
Discovery of Sulforaphane as an Inducer of Ferroptosis in U-937 Leukemia Cells: Expanding Its Anticancer Potential

 , , , , , ,
, , , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Chemicals and Treatments
2.3. Analysis of Nuclear Morphology by Fluorescence Microscopy
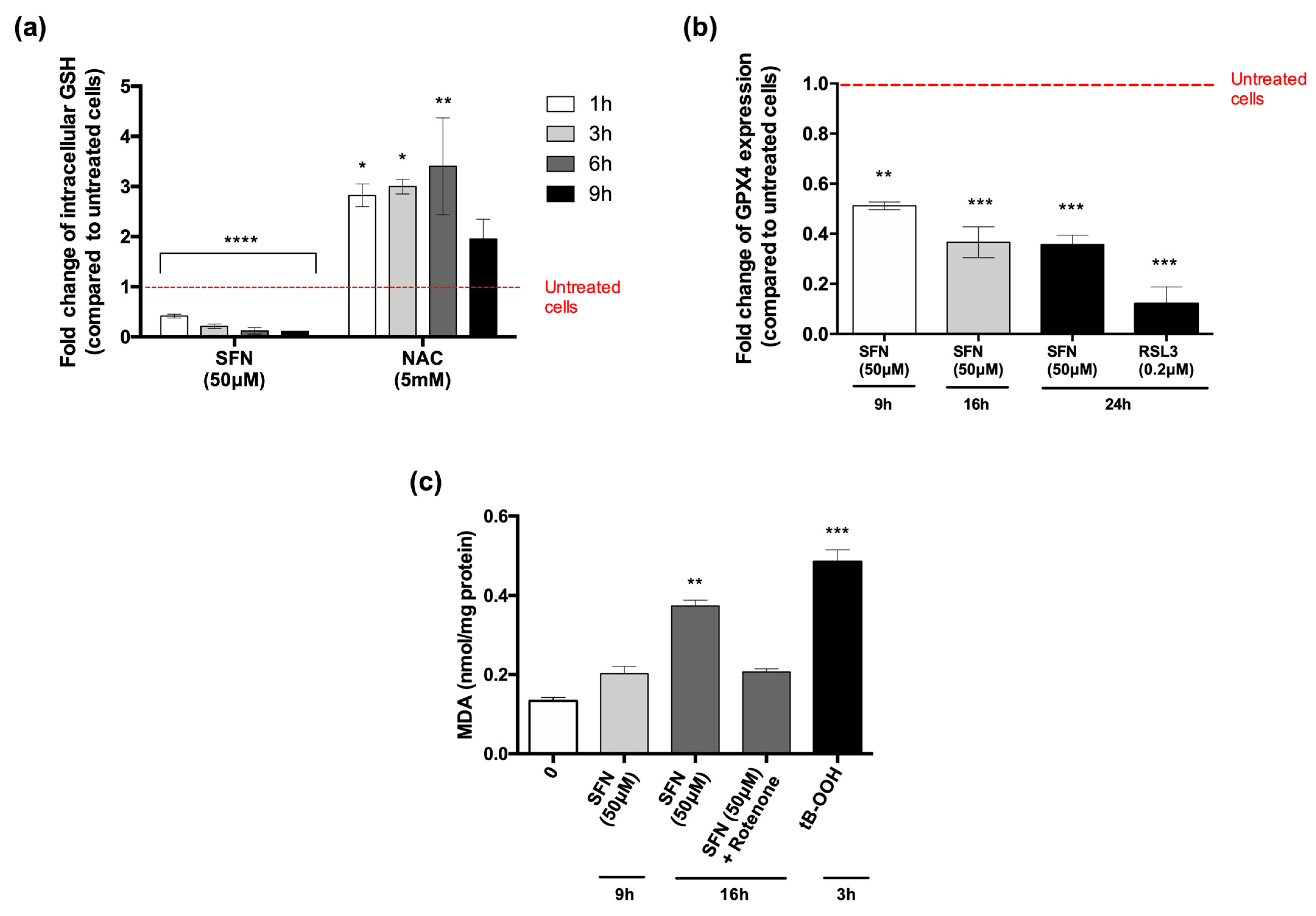
2.4. Measurement of Reduced GSH
2.5. Flow Cytometry Analysis of GPX4 Protein Expression
2.6. Malondialdehyde Assay
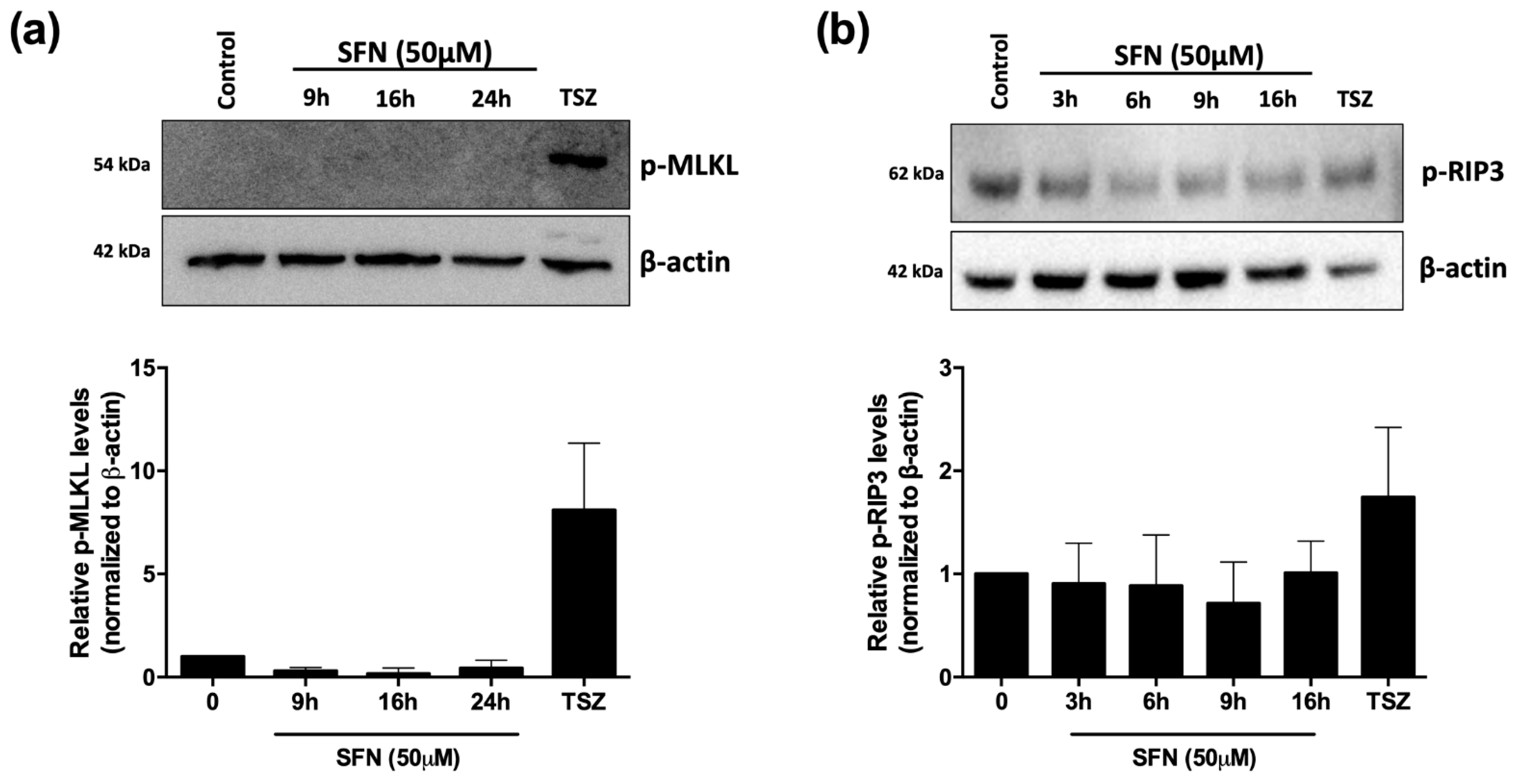
2.7. Whole Cell Extracts and Immunoblotting
2.8. Statistical Analysis
3. Results and Discussion
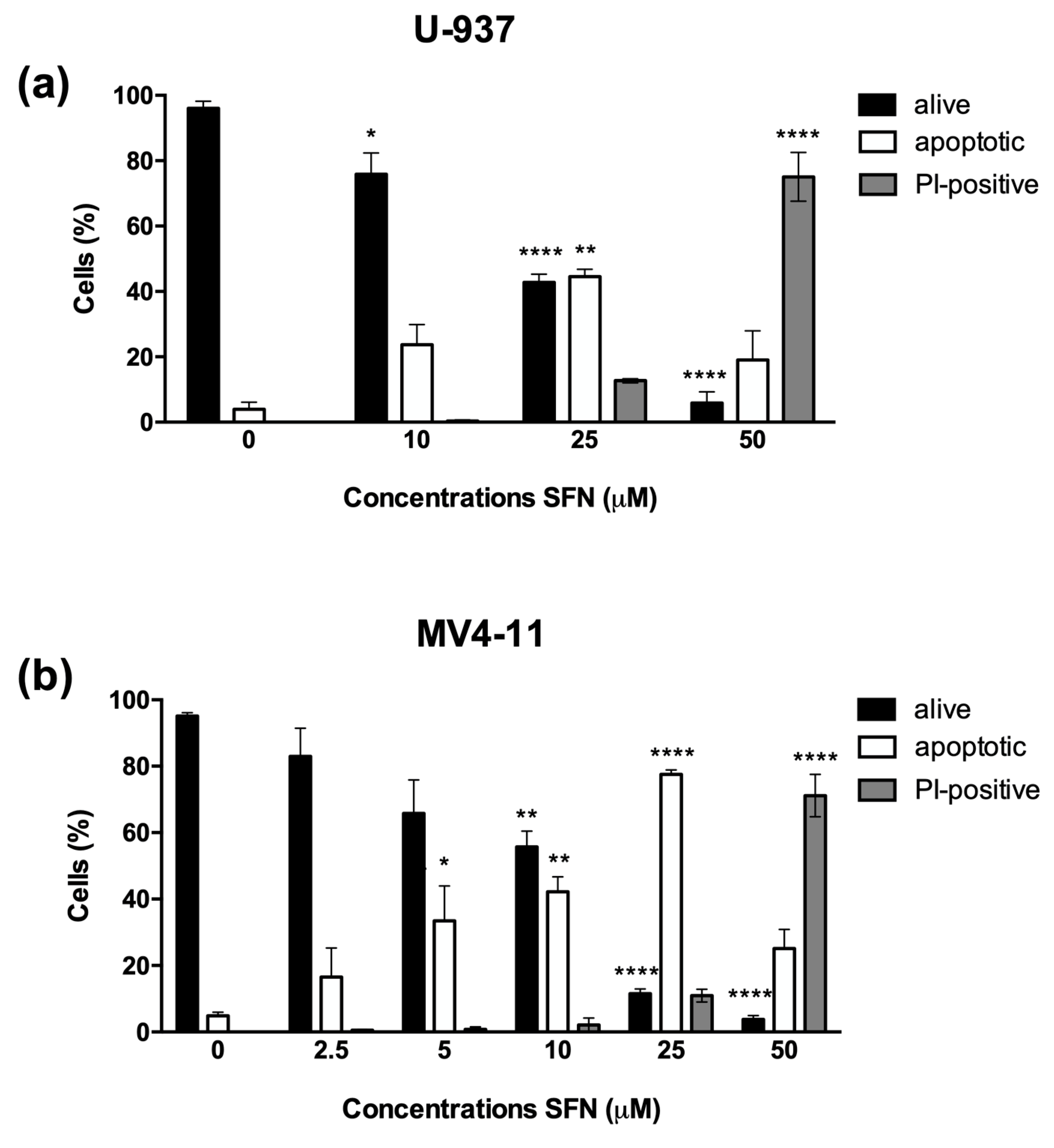
3.1. SFN Induces Apoptosis in U-937 and MV4-11 Cells
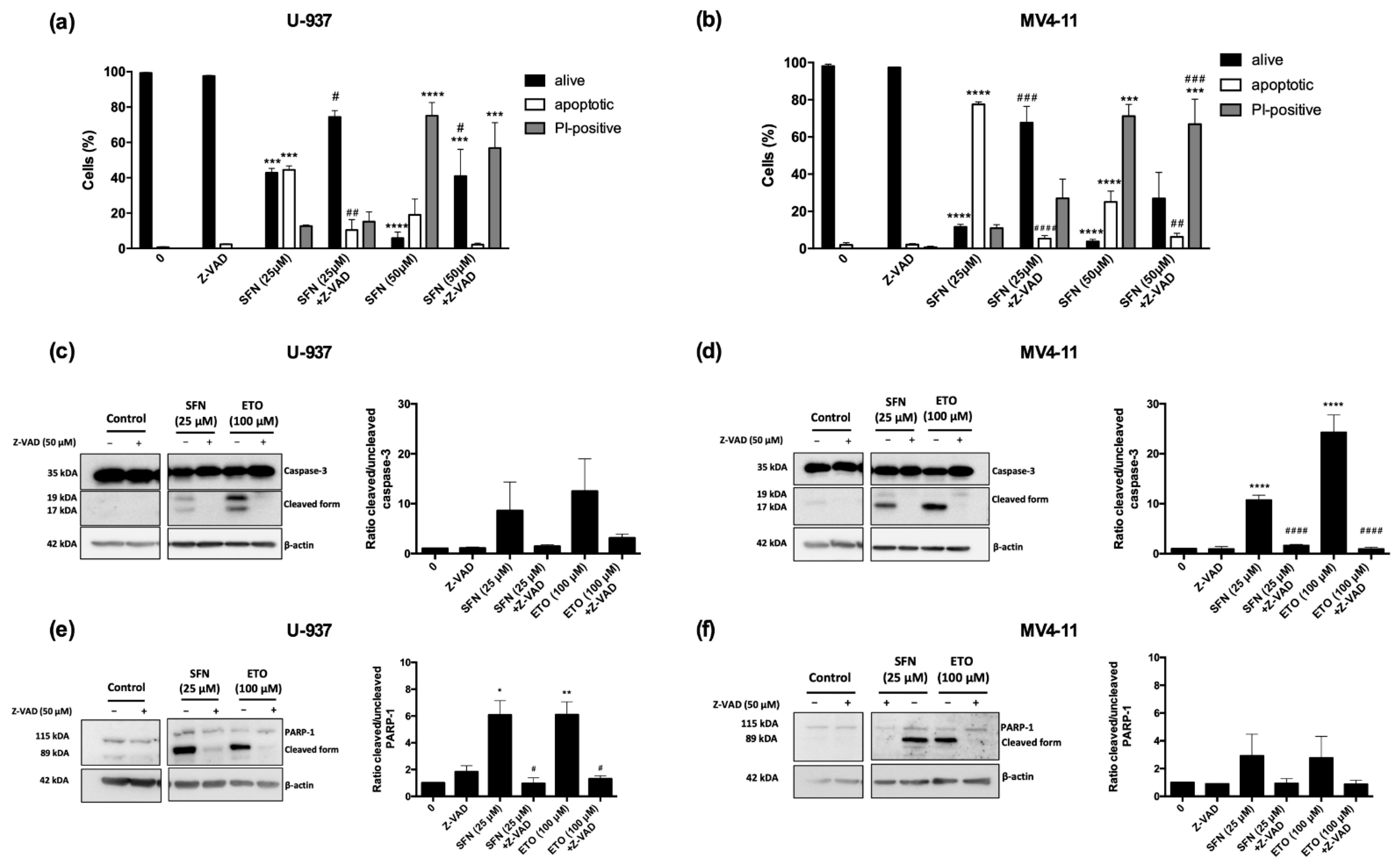
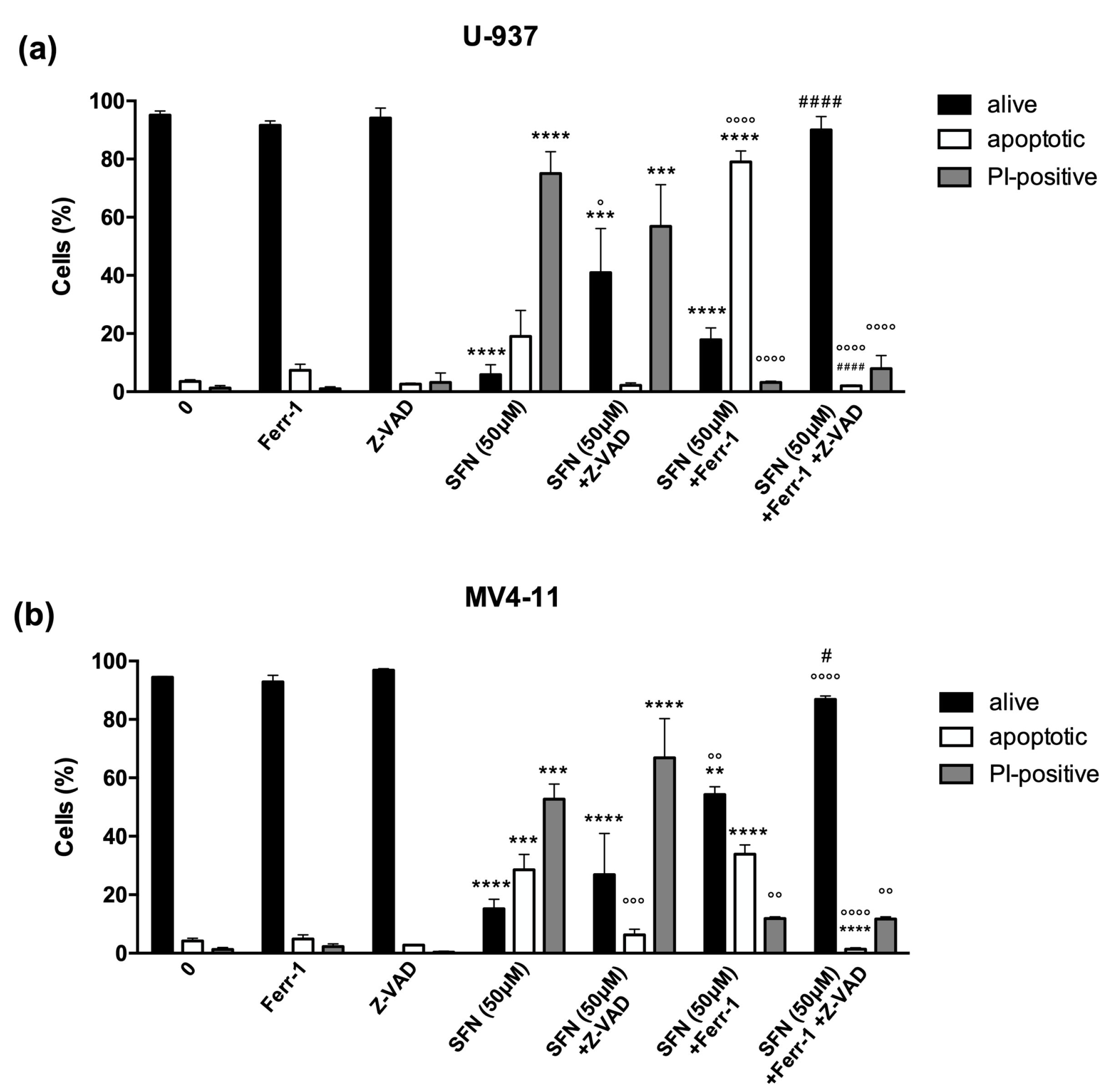
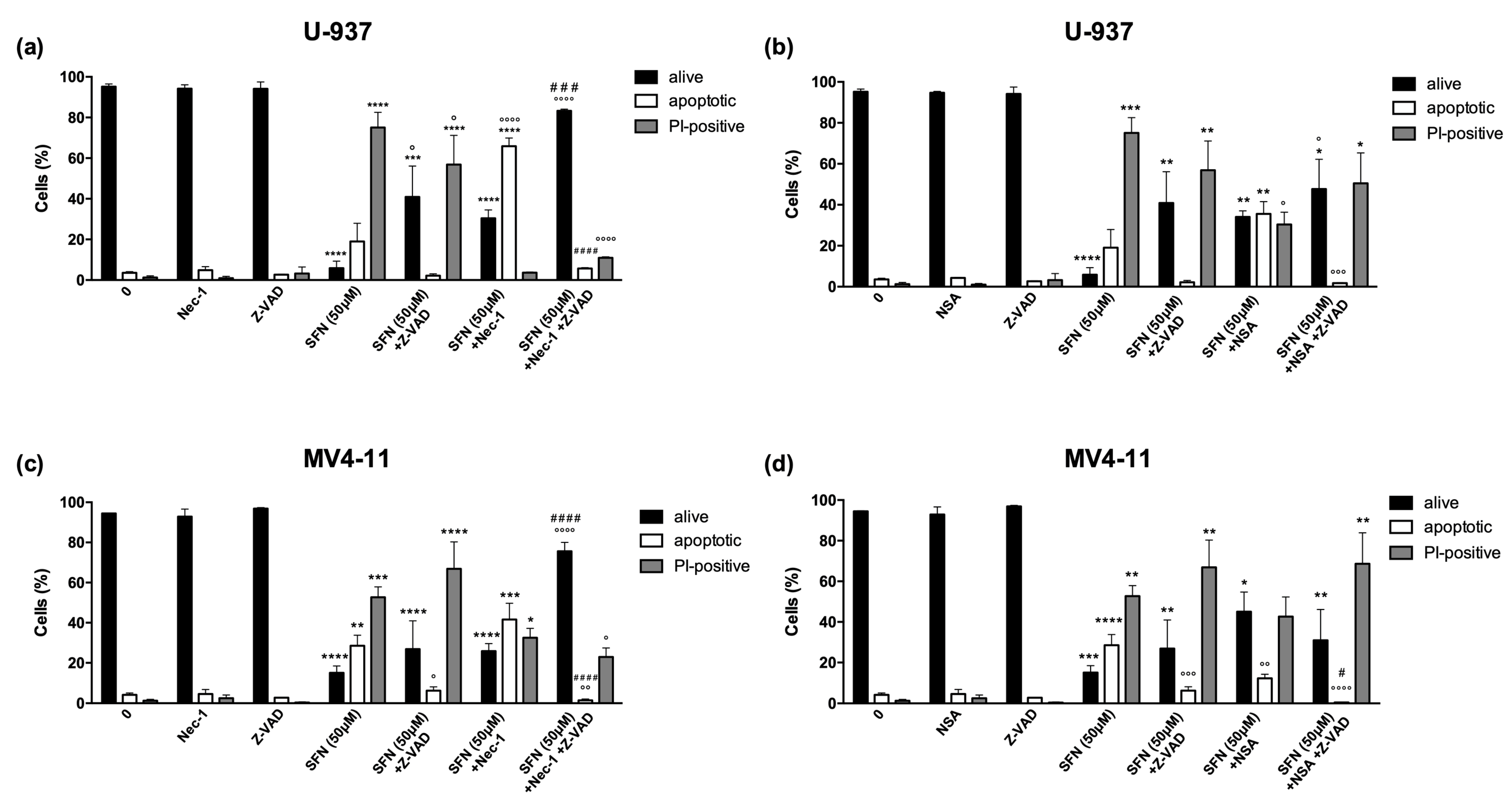
3.2. Ferrostatin-1 and Necrostatin-1 Partially Prevent Cell Death and Switch the Type of Cell Death
3.3. SFN Promotes Ferroptosis but Not Necroptosis in U-937 Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prager, G.W.; Braga, S.; Bystricky, B.; Qvortrup, C.; Criscitiello, C.; Esin, E.; Sonke, G.S.; Martínez, G.A.; Frenel, J.-S.; Karamouzis, M.; et al. Global Cancer Control: Responding to the Growing Burden, Rising Costs and Inequalities in Access. ESMO Open 2018, 3, e000285. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.-Y.; Lin, L.-T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad Targeting of Resistance to Apoptosis in Cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple Cell Death Modalities and Their Key Features (Review). World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco Targets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Chen, J.; Kos, R.; Garssen, J.; Redegeld, F. Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target. Cells 2019, 8, 1486. