
The Role of the Extracellular Matrix and Tumor-Infiltrating Immune Cells in the Prognostication of High-Grade Serous Ovarian Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
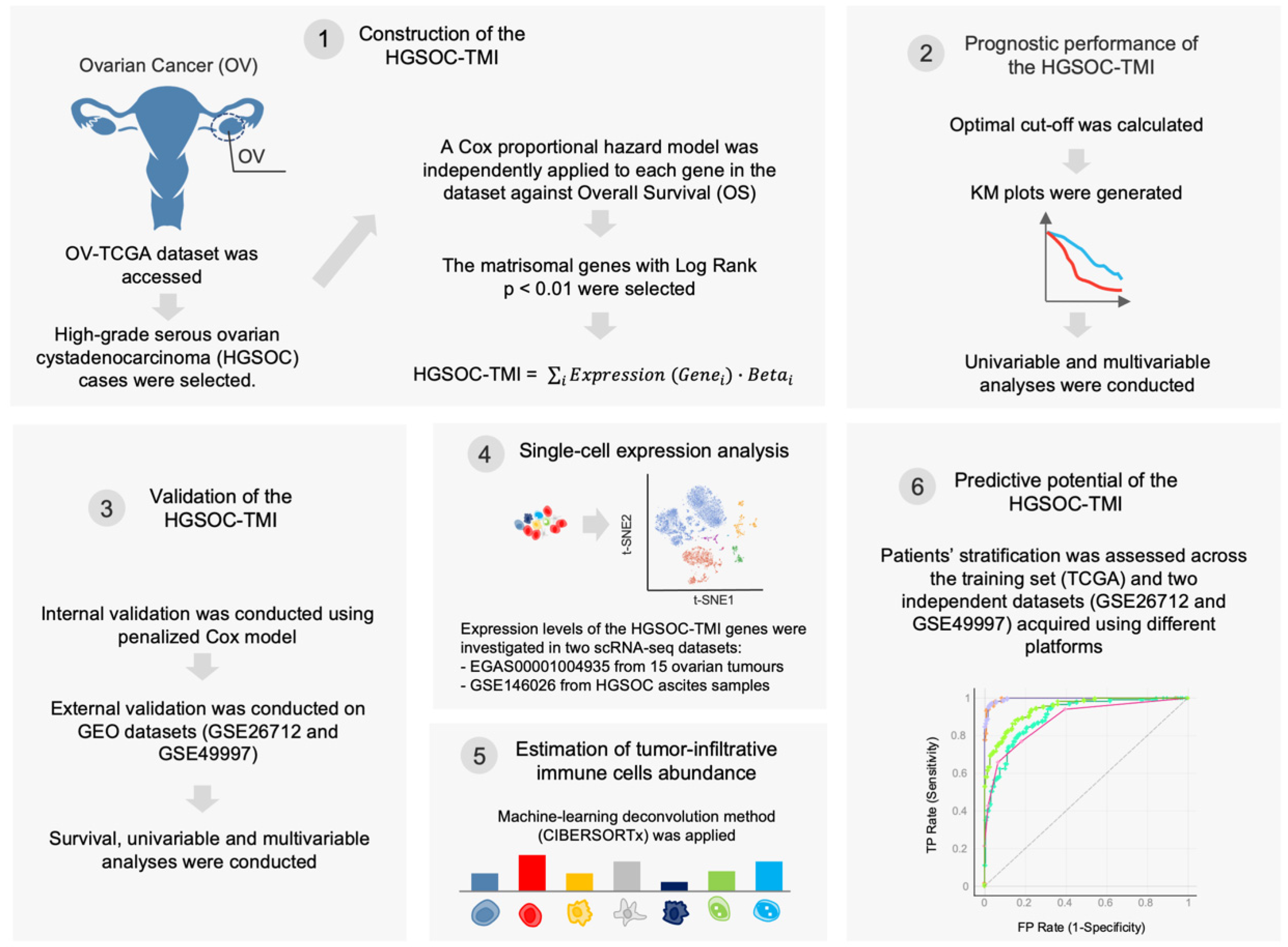
2. Materials and Methods
2.1. Data Retrieval
2.2. Construction of the HGSOC-TMI Risk Score
2.3. Patient Stratification and Survival Analysis
2.4. Internal and External Validation of the HGSOC-TMI Risk Score
2.5. Single-Cell Analysis
2.6. Abundance of Tumor-Infiltrating Immune Cell
2.7. Network Analysis
2.8. Machine-Learning Approach for Risk Group Prediction
3. Results
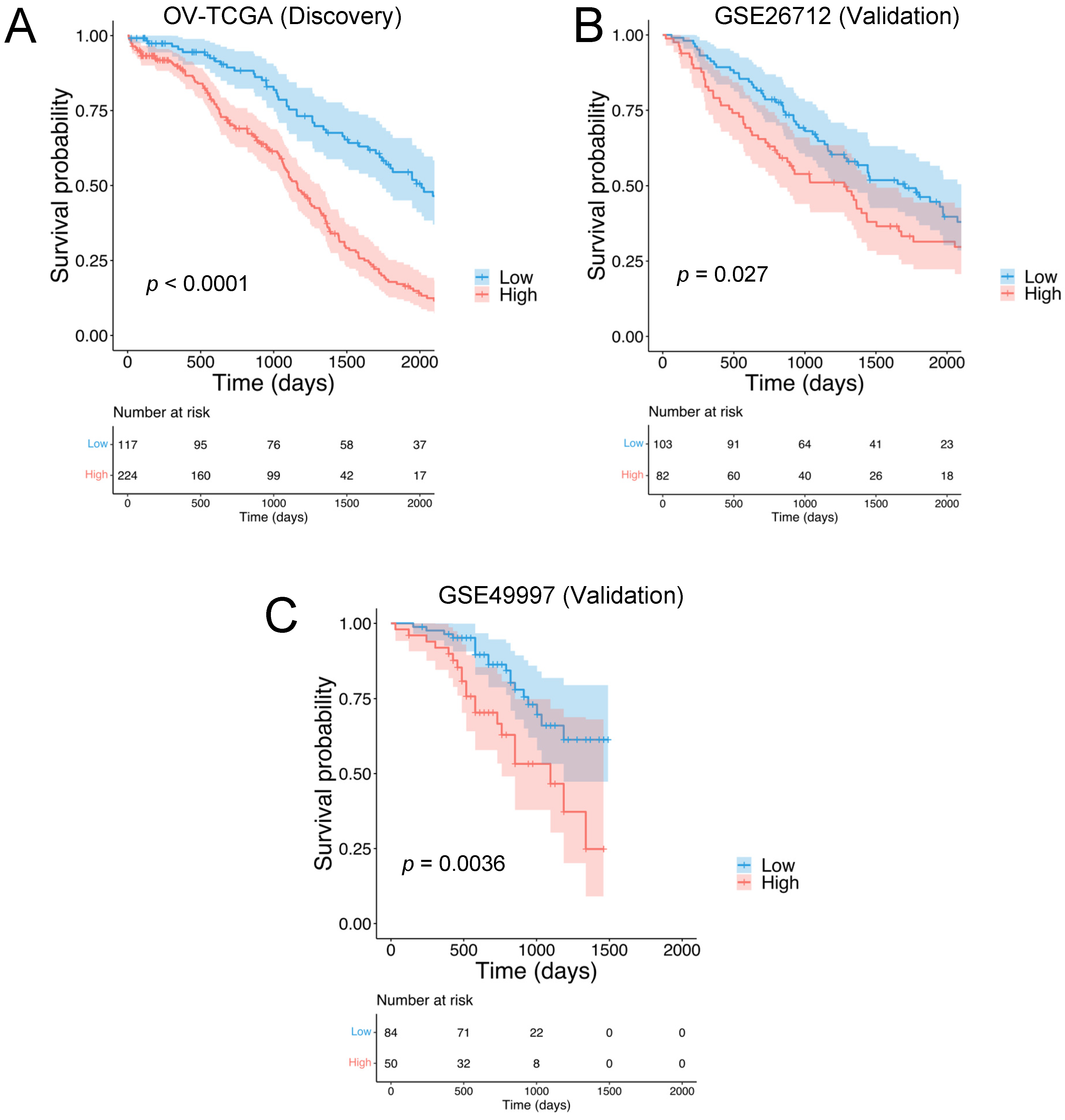
3.1. Construction and Validation of the HGSOC-TMI
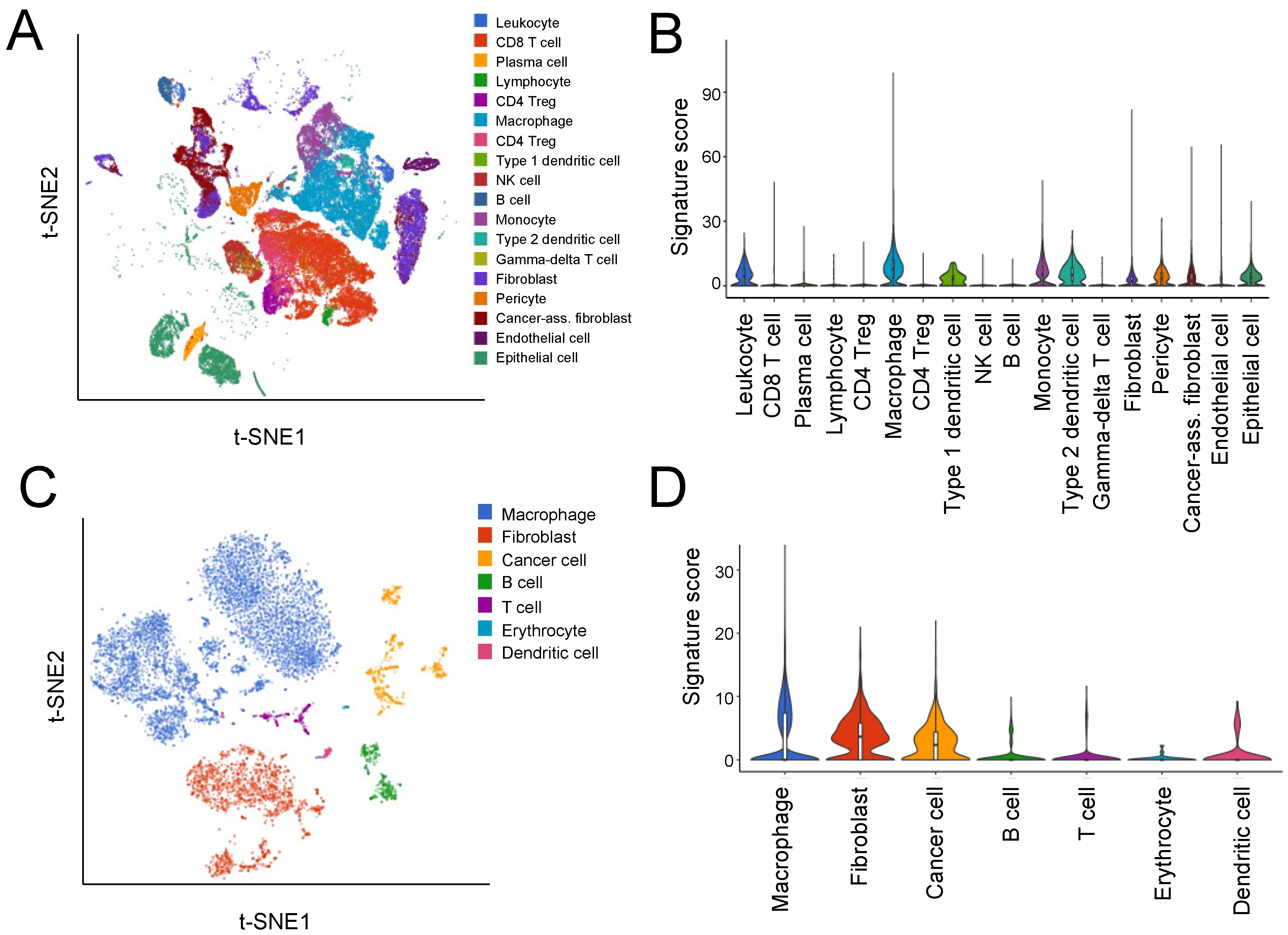
3.2. Single-Cell Analyses
