. Leukemia 2005, 19, 1643–1647. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.; Mori, T.; Reiter, A.; Woessman, W.; Rosolen, A.; Wrobel, G.; Zsiros, J.; Uyttebroeck, A.; Marky, I.; Le Deley, M.C.; et al. Central nervous system involvement in anaplastic large cell lymphoma in childhood: Results from a multicentre European and Japanese study. Pediatr. Blood Cancer 2013, 60, E118–E121. [Google Scholar] [CrossRef]
- Mussolin, L.; Le Deley, M.C.; Carraro, E.; Damm-Welk, C.; Attarbaschi, A.; Williams, D.; Burke, A.; Horibe, K.; Nakazawa, A.; Wrobel, G.; et al. Prognostic factors in childhood anaplastic large cell lymphoma: Long term results of the international ALCL99 trial. Cancers 2020, 12, 2747. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, G.; Mauguen, A.; Rosolen, A.; Reiter, A.; Williams, D.; Horibe, K.; Brugières, L.; Le Deley, M.C. Safety assessment of intensive induction therapy in childhood anaplastic large cell lymphoma: Report of the ALCL99 randomised trial. Pediatr. Blood Cancer 2011, 56, 1071–1077. [Google Scholar] [CrossRef]
- Ruf, R.; Brugieres, L.; Pillon, M.; Zimmermann, M.; Attarbaschi, A.; Melgrenn, K.; Williams, D.; Uyttebroeck, A.; Wrobel, G.; Reiter, A.; et al. Risk-adapted therapy for patients with relapsed or refractory ALCL—Final report of the prospective ALCL-relapse trial of the EICNHL. Br. J. Haematol. 2015, 171, 35. [Google Scholar]
- EMC. Vinblastine Sulphate 1 mg/mL Injection. Published December 2020. Available online: https://www.medicines.org.uk/emc/product/1422/smpc#gref (accessed on 15 May 2021).
- Brugieres, L.; Pacquement, H.; Le Deley, M.C.; Leverger, G.; Lutz, P.; Paillard, C.; Baruchel, A.; Frappaz, D.; Nelken, B.; Lamant, L.; et al. Single-drug vinblastine as salvage treatment for refractory or relapsed anaplastic large-cell lymphoma: A report from the French Society of Pediatric Oncology. J. Clin. Oncol. 2009, 27, 5056–5061. [Google Scholar] [CrossRef]
- Le Deley, M.C.; Rosolen, A.; Williams, D.M.; Horibe, K.; Wrobel, G.; Attarbaschi, A.; Zsiros, J.; Uyttebroeck, A.; Marky, I.M.; Lamant, L.; et al. Vinblastine in children and adolescents with high-risk anaplastic large-cell lymphoma: Results of the randomized ALCL99-vinblastine trial. J. Clin. Oncol. 2010, 28, 3987–3993. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.; Kraveka, J.M.; Weitzman, S.; Lowe, E.; Smith, L.; Lynch, J.C.; Chang, M.; Kinney, M.C.; Perkins, S.L.; Laver, J.; et al. Advanced stage anaplastic large cell lymphoma in children and adolescents: Results of ANHL0131, a randomized phase III trial of APO versus a modified regimen with vinblastine: A report from the children’s oncology group. Pediatr. Blood Cancer 2014, 61, 2236–2242. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yang, J.; Wang, J.; Gao, W.; Ding, Y.; Jia, Z. Down-regulation of miR-210-3p encourages chemotherapy resistance of renal cell carcinoma via modulating ABCC1. Cell Biosci. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Mickisch, G.H.; Roehrich, K.; Koessig, J.; Forster, S.; Tschada, R.K.; Alken, P.M. Mechanisms and modulation of multidrug resistance in primary human renal cell carcinoma. J. Urol. 1990, 144, 755–759. [Google Scholar] [CrossRef]
- Struski, S.; Cornillet-Lefebvre, P.; Doco-Fenzy, M.; Dufer, J.; Ulrich, E.; Masson, L.; Michel, N.; Gruson, N.; Potron, G. Cytogenetic characterization of chromosomal rearrangement in a human vinblastine-resistant CEM cell line: Use of comparative genomic hybridization and fluorescence in situ hybridization. Cancer Genet. Cytogenet. 2002, 132, 51–54. [Google Scholar] [CrossRef]
- Holmes, J.; Jacobs, A.; Carter, G.; Janowska-Wieczorek, A.; Padua, R.A. Multidrug resistance in haemopoietic cell lines, myelodysplastic syndromes and acute myeloblastic leukaemia. Br. J. Haematol. 1989, 72, 40–44. [Google Scholar] [CrossRef]
- Zamora, J.M.; Beck, W.T. Chloroquine enhancement of anticancer drug cytotoxicity in multiple drug resistant human leukemic cells. Biochem. Pharmacol. 1986, 35, 4303–4310. [Google Scholar] [CrossRef]
- Syed, S.K.; Christopherson, R.I.; Roufogalis, B.D. Reversal of vinblastine transport by chlorpromazine in membrane vesicles from multidrug-resistant human CCRF-CEM leukaemia cells. Br. J. Cancer 1998, 78, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci. Rep. 2018, 8, 967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathawala, R.J.; Gupta, P.; Ashby, C.R.; Chen, Z.S., Jr. The modulation of ABC transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist. Updat. 2015, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Obey, T.B.; Lyle, C.S.; Chambers, T.C. Role of c-Jun in cellular sensitivity to the microtubule inhibitor vinblastine. Biochem. Biophys. Res. Commun. 2005, 335, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jaffrezou, J.P.; Tsuchiya, E.; Duran, G.E.; Chen, K.G.; Derry, W.B.; Wilson, L.; Jordan, M.A.; Sikic, B.I. Resistance to microtubule-targeted cytotoxins in a K562 leukemia cell variant associated with altered tubulin expression and polymerization. Bull. Cancer 2004, 91, E81–E112. [Google Scholar] [PubMed]
- Balis, F.M.; Thompson, P.A.; Mosse, Y.P.; Blaney, S.M.; Minard, C.G.; Weigel, B.J.; Fox, E. First-dose and steady-state pharmacokinetics of orally administered crizotinib in children with solid tumors: A report on ADVL0912 from the Children’s Oncology Group Phase 1/Pilot Consortium. Cancer Chemother. Pharmacol. 2017, 79, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.W.; Naeve, C.; Mathew, P.; James, P.L.; Kirstein, M.N.; Cui, X.; Witte, D.P. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997, 14, 2175–2188. [Google Scholar] [CrossRef] [Green Version]
- Orthofer, M.; Valsesia, A.; Mägi, R.; Wang, Q.P.; Kaczanowska, J.; Kozieradzki, I.; Leopoldi, A.; Cikes, D.; Zopf, L.M.; Tretiakov, E.O.; et al. Identification of ALK in Thinness. Cell 2020, 181, 1246–1262.e22. [Google Scholar] [CrossRef]
- Bilsland, J.G.; Wheeldon, A.; Mead, A.; Znamenskiy, P.; Almond, S.; Waters, K.A.; Thakur, M.; Beaumont, V.; Bonnert, T.P.; Heavens, R.; et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology 2008, 33, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- FDA. FDA Approves Crizotinib for Children and Young Adults with Relapsed or Refractory, Systemic Anaplastic Large Cell Lymphoma. Published January 2021. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-crizotinib-children-and-young-adults-relapsed-or-refractory-systemic-anaplastic-large (accessed on 15 May 2021).