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The Role of Necroptosis in Cancer Biology and Therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Greco, G.; Catanzaro, E.; Fimognari, C. Natural Products as Inducers of Non-Canonical Cell Death: A Weapon against Cancer. Cancers 2021, 13, 304. [Google Scholar] [CrossRef] [PubMed]
- Palliyaguru, D.L.; Yuan, J.-M.; Kensler, T.W.; Fahey, J.W. Isothiocyanates: Translating the Power of Plants to People. Mol. Nutr. Food Res. 2018, 62, 1700965. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Kelleher, M.O.; Eggleston, I.M. The Cancer Chemopreventive Actions of Phytochemicals Derived from Glucosinolates. Eur. J. Nutr. 2008, 47, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C.; Nüsse, M.; Berti, F.; Iori, R.; Cantelli-Forti, G.; Hrelia, P. Isothiocyanates as Novel Cytotoxic and Cytostatic Agents: Molecular Pathway on Human Transformed and Non-Transformed Cells. Biochem. Pharmacol. 2004, 68, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C.; Hrelia, P. Sulforaphane as a Promising Molecule for Fighting Cancer. Mutat. Res. Rev. Mutat. Res. 2007, 635, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Plant-Derived Epigenetic Modulators for Cancer Treatment and Prevention. Biotechnol. Adv. 2014, 32, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C.; Nüsse, M.; Berti, F.; Iori, R.; Cantelli-Forti, G.; Hrelia, P. Cyclin D3 and p53 Mediate Sulforaphane-Induced Cell Cycle Delay and Apoptosis in Non-Transformed Human T Lymphocytes. Cell. Mol. Life Sci. 2002, 59, 2004–2012. [Google Scholar] [CrossRef]