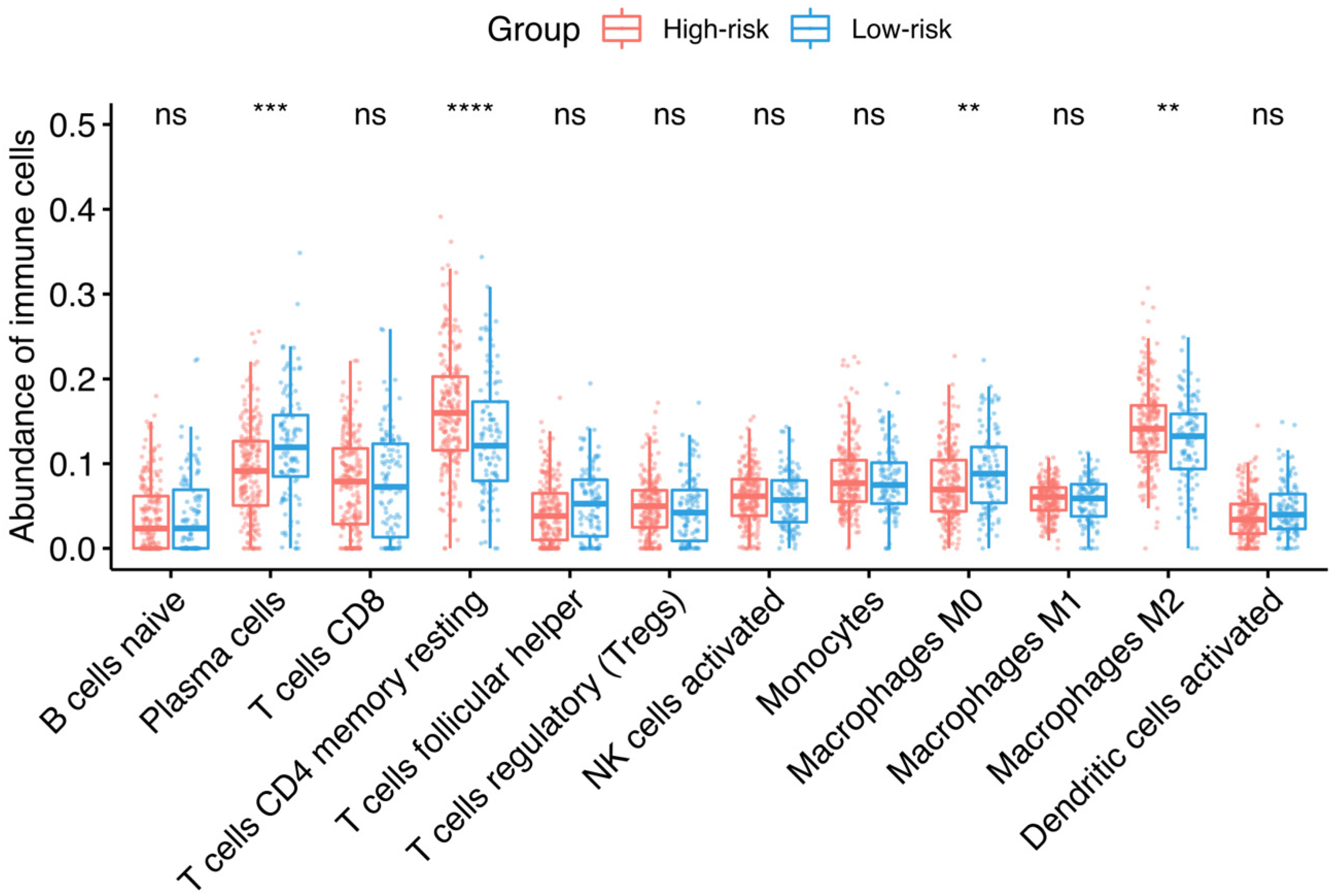
3.3. Association between Risk Groups and Tumor-Infiltrating Immune Cells
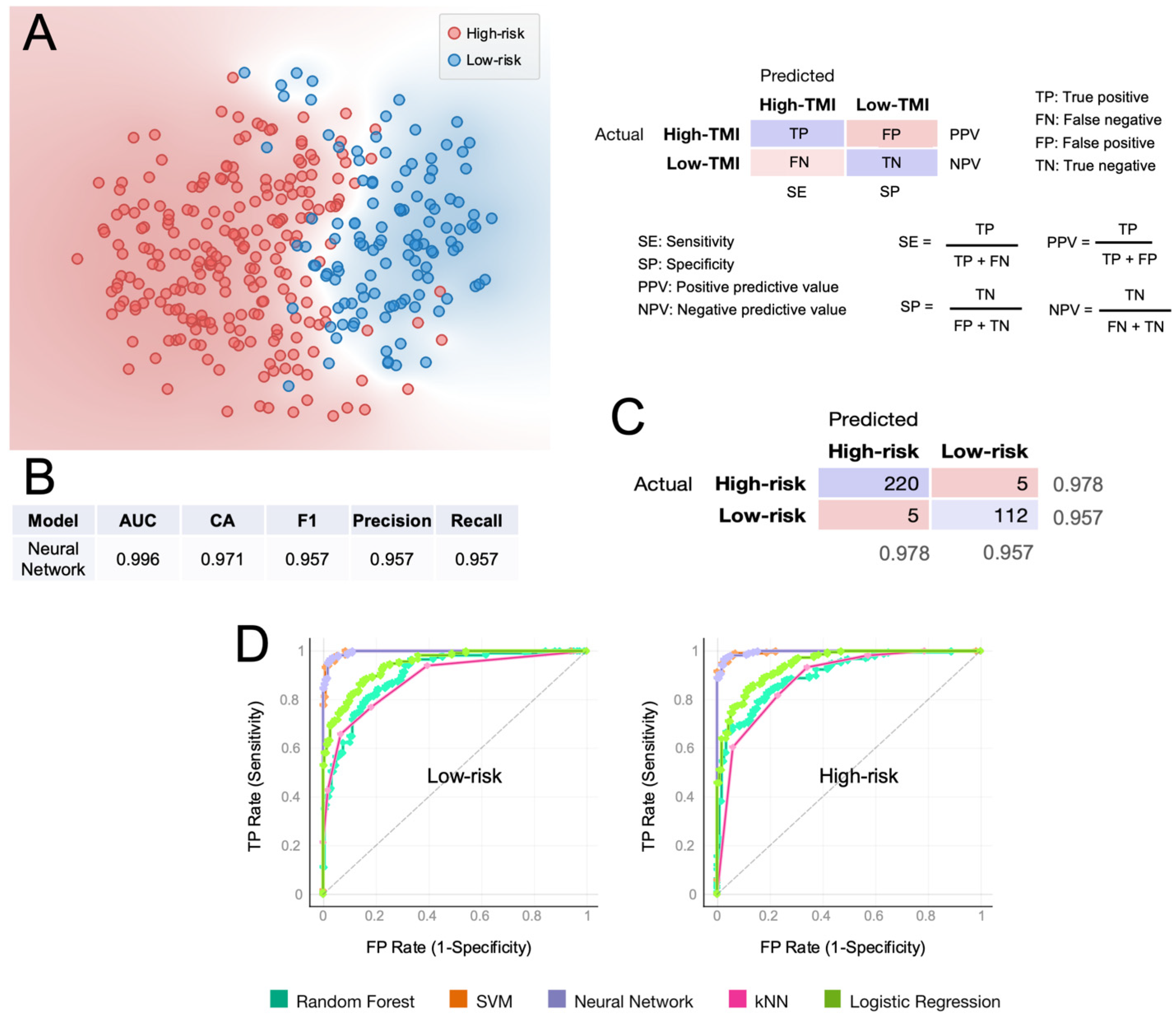
3.4. Machine-Learning Approach for Risk Group Prediction
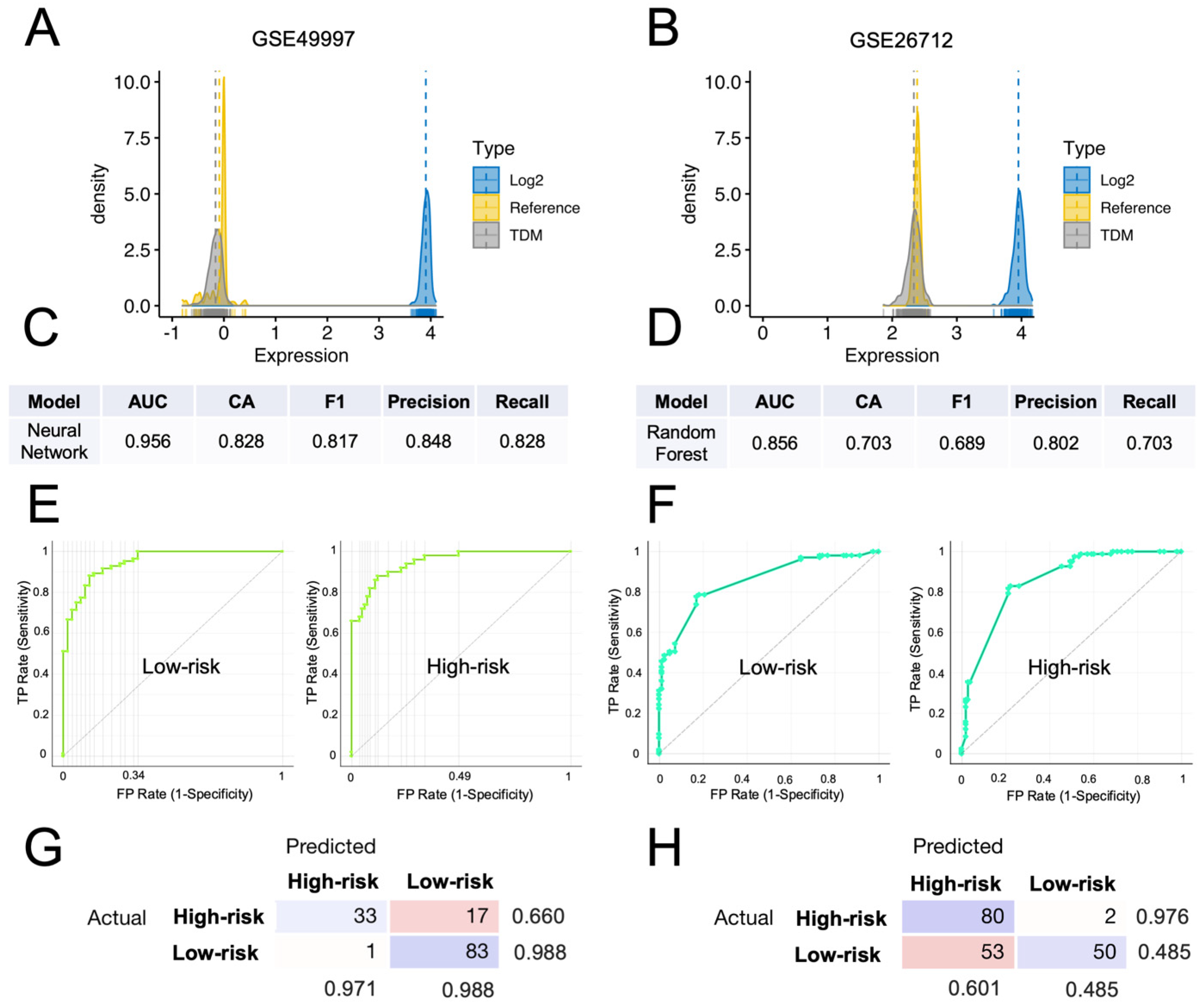
3.5. Comparison with Other Prognostic Gene Signatures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Vaughan, S.; Coward, J.I.; Bast, R.C.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Sawada, K.; Yoshimura, A.; Kinose, Y.; Nakatsuka, E.; Kimura, T. Clinical relevance of circulating cell-free microRNAs in ovarian cancer. Mol. Cancer 2016, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-grade serous ovarian cancer: Basic sciences, clinical and therapeutic standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef]