- Mossé, Y.P.; Voss, S.D.; Lim, M.S.; Rolland, D.; Minard, C.G.; Fox, E.; Adamson, P.; Wilner, K.; Blaney, S.M.; Weigel, B.J. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: A Children’s Oncology Group study. J. Clin. Oncol. 2017, 35, 3215–3221. [Google Scholar] [CrossRef] [PubMed]
- Brugières, L.; Houot, R.; Cozic, N.; De La Fouchardière, C.; Morschhauser, F.; Brice, P.; Laboure, N.A.; Auvrignon, A.; Aladjidi, N.; Kolb, B.; et al. Crizotinib in advanced ALK+ anaplastic large cell lymphoma in children and adults: Results of the Acs© phase II trial. Blood 2017, 130 (Suppl. 1), 2831. [Google Scholar]
- Fukano, R.; Mori, T.; Sekimizu, M.; Choi, I.; Kada, A.; Saito, A.M.; Asada, R.; Takeuchi, K.; Terauchi, T.; Tateishi, U.; et al. Alectinib for relapsed or refractory anaplastic lymphoma kinase-positive anaplastic large cell lymphoma: An open-label phase II trial. Cancer Sci. 2020, 111, 4540–4547. [Google Scholar] [CrossRef] [PubMed]
- Greengard, E.; Mosse, Y.P.; Liu, X.; Minard, C.G.; Reid, J.M.; Voss, S.; Wilner, K.; Fox, E.; Balis, F.; Blaney, S.M.; et al. Safety, tolerability and pharmacokinetics of crizotinib in combination with cytotoxic chemotherapy for pediatric patients with refractory solid tumors or anaplastic large cell lymphoma (ALCL): A Children’s Oncology Group phase 1 consortium study (ADVL1212). Cancer Chemother. Pharmacol. 2020, 86, 829–840. [Google Scholar] [CrossRef] [PubMed]
- EU Clinical Trials Register. A phase 1B of Crizotinib Either in Combination or as Single Agent in Pediatric Patients with ALK, ROS1 or MET Positive Malignancies. Published May 2016. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2015-005437-53/NL (accessed on 31 May 2021).
- ClinicalTrials.gov. Brentuximab Vedotin or Crizotinib and Combination Chemotherapy in Treating Patients with Newly Diagnosed Stage II-IV Anaplastic Large Cell Lymphoma. Published May 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT01979536 (accessed on 31 May 2021).
- Huber, R.M.; Hansen, K.H.; Paz-Ares Rodríguez, L.; West, H.L.; Reckamp, K.L.; Leighl, N.B.; Tiseo, M.; Smit, E.F.; Kim, D.W.; Gettinger, S.N.; et al. Brigatinib in crizotinib-refractory ALK+ NSCLC: 2-year follow-up on systemic and intracranial outcomes in the phase 2 ALTA trial. J. Thorac. Oncol. 2020, 15, 404–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, E.J.; Reilly, A.F.; Lim, M.S.; Gross, T.G.; Saguilig, L.; Barkauskas, D.A.; Wu, R.; Alexander, S.; Bollard, C.M. Brentuximab vedotin in combination with chemotherapy for pediatric patients with ALK+ ALCL: Results of COG trial ANHL12P1. Blood 2021, 137, 3595–3603. [Google Scholar] [CrossRef]
- Chihara, D.; Miljkovic, M.; Iyer, S.P.; Vega, F. Targeted based therapy in nodal T-cell lymphomas. Leukemia 2021, 35, 956–967. [Google Scholar] [CrossRef]
- Forero-Torres, A.; Leonard, J.P.; Younes, A.; Rosenblatt, J.D.; Brice, P.; Bartlett, N.L.; Bosly, A.; Pinter-Brown, L.; Kennedy, D.; Sievers, E.L.; et al. A Phase II study of SGN-30 (anti-CD30 mAb) in Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Br. J. Haematol. 2009, 146, 171–179. [Google Scholar] [CrossRef]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [Green Version]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 2017, 130, 2709–2717. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, S.; O’Connor, O.A.; Pro, B.; Illidge, T.; Fanale, M.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): A global, double-blind, randomised, phase 3 trial. Lancet 2019, 393, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, F.; Mauz-Koerholz, C.; Neville, K.; Llort, A.; Beishuizen, A.; Daw, S.; Pillon, M.; Aladjidi, N.; Klingebiel, T.; Landman-Parker, J.; et al. Brentuximab vedotin for paediatric relapsed or refractory Hodgkin’s lymphoma and anaplastic large-cell lymphoma: A multicentre, open-label, phase 1/2 study. Lancet Haematol. 2018, 5, e450–e461. [Google Scholar] [CrossRef]
- Burke, G.A.A. Brentuximab vedotin: Frontline help in ALCL. Blood 2021, 137, 3581–3582. [Google Scholar] [CrossRef]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5, eaay1863. [Google Scholar] [CrossRef]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 pathway. Sci. Adv. 2020, 6, eabd2712. [Google Scholar] [CrossRef] [PubMed]
- Hebart, H.; Lang, P.; Woessmann, W. Nivolumab for refractory anaplastic large cell lymphoma: A case report. Ann. Intern. Med. 2016, 165, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Rigaud, C.; Abbou, S.; Minard-Colin, V.; Geoerger, B.; Scoazec, J.Y.; Vassal, G.; Jaff, N.; Heuberger, L.; Valteau-Couanet, D.; Brugieres, L. Efficacy of nivolumab in a patient with systemic refractory ALK+ anaplastic large cell lymphoma. Pediatr. Blood Cancer 2018, 65, e26902. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Mussolin, L.; Brugieres, L. Abrupt relapse of ALK-positive lymphoma after discontinuation of crizotinib. N. Engl. J. Med. 2016, 374, 95–96. [Google Scholar] [CrossRef]
- Zdzalik, D.; Dymek, B.; Grygielewicz, P.; Gunerka, P.; Bujak, A.; Lamparska-Przybysz, M.; Wieczorek, M.; Dzwonek, K. Activating mutations in ALK kinase domain confer resistance to structurally unrelated ALK inhibitors in NPM-ALK-positive anaplastic large-cell lymphoma. J. Cancer Res. Clin. Oncol. 2014, 140, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Andraos, E.; Dignac, J.; Meggetto, F. NPM-ALK: A driver of lymphoma pathogenesis and a therapeutic target. Cancers 2021, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med. 2012, 4, 120ra17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [Green Version]
- Katayama, R.; Khan, T.M.; Benes, C.; Lifshits, E.; Ebi, H.; Rivera, V.M.; Shakespeare, W.C.; Iafrate, A.J.; Engelman, J.A.; Shaw, A.T. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. USA 2011, 108, 7535–7540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, T.; Koivunen, J.; Ogino, A.; Yanagita, M.; Nikiforow, S.; Zheng, W.; Lathan, C.; Marcoux, J.P.; Du, J.; Okuda, K.; et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011, 71, 6051–6060. [Google Scholar] [CrossRef] [Green Version]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–4782. [Google Scholar] [CrossRef] [Green Version]