- Sestili, P.; Paolillo, M.; Lenzi, M.; Colombo, E.; Vallorani, L.; Casadei, L.; Martinelli, C.; Fimognari, C. Sulforaphane Induces DNA Single Strand Breaks in Cultured Human Cells. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2010, 689, 65–73. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, Y.; Ma, L.; Ji, R.; Qu, Y.; Xin, Y.; Lv, G. Chemopreventive Activity of Sulforaphane. Drug Des. Dev. Ther. 2018, 12, 2905–2913. [Google Scholar] [CrossRef] [Green Version]
- Briones-Herrera, A.; Eugenio-Pérez, D.; Reyes-Ocampo, J.G.; Rivera-Mancía, S.; Pedraza-Chaverri, J. New Highlights on the Health-Improving Effects of Sulforaphane. Food Funct. 2018, 9, 2589–2606. [Google Scholar] [CrossRef]
- Arumugam, A.; Abdull Razis, A.F. Apoptosis as a Mechanism of the Cancer Chemopreventive Activity of Glucosinolates: A Review. Asian Pac. J. Cancer Prev. 2018, 19, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.Y.; Choi, B.T.; Lee, W.H.; Choi, Y.H. Sulforaphane Generates Reactive Oxygen Species Leading to Mitochondrial Perturbation for Apoptosis in Human Leukemia U937 Cells. Biomed. Pharmacother. 2008, 62, 637–644. [Google Scholar] [CrossRef]
- Koolivand, M.; Ansari, M.; Piroozian, F.; Moein, S.; MalekZadeh, K. Alleviating the Progression of Acute Myeloid Leukemia (AML) by Sulforaphane through Controlling MiR-155 Levels. Mol. Biol. Rep. 2018, 45, 2491–2499. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.J.T.; Singletary, K.W. Sulforaphane Inhibits Human MCF-7 Mammary Cancer Cell Mitotic Progression and Tubulin Polymerization. J. Nutr. 2004, 134, 2229–2236. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.J.T. Sulforaphane: A Naturally Occurring Mammary Carcinoma Mitotic Inhibitor, Which Disrupts Tubulin Polymerization. Carcinogenesis 2003, 25, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.J.T.; Singletary, K.W.; Venema, R.C. Sulforaphane Suppresses Angiogenesis and Disrupts Endothelial Mitotic Progression and Microtubule Polymerization. Vascul. Pharmacol. 2007, 46, 77–84. [Google Scholar] [CrossRef]