- Yuan, Z.; Chen, D.; Chen, X.; Yang, H.; Wei, Y. Overexpression of trefoil factor 3 (TFF3) contributes to the malignant progression in cervical cancer cells. Cancer Cell Int. 2017, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.K. Personalized medicine in lung cancer: What we need to know. Nat. Rev. Clin. Oncol. 2011, 8, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Lim, S.B.; Tan, S.J.; Lim, W.T.; Lim, C.T. An extracellular matrix-related prognostic and predictive indicator for early-stage non-small cell lung cancer. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Lim, S.B.; Tan, S.J.; Lim, W.T.; Lim, C.T. A merged lung cancer transcriptome dataset for clinical predictive modeling. Sci. data 2018, 5, 1–8. [Google Scholar] [CrossRef]
- Lim, S.B.; Chua, M.L.K.; Yeong, J.P.S.; Tan, S.J.; Lim, W.T.; Lim, C.T. Pan-cancer analysis connects tumor matrisome to immune response. npj Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef]
- Lim, S.B.; Tan, S.J.; Lim, W.T.; Lim, C.T. Compendiums of cancer transcriptomes for machine learning applications. Sci. Data 2019, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.B.; Yeo, T.; Di Lee, W.; Bhagat, A.A.S.; Tan, S.J.; Tan, D.S.W.; Lim, W.-T.T.; Lim, C.T. Addressing cellular heterogeneity in tumor and circulation for refined prognostication. Proc. Natl. Acad. Sci. USA 2019, 116, 17957–17962. [Google Scholar] [CrossRef]