- Jamme, P.; Descarpentries, C.; Gervais, R.; Dansin, E.; Wislez, M.; Grégoire, V.; Richard, N.; Baldacci, S.; Rabbe, N.; Kyheng, M.; et al. Relevance of detection of mechanisms of resistance to ALK inhibitors in ALK-rearranged NSCLC in routine practice. Clin. Lung Cancer 2019, 20, 297–304.e1. [Google Scholar] [CrossRef]
- Ceccon, M.; Mologni, L.; Giudici, G.; Piazza, R.; Pirola, A.; Fontana, D.; Gambacorti-Passerini, C. Treatment efficacy and resistance mechanisms using the second-generation ALK inhibitor AP26113 in human NPM-ALK-positive anaplastic large cell lymphoma. Mol. Cancer Res. 2015, 13, 775–783. [Google Scholar] [CrossRef] [Green Version]
- Amin, A.D.; Rajan, S.S.; Liang, W.S.; Pongtornpipat, P.; Groysman, M.J.; Tapia, E.O.; Peters, T.L.; Cuyugan, L.; Adkins, J.; Rimsza, L.M.; et al. Evidence suggesting that discontinuous dosing of ALK kinase inhibitors may prolong control of ALK+ tumors. Cancer Res. 2015, 75, 2916–2927. [Google Scholar] [CrossRef] [Green Version]
- Ceccon, M.; Merlo, M.E.B.; Mologni, L.; Poggio, T.; Varesio, L.M.; Menotti, M.; Bombelli, S.; Rigolio, R.; Manazza, A.D.; Di Giacomo, F.; et al. Excess of NPM-ALK oncogenic signaling promotes cellular apoptosis and drug dependency. Oncogene 2016, 35, 3854–6865. [Google Scholar] [CrossRef] [Green Version]
- Rajan, S.S.; Amin, A.D.; Li, L.; Rolland, D.C.; Li, H.; Kwon, D.; Kweh, M.F.; Arumov, A.; Roberts, E.R.; Yan, A.; et al. The mechanism of cancer drug addiction in ALK-positive T-cell lymphoma. Oncogene 2020, 39, 2103–2117. [Google Scholar] [CrossRef]
- Singh, A.; Chen, H. Optimal care for patients with anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer: A review on the role and utility of ALK inhibitors. Cancer Manag. Res. 2020, 12, 6615–6628. [Google Scholar] [CrossRef]
- Bui, K.T.; Cooper, W.A.; Kao, S.; Boyer, M. Targeted molecular treatments in non-small cell lung cancer: A clinical guide for oncologists. J. Clin. Med. 2018, 7, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.J.; Riely, G.J.; Shaw, A.T. Targeting ALK: Precision medicine takes on drug resistance. Cancer Discov. 2017, 7, 137–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, K.; Araki, M.; Sakashita, T.; Ma, B.; Kanada, R.; Yanagitani, N.; Horiike, A.; Koike, S.; Oh-Hara, T.; Watanabe, K.; et al. Prediction of ALK mutations mediating ALK-TKIs resistance and drug re-purposing to overcome the resistance. EBioMedicine 2019, 41, 105–119. [Google Scholar] [CrossRef] [Green Version]
- Yanagitani, N.; Uchibori, K.; Koike, S.; Tsukahara, M.; Kitazono, S.; Yoshizawa, T.; Horiike, A.; Ohyanagi, F.; Tambo, Y.; Nishikawa, S.; et al. Drug resistance mechanisms in Japanese anaplastic lymphoma kinase-positive non-small cell lung cancer and the clinical responses based on the resistant mechanisms. Cancer Sci. 2020, 111, 932–939. [Google Scholar] [CrossRef]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1749. [Google Scholar] [CrossRef]
- Fontana, D.; Ceccon, M.; Gambacorti-Passerini, C.; Mologni, L. Activity of second-generation ALK inhibitors against crizotinib-resistant mutants in an NPM-ALK model compared to EML4-ALK. Cancer Med. 2015, 4, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, W.; Sun, H.; Pang, L.; Yin, B. Mutation-mediated influences on binding of anaplastic lymphoma kinase to crizotinib decoded by multiple replica Gaussian accelerated molecular dynamics. J. Comput. Aided. Mol. Des. 2020, 34, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.D.; Madhavan, S. A Computational approach for prioritizing selection of therapies targeting drug resistant variation in anaplastic lymphoma kinase. AMIA Summits Transl. Sci. Proc. 2018, 2017, 160–167. [Google Scholar] [PubMed]
- Heuckmann, J.M.; Hölzel, M.; Sos, M.L.; Heynck, S.; Balke-Want, H.; Koker, M.; Peifer, M.; Weiss, J.; Lovly, C.M.; Grütter, C.; et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin. Cancer Res. 2011, 17, 7394–7401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.T.; Chiang, C.L.; Hung, J.Y.; Lee, M.H.; Su, W.C.; Wu, S.Y.; Wei, Y.F.; Lee, K.Y.; Tseng, Y.H.; Su, J.; et al. Resistance profiles of anaplastic lymphoma kinase tyrosine kinase inhibitors in advanced non-small-cell lung cancer: A multicenter study using targeted next-generation sequencing. Eur. J. Cancer 2021, 156, 1–11. [Google Scholar] [CrossRef]
- McCoach, C.E.; Le, A.T.; Gowan, K.; Jones, K.; Schubert, L.; Doak, A.; Estrada-Bernal, A.; Davies, K.D.; Merrick, D.T.; Bunn, P.A., Jr.; et al. Resistance mechanisms to targeted therapies in ROS1(+) and ALK(+) non-small cell lung cancer. Clin. Cancer Res. 2018, 24, 3334–3347. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Okuda, K.; Zheng, W.; Butrynski, J.; Capelletti, M.; Wang, L.; Gray, N.S.; Wilner, K.; Christensen, J.G.; Demetri, G.; et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010, 70, 10038–10043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Merguerian, M.D.; Rowe, S.P.; Pratilas, C.A.; Chen, A.R.; Ladle, B.H. Exceptional response to the ALK and ROS1 inhibitor lorlatinib and subsequent mechanism of resistance in relapsed ALK F1174L-mutated neuroblastoma. Mol. Case Stud. 2021, 7, a006064. [Google Scholar] [CrossRef]
- Ai, X.; Niu, X.; Chang, L.; Chen, R.; Ou, S.I.; Lu, S. Next generation sequencing reveals a novel ALK G1128A mutation resistant to crizotinib in an ALK-Rearranged NSCLC patient. Lung Cancer 2018, 123, 83–86. [Google Scholar] [CrossRef]
- Gristina, V.; La Mantia, M.; Iacono, F.; Galvano, A.; Russo, A.; Bazan, V. The emerging therapeutic landscape of ALK inhibitors in non-small cell lung cancer. Pharmaceuticals 2020, 13, 474. [Google Scholar] [CrossRef]
- Ceccon, M.; Mologni, L.; Bisson, W.; Scapozza, L.; Gambacorti-Passerini, C. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol. Cancer Res. 2013, 11, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Gambacorti Passerini, C.; Farina, F.; Stasia, A.; Redaelli, S.; Ceccon, M.; Mologni, L.; Messa, C.; Guerra, L.; Giudici, G.; Sala, E.; et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J. Natl. Cancer Inst. 2014, 106, djt378. [Google Scholar] [CrossRef]