- Iida, Y.; Okamoto-Κatsuyama, M.; Maruoka, S.; Mizumura, K.; Shimizu, T.; Shikano, S.; Hikichi, M.; Takahashi, M.; Tsuya, K.; Okamoto, S.; et al. Effective Ferroptotic Small-Cell Lung Cancer Cell Death from SLC7A11 Inhibition by Sulforaphane. Oncol. Lett. 2020, 21, 71. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for Quantitative Determination of Glutathione and Glutathione Disulfide Levels Using Enzymatic Recycling Method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef]
- Betti, M.; Minelli, A.; Ambrogini, P.; Ciuffoli, S.; Viola, V.; Galli, F.; Canonico, B.; Lattanzi, D.; Colombo, E.; Sestili, P.; et al. Dietary Supplementation with α-Tocopherol Reduces Neuroinflammation and Neuronal Degeneration in the Rat Brain after Kainic Acid-Induced Status Epilepticus. Free Rad. Res. 2011, 45, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Quentmeier, H.; Reinhardt, J.; Zaborski, M.; Drexler, H.G. FLT3 Mutations in Acute Myeloid Leukemia Cell Lines. Leukemia 2003, 17, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.A.; Berger-Becvar, A.; de Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic Inhibition of STAT5 in Acute Myeloid Leukemia. Leukemia 2018, 32, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.-T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal Tandem Duplication of FLT3 (FLT3/ITD) Induces Increased ROS Production, DNA Damage, and Misrepair: Implications for Poor Prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef] [Green Version]
- Pinz, S.; Unser, S.; Rascle, A. The Natural Chemopreventive Agent Sulforaphane Inhibits STAT5 Activity. PLoS ONE 2014, 9, e99391. [Google Scholar] [CrossRef] [Green Version]
