
Therapeutic Targeting of Exportin-1 in Childhood Cancer
by

Basia Galinski
1,†, 
Thomas B. Alexander
2,†,
Daniel A. Mitchell
1,
Hannah V. Chatwin
3,
Chidiebere Awah
1,
Adam L. Green
3,* and
Daniel A. Weiser
1,* 1
Department of Pediatrics, Albert Einstein College of Medicine, Bronx, NY 10461, USA
2
Department of Pediatrics, University of North Carolina, Chapel Hill, NC 27599, USA
3
Morgan Adams Foundation Pediatric Brain Tumor Research Program, University of Colorado School of Medicine, Aurora, CO 80045, USA
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cancers 2021, 13(24), 6161; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13246161
Submission received: 29 September 2021
/
Revised: 16 November 2021
/
Accepted: 1 December 2021
/
Published: 7 December 2021
(This article belongs to the Collection The Development of Anti-cancer Agents)
Abstract
:Simple Summary
Exportin-1 is a nuclear transport protein that is overexpressed in cancer cells and associated with inferior outcomes across a range of malignancies. Selinexor is a novel FDA-approved inhibitor of Exportin-1 (XPO1). Although significant research has focused on integration of selinexor into the treatment regimens of adult cancers, it is increasingly recognized that XPO1-directed therapy may be effective as part of management of childhood cancers. We therefore summarize the history of, and latest knowledge about, the function and therapeutic inhibition of XPO1 as it relates to childhood cancer pathogenesis and treatment.
Abstract
Overexpression of Exportin-1 (XPO1), a key regulator of nuclear-to-cytoplasmic transport, is associated with inferior patient outcomes across a range of adult malignancies. Targeting XPO1 with selinexor has demonstrated promising results in clinical trials, leading to FDA approval of its use for multiple relapsed/refractory cancers. However, XPO1 biology and selinexor sensitivity in childhood cancer is only recently being explored. In this review, we will focus on the differential biology of childhood and adult cancers as it relates to XPO1 and key cargo proteins. We will further explore the current state of pre-clinical and clinical development of XPO1 inhibitors in childhood cancers. Finally, we will outline potentially promising future therapeutic strategies for, as well as potential challenges to, integrating XPO1 inhibition to improve outcomes for children with cancer.
1. Introduction
Childhood cancer, affecting more than 15,000 children per year in the United States and over 300,000 per year globally, is the result of tumor and host biology that is different than adult cancers [1]. Childhood cancers tend to arise spontaneously, have varying degrees of hereditary contribution, and are generally not the result of an accumulation of genetic mutations over a lifetime [2]. Therapy for childhood cancer is often multi-modal, including chemotherapy, surgery, radiotherapy, and/or immunotherapy. Five-year overall survival for children with cancer currently exceeds 80% in many high-income countries, yet a tremendous unmet need exists to improve survival and reduce long-term sequelae of treatment. First, we must better identify subsets of patients who have high-risk disease with inferior outcomes and develop improved therapeutic strategies for their resistant disease [3,4,5]. Second, therapy de-intensification may be considered for cancers with favorable clinical and biological features and a relatively high overall survival rate, so that treatment-related morbidity can be minimized [6]. Third, identifying therapeutically targetable oncogenic vulnerabilities may allow for personalized and more effective approaches to care. Recent advances in the understanding of adult malignancies have demonstrated the relevance of Exportin-1 (XPO1) overexpression in highly aggressive cancers and the potential for precise therapeutic targeting, yet the overall understanding of this protein in childhood cancer is less well established. In this review, we will focus on clinical implications of overexpression of XPO1, including pharmacologic targeting with commercially available XPO1 inhibitors such as selinexor.
2. XPO1 Biological Function and Relevance
XPO1, historically referred to as CRM1 (Chromosomal Region Maintenance 1), was initially characterized for its role in mitotic spindle activity and chromosome assembly [7,8]. During development, XPO1 is ubiquitously expressed, and Xenopus and Drosophila studies demonstrated the requirement for early expression that continues through development; XPO1 is embryonic lethal if genetically knocked out [9]. Studies in yeast have demonstrated that mutations in XPO1 lead to a reduction in cytoplasmic microtubules and improper spindle formation [10,11]. Microdeletions of XPO1 in autism spectrum disorders have also been identified and result in disruption of mitosis leading to apoptosis in neural progenitors [12].
2.1. Regulation of Homeostasis by Exportins
Cellular homeostasis depends heavily on the balance of activity and localization of proteins, as together they affect signaling pathways that can either promote cellular survival or cell death. This balance is tightly regulated to maintain healthy cellular conditions, but in cancer, changes to the activity and localization of proteins can promote survival, growth, and metastasis [13,14]. The karyopherin protein family, which is comprised of importins, transportins, and exportins, shuttles cargo (proteins, tRNA, and microRNA) between the nucleus and cytoplasm through the nuclear pore complex [13,15,16,17]. These cargos, sized 30 kDa or greater, are not able to diffuse freely from one compartment to another. They rely on the gradients of high RAN-GTP in the nuclear compartment and high RAN-GDP in the cytoplasmic compartment to facilitate this energy-dependent translocation [18,19,20,21,22]. This gradient, along with RCC1 and RanGAP to catalyze the exchange of nucleotides, creates a cycling of RAN ensuring proper export and import of cargos [23].
XPO1 is one of seven exportins in humans that regulate the unidirectional export of proteins, tRNAs, and microRNAs from the nucleus to the cytoplasm using the nuclear pore complex [24]. Exportins recognize and bind to a leucine rich hydrophobic region, known as a nuclear exportation signal (NES), on cargo (with RNA species using adapter proteins) and, with RAN-GTP in a complex, translocate to the cytoplasm [25,26,27]. The hydrolysis of RAN-GTP to RAN-GDP releases the complex, allowing the cargo to diffuse freely in the cytoplasm, while the exportin returns to the nucleus through the nuclear pore [19]. While certain exportins recognize few proteins or microRNAs exclusively, XPO1 recognizes the nuclear export signals of over 200 proteins and microRNAs. Characterization of the numerous cargos with NES binding motifs was initially established through use of leptomycin B, an antifungal agent that recognizes the NES site of XPO1 and blocks cargos from forming the exportation complex, leading to nuclear accumulation of cargos [8,28]. Current approaches apply in silico modeling to identify and validate XPO1 cargos (http://prodata.swmed.edu/LRNes, accessed on 30 June 2021), many of which function in signaling pathways and cellular regulation, with implications for understanding the pathogenesis of childhood cancer and the effect of pharmacologic inhibition of XPO1 (Table 1). The promiscuity in cargo exportation by XPO1, which is universally expressed across human cells, is consistent with its vital, non-redundant intermediary role in normal homeostatic processes [27,29].
At centrosomes and kinetochores, XPO1 colocalizes with RAN-GTP and cargos with NES that are important for normal cell division [40,41]. Recruitment of XPO1 to these sites is via phosphorylation by the CDK1/Cyclin B complex at the serine 391 site of XPO1, which is a different site than where it binds to various cargo NES [42,43]. One cargo, Survivin, which is upregulated during cellular development and is a member of the inhibitor of apoptosis family, complexes with XPO1 to facilitate normal cellular division through assembly of the chromosome passenger complex during mitosis [44,45,46]. Increased expression of Survivin is also associated with cancer cell survival; specifically, cytoplasmic Survivin is more abundant when XPO1 is overexpressed and promotes chemoresistance through inhibition of apoptosis [47,48,49,50,51]. While XPO1 and its role in regulating mitosis is important, in the context of cancer targeting and treatment; the focus has been on its role as a nuclear export protein and the disruption of downstream signaling pathways.
2.2. XPO1 Function in Human Disease
Across nearly all cancers, including those affecting children, adolescents, and young adults, XPO1 is expressed (Figure 1) [52,53,54,55,56,57,58], and higher expression has been correlated with inferior outcome. XPO1 is located on chromosome arm 2p, a region frequently associated with somatic copy number changes in cancer (e.g., gastric cancer, CLL, lymphomas, neuroblastoma, and rhabdomyosarcoma), and a region that includes putative proto-oncogenes N-MYC, REL, and BCL11A [7,59,60,61,62]. This region may be key to understanding how XPO1 regulation is coopted to promote oncogenesis and to distinguish between sufficiency and necessity of overexpression to promote aggressive disease.
For example, N-MYC is amplified in approximately 20% of neuroblastomas, expressed in other pediatric cancers, and associated with more aggressive disease [63,64,65]. It is expressed primarily in neural tissues, in contrast to ubiquitously expressed C-MYC, sharing redundant functions and cross regulation in normal development [66,67,68]. In adult cancers, pharmacologic targeting of XPO1 decreases C-MYC levels [69]. Though there is not a clear understanding of what drives XPO1 overexpression as a primary or secondary event in tumor development, it has been shown that knockdown of the tumor suppressor TP53 results in increased XPO1 expression, while knockdown of the proto-oncogene C-MYC results in decreased XPO1 expression [70]. Dysregulation of TP53 and C-MYC could be an initiating event that leads to overexpression of XPO1 [70,71,72,73].
XPO1 mutations have been identified, particularly in patients with hematologic malignancies [74]. The prevalence ranges from 8% in chronic lymphocytic leukemia (CLL) (primarily D624 mutation) to 25% in primary mediastinal B-cell lymphoma (PMBL) and classical Hodgkin lymphoma (cHL) (primarily E571 mutations). XPO1 R749 mutations have also been identified in solid tumors [75,76,77]. In limited studies of CLL, XPO1 mutations correlate with inferior outcome and the need for early aggressive therapeutic intervention [78,79,80,81]. It is unclear if this correlation is due to co-occurrence with TP53 mutations, or a direct effect of the mutation leading to pathologically increased cargo binding affinity and depletion of regulatory cargos from the nucleus [76]. Leukemia and lymphoma cells with E571K mutations have increased sensitivity to selinexor in vitro, suggesting that upfront screening for mutations may have utility for precision medicine approaches utilizing XPO1-based management strategies [75].
3. Development of Selective Inhibitors of Nuclear Export for Cancer Treatment
Selinexor was FDA approved in 2019 as part of combination therapy for relapsed multiple myeloma, an exclusively adult malignancy [82]. It was approved in 2020 as a single agent for adults with relapsed/refractory diffuse large B cell lymphoma (DLBCL) [83]. While adolescents can develop DLBCL, the currently approved indications for selinexor do not directly translate to most pediatric tumors. However, parallel preclinical and early clinical trial efforts have been ongoing, focused on tumor biology related to XPO1 and safety of selinexor for children with cancer.
3.1. Preclinical Development of XPO1-Directed Therapeutics for Childhood Cancers
3.1.1. XPO1 Inhibitors as Single Agent Treatment
Many early pharmacologic compounds that inhibit XPO1, such as Ratjadone C (myxobacterial metabolite), KOS-2464 (leptomycin B derivative), N-azolylacrylate analogs, valtrate, and acetoxychavicol acetate have been identified in vitro to bind competitively to the cystine 528 residue on XPO1, leading to nuclear retention of cargos such as TOPO2A and the HIV Rev protein [84,85,86]. These agents were not tested in vivo and safety and tolerability have not been established; therefore there are no data to encourage clinical testing. Other compounds that inhibit XPO1, such as the antifungal agent leptomycin B (elactocin), showed numerous irreversible off-target effects, including severe anorexia and malaise, that limited its clinical advancement [87,88].