- Sehgal, K.; Peters, M.L.B.; VanderLaan, P.A.; Rangachari, D.; Kobayashi, S.S.; Costa, D.B. Activity of brigatinib in the setting of alectinib resistance mediated by ALK I1171S in ALK-rearranged lung cancer. J. Thorac. Oncol. 2019, 14, e1–e3. [Google Scholar] [CrossRef] [Green Version]
- Golding, B.; Luu, A.; Jones, R.; Viloria-Petit, A.M. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol. Cancer 2018, 17, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, A.D.; Li, L.; Rajan, S.S.; Gokhale, V.; Groysman, M.J.; Pongtornpipat, P.; Tapia, E.O.; Wang, M.; Schatz, J.H. TKI sensitivity patterns of novel kinase-domain mutations suggest therapeutic opportunities for patients with resistant ALK+ tumors. Oncotarget 2016, 7, 23715–23729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, V.W.; Cui, J.J.; Fernandez-Rocha, M.; Schrock, A.B.; Ali, S.M.; Ou, S.I. Identification of a novel T1151K ALK mutation in a patient with ALK-rearranged NSCLC with prior exposure to crizotinib and ceritinib. Lung Cancer 2017, 110, 32–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, V.W.; Schrock, A.B.; Bosemani, T.; Benn, B.S.; Ali, S.M.; Ou, S.I. Dramatic response to alectinib in a lung cancer patient with a novel VKORC1L1-ALK fusion and an acquired ALK T1151K mutation. Lung Cancer 2018, 9, 111–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suryavanshi, M.; Chaudhari, K.; Nathany, S.; Talwar, V. Identification of a novel resistance ALK p.(Q1188_L1190del) deletion in a patient with ALK-rearranged non–small-cell lung cancer. Cancer Genet. 2021, 256, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, M.G.; Di Noia, V.; D’Argento, E.; Vita, E.; Damiano, P.; Cannella, A.; Ribelli, M.; Pilotto, S.; Milella, M.; Tortora, G.; et al. Oncogene-addicted non-small-cell lung cancer: Treatment opportunities and future perspectives. Cancers 2020, 12, 1196. [Google Scholar] [CrossRef]
- Kodityal, S.; Elvin, J.A.; Squillace, R.; Agarwal, N.; Miller, V.A.; Ali, S.M.; Klempner, S.J.; Ou, S.H.I. A novel acquired ALK F1245C mutation confers resistance to crizotinib in ALK-positive NSCLC but is sensitive to ceritinib. Lung Cancer 2016, 92, 19–21. [Google Scholar] [CrossRef]
- Toyokawa, G.; Inamasu, E.; Shimamatsu, S.; Yoshida, T.; Nosaki, K.; Hirai, F.; Yamaguchi, M.; Seto, T.; Takenoyama, M.; Ichinose, Y. Identification of a novel ALK G1123S mutation in a patient with ALK-rearranged non-small-cell lung cancer exhibiting resistance to ceritinib. J. Thorac. Oncol. 2015, 10, e55–e57. [Google Scholar] [CrossRef] [Green Version]
- Ceccon, M. Ceritinib as a promising therapy for ALK related diseases. Transl. Lung Cancer Res. 2014, 3, 376–378. [Google Scholar]
- Takahashi, K.; Seto, Y.; Okada, K.; Uematsu, S.; Uchibori, K.; Tsukahara, M.; Oh-Hara, T.; Fujita, N.; Yanagitani, N.; Nishio, M.; et al. Overcoming resistance by ALK compound mutation (I1171S + G1269A) after sequential treatment of multiple ALK inhibitors in non-small cell lung cancer. Thorac. Cancer 2020, 11, 581–587. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Guo, L.; Sun, L.; Wu, Z.; Ye, J.; Liu, J.; Zuo, Q. Capture-based ultra-deep sequencing in plasma ctDNA reveals the resistance mechanism of ALK inhibitors in a patient with advanced ALK-positive NSCLC. Cancer Biol. Ther. 2018, 19, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Tsukaguchi, T.; Yoshida, M.; Kondoh, O.; Sakamoto, H. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014, 351, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Cao, R.; Bao, H.; Wu, X.; Yang, L.; Zhu, D.; Zhang, L.; Peng, L.; Cai, Y.; Zhang, W.; et al. Identification of novel Alectinib-resistant ALK mutation G1202K with sensitization to lorlatinib: A case report and in silico structural modelling. Onco Targets Ther. 2021, 14, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Li, T.; Wang, P.; Lizaso, A.; Huang, D. The efficacy of lorlatinib in a lung adenocarcinoma patient with a novel ALK G1202L mutation: A case report. Cancer Biol. Ther. 2021, 22, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.G.; Cortinovis, D.; Agustoni, F.; Arosio, G.; Villa, M.; Cordani, N.; Bidoli, P.; Bisson, W.H.; Pagni, F.; Piazza, R.; et al. A compound L1196M/G1202R ALK mutation in a patient with ALK-positive lung cancer with acquired resistance to brigatinib also confers primary resistance to lorlatinib. J. Thorac. Oncol. 2019, 14, e257–e259. [Google Scholar] [CrossRef]
- Mologni, L.; Ceccon, M.; Pirola, A.; Chiriano, G.; Piazza, R.; Scapozza, L.; Gambacorti-Passerini, C. NPM/ALK mutants resistant to ASP3026 display variable sensitivity to alternative ALK inhibitors but succumb to the novel compound PF-06463922. Oncotarget 2015, 6, 5720–5734. [Google Scholar] [CrossRef] [Green Version]
- Redaelli, S.; Ceccon, M.; Zappa, M.; Sharma, G.G.; Mastini, C.; Mauri, M.; Nigoghossian, M.; Massimino, L.; Cordani, N.; Farina, F.; et al. Lorlatinib treatment elicits multiple on- and off-target mechanisms of resistance in ALK-driven cancer. Cancer Res. 2018, 78, 6866–6880. [Google Scholar] [CrossRef] [Green Version]
- Yoda, S.; Lin, J.J.; Lawrence, M.S.; Burke, B.J.; Friboulet, L.; Langenbucher, A.; Dardaei, L.; Prutisto-Chang, K.; Dagogo-Jack, I.; Timofeevski, S.; et al. Sequential ALK inhibitors can select for lorlatinib-resistant compound ALK mutations in ALK-positive lung cancer. Cancer Discov. 2018, 8, 714–729. [Google Scholar] [CrossRef] [Green Version]
- Pastor, E.R.; Mousa, S.A. Current management of neuroblastoma and future direction. Crit. Rev. Oncol. Hematol. 2019, 138, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Recondo, G.; Mezquita, L.; Facchinetti, F.; Planchard, D.; Gazzah, A.; Bigot, L.; Rizvi, A.Z.; Frias, R.L.; Thiery, J.P.; Scoazec, J.Y.; et al. Diverse resistance mechanisms to the third-generation ALK inhibitor lorlatinib in ALK-rearranged lung cancer. Clin. Cancer Res. 2020, 26, 242–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.T.; Felip, E.; Bauer, T.M.; Besse, B.; Navarro, A.; Postel-Vinay, S.; Gainor, J.F.; Johnson, M.; Dietrich, J.; James, L.P.; et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: An international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017, 18, 1590–1599. [Google Scholar] [CrossRef]