- Annageldiyev, C.; Tan, S.-F.; Thakur, S.; Dhanyamraju, P.K.; Ramisetti, S.R.; Bhadauria, P.; Schick, J.; Zeng, Z.; Sharma, V.; Dunton, W.; et al. The PI3K/AKT Pathway Inhibitor ISC-4 Induces Apoptosis and Nhibits Growth of Leukemia in Preclinical Models of Acute Myeloid Leukemia. Front. Oncol. 2020, 10, 393. [Google Scholar] [CrossRef] [Green Version]
- Sestili, P.; Fimognari, C. Cytotoxic and Antitumor Activity of Sulforaphane: The Role of Reactive Oxygen Species. BioMed Res. Int. 2015, 2015, 402386. [Google Scholar] [CrossRef] [PubMed]
- Ferreira de Oliveira, J.M.P.; Costa, M.; Pedrosa, T.; Pinto, P.; Remédios, C.; Oliveira, H.; Pimentel, F.; Almeida, L.; Santos, C. Sulforaphane Induces Oxidative Stress and Death by P53-Independent Mechanism: Implication of Impaired Glutathione Recycling. PLoS ONE 2014, 9, e92980. [Google Scholar] [CrossRef]
- Jo, G.H.; Kim, G.-Y.; Kim, W.-J.; Park, K.Y.; Choi, Y.H. Sulforaphane Induces Apoptosis in T24 Human Urinary Bladder Cancer Cells through a Reactive Oxygen Species-Mediated Mitochondrial Pathway: The Involvement of Endoplasmic Reticulum Stress and the Nrf2 Signaling Pathway. Int. J. Oncol. 2014, 45, 1497–1506. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Han, M.H.; Kim, G.-Y.; Moon, S.-K.; Kim, W.-J.; Hwang, H.J.; Park, K.Y.; Choi, Y.H. Sulforaphane Induces Reactive Oxygen Species-Mediated Mitotic Arrest and Subsequent Apoptosis in Human Bladder Cancer 5637 Cells. Food Chem. Toxicol. 2014, 64, 157–165. [Google Scholar] [CrossRef]
- Cho, S.-D.; Li, G.; Hu, H.; Jiang, C.; Kang, K.-S.; Lee, Y.-S.; Kim, S.-H.; Lu, J. Involvement of C-Jun N-Terminal Kinase in G2/M Arrest and Caspase-Mediated Apoptosis Induced by Sulforaphane in DU145 Prostate Cancer Cells. Nutr. Cancer 2005, 52, 213–224. [Google Scholar] [CrossRef]