Four selective inhibitors of nuclear export (SINE) compounds that reversibly bind XPO1, KPT-185, KPT-251, KPT-330 (selinexor), and SL-801 (felezonexor), have been explored across a range of malignancies. Preclinical evaluation of SINE compounds in specific childhood cancers dates back to at least 2012 with agents demonstrating low single-agent IC50 concentrations across various pediatric leukemia subtypes [89,90,91,92], high-grade gliomas [92,93], atypical teratoid/rhabdoid tumor (ATRT) [92], malignant rhabdoid tumor (MRT) [92], sarcoma [92,94], neuroblastoma [30,92], and Wilms tumor [92]. Patient-derived xenograft (PDX) mouse models have shown encouraging anti-tumor activity and survival advantages in treated animals [89,90,91,92]. A study by the Pediatric Preclinical Testing Consortium utilizing pediatric xenografts, including gliomas, leukemias, and sarcomas showed improvement in event-free survival for 29 out of 38 (76%) solid tumor models and 5 out of 8 (63%) leukemia models [92].
Similar to findings in adult malignancies, XPO1 inhibitors in pediatric cancers have been found to induce a G1 cell cycle arrest, leading to a decrease in cells entering S/G2, in leukemia, neuroblastoma, MRT, and ATRT [30,89,90,91,95]. Further, XPO1-directed therapy has been shown to upregulate apoptosis in leukemia, high-grade glioma, ATRT, neuroblastoma, MRT, and sarcoma [30,89,90,91,93,94,95]. In acute leukemia, Etchin et al. demonstrated the effect of KPT-185 on G1 cell cycle arrest and showed that the majority of apoptosis is induced at G1 [89]. These results provide insight into potential combination strategies that would promote a synergistic anti-cancer activity based on convergence of anti-cancer mechanisms.
3.1.2. Selinexor-Based Combination Strategies with Mechanistic Rationale for Use in Pediatrics
The success of the reversible SINE compounds as single agents prompted further investigation of combinatorial strategies to improve efficacy and offset potential dose-related toxicity concerns through lower dosing. The results demonstrating that selinexor-induced apoptosis occurs in the G1 phase of the cell cycle suggests potential for synergy when combined with conventional cytotoxic chemotherapy. Many commonly used chemotherapeutics have their largest effects in the cell cycle phases S, G2, and M. Thus, combining SINE compounds with fludarabine and cytarabine (primarily S phase effects) for acute myeloid leukemia (AML), anthracyclines (primarily S/G2 effects) for soft tissue sarcomas, or topotecan (S/G2 effects) or paclitaxel (M phase effects) for solid tumors are rational approaches for further study [96,97,98,99,100,101,102].
Selinexor has been combined with proteasome inhibition. The most well-studied combination in adults is selinexor in combination with bortezomib and dexamethasone for the treatment of multiple myeloma. Based on these data, the combination of selinexor and proteasome inhibition is being explored in pediatric tumors, including neuroblastoma. Researchers demonstrated synergistic anti-cancer activity and increases in total apoptosis that were mediated by IkB nuclear localization induced by selinexor and diminished IkB degradation with bortezomib; they also identified synergistic functional inhibition of NF-kB transcriptional activity [30].
Combinations of decitabine, a DNA methyltransferase inhibitor, and selinexor have also been investigated. In vitro studies in AML showed no benefit of concurrent treatment, but pre-treatment with decitabine followed by selinexor showed synergy. The findings were recapitulated in a PDX murine model, where pre-treating with decitabine followed by sub-IC50 selinexor dosing, lead to a survival advantage over single agent selinexor [103]. Priming with decitabine may increase selinexor tolerability and effectiveness by 30–47% as increasing DNA methylation induces re-expression of tumor suppressor proteins, which are later trafficked by XPO1 [104].
Selinexor for use in children with cancer is also being explored with other novel therapies, including small molecules targeting CDK 4/6 (palbociclib) and Wee1 (AZD1775). In a study focusing on a single pediatric patient-derived cell line from a rare undifferentiated sarcoma, selinexor was effective alone, but combination with palbociclib demonstrated synergy [94]. Other combinations that have been studied include BCL-2 inhibitors [105,106,107,108], PARP inhibitors [109,110], and CAR-T cells [111], with potential implications for many pediatric cancer types, including AML, ALL, anaplastic large cell lymphomas, Ewing sarcoma, neuroblastoma, osteosarcoma and glioblastoma. Immune targeting approaches with pembrolizumab and rituximab, as well as targeting JAK/STAT and tyrosine kinase signaling pathways may be of value. An additional treatment strategy involves the combination of selinexor with radiotherapy, an essential component of pediatric high-grade glioma management, where selinexor improves sensitivity to ionizing radiation and increases anti-tumor activity in adult patients [93,112].
Selinexor has potential to be rationally combined with numerous established and novel therapeutics agents to create critical synergy against pediatric malignancies. The drug has been shown to offset resistance to conventional chemotherapeutics via numerous pathways related to nuclear transport of key cargo, suggesting that patients with relapsed and refractory cancers might benefit from incorporating XPO1 inhibitors into treatment regimens [113,114,115,116]. Continued exploration of combination strategies and timing of drug administration will provide opportunities to define optimal management approaches that can be developed further as clinical trials across a range of cancers that affect children.
3.1.3. Therapeutic Resistance to Selinexor
Specific mechanisms that promote resistance to XPO1 inhibition have not yet been fully elucidated. One group was able to create selinexor-resistant leukemia models through a CRISPR-induced heterozygous mutation in the XPO1 cargo-binding pocket (C528S), rendering XPO1 resistant to degradation in these cells [86]. The unique ability of C528S to induce resistance has only been observed preclinically whereas a clinically described E571K mutation in B-cell lymphoma cell lines has no effect on selinexor efficacy [75,106]. Other studies of resistance mechanisms have focused on possibilities outside of XPO1 mutations. As part of preclinical investigation in hematologic malignancies, one group chronically treated cells with increasing concentrations of selinexor and was able to create a resistant line with upregulated NF-kB activity [117,118]. They found that combination with proteasome inhibitors re-sensitized cells to selinexor, indicating that resistance might be overcome through drug combination regimens [119]. Ongoing work is focused on identifying molecular pathways involved in resistance to XPO1 inhibition, such as increased NF-kB signaling, TGF-β/SMAD pathway, and ESF1 transcription activity of G1/S cell cycle [120,121,122,123].
3.1.4. Toxicity Challenges and Next Generation SINE Compounds
Experiments in PDX models [89,90] and clinical trials in adults and children have identified mild and serious selinexor-induced adverse effects. In adults, the most common noted side effects are nausea, emesis, decreased appetite/weight, confusion, neutropenia, thrombocytopenia, anemia, fatigue, infections, hyponatremia and blurred vision [124,125]. The most common selinexor-related toxicities in children have been electrolyte abnormalities (hyponatremia and hypokalemia), elevated liver function enzymes, nausea, and diarrhea [96,126]. Grade 4 tumor lysis syndrome after selinexor administration has also been seen [126]. Dose-limiting toxicities (DLT) include pancreatitis, cognitive disturbance, and reversible cerebellar toxicity [96,126].
A second-generation XPO1 inhibitor, KPT-8602, has been rationally designed to have increased reversibility and limited blood-brain barrier penetration with the intention to reduce toxicities. This toxicity reduction was clear in murine models using KPT-8602, where mice tolerated increased dosing of KPT-8602 without the anorexia and weight loss that was seen with selinexor dosing [127]. This design would not be appropriate for CNS malignancies [128]. Thus far, KPT-8602 therapeutic activity has been studied preclinically in pediatric leukemias, where IC50s were found to be even lower than selinexor [127,129]. Similar to selinexor, KPT-8602 leads to increased apoptosis and nuclear retention of TP53 [129], while causing minimal toxicity to normal hematopoietic precursors [127]. In murine PDX models of ALL and AML, KPT-8602 successfully reduced disease burden and extended survival, with evidence indicating that its anti-leukemic activity and elimination of leukemia-inducing cells are superior to selinexor [127,129]. Similar to selinexor, KPT-8602 has demonstrated synergy with other chemotherapy agents. Dexamethasone and KPT-8602 synergistically increase apoptosis in multiple ALL cell lines. In T-ALL and B-ALL mouse models, combination treatment resulted in increased survival compared to single agents [100]. In addition, some AML cell lines were found to be synergistically vulnerable to KPT-8602 and venetoclax, a BCL2 inhibitor that promotes apoptosis. The combination resulted in greater nuclear TP53, decreased MCL1, and increased apoptosis than with either drug alone; this combination also resulted in greater reduction of leukemic cells in an AML xenograft model [105].
3.2. Clinical Development of XPO1-Directed Therapeutics for Childhood Cancers
As has been the case for many agents in development, the early phase selinexor clinical trials technically allowed for enrollment of older children, yet accrual goals have rarely been achieved. In 2015, a clinical trial designed to assess the tolerability and efficacy of selinexor in patients with AML was open to enrolling children, adults, and older adults, but only enrolled patients 44 to 76 years old, with a median age of 61 [130]. In 2019, researchers began a clinical trial to assess the adverse event rate, MTD, and ORR of selinexor and ixazomib combination treatment in patients with sarcomas aged 14 years or older, but the trial was discontinued after 9 months due to failure to enroll any participants [131].
Two studies on the use of selinexor in pediatric patients have been completed [96,126]. The first was a breakthrough phase 1 study of 19 patients examining the use of selinexor in a combination with fludarabine and cytarabine in relapsed or refractory AML or mixed phenotype acute leukemia (MPAL) [96]. Based on preclinical research and early clinical studies in adults, researchers believed that the DLTs from selinexor were related to anorexia and weight loss, so they chose to use a chemotherapeutic regimen known to have different toxicity related to myelosuppression [96]. For the first two weeks of treatment, patients received single agent selinexor to evaluate safety and efficacy, followed by fludarabine and cytarabine on days 15–19 [96]. Following the treatment schedule prescribed, severe nausea/vomiting and fatigue were uncommon [96].
The treatment responses were encouraging. On day 15, after only receiving single agent selinexor, three (out of 15) patients experienced complete response with undetectable MRD (<0.1% based upon flow cytometric analysis) [96]. After completing one cycle of combination treatment, 7/15 patients (47%) reached complete response or complete response with incomplete count recovery, with five of these patients MRD negative [96]. Six of those seven patients were in remission at a median time of 9.5 months from enrollment [96]. One of these six patients with durable response had AML with t (6;9) translocation, which has been shown to have a high risk of relapse and poor outcomes [96,132,133]. This translocation forms a chimeric DEK-NUP214 gene, which creates an altered nucleoporin fusion protein known to interact tightly with XPO1 [134]. There have been other studies evaluating the response of known oncogenic mutations to XPO1 inhibition, but to our knowledge, this is the first pediatric mutation that may predict therapeutic response to selinexor [76,77,96,135].