- Lovly, C.M.; McDonald, N.T.; Chen, H.; Ortiz-Cuaran, S.; Heukamp, L.C.; Yan, Y.; Florin, A.; Ozretić, L.; Lim, D.; Wang, L.; et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat. Med. 2014, 20, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Shi, P.; Lai, R.; Lin, Q.; Iqbal, A.S.; Young, L.C.; Kwak, L.W.; Ford, R.J.; Amin, H.M. IGF-IR tyrosine kinase interacts with NPM-ALK oncogene to induce survival of T-cell ALK+ anaplastic large-cell lymphoma cells. Blood 2009, 114, 360–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossing, H.H.; Grauslund, M.; Urbanska, E.M.; Melchior, L.C.; Rask, C.K.; Costa, J.C.; Skov, B.G.; Sørensen, J.B.; Santoni-Rugiu, E. Concomitant occurrence of EGFR (epidermal growth factor receptor) and KRAS (V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) mutations in an ALK (anaplastic lymphoma kinase)-positive lung adenocarcinoma patient with acquired resistance to crizotinib: A case report. BMC Res. Notes 2013, 6, 489. [Google Scholar]
- Sánchez-Herrero, E.; Serna-Blasco, R.; Ivanchuk, V.; García-Campelo, R.; Dómine Gómez, M.; Sánchez, J.M.; Massutí, B.; Reguart, N.; Camps, C.; Sanz-Moreno, S.; et al. NGS-based liquid biopsy profiling identifies mechanisms of resistance to ALK inhibitors: A step toward personalized NSCLC treatment. Mol. Oncol. 2021, 15, 2363–2376. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, M.; Yasuda, H.; Tani, T.; Hamamoto, J.; Arai, D.; Ishioka, K.; Ohgino, K.; Nukaga, S.; Hirano, T.; Kawada, I.; et al. Overcoming EGFR bypass signal-induced acquired resistance to ALK tyrosine kinase inhibitors in ALK-translocated lung cancer. Mol. Cancer Res. 2017, 15, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanimoto, A.; Yamada, T.; Nanjo, S.; Takeuchi, S.; Ebi, H.; Kita, K.; Matsumoto, K.; Seiji, Y. Receptor ligand-triggered resistance to alectinib and its circumvention by Hsp90 inhibition in EML4-ALK lung cancer cells. Oncotarget 2014, 5, 4920–4928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minari, R.; Gnetti, L.; Lagrasta, C.A.; Squadrilli, A.; Bordi, P.; Azzoni, C.; Bottarelli, L.; Cosenza, A.; Ferri, L.; Caruso, G.; et al. Emergence of a HER2-amplified clone during disease progression in an ALK-rearranged NSCLC patient treated with ALK-inhibitors: A case report. Transl. Lung Cancer Res. 2020, 9, 787–792. [Google Scholar] [CrossRef]
- Dong, X.; Fernandez-Salas, E.; Li, E.; Wang, S. Elucidation of resistance mechanisms to second-generation ALK inhibitors alectinib and ceritinib in non-small cell lung cancer cells. Neoplasia 2016, 18, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Kogita, A.; Togashi, Y.; Hayashi, H.; Banno, E.; Terashima, M.; De Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Takeyama, Y.; et al. Activated MET acts as a salvage signal after treatment with alectinib, a selective ALK inhibitor, in ALK-positive non-small cell lung cancer. Int. J. Oncol. 2015, 46, 1025–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, T.; Ozasa, H.; Aoki, W.; Aburaya, S.; Funazo, T.; Furugaki, K.; Yoshimura, Y.; Ajimizu, H.; Okutani, R.; Yasuda, Y.; et al. Alectinib resistance in ALK-rearranged lung cancer by dual salvage signaling in a clinically paired resistance model. Mol. Cancer Res. 2019, 17, 212–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Lin, C.; Peng, T.; Hu, C.; Lu, C.; Li, L.; Wang, Y.; Han, R.; Feng, M.; Sun, F.; et al. Metformin reduces HGF-induced resistance to alectinib via the inhibition of Gab1. Cell Death Dis. 2020, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Filho, S.N.M.; Li, M.; Fares, A.; Weiss, J.; Pham, N.A.; Ludkovski, O.; Raghavan, V.; Li, Q.; Ravi, D.; et al. BRAF V600E mutation and MET amplification as resistance pathways of the second-generation anaplastic lymphoma kinase (ALK) inhibitor alectinib in lung cancer. Lung Cancer 2020, 146, 78–85. [Google Scholar] [CrossRef]
- Fan, P.D.; Narzisi, G.; Jayaprakash, A.D.; Venturini, E.; Robine, N.; Smibert, P.; Germer, S.; Yu, H.A.; Jordan, E.J.; Paik, P.K.; et al. YES1 amplification is a mechanism of acquired resistance to EGFR inhibitors identified by transposon mutagenesis and clinical genomics. Proc. Natl. Acad. Sci. USA 2018, 115, E6030–E6038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokoph, N.; Probst, N.A.; Lee, L.C.; Monahan, J.M.; Matthews, J.D.; Liang, H.C.; Bahnsen, K.; Montes-Mojarro, I.A.; Karaca-Atabay, E.; Sharma, G.G.; et al. IL10RA modulates crizotinib sensitivity in NPM1-ALK+ anaplastic large cell lymphoma. Blood 2020, 136, 1657–1669. [Google Scholar] [CrossRef]
- Karaca-Atabay, E.; Mecca, C.; Wang, Q.; Ambrogio, C.; Mota, I.; Prokoph, N.; Mura, G.; Martinengo, C.; Patrucco, E.; Leonardi, G.; et al. Tyrosine phosphatases regulate resistance to ALK inhibitors in ALK+ anaplastic large cell lymphoma. Blood 2021, in press. [Google Scholar] [CrossRef]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.; et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef]
- Mengoli, M.C.; Barbieri, F.; Bertolini, F.; Tiseo, M.; Rossi, G. K-RAS mutations indicating primary resistance to crizotinib in ALK-rearranged adenocarcinomas of the lung: Report of two cases and review of the literature. Lung Cancer 2016, 93, 55–58. [Google Scholar] [CrossRef]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [Green Version]
- Debruyne, D.N.; Bhatnagar, N.; Sharma, B.; Luther, W.; Moore, N.F.; Cheung, N.K.; Gray, N.S.; George, R.E. ALK inhibitor resistance in ALK(F1174L)-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene 2016, 35, 3681–3691. [Google Scholar] [CrossRef] [Green Version]