- Pledgie-Tracy, A.; Sobolewski, M.D.; Davidson, N.E. Sulforaphane Induces Cell Type–Specific Apoptosis in Human Breast Cancer Cell Lines. Mol. Cancer Ther. 2007, 6, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Qu, Z.; Fang, Y.; Shi, X.; Ji, Y. Endoplasmic Reticulum Stress Mediates Sulforaphane-Induced Apoptosis of HepG2 Human Hepatocellular Carcinoma Cells. Mol. Med. Rep. 2017, 15, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Park, K.-S.; Baek, B.J.; Lee, K.-A.; Lee, S.-H. Apoptosis and Necroptosis-Inducing Effects of Arctigenin on Nasal Septum Carcinoma RPMI-2650 Cells in 2D and 3D Culture. Mol. Cell. Toxicol. 2020, 16, 1–11. [Google Scholar] [CrossRef]
- Deng, Q.; Yu, X.; Xiao, L.; Hu, Z.; Luo, X.; Tao, Y.; Yang, L.; Liu, X.; Chen, H.; Ding, Z.; et al. Neoalbaconol Induces Energy Depletion and Multiple Cell Death in Cancer Cells by Targeting PDK1-PI3-K/Akt Signaling Pathway. Cell Death Dis. 2013, 4, e804. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-Y.; Chang, T.-W.; Hsieh, W.-H.; Hung, M.-C.; Lin, I.-H.; Lai, S.-C.; Tzeng, Y.-J. Simultaneous Induction of Apoptosis and Necroptosis by Tanshinone IIA in Human Hepatocellular Carcinoma HepG2 Cells. Cell Death Discov. 2016, 2, 16065. [Google Scholar] [CrossRef] [Green Version]
- Longato, G.B.; Fiorito, G.F.; Vendramini-Costa, D.B.; de Oliviera Sousa, I.M.; Tinti, S.V.; Ruiz, A.L.T.G.; de Almeida, S.M.V.; Padilha, R.J.R.; Foglio, M.A.; de Carvalho, J.E. Different Cell Death Responses Induced by Eupomatenoid-5 in MCF-7 and 786-0 Tumor Cell Lines. Toxicol. In Vitro 2015, 29, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.-L.; Cui, Z.