Another clinical trial evaluated single agent selinexor in relapsed/refractory AML, ALL, BC-CML, and MPAL [126]. This trial found that single agent selinexor was well tolerated in children apart from two DLTs of pancreatitis and cognitive disturbance [126]. The outcomes of this trial included a ORR of 12.5%, with one patient experiencing complete remission with incomplete platelet recovery and 11/16 patients having a clinical benefit defined by reduction in transfusions, clearance of peripheral blasts, and decreased pain [126]. In addition to these two completed trials, there are five active clinical trials using selinexor open to enrolling pediatric patients, with two in the process of recruiting (Table 2, based on clinicaltrials.gov, accessed on 30 June 2021) [136]. Phase 1 study results from the first trial of selinexor in recurrent pediatric brain and solid tumors (NCT02323880) were recently presented [137]. Primary toxicities in this population were hematological and gastrointestinal, with DLTs on a weekly treatment schedule of thrombocytopenia (2 subjects) and seizure (1 CNS tumor patient). The recommended phase 2 dose was established as 35 mg/m2/dose weekly. Treatment response results are still pending from this trial.
4. Future Potential of XPO1 Inhibition in Pediatric Oncology
4.1. Precision Medicine Using XPO1 Inhibition
An ideal biomarker facilitates upfront risk stratification and accurate prediction of which patients are most likely to benefit from a specific therapy. Overexpression of XPO1 portends a poor prognosis in some diseases, which implies XPO1 activity drives or is the result of aggressive disease. This has been an attractive area of investigation in the search for a predictive biomarker of selinexor effectiveness [53,55,56,57,80,117,138]. However, XPO1 transcript expression levels have not been generally predictive of response to targeted inhibition. Specifically in neuroblastoma, XPO1 protein expression and RNA expression do not correlate with sensitivity to selinexor [30]. However, recent efforts have explored XPO1 cargo localization and function, such as those listed in Table 1, as predictors of response to XPO1 directed therapy [121,139]. For example, in gastric cancer, the presence of functional TP53 in the nuclear compartment seems to be crucial to promoting TP53-dependent apoptosis with XPO1/proteasome co-inhibition [140]. The anti-apoptotic protein Survivin could be a reasonable biomarker of sensitivity to XPO1 inhibition. In pediatric cancers, inferior outcome correlates with high levels of Survivin expression, as it functions as an inhibitor of apoptotic signaling [141,142]. Nuclear accumulation of Survivin blocks STAT3 activation, leads to its own degradation, and therefore promotes apoptosis [49,143]. Additional research is focused on therapeutic predictors by identifying molecular pathways involved in resistance to XPO1 inhibition [120,121,122,123]. Development of biomarkers predictive of selinexor response will be key to future application of selinexor in children with cancer.
4.2. New Translational Opportunities
Neuroblastoma, osteosarcoma, and high-grade gliomas (HGG) are particularly difficult to treat cancers that affect children, and integration of XPO1 inhibition into therapy has potential to transform management. In neuroblastoma, multimodal therapy is highly toxic and results in a cure rate for those with high-risk disease of approximately 50%, with substantial treatment-associated morbidity in survivors. Numerous publicly available gene expression datasets support the recently published findings from a proteomics screen in neuroblastoma that XPO1 is most highly expressed in patients with inferior outcome [30]. The numerous cargos transported by XPO1, including many affected by combination treatment and implicated in augmenting selinexor activity, reinforces current understand that no single agent or directed therapy can fully undermine the propensity of a cancer cell to survive. Given its broad sensitivity across neuroblastoma cell lines, and its safety when used in children, selinexor is a unique agent that may introduce novel oncogenic vulnerabilities, in particular once there is an improved mechanistic understanding of the role of XPO1 in cancer development and aggressiveness.
Similar to most malignant tissues, osteosarcoma shows increased XPO1 protein expression compared with normal tissues, and its expression is associated with worse prognosis, unrelated to important clinical features such as presence of metastatic disease [58]. Selinexor has shown activity in preclinical testing of osteosarcoma through different mechanisms. XPO1 inhibition blocks nuclear export of CDKN1B, an oncoprotein, leading to growth arrest in vitro [33]. Therefore a rational combination of selinexor with Wee1 inhibition was studied and showed greater inhibition of cell proliferation than either drug alone, with Wee1 inhibition of CDK2 activity and selinexor-induced stabilization of CDKN1B in the nucleus promoting G2/M arrest [33]. In osteosarcoma cell lines, XPO1 inhibition decreases hypoxia inducible factor, a key transcriptional regulator of tumor growth and therapy (including radiotherapy) resistance, emphasizing the potential role of selinexor as a radiation sensitizer [144]. In separate preclinical work in osteosarcoma, neuroblastoma, and other malignancies, selinexor was shown to act partially through inhibition of NF-kB. Resistance to selinexor through high NF-kB expression can be overcome with proteasome inhibition, again pointing to this combination as a rational therapeutic approach similar to what is used for multiple myeloma [119].
HGGs, which include multiple subtypes, such as diffuse midline gliomas, lack effective treatment options and are nearly uniformly fatal [145]. These tumors have been of high interest for XPO1 inhibition due to selinexor’s favorable blood-brain barrier penetrance and preclinical activity in patient-derived models of adult and pediatric HGG [93]. A phase 2 trial of selinexor in recurrent adult HGG showed a promising six-cycle progression-free survival rate and some objective responses [146]. As reviewed above, a phase 1 trial of selinexor in recurrent pediatric brain and solid tumors, with a special focus on HGG, has presented initial results and is ongoing. Clinical combination use with radiation and proteasome inhibition is of interest as well, particularly given promising preclinical data and the ability of both selinexor and proteasome inhibitors to target the NF-kB pathway, which may be a key activating pathway in pediatric HGG. NF-κB targeting by selinexor in pediatric HGG appears to be via inducing NGFR expression and increasing nuclear presence of IkB-a [147].
One current approach to defining response prediction is genome-scale CRISPR-Cas9a-mediated knock out screens, which was recently applied to assess genes and pathways associated with HGG response and resistance to etoposide and temozolomide. This can be readily applied as part of testing of selinexor combination strategies across all childhood cancers [148]. A similar functional screening assay led to development of a cancer dependency map in MYCN-amplified neuroblastoma that identified a novel protein, Nuclear Transport Factor 2 Like Export Factor 1 (NXT1, p15), that is essential for mRNA nuclear export and cellular integrity [149]. Targeting of NXT1, like XPO1, promotes cell lethality and reinforces the importance of additional research focused on nuclear export machinery in childhood cancer.
5. Conclusions
XPO1 overexpression is nearly universally associated with inferior outcome in cancer. Development of XPO1-directed treatment strategies has been brisk, though pediatric-focused studies have lagged. Our review of the current knowledge about XPO1 and XPO1 inhibition in childhood cancer offers an enthusiastic outlook about the potential for integrating XPO1-directed therapy as part of combination treatment to improve outcomes for children with highly aggressive malignancies. Selinexor, the lead clinical-grade XPO1 inhibitor, holds promise to improve treatment efficacy while minimizing toxic effects of conventional therapy. Improved understanding of regulated cargos, functionally related pathways, mechanistic implications, and biomarkers of efficacy will help guide optimal clinical incorporation of XPO1 inhibition.
Author Contributions
Conceptualization, B.G. and D.A.W.; writing—original draft preparation, B.G., T.B.A., D.A.M., H.V.C. and C.A.; writing—review and editing, B.G., T.B.A., A.L.G. and D.A.W.; visualization, B.G.; supervision, B.G., T.B.A., A.L.G. and D.A.W.; project administration, A.L.G. and D.A.W.; funding acquisition, D.A.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Hyundai Hope on Wheels (D.A.W.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pui, C.-H.; Gajjar, A.J.; Kane, J.R.; Qaddoumi, I.A.; Pappo, A.S. Challenging issues in pediatric oncology. Nat. Rev. Clin. Oncol. 2011, 8, 540–549. [Google Scholar] [CrossRef]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Stachowicz-Stencel, T.; Synakiewicz, A. Biomarkers for pediatric cancer detection: Latest advances and future perspectives. Biomark. Med. 2020, 14, 391–400. [Google Scholar] [CrossRef]
- Wasilewski-Masker, K.; Liu, Q.; Yasui, Y.; Leisenring, W.; Meacham, L.R.; Hammond, S.; Meadows, A.T.; Robison, L.L.; Mertens, A.C. Late recurrence in pediatric cancer: A report from the Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2009, 101, 1709–1720. [Google Scholar] [CrossRef]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975-2014, Featuring Survival. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Leahy, A.B.; Steineck, A. Patient-Reported Outcomes in Pediatric Oncology: The Patient Voice as a Gold Standard. JAMA Pediatr. 2020, 174, e202868. [Google Scholar] [CrossRef]
- Kudo, N.; Khochbin, S.; Nishi, K.; Kitano, K.; Yanagida, M.; Yoshida, M.; Horinouchi, S. Molecular cloning and cell cycle-dependent expression of mammalian CRM1, a protein involved in nuclear export of proteins. J. Biol. Chem. 1997, 272, 29742–29751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, N.; Matsumori, N.; Taoka, H.; Fujiwara, D.; Schreiner, E.P.; Wolff, B.; Yoshida, M.; Horinouchi, S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA 1999, 96, 9112–9117. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.; Braggio, E.; Kortuem, K.M.; Egan, J.B.; Zhu, Y.X.; Xin, C.S.; Tiedemann, R.E.; Palmer, S.E.; Garbitt, V.M.; McCauley, D.; et al. Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT-276. Leukemia 2013, 27, 2357–2365. [Google Scholar] [CrossRef]