- Larose, H.; Prokoph, N.; Matthews, J.D.; Schlederer, M.; Högler, S.; Alsulami, A.F.; Ducray, S.P.; Nuglozeh, E.; Fazaludeen, F.M.S.; Elmouna, A.; et al. Whole exome sequencing reveals NOTCH1 mutations in anaplastic large cell lymphoma and points to Notch both as a key pathway and a potential therapeutic target. Haematologica 2021, 106, 1693–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trigg, R.M.; Lee, L.C.; Prokoph, N.; Jahangiri, L.; Reynolds, C.P.; Amos Burke, G.A.; Probst, N.A.; Han, M.; Matthews, J.D.; Lim, H.K.; et al. The targetable kinase PIM1 drives ALK inhibitor resistance in high-risk neuroblastoma independent of MYCN status. Nat. Commun. 2019, 10, 5428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogita, A.; Togashi, Y.; Hayashi, H.; Sogabe, S.; Terashima, M.; De Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Takeyama, Y.; et al. Hypoxia induces resistance to ALK inhibitors in the H3122 non-small cell lung cancer cell line with an ALK rearrangement via epithelial-mesenchymal transition. Int. J. Oncol. 2014, 45, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Urbanska, E.M.; Sørensen, J.B.; Melchior, L.C.; Costa, J.C.; Santoni-Rugiu, E. Changing ALK-TKI-resistance mechanisms in rebiopsies of ALK-rearranged NSCLC: ALK- and BRAF-mutations followed by epithelial-mesenchymal transition. Int. J. Mol. Sci. 2020, 21, 2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, Y.J.; Cho, B.C.; Kim, H.R.; Lee, H.J.; Shim, H.S. A case of ALK-rearranged adenocarcinoma with small cell carcinoma-like transformation and resistance to crizotinib. J. Thorac. Oncol. 2016, 11, e55–e58. [Google Scholar] [CrossRef] [Green Version]
- Fujita, S.; Masago, K.; Katakami, N.; Yatabe, Y. Transformation to SCLC after treatment with the ALK inhibitor alectinib. J. Thorac. Oncol. 2016, 11, e67–e72. [Google Scholar] [CrossRef] [Green Version]
- Levacq, D.; D’Haene, N.; de Wind, R.; Remmelink, M.; Berghmans, T. Histological transformation of ALK rearranged adenocarcinoma into small cell lung cancer: A new mechanism of resistance to ALK inhibitors. Lung Cancer 2016, 102, 38–41. [Google Scholar] [CrossRef]
- Coleman, N.; Wotherspoon, A.; Yousaf, N.; Popat, S. Transformation to neuroendocrine carcinoma as a resistance mechanism to lorlatinib. Lung Cancer 2019, 134, 117–120. [Google Scholar] [CrossRef]
- Debruyne, D.N.; Dries, R.; Sengupta, S.; Seruggia, D.; Gao, Y.; Sharma, B.; Huang, H.; Moreau, L.; McLane, M.; Day, D.S.; et al. BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells. Nature 2019, 572, 676–680. [Google Scholar] [CrossRef]
- Berko, E.R.; Mossé, Y.P. Thrown for a loop: Awakening BORIS to evade ALK inhibition therapy. Cancer Cell 2019, 36, 345–347. [Google Scholar] [CrossRef]
- Cai, C.; Long, Y.; Li, Y.; Huang, M. Coexisting of COX7A2L-ALK, LINC01210-ALK, ATP13A4-ALK and acquired SLCO2A1-ALK in a lung adenocarcinoma with rearrangements loss during the treatment of crizotinib and ceritinib: A case report. Onco Targets Ther. 2020, 13, 8313–8316. [Google Scholar] [CrossRef] [PubMed]
- Mitou, G.; Frentzel, J.; Desquesnes, A.; Le Gonidec, S.; AlSaati, T.; Beau, I.; Lamant, L.; Meggetto, F.; Espinos, E.; Codogno, P.; et al. Targeting autophagy enhances the anti-tumoral action of crizotinib in ALK-positive anaplastic large cell lymphoma. Oncotarget 2015, 6, 30149–30164. [Google Scholar] [CrossRef]
- Ji, C.; Zhang, L.; Cheng, Y.; Patel, R.; Wu, H.; Zhang, Y.; Wang, M.; Ji, S.; Belani, C.P.; Yang, J.M.; et al. Induction of autophagy contributes to crizotinib resistance in ALK-positive lung cancer. Cancer Biol. Ther. 2014, 15, 570–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moia, R.; Boggione, P.; Mahmoud, A.M.; Kodipad, A.A.; Adhinaveni, R.; Sagiraju, S.; Patriarca, A.; Gaidano, G. Targeting p53 in chronic lymphocytic leukemia. Expert Opin. Ther. Targets 2020, 24, 1239–1250. [Google Scholar] [CrossRef]
- Moia, R.; Patriarca, A.; Schipani, M.; Ferri, V.; Favini, C.; Sagiraju, S.; Al Essa, W.; Gaidano, G. Precision medicine management of chronic lymphocytic leukemia. Cancers 2020, 12, 642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, M.; Otomo, R.; Matsushima-Hibiya, Y.; Suzuki, H.; Nakajima, A.; Abe, N.; Tomiyama, A.; Ichimura, K.; Matsuda, K.; Watanabe, T.; et al. The p53 activator overcomes resistance to ALK inhibitors by regulating p53-target selectivity in ALK-driven neuroblastomas. Cell Death Discov. 2018, 4, 56. [Google Scholar] [CrossRef]
- Rassidakis, G.Z.; Thomaides, A.; Wang, S.; Jiang, Y.; Fourtouna, A.; Lai, R.; Medeiros, L.J. p53 gene mutations are uncommon but p53 is commonly expressed in anaplastic large-cell lymphoma. Leukemia 2005, 19, 1663–1669. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.-X.; Kerby, A.; McDuff, F.K.E.; Ye, H.; Turner, S.D. NPM-ALK inhibits the p53 tumor suppressor pathway in an MDM2 and JNK-dependent manner. Blood 2009, 113, 5217–5227. [Google Scholar] [CrossRef] [Green Version]
- Drakos, E.; Atsaves, V.; Schlette, E.; Li, J.; Papanastasi, I.; Rassidakis, G.Z.; Medeiros, L.J. The therapeutic potential of p53 reactivation by nutlin-3a in ALK+ anaplastic large cell lymphoma with wild-type or mutated p53. Leukemia 2009, 23, 2290–2299. [Google Scholar] [CrossRef]
- Redaelli, S.; Ceccon, M.; Antolini, L.; Rigolio, R.; Pirola, A.; Peronaci, M.; Gambacorti-Passerini, C.; Mologni, L. Synergistic activity of ALK and mTOR inhibitors for the treatment of NPM-ALK positive lymphoma. Oncotarget 2016, 7, 72886–72897. [Google Scholar] [CrossRef] [Green Version]
- Dagogo-Jack, I.; Yoda, S.; Lennerz, J.K.; Langenbucher, A.; Lin, J.J.; Rooney, M.M.; Prutisto-Chang, K.; Oh, A.; Adams, N.A.; Yeap, B.Y.; et al. MET alterations are a recurring and actionable resistance mechanism in ALK-positive lung cancer. Clin. Cancer Res. 2020, 26, 2535–2545. [Google Scholar] [CrossRef] [Green Version]
- Chihara, D.; Wong, S.; Feldman, T.; Fanale, M.A.; Sanchez, L.; Connors, J.M.; Savage, K.J.; Oki, Y. Outcome of patients with relapsed or refractory anaplastic large cell lymphoma who have failed brentuximab vedotin. Hematol. Oncol. 2019, 37, 35–38. [Google Scholar] [CrossRef] [Green Version]
- Arai, H.; Furuichi, S.; Nakamura, Y.; Ichikawa, M.; Mitani, K. ALK-negative anaplastic large cell lymphoma with loss of CD30 expression during treatment with brentuximab vedotin. Rinsho Ketsueki 2016, 57, 634–637. [Google Scholar]