- Webb, J.R.; Milne, K.; Watson, P.; DeLeeuw, R.J.; Nelson, B.H. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker cd103 are associated with increased survival in high-grade serous ovarian cancer. Clin. Cancer Res. 2014, 20, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Izar, B.; Tirosh, I.; Stover, E.H.; Wakiro, I.; Cuoco, M.S.; Alter, I.; Rodman, C.; Leeson, R.; Su, M.J.; Shah, P.; et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 2020, 26, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Hornburg, M.; Desbois, M.; Lu, S.; Guan, Y.; Lo, A.A.; Kaufman, S.; Elrod, A.; Lotstein, A.; DesRochers, T.M.; Munoz-Rodriguez, J.L.; et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell 2021, 39, 928–944.e6. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.T.; Knops, A.; Swendseid, B.; Martinez-Outschoom, U.; Harshyne, L.; Philp, N.; Rodeck, U.; Luginbuhl, A.; Cognetti, D.; Johnson, J.; et al. Prognostic Significance of Tumor-Associated Macrophage Content in Head and Neck Squamous Cell Carcinoma: A Meta-Analysis. Front. Oncol. 2019, 9, 656. [Google Scholar] [CrossRef]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wohrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro. Oncol. 2015, 17, 1064–1075. [Google Scholar] [CrossRef]
- Nielsen, J.S.; Sahota, R.A.; Milne, K.; Kost, S.E.; Nesslinger, N.J.; Watson, P.H.; Nelson, B.H. CD20+ tumor-infiltrating lymphocytes have an atypical CD27—Memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin. Cancer Res. 2012, 18, 3281–3292. [Google Scholar] [CrossRef]
- Kroeger, D.R.; Milne, K.; Nelson, B.H. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic T-cell responses, and superior prognosis in ovarian cancer. Clin. Cancer Res. 2016, 22, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Belotti, Y.; Lim, S.B.; Iyer, N.G.; Lim, W.; Lim, C.T. Prognostic Matrisomal Gene Panel and Its Association with Immune Cell Infiltration in Head and Neck Carcinomas. Cancers 2021, 13, 5761. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Prim. 2016, 2, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteomics 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M. Survival Analysis [R Package Survival Version 3.2-13]. Available online: https://cran.r-project.org/package=survival (accessed on 12 January 2022).