- Bembenek, J.; Kang, J.; Kurischko, C.; Li, B.; Raab, J.R.; Belanger, K.D.; Luca, F.C.; Yu, H. Crm1-mediated nuclear export of Cdc14 is required for the completion of cytokinesis in budding yeast. Cell Cycle 2005, 4, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Neuber, A.; Franke, J.; Wittstruck, A.; Schlenstedt, G.; Sommer, T.; Stade, K. Nuclear export receptor Xpo1/Crm1 is physically and functionally linked to the spindle pole body in budding yeast. Mol. Cell. Biol. 2008, 28, 5348–5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Feng, Y.; Yan, M.; Tu, X.; Xie, B.; Ni, F.; Qu, C.; Chen, J.-G. Inhibition of Autism-Related Crm1 Disrupts Mitosis and Induces Apoptosis of the Cortical Neural Progenitors. Cereb. Cortex 2020, 30, 3960–3976. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.T.; Holloway, M.P.; Altura, R.A. The CRM1 nuclear export protein in normal development and disease. Int. J. Biochem. Mol. Biol. 2012, 3, 137–151. [Google Scholar] [PubMed]
- Ishizawa, J.; Kojima, K.; Hail, N.; Tabe, Y.; Andreeff, M. Expression, function, and targeting of the nuclear exporter chromosome region maintenance 1 (CRM1) protein. Pharmacol. Ther. 2015, 153, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Chook, Y.M.; Süel, K.E. Nuclear import by karyopherin-βs: Recognition and inhibition. Biochim. Biophys. Acta 2011, 1813, 1593–1606. [Google Scholar] [CrossRef] [Green Version]
- Güttler, T.; Görlich, D. Ran-dependent nuclear export mediators: A structural perspective. EMBO J. 2011, 30, 3457–3474. [Google Scholar] [CrossRef] [Green Version]
- Chook, Y.M.; Blobel, G. Karyopherins and nuclear import. Curr. Opin. Struct. Biol. 2001, 11, 703–715. [Google Scholar] [CrossRef]
- Kalab, P.; Weis, K.; Heald, R. Visualization of a Ran-GTP gradient in interphase and mitotic Xenopus egg extracts. Science 2002, 295, 2452–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlberg, J.E.; Lund, E. Functions of the GTPase Ran in RNA export from the nucleus. Curr. Opin. Cell Biol. 1998, 10, 400–408. [Google Scholar] [CrossRef]
- Hamada, M.; Haeger, A.; Jeganathan, K.B.; van Ree, J.H.; Malureanu, L.; Wälde, S.; Joseph, J.; Kehlenbach, R.H.; van Deursen, J.M. Ran-dependent docking of importin-beta to RanBP2/Nup358 filaments is essential for protein import and cell viability. J. Cell Biol. 2011, 194, 597–612. [Google Scholar] [CrossRef] [Green Version]
- Fung, H.Y.J.; Fu, S.-C.; Chook, Y.M. Nuclear export receptor CRM1 recognizes diverse conformations in nuclear export signals. eLife 2017, 6, e23961. [Google Scholar] [CrossRef]
- Roth, P.; Xylourgidis, N.; Sabri, N.; Uv, A.; Fornerod, M.; Samakovlis, C. The Drosophila nucleoporin DNup88 localizes DNup214 and CRM1 on the nuclear envelope and attenuates NES-mediated nuclear export. J. Cell Biol. 2003, 163, 701–706. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.; Dasso, M. Cellular roles of the ran gtpase. In Handbook of Cell Signaling; Elsevier: Amsterdam, The Netherlands, 2003; pp. 695–699. [Google Scholar]
- Mosammaparast, N.; Pemberton, L.F. Karyopherins: From nuclear-transport mediators to nuclear-function regulators. Trends Cell Biol. 2004, 14, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.B.; Patel, H.H.; Roepman, R.; Schick, D.; Ferreira, P.A. The zinc finger cluster domain of RanBP2 is a specific docking site for the nuclear export factor, exportin-1. J. Biol. Chem. 1999, 274, 37370–37378. [Google Scholar] [CrossRef] [Green Version]
- Kutay, U.; Güttinger, S. Leucine-rich nuclear-export signals: Born to be weak. Trends Cell Biol. 2005, 15, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Ossareh-Nazari, B.; Bachelerie, F.; Dargemont, C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science 1997, 278, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Wolff, B.; Sekimoto, T.; Schreiner, E.P.; Yoneda, Y.; Yanagida, M.; Horinouchi, S.; Yoshida, M. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 1998, 242, 540–547. [Google Scholar] [CrossRef]
- Stade, K.; Ford, C.S.; Guthrie, C.; Weis, K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 1997, 90, 1041–1050. [Google Scholar] [CrossRef] [Green Version]
- Galinski, B.; Luxemburg, M.; Landesman, Y.; Pawel, B.; Johnson, K.J.; Master, S.R.; Freeman, K.W.; Loeb, D.M.; Hébert, J.M.; Weiser, D.A. XPO1 inhibition with selinexor synergizes with proteasome inhibition in neuroblastoma by targeting nuclear export of IkB. Transl. Oncol. 2021, 14, 101114. [Google Scholar] [CrossRef]
- Stauber, R.H.; Mann, W.; Knauer, S.K. Nuclear and cytoplasmic survivin: Molecular mechanism, prognostic, and therapeutic potential. Cancer Res. 2007, 67, 5999–6002. [Google Scholar] [CrossRef] [Green Version]
- Alt, J.R.; Gladden, A.B.; Diehl, J.A. p21(Cip1) Promotes cyclin D1 nuclear accumulation via direct inhibition of nuclear export. J. Biol. Chem. 2002, 277, 8517–8523. [Google Scholar] [CrossRef] [Green Version]
- Currier, A.W.; Kolb, E.A.; Gorlick, R.G.; Roth, M.E.; Gopalakrishnan, V.; Sampson, V.B. p27/Kip1 functions as a tumor suppressor and oncoprotein in osteosarcoma. Sci. Rep. 2019, 9, 6161. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Ostermeyer, A.G.; Haladay, R.; Winkfield, B.; Frazier, M.; Zambetti, G. Cytoplasmic sequestration of wild-type p53 protein impairs the G1 checkpoint after DNA damage. Mol. Cell. Biol. 1996, 16, 1126–1137. [Google Scholar] [CrossRef] [Green Version]
- Baek, H.B.; Lombard, A.P.; Libertini, S.J.; Fernandez-Rubio, A.; Vinall, R.; Gandour-Edwards, R.; Nakagawa, R.; Vidallo, K.; Nishida, K.; Siddiqui, S.; et al. XPO1 inhibition by selinexor induces potent cytotoxicity against high grade bladder malignancies. Oncotarget 2018, 9, 34567–34581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corno, C.; Stucchi, S.; De Cesare, M.; Carenini, N.; Stamatakos, S.; Ciusani, E.; Minoli, L.; Scanziani, E.; Argueta, C.; Landesman, Y.; et al. FoxO-1 contributes to the efficacy of the combination of the XPO1 inhibitor selinexor and cisplatin in ovarian carcinoma preclinical models. Biochem. Pharmacol. 2018, 147, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bill, M.A.; Young, G.S.; La Perle, K.; Landesman, Y.; Shacham, S.; Kauffman, M.; Senapedis, W.; Kashyap, T.; Saint-Martin, J.-R.; et al. Novel small molecule XPO1/CRM1 inhibitors induce nuclear accumulation of TP53, phosphorylated MAPK and apoptosis in human melanoma cells. PLoS ONE 2014, 9, e102983. [Google Scholar] [CrossRef] [Green Version]
- Henderson, B.R. Nuclear-cytoplasmic shuttling of APC regulates beta-catenin subcellular localization and turnover. Nat. Cell Biol. 2000, 2, 653–660. [Google Scholar] [CrossRef]
- Rosin-Arbesfeld, R.; Cliffe, A.; Brabletz, T.; Bienz, M. Nuclear export of the APC tumour suppressor controls beta-catenin function in transcription. EMBO J. 2003, 22, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Hauk, G.; Bowman, G.D. Formation of a Trimeric Xpo1-Ran[GTP]-Ded1 Exportin Complex Modulates ATPase and Helicase Activities of Ded1. PLoS ONE 2015, 10, e0131690. [Google Scholar] [CrossRef]
- Arnaoutov, A.; Azuma, Y.; Ribbeck, K.; Joseph, J.; Boyarchuk, Y.; Karpova, T.; McNally, J.; Dasso, M. Crm1 is a mitotic effector of Ran-GTP in somatic cells. Nat. Cell Biol. 2005, 7, 626–632. [Google Scholar] [CrossRef]
- Yang, J.; Song, H.; Walsh, S.; Bardes, E.S.; Kornbluth, S. Combinatorial control of cyclin B1 nuclear trafficking through phosphorylation at multiple sites. J. Biol. Chem. 2001, 276, 3604–3609. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Jiang, Q.; Clarke, P.R.; Zhang, C. Phosphorylation of Crm1 by CDK1-cyclin-B promotes Ran-dependent mitotic spindle assembly. J. Cell Sci. 2013, 126, 3417–3428. [Google Scholar] [CrossRef] [Green Version]
- Labarrade, F.; Botto, J.M.; Domloge, N. CRM1 and chromosomal passenger complex component survivin are essential to normal mitosis progress and to preserve keratinocytes from mitotic abnormalities. Int. J. Cosmet. Sci. 2016, 38, 452–461. [Google Scholar] [CrossRef]
- Knauer, S.K.; Bier, C.; Habtemichael, N.; Stauber, R.H. The Survivin-Crm1 interaction is essential for chromosomal passenger complex localization and function. EMBO Rep. 2006, 7, 1259–1265. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Holloway, M.P.; Nguyen, K.; McCauley, D.; Landesman, Y.; Kauffman, M.G.; Shacham, S.; Altura, R.A. XPO1 (CRM1) inhibition represses STAT3 activation to drive a survivin-dependent oncogenic switch in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angell, H. A study into the potential role of Survivin localization in resistance to drug-induced apoptosis. Biosci. Horiz. 2008, 1, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.-P.; Dong, X.; Lin, L.; Jiang, X.; Wei, Z.; Zhai, B.; Sun, B.; Zhang, Q.; Wang, X.; Jiang, H.; et al. Up-regulation of survivin by AKT and hypoxia-inducible factor 1α contributes to cisplatin resistance in gastric cancer. FEBS J. 2014, 281, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Knauer, S.K.; Krämer, O.H.; Knösel, T.; Engels, K.; Rödel, F.; Kovács, A.F.; Dietmaier, W.; Klein-Hitpass, L.; Habtemichael, N.; Schweitzer, A.; et al. Nuclear export is essential for the tumor-promoting activity of survivin. FASEB J. 2007, 21, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Caldas, H.; Jiang, Y.; Holloway, M.P.; Fangusaro, J.; Mahotka, C.; Conway, E.M.; Altura, R.A. Survivin splice variants regulate the balance between proliferation and cell death. Oncogene 2005, 24, 1994–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamm, I.; Wang, Y.; Sausville, E.; Scudiero, D.A.; Vigna, N.; Oltersdorf, T.; Reed, J.C. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998, 58, 5315–5320. [Google Scholar]
- Saulino, D.M.; Younes, P.S.; Bailey, J.M.; Younes, M. CRM1/XPO1 expression in pancreatic adenocarcinoma correlates with survivin expression and the proliferative activity. Oncotarget 2018, 9, 21289–21295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Qiu, W.; Yao, R.; Xiang, J.; Sun, X.; Liu, S.; Lv, J.; Yue, L. CRM1 is a novel independent prognostic factor for the poor prognosis of gastric carcinomas. Med. Oncol. 2013, 30, 726. [Google Scholar] [CrossRef]