- Fordham, A.M.; Xie, J.; Gifford, A.J.; Wadham, C.; Morgan, L.T.; Mould, E.V.A.; Fadia, M.; Zhai, L.; Massudi, H.; Ali, Z.S.; et al. CD30 and ALK combination therapy has high therapeutic potency in RANBP2-ALK-rearranged epithelioid inflammatory myofibroblastic sarcoma. Br. J. Cancer 2020, 123, 1101–1113. [Google Scholar] [CrossRef]
- Chen, R.; Hou, J.; Newman, E.; Kim, Y.; Donohue, C.; Liu, X.; Thomas, S.H.; Forman, S.J.; Kane, S.E. CD30 Downregulation, MMAE resistance, and MDR1 upregulation are all associated with resistance to brentuximab vedotin. Mol. Cancer Ther. 2015, 14, 1376–1384. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Lin, Y.; Song, Z.; Xiao, W.; Chen, L.; Yin, J.; Zhou, Y.; Barta, S.K.; Petrus, M.; Waldmann, T.A.; et al. A20 and RBX1 regulate brentuximab vedotin sensitivity in hodgkin lymphoma models. Clin. Cancer Res. 2020, 26, 4093–4106. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Herrera, A.F.; Hou, J.; Chen, L.; Wu, J.; Guo, Y.; Synold, T.W.; Ngo, V.N.; Puverel, S.; Mei, M.; et al. Inhibition of MDR1 overcomes resistance to brentuximab vedotin in hodgkin lymphoma. Clin. Cancer Res. 2020, 26, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Oh, M.S.; Giles, F.J. Molecular biomarkers of primary and acquired resistance to T-cell-mediated immunotherapy in cancer: Landscape, clinical implications, and future directions. Oncologist 2018, 23, 410–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Wu, Z.X.; Assaraf, Y.G.; Chen, Z.S.; Wang, L. Overcoming anti-cancer drug resistance via restoration of tumor suppressor gene function. Drug Resist. Updat. 2021, 57, 100770. [Google Scholar] [CrossRef]
- Cretella, D.; Digiacomo, G.; Giovannetti, E.; Cavazzoni, A. PTEN alterations as a potential mechanism for tumor cell escape from PD-1/PD-L1 inhibition. Cancers 2019, 11, 1318. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zhou, Y.; Zhang, J.; Jin, X.; Wu, H.; Huang, H. Fructose-1,6-bisphosphatase loss modulates STAT3-dependent expression of PD-L1 and cancer immunity. Theranostics 2020, 10, 1033–1045. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [Green Version]
- De Souza, A. Finding the hot spot: Identifying immune sensitive gastrointestinal tumors. Transl. Gastroenterol. Hepatol. 2020, 5, 48. [Google Scholar] [CrossRef]
- Ross-Macdonald, P.; Walsh, A.M.; Chasalow, S.D.; Ammar, R.; Papillon-Cavanagh, S.; Szabo, P.M.; Choueiri, T.K.; Sznol, M.; Wind-Rotolo, M. Molecular correlates of response to nivolumab at baseline and on treatment in patients with RCC. J. Immunother. Cancer 2021, 9, e001506. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Peggs, K.S.; Curran, M.A.; Allison, J.P. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Investig. 2006, 116, 1935–1945. [Google Scholar] [CrossRef] [Green Version]
- Restifo, N.P.; Marincola, F.M.; Kawakami, Y.; Taubenberger, J.; Yannelli, J.R.; Rosenberg, S.A. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J. Natl. Cancer Inst. 1996, 88, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Vagia, E.; Viveiros, P.; Kang, C.Y.; Lee, J.Y.; Gim, G.; Cho, S.; Choi, H.; Kim, L.; Park, I.; et al. Overcoming acquired resistance to PD-1 inhibitor with the addition of metformin in small cell lung cancer (SCLC). Cancer Immunol. Immunother. 2021, 70, 961–965. [Google Scholar] [CrossRef] [PubMed]
- De Wispelaere, W.; Annibali, D.; Tuyaerts, S.; Lambrechts, D.; Amant, F. Resistance to immune checkpoint blockade in uterine leiomyosarcoma: What can we learn from other cancer types? Cancers 2021, 13, 2040. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Tsuchihashi, K.; Tsuji, K.; Kito, Y.; Tanoue, K.; Ohmura, H.; Ito, M.; Isobe, T.; Ariyama, H.; Kusaba, H.; et al. Prominent PD-L1-positive M2 macrophage infiltration in gastric cancer with hyper-progression after anti-PD-1 therapy: A case report. Medicine 2021, 100, e25773. [Google Scholar] [CrossRef]
- Viveiros, P.; Burns, M.; Davis, A.; Oh, M.; Park, K.; Jain, S.; Chae, Y.K. EP1.04-12 Response to combination of metformin and nivolumab in a NSCLC patient whose disease previously progressed on nivolumab. J. Thorac. Oncol. 2019, 14, S976. [Google Scholar] [CrossRef]
- Cabrera, C.M.; Jiménez, P.; Cabrera, T.; Esparza, C.; Ruiz-Cabello, F.; Garrido, F. Total loss of MHC class I in colorectal tumors can be explained by two molecular pathways: Beta2-microglobulin inactivation in MSI-positive tumors and LMP7/TAP2 downregulation in MSI-negative tumors. Tissue Antigens 2003, 61, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
The NPM1-ALK fusion protein produced due to a t(2;5)(p23;q35) chromosomal translocation. The kinase domain, depicted in red, is the site within the ALK portion of the fusion protein where ALK tyrosine kinase inhibitors (ALK TKIs) bind. Mutations here can lead to ALK TKI resistance [15,16]. AAs = amino acids, TM = transmembrane, TK = tyrosine kinase.
Figure 1.
The NPM1-ALK fusion protein produced due to a t(2;5)(p23;q35) chromosomal translocation. The kinase domain, depicted in red, is the site within the ALK portion of the fusion protein where ALK tyrosine kinase inhibitors (ALK TKIs) bind. Mutations here can lead to ALK TKI resistance [15,16]. AAs = amino acids, TM = transmembrane, TK = tyrosine kinase.

Figure 2.
Summary of the signalling pathways activated by NPM1-ALK. The hyperactive intracellular tyrosine kinase NPM1-ALK activates a plethora of signalling pathways including MAPK, PI3K-Akt, JAK-STAT and PLCγ. These collectively drive ALCL through conferring the hallmarks of cancer including increasing cell survival and reducing apoptosis [11,12,13,14,17,18].
Figure 2.
Summary of the signalling pathways activated by NPM1-ALK. The hyperactive intracellular tyrosine kinase NPM1-ALK activates a plethora of signalling pathways including MAPK, PI3K-Akt, JAK-STAT and PLCγ. These collectively drive ALCL through conferring the hallmarks of cancer including increasing cell survival and reducing apoptosis [11,12,13,14,17,18].