-G.; Furusawa, Y.; Ahmed, K.; Rehman, M.U.; Tabuchi, Y.; Kadowaki, M.; Kondo, T. The Molecular Mechanisms and Gene Expression Profiling for Shikonin-Induced Apoptotic and Necroptotic Cell Death in U937 Cells. Chem. Biol. Interact. 2013, 205, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.I.; Hong, J.-Y.; Choi, J.S.; Lee, S.K. Columbianadin Inhibits Cell Proliferation by Inducing Apoptosis and Necroptosis in HCT116 Colon Cancer Cells. Biomol. Ther. 2016, 24, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.-J.; Chen, T.-S.; Wang, X.-P.; Chen, R. Taxol Induces Concentration-Dependent Apoptotic and Paraptosis-like Cell Death in Human Lung Adenocarcinoma (ASTC-a-1) Cells. J. X-ray Sci. Technol. 2010, 18, 293–308. [Google Scholar] [CrossRef]
- Kaminski, B.M.; Weigert, A.; Brüne, B.; Schumacher, M.; Wenzel, U.; Steinhilber, D.; Stein, J.; Ulrich, S. Sulforaphane Potentiates Oxaliplatin-Induced Cell Growth Inhibition in Colorectal Cancer Cells via Induction of Different Modes of Cell Death. Cancer Chemother. Pharmacol. 2011, 67, 1167–1178. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Fan, J.; Ai, G.; Liu, J.; Luo, N.; Li, C.; Cheng, Z. Berberine in Combination with Cisplatin Induces Necroptosis and Apoptosis in Ovarian Cancer Cells. Biol. Res. 2019, 52, 37. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Badireenath Konkimalla, V. Hormetic Potential of Sulforaphane (SFN) in Switching Cells’ Fate Towards Survival or Death. Mini Rev. Med. Chem. 2016, 16, 980–995. [Google Scholar] [CrossRef]
- Martel, J.; Ojcius, D.M.; Ko, Y.-F.; Ke, P.-Y.; Wu, C.-Y.; Peng, H.-H.; Young, J.D. Hormetic Effects of Phytochemicals on Health and Longevity. Trends Endocrinol. Metab. 2019, 30, 335–346. [Google Scholar] [CrossRef]