- Shen, A.; Wang, Y.; Zhao, Y.; Zou, L.; Sun, L.; Cheng, C. Expression of crm1 in human gliomas and its significance in P27 expression and clinical prognosis. Neurosurgery 2009, 65, 153–160. [Google Scholar] [CrossRef]
- Kojima, K.; Kornblau, S.M.; Ruvolo, V.; Dilip, A.; Duvvuri, S.; Davis, R.E.; Zhang, M.; Wang, Z.; Coombes, K.R.; Zhang, N.; et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood 2013, 121, 4166–4174. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Yue, L.; Qiu, W.; Wang, L.; Zhou, X.; Sun, Y. Prognostic value of CRM1in pancreas cancer. Clin. Investig. Med. 2009, 32, E315. [Google Scholar] [CrossRef] [Green Version]
- Noske, A.; Weichert, W.; Niesporek, S.; Röske, A.; Buckendahl, A.-C.; Koch, I.; Sehouli, J.; Dietel, M.; Denkert, C. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer 2008, 112, 1733–1743. [Google Scholar] [CrossRef]
- Yao, Y.; Dong, Y.; Lin, F.; Zhao, H.; Shen, Z.; Chen, P.; Sun, Y.-J.; Tang, L.-N.; Zheng, S.-E. The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol. Rep. 1994, 21, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Kostopoulou, F.; Gabillaud, C.; Chapiro, E.; Grange, B.; Tran, J.; Bouzy, S.; Degaud, M.; Ghamlouch, H.; Le Garff-Tavernier, M.; Maloum, K.; et al. French Innovative Leukemia Organization (FILO) group Gain of the short arm of chromosome 2 (2p gain) has a significant role in drug-resistant chronic lymphocytic leukemia. Cancer Med. 2019, 8, 3131–3141. [Google Scholar] [CrossRef] [Green Version]
- Cosson, A.; Chapiro, E.; Bougacha, N.; Lambert, J.; Herbi, L.; Cung, H.A.; Algrin, C.; Keren, B.; Damm, F.; Gabillaud, C.; et al. Gain in the short arm of chromosome 2 (2p+) induces gene overexpression and drug resistance in chronic lymphocytic leukemia: Analysis of the central role of XPO1. Leukemia 2017, 31, 1625–1629. [Google Scholar] [CrossRef]
- Bagheri, H.; Badduke, C.; Qiao, Y.; Colnaghi, R.; Abramowicz, I.; Alcantara, D.; Dunham, C.; Wen, J.; Wildin, R.S.; Nowaczyk, M.J.M.; et al. Identifying candidate genes for 2p15p16.1 microdeletion syndrome using clinical, genomic, and functional analysis. JCI Insight 2016, 1, e85461. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, T.; Wirtz, H.C.; Michaelis, S.; Gabbert, H.E.; Mueller, W. Chromosomal imbalances in gastric cancer. Correlation with histologic subtypes and tumor progression. Am. J. Clin. Pathol. 2001, 115, 828–834. [Google Scholar] [CrossRef]
- Olsen, R.R.; Otero, J.H.; García-López, J.; Wallace, K.; Finkelstein, D.; Rehg, J.E.; Yin, Z.; Wang, Y.D.; Freeman, K.W. MYCN induces neuroblastoma in primary neural crest cells. Oncogene 2017, 36, 5075–5082. [Google Scholar] [CrossRef] [Green Version]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, S.S.; Clarke, S.; Veschi, V.; Thiele, C.J. Targeting MYCN in pediatric and adult cancers. Front. Oncol. 2020, 10, 623679. [Google Scholar] [CrossRef]
- Schulte, J.H.; Lindner, S.; Bohrer, A.; Maurer, J.; De Preter, K.; Lefever, S.; Heukamp, L.; Schulte, S.; Molenaar, J.; Versteeg, R.; et al. MYCN and ALKF1174L are sufficient to drive neuroblastoma development from neural crest progenitor cells. Oncogene 2013, 32, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Wey, A.; Knoepfler, P.S. c-myc and N-myc promote active stem cell metabolism and cycling as architects of the developing brain. Oncotarget 2010, 1, 120–130. [Google Scholar] [CrossRef]
- Otte, J.; Dyberg, C.; Pepich, A.; Johnsen, J.I. MYCN function in neuroblastoma development. Front. Oncol. 2020, 10, 624079. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, U.H.; Senapedis, W.; Baloglu, E.; Unger, T.J.; Chari, A.; Vogl, D.; Cornell, R.F. Clinical Implications of Targeting XPO1-mediated Nuclear Export in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2018, 18, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Golomb, L.; Bublik, D.R.; Wilder, S.; Nevo, R.; Kiss, V.; Grabusic, K.; Volarevic, S.; Oren, M. Importin 7 and exportin 1 link c-Myc and p53 to regulation of ribosomal biogenesis. Mol. Cell 2012, 45, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonish-Rouach, E.; Resnitzky, D.; Lotem, J.; Sachs, L.; Kimchi, A.; Oren, M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 1991, 352, 345–347. [Google Scholar] [CrossRef]
- Kastan, M.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R. Participation of p53 Protein in the Cellular Response to DNA Damage1. Cancer Res. 1991, 51, 6304–6311. [Google Scholar]
- Pinto, E.M.; Ribeiro, R.C.; Figueiredo, B.C.; Zambetti, G.P. TP53-Associated Pediatric Malignancies. Genes Cancer 2011, 2, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Miloudi, H.; Bohers, É.; Guillonneau, F.; Taly, A.; Gibouin, V.C.; Viailly, P.-J.; Jego, G.; Grumolato, L.; Jardin, F.; Sola, B. XPO1E571K Mutation Modifies Exportin 1 Localisation and Interactome in B-cell Lymphoma. Cancers 2020, 12, 2829. [Google Scholar] [CrossRef]
- Jardin, F.; Pujals, A.; Pelletier, L.; Bohers, E.; Camus, V.; Mareschal, S.; Dubois, S.; Sola, B.; Ochmann, M.; Lemonnier, F.; et al. Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B-cell lymphoma. Am. J. Hematol. 2016, 91, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.S.; Hing, Z.A.; Harrington, B.; Baumhardt, J.; Ozer, H.G.; Lehman, A.; Giacopelli, B.; Beaver, L.; Williams, K.; Skinner, J.N.; et al. Recurrent XPO1 mutations alter pathogenesis of chronic lymphocytic leukemia. J. Hematol. Oncol. 2021, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Miloudi, H.; Taly, A.; Sola, B.; Jardin, F. XPO1 in B cell hematological malignancies: From recurrent somatic mutations to targeted therapy. J. Hematol. Oncol. 2017, 10, 47. [Google Scholar] [CrossRef] [Green Version]
- Moia, R.; Favini, C.; Sagiraju, S.; Spina, V.; Bruscaggin, A.; Rasi, S.; De Paoli, L.; Deambrogi, C.; Schipani, M.; Kodipad, A.A.; et al. XPO1 Mutations May Identify Binet A Chronic Lymphocytic Leukemia Patients with Shorter Time to First Treatment. Blood 2019, 134, 1743. [Google Scholar] [CrossRef]
- Asad, M.F.B.; Al Hallak, M.N.; Sukari, A.; Baca, Y.; Xiu, J.; Mamdani, H.; Uprety, D.; Kim, C.; Xia, B.; Liu, S.V.; et al. Prognostic impact of XPO1 mutations in metastatic non-small cell lung cancer (NSCLC). JCO 2021, 39, e20533. [Google Scholar] [CrossRef]
- Camus, V.; Stamatoullas, A.; Mareschal, S.; Viailly, P.-J.; Sarafan-Vasseur, N.; Bohers, E.; Dubois, S.; Picquenot, J.M.; Ruminy, P.; Maingonnat, C.; et al. Detection and prognostic value of recurrent exportin 1 mutations in tumor and cell-free circulating DNA of patients with classical Hodgkin lymphoma. Haematologica 2016, 101, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Moia, R. Mutations of the Exportin 1 (XPO1) Gene Predict Shorter Time to First Treatment in 1092 Early Stage Chronic Lymphocytic Leukemia Patients. A Training/Validation Study. Blood 2020, 136, 31–32. [Google Scholar] [CrossRef]
- Syed, Y.Y. Selinexor: First global approval. Drugs 2019, 79, 1485–1494. [Google Scholar] [CrossRef]
- Kalakonda, N.; Maerevoet, M.; Cavallo, F.; Follows, G.; Goy, A.; Vermaat, J.S.P.; Casasnovas, O.; Hamad, N.; Zijlstra, J.M.; Bakhshi, S.; et al. Selinexor in patients with relapsed or refractory diffuse large B-cell lymphoma (SADAL): A single-arm, multinational, multicentre, open-label, phase 2 trial. Lancet Haematol. 2020, 7, e511–e522. [Google Scholar] [CrossRef]
- Fleta-Soriano, E.; Martinez, J.P.; Hinkelmann, B.; Gerth, K.; Washausen, P.; Diez, J.; Frank, R.; Sasse, F.; Meyerhans, A. The myxobacterial metabolite ratjadone A inhibits HIV infection by blocking the Rev/CRM1-mediated nuclear export pathway. Microb. Cell Fact. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köster, M.; Lykke-Andersen, S.; Elnakady, Y.A.; Gerth, K.; Washausen, P.; Höfle, G.; Sasse, F.; Kjems, J.; Hauser, H. Ratjadones inhibit nuclear export by blocking CRM1/exportin 1. Exp. Cell Res. 2003, 286, 321–331. [Google Scholar] [CrossRef]
- Neggers, J.E.; Vanstreels, E.; Baloglu, E.; Shacham, S.; Landesman, Y.; Daelemans, D. Heterozygous mutation of cysteine528 in XPO1 is sufficient for resistance to selective inhibitors of nuclear export. Oncotarget 2016, 7, 68842–68850. [Google Scholar] [CrossRef] [PubMed]
- Wolff, B.; Sanglier, J.J.; Wang, Y. Leptomycin B is an inhibitor of nuclear export: Inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol. 1997, 4, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Newlands, E.S.; Rustin, G.J.; Brampton, M.H. Phase I trial of elactocin. Br. J. Cancer 1996, 74, 648–649. [Google Scholar] [CrossRef] [Green Version]
- Etchin, J.; Sun, Q.; Kentsis, A.; Farmer, A.; Zhang, Z.C.; Sanda, T.; Mansour, M.R.; Barcelo, C.; McCauley, D.; Kauffman, M.; et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia 2013, 27, 66–74. [Google Scholar] [CrossRef]
- Etchin, J.; Sanda, T.; Mansour, M.R.; Kentsis, A.; Montero, J.; Le, B.T.; Christie, A.L.; McCauley, D.; Rodig, S.J.; Kauffman, M.; et al. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br. J. Haematol. 2013, 161, 117–127. [Google Scholar] [CrossRef]
- Ranganathan, P.; Yu, X.; Na, C.; Santhanam, R.; Shacham, S.; Kauffman, M.; Walker, A.; Klisovic, R.; Blum, W.; Caligiuri, M.; et al. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood 2012, 120, 1765–1773. [Google Scholar] [CrossRef]
- Attiyeh, E.F.; Maris, J.M.; Lock, R.; Reynolds, C.P.; Kang, M.H.; Carol, H.; Gorlick, R.; Kolb, E.A.; Keir, S.T.; Wu, J.; et al. Pharmacodynamic and genomic markers associated with response to the XPO1/CRM1 inhibitor selinexor (KPT-330): A report from the pediatric preclinical testing program. Pediatr. Blood Cancer 2016, 63, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Green, A.L.; Ramkissoon, S.H.; McCauley, D.; Jones, K.; Perry, J.A.; Hsu, J.H.-R.; Ramkissoon, L.A.; Maire, C.L.; Hubbell-Engler, B.; Knoff, D.S.; et al. Preclinical antitumor efficacy of selective exportin 1 inhibitors in glioblastoma. Neuro Oncol. 2015, 17, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Hong, A.L.; Tseng, Y.-Y.; Cowley, G.S.; Jonas, O.; Cheah, J.H.; Kynnap, B.D.; Doshi, M.B.; Oh, C.; Meyer, S.C.; Church, A.J.; et al. Integrated genetic and pharmacologic interrogation of rare cancers. Nat. Commun. 2016, 7, 11987. [Google Scholar] [CrossRef]