Figure 3.
The pathogenesis and aetiology of ALCL expressing NPM1-ALK may be dependent on TCR signalling and/or microenvironmental factors. NPM1-ALK mimics low-intensity tonic TCR signalling required for T cell development. The TCRs are ultimately downregulated as they are either surplus to requirement or are prohibitive towards tumour development. If the TCR is downregulated or non-functional soon after emerging into the periphery, antigen-independent inflammatory microenvironmental factors might provide the ‘second hit’ promoting ALCL development. If the TCR is functional after emerging into the periphery, an antigen-presenting cell (APC) might expose the T cell to a ‘second hit’ in the form of a major histocompatibility complex (MHC)-bound ligand that provides additional stimulation and promotes ALCL development. With this additional stimulation, the TCR might then be downregulated to facilitate cell survival by preventing over-stimulation [2,7,11,20,21].
Figure 3.
The pathogenesis and aetiology of ALCL expressing NPM1-ALK may be dependent on TCR signalling and/or microenvironmental factors. NPM1-ALK mimics low-intensity tonic TCR signalling required for T cell development. The TCRs are ultimately downregulated as they are either surplus to requirement or are prohibitive towards tumour development. If the TCR is downregulated or non-functional soon after emerging into the periphery, antigen-independent inflammatory microenvironmental factors might provide the ‘second hit’ promoting ALCL development. If the TCR is functional after emerging into the periphery, an antigen-presenting cell (APC) might expose the T cell to a ‘second hit’ in the form of a major histocompatibility complex (MHC)-bound ligand that provides additional stimulation and promotes ALCL development. With this additional stimulation, the TCR might then be downregulated to facilitate cell survival by preventing over-stimulation [2,7,11,20,21].

Figure 4.
ALK-dependent mechanisms of resistance to ALK TKIs. (A) Mutations in the ALK tyrosine kinase domain prevent the ALK TKI from binding to the receptor and exerting its inhibitory effect on oncogenic ALK signalling. (B) Amplification of the ALK gene provides an excess of drug target outcompeting the inhibitor.
Figure 4.
ALK-dependent mechanisms of resistance to ALK TKIs. (A) Mutations in the ALK tyrosine kinase domain prevent the ALK TKI from binding to the receptor and exerting its inhibitory effect on oncogenic ALK signalling. (B) Amplification of the ALK gene provides an excess of drug target outcompeting the inhibitor.

Figure 5.
ALK-independent ‘bypass’ mechanisms of resistance to ALK TKIs. The effects of inhibited ALK activity are substituted by the upregulation of alternative signalling cascades that activate the same downstream targets as does aberrantly active ALK. The need for ALK is effectively bypassed. Mutations in the downstream targets of ALK, indicated with a lightning symbol, also bypass the need for ALK.
Figure 5.
ALK-independent ‘bypass’ mechanisms of resistance to ALK TKIs. The effects of inhibited ALK activity are substituted by the upregulation of alternative signalling cascades that activate the same downstream targets as does aberrantly active ALK. The need for ALK is effectively bypassed. Mutations in the downstream targets of ALK, indicated with a lightning symbol, also bypass the need for ALK.

Table 2.
Summary of reported bypass tracks conferring resistance to ALK TKIs.
| Protein Alteration (Upregulation Unless Otherwise Specified) | ALK TKI | Disease |
|---|---|---|
| IGF-1R | Crizotinib [129,130] | NSCLC and ALCL |
| Epidermal growth factor receptor (EGFR) | Crizotinib [78,81,98,131,132] Ceritinib [98,133] Alectinib [98,132,134] Lorlatinib [124,132] | NSCLC |
| Lorlatinib [124] | Neuroblastoma | |
| Human epidermal growth factor receptor (HER), including via increased neuregulin 1 ligand | Ceritinib and alectinib [135,136] | NSCLC |
| KIT proto-oncogene receptor tyrosine kinase (KIT), including via increased stem cell factor (SCF) ligand | Crizotinib [78] Ceritinib [98] | NSCLC |
| MET proto-oncogene receptor tyrosine kinase (MET), including via increased hepatocyte growth factor (HGF) ligand | Alectinib [134,137,138,139,140] Ceritinib and lorlatinib [79,98,117] | NSCLC |
| SRC proto-oncogene, non-receptor tyrosine kinase (SRC) | Crizotinib [141] Alectinib [138] | NSCLC |
| Discoidin domain receptor tyrosine kinase 2 (DDR2) | Alectinib [79] | NSCLC |
| Fibroblast growth factor receptor 2 (FGFR2) | Ceritinib [79] | NSCLC |
| ERb-B4 receptor tyrosine kinase 4 (ErbB4) | Lorlatinib [124] | Neuroblastoma |
| Interleukin 10 receptor subunit alpha (IL10RA) | Crizotinib [142] Alectinib [142] Brigatinib [142] Lorlatinib [142] | ALCL |
| Protein tyrosine phosphatase non-receptor tyrosine kinase 1/2 (PTPN1/2) loss | Crizotinib [143] | ALCL |
Table 3.
A skew towards immunosuppression in the tumour microenvironment can drive resistance to immune checkpoint inhibitors.
Table 3.
A skew towards immunosuppression in the tumour microenvironment can drive resistance to immune checkpoint inhibitors.
| Event | Impact on the Tumour Microenvironment |
|---|---|
| Increased AXL receptor tyrosine kinase (AXL) expression | Increases regulatory T cells, MDSCs and M2 macrophages [185] |
| Increased Wnt signalling | Decreases tumour infiltrating lymphocytes [185] |
| Loss of Phosphatase and tensin homolog (PTEN) | Induces vascular endothelial growth factor (VEGF) production and reduces T cell infiltration [176,185] |
| Loss of functional beta 2 microglobulin | Dysfunctional CD8+ T cells [175,188] |
| Hypoxia | Dysfunctional CD8+ T cells [184,187] |
| Upregulation of T cell immunoglobulin and mucin-domain containing-3 (Tim-3) | Dysfunctional T helper 1 (Th1) cells and reduced cytokine expression [189,190] |
| Reduced expression of absent in melanoma 2 (AIM2) | Decreases inflammation [181] |
| Reduced expression of poliovirus receptor-related immunoglobulin domain containing protein (PVRIG) | Dysfunctional CD8+ T cells [181] |
| Increased expression of mannosidase alpha class 2A member 1 (MAN2A1) | Altered Th1/T-helper 2 (Th2) axis towards Th2 expression [181] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hare, L.; Burke, G.A.A.; Turner, S.D. Resistance to Targeted Agents Used to Treat Paediatric ALK-Positive ALCL. Cancers 2021, 13, 6003. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13236003
AMA Style
Hare L, Burke GAA, Turner SD. Resistance to Targeted Agents Used to Treat Paediatric ALK-Positive ALCL. Cancers. 2021; 13(23):6003. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13236003
Chicago/Turabian StyleHare, Lucy, G. A. Amos Burke, and Suzanne D. Turner. 2021. "Resistance to Targeted Agents Used to Treat Paediatric ALK-Positive ALCL" Cancers 13, no. 23: 6003. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13236003
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.