- Zuo, S.; Yu, J.; Pan, H.; Lu, L. Novel Insights on Targeting Ferroptosis in Cancer Therapy. Biomark. Res. 2020, 8, 50. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Hu, F.; Feng, H.; Linkermann, A.; Min, W.; Stockwell, B.R. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. Am. Chem. Soc. Chem. Biol. 2018, 13, 1013–1020. [Google Scholar] [CrossRef]
- Zhan, C.; Huang, M.; Yang, X.; Hou, J. MLKL: Functions beyond Serving as the Executioner of Necroptosis. Theranostics 2021, 11, 4759–4769. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Efimova, I.; Catanzaro, E.; Van der Meeren, L.; Turubanova, V.D.; Hammad, H.; Mishchenko, T.A.; Vedunova, M.V.; Fimognari, C.; Bachert, C.; Coppieters, F.; et al. Vaccination with Early Ferroptotic Cancer Cells Induces Efficient Antitumor Immunity. J. Immunother. Cancer 2020, 8, e001369. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greco, G.; Schnekenburger, M.; Catanzaro, E.; Turrini, E.; Ferrini, F.; Sestili, P.; Diederich, M.; Fimognari, C. Discovery of Sulforaphane as an Inducer of Ferroptosis in U-937 Leukemia Cells: Expanding Its Anticancer Potential. Cancers 2022, 14, 76. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14010076
Greco G, Schnekenburger M, Catanzaro E, Turrini E, Ferrini F, Sestili P, Diederich M, Fimognari C. Discovery of Sulforaphane as an Inducer of Ferroptosis in U-937 Leukemia Cells: Expanding Its Anticancer Potential. Cancers. 2022; 14(1):76. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14010076
Chicago/Turabian StyleGreco, Giulia, Michael Schnekenburger, Elena Catanzaro, Eleonora Turrini, Fabio Ferrini, Piero Sestili, Marc Diederich, and Carmela Fimognari. 2022. "Discovery of Sulforaphane as an Inducer of Ferroptosis in U-937 Leukemia Cells: Expanding Its Anticancer Potential" Cancers 14, no. 1: 76. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14010076