- Marks, L.J.; Diolaiti, D.; Mundi, P.S.; Gaviria, E.; Rainey, A.; Dela Cruz, F.; Califano, A.; Kung, A. Preclinical effect of selinexor (KPT-330), a selective inhibitor of nuclear export, in pediatric rhabdoid tumors. JCO 2018, 36, 10552. [Google Scholar] [CrossRef]
- Alexander, T.B.; Lacayo, N.J.; Choi, J.K.; Ribeiro, R.C.; Pui, C.-H.; Rubnitz, J.E. Phase I study of selinexor, a selective inhibitor of nuclear export, in combination with fludarabine and cytarabine, in pediatric relapsed or refractory acute leukemia. J. Clin. Oncol. 2016, 34, 4094–4101. [Google Scholar] [CrossRef] [Green Version]
- Malone, E.R.; Lewin, J.H.; Al-Ezzi, E.M.; Gupta, A.A.; Pedersen, P.; Ng, M.; Wang, L.; Rodriguez, A.; Abdul Razak, A.R. Phase 1b study of selinexor, a first-in-class selective inhibitor of nuclear export (SINE) compound, in combination with doxorubicin in patients (pts) with locally advanced or metastatic soft tissue sarcoma (STS). JCO 2019, 37, 3123. [Google Scholar] [CrossRef]
- Garg, M.; Kanojia, D.; Mayakonda, A.; Ganesan, T.S.; Sadhanandhan, B.; Suresh, S.; Nagare, R.P.; Said, J.W.; Doan, N.B.; Ding, L.-W.; et al. Selinexor (KPT-330) has antitumor activity against anaplastic thyroid carcinoma in vitro and in vivo and enhances sensitivity to doxorubicin. Sci. Rep. 2017, 7, 9749. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.G.; Kaufmann, S.H. How does doxorubicin work? eLife 2012, 1, e00387. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, D.; Demeyer, S.; Prieto, C.; de Bock, C.E.; De Bie, J.; Gielen, O.; Jacobs, K.; Mentens, N.; Verhoeven, B.M.; Uyttebroeck, A.; et al. The XPO1 Inhibitor KPT-8602 Synergizes with Dexamethasone in Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2020, 26, 5747–5758. [Google Scholar] [CrossRef] [PubMed]
- Arango, N.P.; Yuca, E.; Zhao, M.; Evans, K.W.; Scott, S.; Kim, C.; Gonzalez-Angulo, A.M.; Janku, F.; Ueno, N.T.; Tripathy, D.; et al. Selinexor (KPT-330) demonstrates anti-tumor efficacy in preclinical models of triple-negative breast cancer. Breast Cancer Res. 2017, 19, 93. [Google Scholar] [CrossRef] [PubMed]
- Thein, K.Z.; Piha-Paul, S.A.; Tsimberidou, A.; Karp, D.D.; Janku, F.; Zarifa, A.; Shah, J.; Milton, D.R.; Bean, S.; McQuinn, L.; et al. Selinexor in combination with topotecan in patients with advanced or metastatic solid tumors: Results of an open-label, single-center, multi-arm phase Ib study. Investig. New Drugs 2021, 39, 1357–1365. [Google Scholar] [CrossRef]
- Ranganathan, P.; Yu, X.; Santhanam, R.; Hofstetter, J.; Walker, A.; Walsh, K.; Bhatnagar, B.; Klisovic, R.; Vasu, S.; Phelps, M.A.; et al. Decitabine priming enhances the antileukemic effects of exportin 1 (XPO1) selective inhibitor selinexor in acute myeloid leukemia. Blood 2015, 125, 2689–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatnagar, B.; Zhao, Q.; Mims, A.S.; Vasu, S.; Behbehani, G.K.; Larkin, K.; Blachly, J.S.; Blum, W.; Klisovic, R.B.; Ruppert, A.S.; et al. Selinexor in combination with decitabine in patients with acute myeloid leukemia: Results from a phase 1 study. Leuk. Lymphoma 2020, 61, 387–396. [Google Scholar] [CrossRef]
- Fischer, M.A.; Friedlander, S.Y.; Arrate, M.P.; Chang, H.; Gorska, A.E.; Fuller, L.D.; Ramsey, H.E.; Kashyap, T.; Argueta, C.; Debler, S.; et al. Venetoclax response is enhanced by selective inhibitor of nuclear export compounds in hematologic malignancies. Blood Adv. 2020, 4, 586–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.-C.; Liu, J.-W.; Yang, C.; Zhao, M.; Xiong, Z.-Q. XPO1 inhibitor KPT-330 synergizes with Bcl-xL inhibitor to induce cancer cell apoptosis by perturbing rRNA processing and Mcl-1 protein synthesis. Cell Death Dis. 2019, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.A.; Friedlander, S.; Hogdal, L.; Arrate, P.; Chang, H.; Kashyap, T.; Argueta, C.; Klebanov, B.; Senapedis, W.; Baloglu, E.; et al. Combination of Selective Inhibitor of Nuclear Export (SINE) Compounds, Selinexor and KPT-8602, with Venetoclax (ABT-199) Displays Enhanced Activity in Leukemia and Large Cell Lymphoma. Blood 2016, 128, 3949. [Google Scholar] [CrossRef]
- Luedtke, D.A.; Su, Y.; Liu, S.; Edwards, H.; Wang, Y.; Lin, H.; Taub, J.W.; Ge, Y. Inhibition of XPO1 enhances cell death induced by ABT-199 in acute myeloid leukaemia via Mcl-1. J. Cell Mol. Med. 2018, 22, 6099–6111. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.H.; Li, Y.; Khan, H.Y.; Muqbil, I.; Aboukameel, A.; Sexton, R.E.; Reddy, S.; Landesman, Y.; Kashyap, T.; Azmi, A.S.; et al. Nuclear Export Inhibitor KPT-8602 Synergizes with PARP Inhibitors in Escalating Apoptosis in Castration Resistant Cancer Cells. Int. J. Mol. Sci. 2021, 22, 6676. [Google Scholar] [CrossRef]
- Wang, J.; Sun, T.; Meng, Z.; Wang, L.; Li, M.; Chen, J.; Qin, T.; Yu, J.; Zhang, M.; Bie, Z.; et al. XPO1 inhibition synergizes with PARP1 inhibition in small cell lung cancer by targeting nuclear transport of FOXO3a. Cancer Lett. 2021, 503, 197–212. [Google Scholar] [CrossRef]
- Wang, S.; Sellner, L.; Wang, L.; Sauer, T.; Neuber, B.; Gong, W.; Stock, S.; Ni, M.; Yao, H.; Kleist, C.; et al. Combining selective inhibitors of nuclear export (SINEs) with chimeric antigen receptor (CAR) T cells for CD19-positive malignancies. Oncol. Rep. 2021, 46, 170. [Google Scholar] [CrossRef]
- Ferreiro-Neira, I.; Torres, N.E.; Liesenfeld, L.F.; Chan, C.H.F.; Penson, T.; Landesman, Y.; Senapedis, W.; Shacham, S.; Hong, T.S.; Cusack, J.C. XPO1 inhibition enhances radiation response in preclinical models of rectal cancer. Clin. Cancer Res. 2016, 22, 1663–1673. [Google Scholar] [CrossRef] [Green Version]
- Ming, M.; Wu, W.; Xie, B.; Sukhanova, M.; Wang, W.; Kadri, S.; Sharma, S.; Lee, J.; Shacham, S.; Landesman, Y.; et al. XPO1 inhibitor selinexor overcomes intrinsic ibrutinib resistance in mantle cell lymphoma via nuclear retention of iκb. Mol. Cancer Ther. 2018, 17, 2564–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, D.M.; Kashyap, T.; Dawson, J.L.; Landesman, Y.; Grant, S.; Shain, K.H.; Dai, Y.; Kauffman, M.; Shacham, S.; Turner, J.G. Combination Therapy of Selinexor with Bortezomib or Carfilzomib Overcomes Drug Resistance to Proteasome Inhibitors (PI) in Human Multiple Myeloma. Blood 2015, 126, 3048. [Google Scholar] [CrossRef]
- Muz, B.; Azab, F.; de la Puente, P.; Landesman, Y.; Azab, A.K. Selinexor Overcomes Hypoxia-Induced Drug Resistance in Multiple Myeloma. Transl. Oncol. 2017, 10, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.G.; Kashyap, T.; Dawson, J.L.; Gomez, J.; Bauer, A.A.; Grant, S.; Dai, Y.; Shain, K.H.; Meads, M.; Landesman, Y.; et al. XPO1 inhibitor combination therapy with bortezomib or carfilzomib induces nuclear localization of IκBα and overcomes acquired proteasome inhibitor resistance in human multiple myeloma. Oncotarget 2016, 7, 78896–78909. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.J.; Chang, H.; Liu, J.; Vincenzi, B.; Napolitano, A.; Bolognese, B.; Shah, J.J.; Kauffman, M.G.; Shacham, S.; Landesman, Y. Molecular predictors of response to selinexor in advanced unresectable de-differentiated liposarcoma (DDLS). JCO 2021, 39, 11509. [Google Scholar] [CrossRef]
- Garg, M.; Kanojia, D.; Mayakonda, A.; Said, J.W.; Doan, N.B.; Chien, W.; Ganesan, T.S.; Chuang, L.S.H.; Venkatachalam, N.; Baloglu, E.; et al. Molecular mechanism and therapeutic implications of selinexor (KPT-330) in liposarcoma. Oncotarget 2017, 8, 7521–7532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashyap, T.; Argueta, C.; Aboukameel, A.; Unger, T.J.; Klebanov, B.; Mohammad, R.M.; Muqbil, I.; Azmi, A.S.; Drolen, C.; Senapedis, W.; et al. Selinexor, a Selective Inhibitor of Nuclear Export (SINE) compound, acts through NF-κB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death. Oncotarget 2016, 7, 78883–78895. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, T.; Klebanov, B.; Argueta, C.; Lee, M.S.; Shacham, S.; Kauffman, M.; Landesman, Y.; Senapedis, W. Efficacy of Selinexor Is Dependent on IκB-α Expression and NF-Kb Deactivation in Multiple Myeloma Cells. Blood 2016, 128, 5660. [Google Scholar] [CrossRef]
- Kwanten, B.; Landesman, Y.; Daelemans, D. Crispr/cas9 chemogenetic profiling identifies candidate biomarker genes that modulate sensitivity to selinexor. Blood 2020, 136, 14. [Google Scholar] [CrossRef]
- Lagana, A.; Park, S.; Edwards, D.; Leshchenko, V.; Crochiere, M.; Landesman, Y.; Melnekoff, D.; Beno, I.; Perumal, D.; Madduri, D.; et al. E2F1 is a biomarker of selinexor resistance in relapsed/refractory multiple myeloma patients. Blood 2018, 132, 3216. [Google Scholar] [CrossRef]
- Burke, R.T.; Marcus, J.M.; Orth, J.D. Inhibition of exportin-1 function results in rapid cell cycle-associated DNA damage in cancer cells. Oncotarget 2017, 8, 39460–39475. [Google Scholar] [CrossRef]
- Vogl, D.T.; Dingli, D.; Cornell, R.F.; Huff, C.A.; Jagannath, S.; Bhutani, D.; Zonder, J.; Baz, R.; Nooka, A.; Richter, J.; et al. Selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J. Clin. Oncol. 2018, 36, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinexor-Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Place, A.E.; Blonquist, T.M.; Stieglitz, E.; Cooper, T.M.; Gore, L.; Aplenc, R.; Loh, M.L.; Pauly, M.; Rau, R.E.; Burke, M.J.; et al. Phase I study of the selinexor in relapsed/refractory childhood acute leukemia. Blood 2018, 132, 1405. [Google Scholar] [CrossRef]
- Etchin, J.; Berezovskaya, A.; Conway, A.S.; Galinsky, I.A.; Stone, R.M.; Baloglu, E.; Senapedis, W.; Landesman, Y.; Kauffman, M.; Shacham, S.; et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia 2017, 31, 143–150. [Google Scholar] [CrossRef]
- Hing, Z.A.; Fung, H.Y.J.; Ranganathan, P.; Mitchell, S.; El-Gamal, D.; Woyach, J.A.; Williams, K.; Goettl, V.M.; Smith, J.; Yu, X.; et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia 2016, 30, 2364–2372. [Google Scholar] [CrossRef] [Green Version]
- Vercruysse, T.; De Bie, J.; Neggers, J.E.; Jacquemyn, M.; Vanstreels, E.; Schmid-Burgk, J.L.; Hornung, V.; Baloglu, E.; Landesman, Y.; Senapedis, W.; et al. The Second-Generation Exportin-1 Inhibitor KPT-8602 Demonstrates Potent Activity against Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2017, 23, 2528–2541. [Google Scholar] [CrossRef] [Green Version]
- Selinexor with Combination Chemotherapy in Treating Patients with Acute Myeloid Leukemia. Available online: https://clinicaltrials.gov/ct2/show/NCT02573363?term=KPT330&age=0&draw=2&rank=9 (accessed on 14 July 2021).
- Selinexor in Combination with Ixazomib for the Treatment of Advanced Sarcoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03880123?term=selinexor&age=0&draw=2 (accessed on 14 July 2021).
- Sandahl, J.D.; Coenen, E.A.; Forestier, E.; Harbott, J.; Johansson, B.; Kerndrup, G.; Adachi, S.; Auvrignon, A.; Beverloo, H.B.; Cayuela, J.-M.; et al. t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: An international study of 62 patients. Haematologica 2014, 99, 865–872. [Google Scholar] [CrossRef]
- Tarlock, K.; Alonzo, T.A.; Moraleda, P.P.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Ravindranath, Y.; Lange, B.; Woods, W.G.; Gamis, A.S.; et al. Acute myeloid leukaemia (AML) with t(6;9)(p23;q34) is associated with poor outcome in childhood AML regardless of FLT3-ITD status: A report from the Children’s Oncology Group. Br. J. Haematol. 2014, 166, 254–259. [Google Scholar] [CrossRef] [Green Version]
- Port, S.A.; Monecke, T.; Dickmanns, A.; Spillner, C.; Hofele, R.; Urlaub, H.; Ficner, R.; Kehlenbach, R.H. Structural and Functional Characterization of CRM1-Nup214 Interactions Reveals Multiple FG-Binding Sites Involved in Nuclear Export. Cell Rep. 2015, 13, 690–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, P.; Kanagal-Shamanna, R.; Wierda, W.; Keating, M.; Sarwari, N.; Rozovski, U.; Thompson, P.; Burger, J.; Kantarjian, H.; Patel, K.P.; et al. Clinical and molecular characteristics of XPO1 mutations in patients with chronic lymphocytic leukemia. Am. J. Hematol. 2016, 91, E478–E479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul Razak, A.R.; Mau-Soerensen, M.; Gabrail, N.Y.; Gerecitano, J.F.; Shields, A.F.; Unger, T.J.; Saint-Martin, J.R.; Carlson, R.; Landesman, Y.; McCauley, D.; et al. First-in-Class, First-in-Human Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4142–4150. [Google Scholar] [CrossRef] [Green Version]
- Green, A.L.; Minard, C.G.; Liu, X.; Reid, J.M.; Pinkney, K.; Voss, S.; Nelson, M.D.; Fox, E.; Weigel, B.J.; Glade Bender, J. Phase 1 trial of selinexor in children and adolescents withrecurrent/refractory solid and CNS tumors (ADVL1414): A Children’s OncologyGroup Phase 1 Consortium trial. In Proceedings of the AACR Proceedings, London, UK, 9–14 April 2021. [Google Scholar]
- Deng, M.; Zhang, M.; Xu-Monette, Z.Y.; Pham, L.V.; Tzankov, A.; Visco, C.; Fang, X.; Bhagat, G.; Zhu, F.; Dybkaer, K.; et al. XPO1 expression worsens the prognosis of unfavorable DLBCL that can be effectively targeted by selinexor in the absence of mutant p53. J. Hematol. Oncol. 2020, 13, 148. [Google Scholar] [CrossRef] [PubMed]
- Muqbil, I.; Kauffman, M.; Shacham, S.; Mohammad, R.M.; Azmi, A.S. Understanding XPO1 target networks using systems biology and mathematical modeling. Curr. Pharm. Des. 2014, 20, 56–65. [Google Scholar] [CrossRef]
- Subhash, V.V.; Yeo, M.S.; Wang, L.; Tan, S.H.; Wong, F.Y.; Thuya, W.L.; Tan, W.L.; Peethala, P.C.; Soe, M.Y.; Tan, D.S.P.; et al. Anti-tumor efficacy of Selinexor (KPT-330) in gastric cancer is dependent on nuclear accumulation of p53 tumor suppressor. Sci. Rep. 2018, 8, 12248. [Google Scholar] [CrossRef] [PubMed]
- Azuhata, T.; Scott, D.; Takamizawa, S.; Wen, J.; Davidoff, A.; Fukuzawa, M.; Sandler, A. The inhibitor of apoptosis protein survivin is associated with high-risk behavior of neuroblastoma. J. Pediatr. Surg. 2001, 36, 1785–1791. [Google Scholar] [CrossRef]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A molecular biomarker in cancer. Indian J. Med. Res. 2015, 141, 389–397. [Google Scholar] [CrossRef]
- Chen, X.; Duan, N.; Zhang, C.; Zhang, W. Survivin and tumorigenesis: Molecular mechanisms and therapeutic strategies. J. Cancer 2016, 7, 314–323. [Google Scholar] [CrossRef]
- Von Fallois, M.; Kosyna, F.K.; Mandl, M.; Landesman, Y.; Dunst, J.; Depping, R. Selinexor decreases HIF-1α via inhibition of CRM1 in human osteosarcoma and hepatoma cells associated with an increased radiosensitivity. J. Cancer Res. Clin. Oncol. 2021, 147, 2025–2033. [Google Scholar] [CrossRef]
- Fangusaro, J. Pediatric high-grade gliomas and diffuse intrinsic pontine gliomas. J. Child Neurol. 2009, 24, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.B.; Wen, P.Y.; Van Den Bent, M.J.; Plotkin, S.R.; Walenkamp, A.M.E.; Huang, X.; Rodriguez-Lopez, K.; Kauffman, M.G.; Shacham, S.; Mau-Sørensen, M. Efficacy and safety of selinexor in recurrent glioblastoma. J. Clin. Oncol. 2019, 37, 2005. [Google Scholar] [CrossRef]
- DeSisto, J.A.; Flannery, P.; Lemma, R.; Pathak, A.; Mestnik, S.; Philips, N.; Bales, N.J.; Kashyap, T.; Moroze, E.; Venkataraman, S.; et al. Exportin 1 Inhibition Induces Nerve Growth Factor Receptor Expression to Inhibit the NF-κB Pathway in Preclinical Models of Pediatric High-Grade Glioma. Mol. Cancer Ther. 2020, 19, 540–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awah, C.U.; Chen, L.; Bansal, M.; Mahajan, A.; Winter, J.; Lad, M.; Warnke, L.; Gonzalez-Buendia, E.; Park, C.; Zhang, D.; et al. Ribosomal protein S11 influences glioma response to TOP2 poisons. Oncogene 2020, 39, 5068–5081. [Google Scholar] [CrossRef]
- Malone, C.F.; Dharia, N.V.; Kugener, G.; Forman, A.B.; Rothberg, M.V.; Abdusamad, M.; Gonzalez, A.; Kuljanin, M.; Robichaud, A.L.; Conway, A.S.; et al. Selective Modulation of a Pan-Essential Protein as a Therapeutic Strategy in Cancer. Cancer Discov. 2021, 11, 2282–2299. [Google Scholar] [CrossRef]
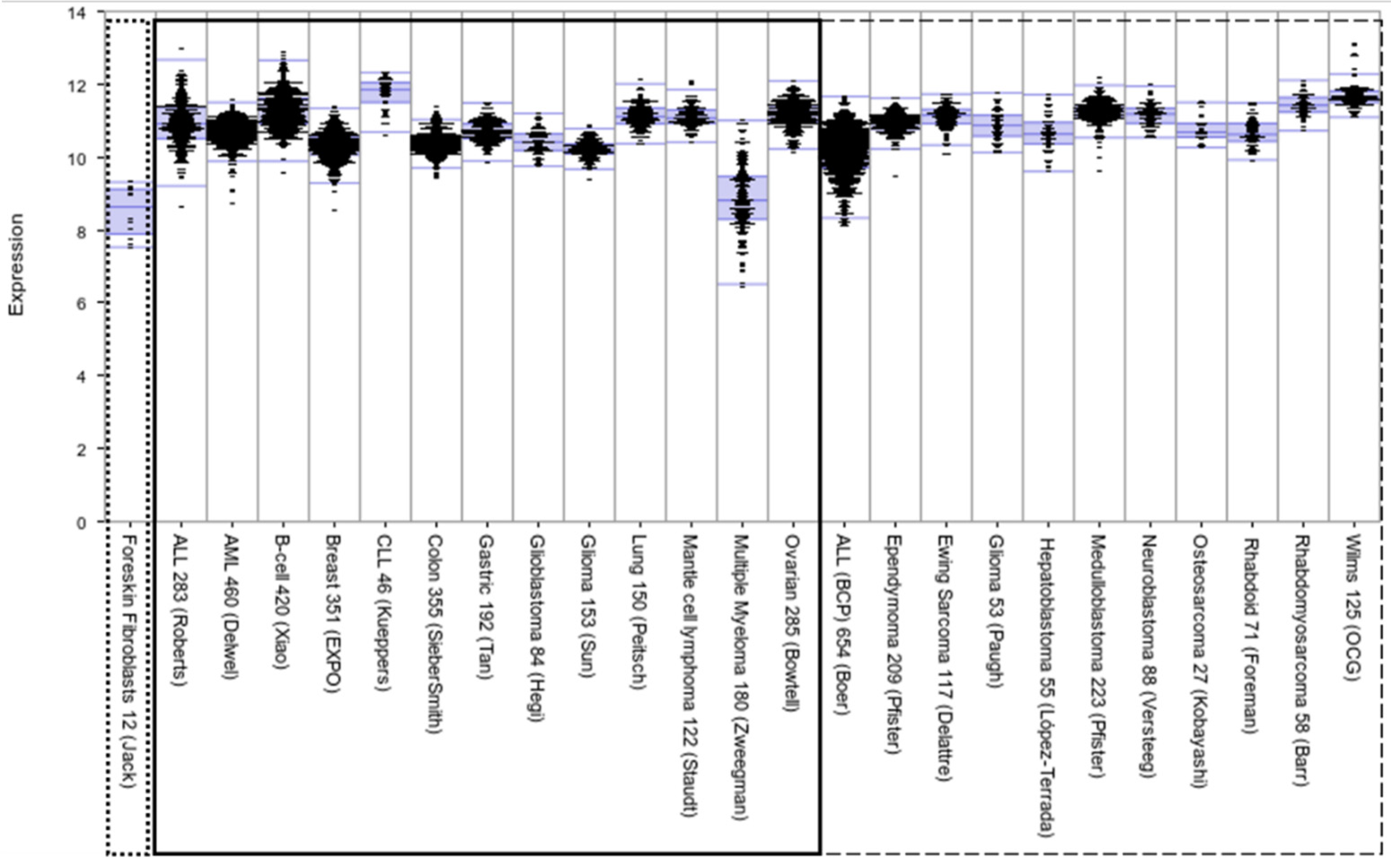
Figure 1.
Comparative expression levels of XPO1 in childhood cancers (right panel, dashed box) and select adult malignancies (left panel, solid box). XPO1 is expressed highly across malignancies, including adult and pediatric cancers, reflecting the broad clinical implications of XPO1-focused research. Data are from R2 database (http://r2.amc.nl, accessed on 30 June 2021), including foreskin fibroblast (in the dotted box) as representative of baseline expression in normal tissue.
Figure 1.
Comparative expression levels of XPO1 in childhood cancers (right panel, dashed box) and select adult malignancies (left panel, solid box). XPO1 is expressed highly across malignancies, including adult and pediatric cancers, reflecting the broad clinical implications of XPO1-focused research. Data are from R2 database (http://r2.amc.nl, accessed on 30 June 2021), including foreskin fibroblast (in the dotted box) as representative of baseline expression in normal tissue.


{kind=link}
Table 1.
Select XPO1 cargos implicated in childhood cancer pathogenesis.
| Cargo | Normal Function | Result of XPO1 Inhibition | Citation |
|---|---|---|---|
| IkB | NF-kB transcription factor signaling | Inhibition of cell survival-promoting NF-kB transcription factor activity | [30] |
| Survivin (BIRC5) | Inhibition of apoptosis | Degraded to release inhibition of apoptotic pathway | [31] |
| CDKN1A (p21) | Cell cycle kinase | Halts cell cycle progression, leading to cell cycle arrest | [32] |
| CDKN1B (p27) | Cell cycle inhibitor | Halts cell cycle progression, leading to cell cycle arrest | [33] |
| p53 | DNA damage recognition | TP53-dependent apoptosis proceeds | [34] |
| pRB | Cell cycle regulator | Halts cell cycle progression, leading to cell cycle arrest | [35] |
| FOXO1 | Transcription factor-Differentiation | Promotes sensitivity to selinexor in cisplatin-based combination | [36] |
| pMAPK | MAPK/Developmental signaling processes | Promotes apoptosis by decreasing pro-survival ERK pathways | [37] |
| APC | β-catenin signaling | Reduction of β-catenin levels by binding and leading to destruction; halts stemness-inducing Wnt signaling | [38,39] |
Table 2.
Overview of clinical trials involving selinexor in childhood cancer.
| Diseases | Treatment(s) | Status of Study | Study Phase | Age Range | # of Pts | Outcome Measure | NCT Number |
|---|---|---|---|---|---|---|---|
| Malignant Glioma, Recurrent or Refractory Solid Tumors | Selinexor | Recruiting | I | 12 mo to 21 yo | 68 | Frequency of DLT, AE Rate, Antitumor Effect of Selinexor, 6-mo PFS | NCT02323880 https://clinicaltrials.gov/ct2/show/NCT02323880?term=selinexor&type=Intr&age=0&draw=2&rank=1, accessed on 30 June 2021 |
| Relapsed or Refractory Childhood ALL, AML, Mixed Lineage Leukemia, Biphenotypic Leukemia, CML in Blast Crisis | Selinexor | Active, not recruiting | I | 12 mo to 21 yo | 16 | Toxicity Profile, MTD, ORR | NCT02091245 https://clinicaltrials.gov/ct2/show/NCT02091245?term=selinexor&age=0&draw=1&rank=2, accessed on 30 June 2021 |
| Dedifferentiated Liposarcoma | Selinexor or Placebo (Double-Blinded Study) | Active, not recruiting | II–III | 12 yo and older | 342 | PFS of patients receiving 60mg of Selinexor vs. Placebo | NCT02606461 https://clinicaltrials.gov/ct2/show/NCT02606461?term=selinexor&age=0&draw=1, accessed on 30 June 2021 |
| AML, de Novo MDS, MDS, Secondary AML, Secondary MDS | Selinexor after allogeneic stem cell transplant | Completed | I | Child, adult, older adult | 12 | MTD, DLT, 2-yr PFS, incidence of GVHD, incidence of AE, incidence of non-relapse mortality, assess lymphoid and myeloid chimerism, Overall Survival | NCT02485535 https://clinicaltrials.gov/ct2/show/NCT02485535?term=selinexor&age=0&draw=1, accessed on 30 June 2021 |
| Malignant Glioma, Glioblastoma, Diffuse Midline Glioma/ Intrinsic Pontine Glioma, Anaplastic Astrocytoma | Selinexor and Radiation Therapy | Not Yet Recruiting | I–II | 12 mo to 21 yo | 36 | MTD, ORR, Event free survival, Overall survival | NCT05099003 https://clinicaltrials.gov/ct2/show/NCT05099003?term=selinexor&age=0&draw=2&rank=4, accessed on 30 June 2021 |
| Relapsed and Refractory Aggressive B-Cell Lymphoma | Selinexor in combination with Rituximab, Gemcitabine, Dexamethasone, and Cisplatin vs. 2 other experimental treatment arms vs. active comparator arm | Recruiting | II | 16 to 65 yo | 320 | ORR, AE Rate, Transplantation rate, Stem cell collection rate, Event free survival, Survival | NCT02436707 https://clinicaltrials.gov/ct2/show/NCT02436707?term=selinexor&age=0&draw=2, accessed on 30 June 2021 |
| Relapsed or Refractory AML, Relapsed or Refractory Acute Leukemia of Ambiguous Lineage | Selinexor and Venetoclax with and without chemotherapy | Recruiting | I–II | Up to 30 yo | 42 | RP2D, Hematologic and Non-Hematologic DLT, CR, Survival | NCT04898894 https://clinicaltrials.gov/ct2/show/NCT04898894?term=KPT330&age=0&draw=2, accessed on 30 June 2021 |
| Refractory or Relapsed AML | Selinexor in combination with Fludarabine, Cytarabine, and Methotrexate/Hydrocortisone/Cytarabine | Terminated (due to slow enrollment) | I–II | Up to 24 yo | 37 | ORR, CRR, CR with Incomplete Count Recovery | NCT03071276 https://clinicaltrials.gov/ct2/show/NCT03071276?term=selinexor&age=0&draw=1, accessed on 30 June 2021 |
| AML, ALL, MDS, Mixed Phenotype Acute Leukemia | Selinexor in combination with Fludarabine, Cytarabine, and Methotrexate/Hydrocortisone/Cytarabine | Completed | I–II | Up to 24 yo | 19 | MTD, DLT, Maximum Plasma Concentration, CRR, Overall Response Rate, AUC of Selinexor | NCT02212561 https://clinicaltrials.gov/ct2/show/NCT02212561?term=selinexor&age=0&draw=1&rank=3, accessed on 30 June 2021 |
| Liposarcoma, Malignant Peripheral Nerve Sheath Tumors, Alveolar Soft Part Sarcoma, Ewing Sarcoma, Sarcoma | Selinexor and Ixazomib combination | Withdrawn | II | 14 yo and older | 0 | MTD, AE Rate, ORR | NCT03880123 https://clinicaltrials.gov/ct2/show/NCT03880123?term=selinexor&age=0&draw=1, accessed on 30 June 2021 |
mo = months old, yo = years old, RP2D = Recommended Phase 2 Dose, DLT = Dose-Limiting Toxicity, %DLT = % of patients experiencing a dose-limiting toxicity at least possibly attributable to Selinexor, PFS = Progression Free Survival, ALL = Acute, Lymphoblastic Leukemia, AML = Acute Myelogenous Leukemia, CML = Chronic Myelogenous Leukemia, MDS = Myelodysplastic Syndrome, ORR = Objective Response Rate, CRR = Complete Response Rate, MTD = Maximum Tolerated Dose, AUC = Area Under the Curve, AE = Adverse Event, GVHD = Graft vs. Host Disease.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Galinski, B.; Alexander, T.B.; Mitchell, D.A.; Chatwin, H.V.; Awah, C.; Green, A.L.; Weiser, D.A. Therapeutic Targeting of Exportin-1 in Childhood Cancer. Cancers 2021, 13, 6161. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13246161
AMA Style
Galinski B, Alexander TB, Mitchell DA, Chatwin HV, Awah C, Green AL, Weiser DA. Therapeutic Targeting of Exportin-1 in Childhood Cancer. Cancers. 2021; 13(24):6161. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13246161
Chicago/Turabian StyleGalinski, Basia, Thomas B. Alexander, Daniel A. Mitchell, Hannah V. Chatwin, Chidiebere Awah, Adam L. Green, and Daniel A. Weiser. 2021. "Therapeutic Targeting of Exportin-1 in Childhood Cancer" Cancers 13, no. 24: 6161. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13246161
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.