- Lausen, B.; Hothorn, T.; Bretz, F.; Schumacher, M. Assessment of Optimal Selected Prognostic Factors. Biometrical J. 2004, 46, 364–374. [Google Scholar] [CrossRef]
- Le, T.; Gill, T.; Pham, S. BBrowser: Making single-cell data easily accessible. bioRxiv 2020. [Google Scholar] [CrossRef]
- Belotti, Y.; Jokhun, D.S.; Ponnambalam, J.S.; Valerio, V.L.M.; Lim, C.T. Machine learning based approach to pH imaging and classification of single cancer cells. APL Bioeng. 2021, 5, 016105. [Google Scholar] [CrossRef]
- NCI. NHGRI The Cancer Genome Atlas Program—National Cancer Institute. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 14 April 2020).
- Thompson, J.A.; Tan, J.; Greene, C.S. Cross-platform normalization of microarray and RNA-seq data for machine learning applications. PeerJ 2016, 4, e1621. [Google Scholar] [CrossRef]
- Schulz, H.; Schmoeckel, E.; Kuhn, C.; Hofmann, S.; Mayr, D.; Mahner, S.; Jeschke, U. Galectins-1, -3, and-7 are prognostic markers for survival of ovarian cancer patients. Int. J. Mol. Sci. 2017, 18, 1230. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Sun, H.; Liu, S.; Liang, B.; Bai, H.; Wang, S.; Li, S. Identification of a Prognostic Signature for Ovarian Cancer Based on the Microenvironment Genes. Front. Genet. 2021, 12, 272. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bie, L.; Sun, L.; Yue, Y. Neural activities are unfavorable for the prognosis of ovarian cancer through mRNA expression analysis. Biomark. Med. 2019, 13, 663–674. [Google Scholar] [CrossRef]
- Reinartz, S.; Finkernagel, F.; Adhikary, T.; Rohnalter, V.; Schumann, T.; Schober, Y.; Nockher, W.A.; Nist, A.; Stiewe, T.; Jansen, J.M.; et al. A transcriptome-based global map of signaling pathways in the ovarian cancer microenvironment associated with clinical outcome. Genome Biol. 2016, 17, 108. [Google Scholar] [CrossRef]
- Davies, E.J.; Blackhall, F.H.; Shanks, J.H.; David, G.; McGown, A.T.; Swindell, R.; Slade, R.J.; Martin-Hirsch, P.; Gallagher, J.T.; Jayson, G.C. Distribution and clinical significance of heparan sulfate proteoglycans in ovarian cancer. Clin. Cancer Res. 2004, 10, 5178–5186. [Google Scholar] [CrossRef]
- TGFBI Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000120708-TGFBI (accessed on 30 December 2021).
- Kang, S.; Dong, S.M.; Park, N.H. Frequent promoter hypermethylation of TGFBI in epithelial ovarian cancer. Gynecol. Oncol. 2010, 118, 58–63. [Google Scholar] [CrossRef]
- Ween, M.P.; Oehler, M.K.; Ricciardelli, C. Transforming Growth Factor-Beta-Induced Protein (TGFBI)/(βig-H3): A Matrix Protein with Dual Functions in Ovarian Cancer. Int. J. Mol. Sci. 2012, 13, 10461–10477. [Google Scholar] [CrossRef] [PubMed]
- Lecker, L.S.M.; Berlato, C.; Maniati, E.; Delaine-Smith, R.; Pearce, O.M.T.; Heath, O.; Nichols, S.J.; Trevisan, C.; Novak, M.; McDermott, J.; et al. TGFBI Production by Macrophages Contributes to an Immunosuppressive Microenvironment in Ovarian Cancer. Cancer Res. 2021, 81, 5706–5719. [Google Scholar] [CrossRef] [PubMed]
- Steitz, A.M.; Steffes, A.; Finkernagel, F.; Unger, A.; Sommerfeld, L.; Jansen, J.M.; Wagner, U.; Graumann, J.; Müller, R.; Reinartz, S. Tumor-associated macrophages promote ovarian cancer cell migration by secreting transforming growth factor beta induced (TGFBI) and tenascin C. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Xiang, G.; Cheng, Y. MiR-126-3p inhibits ovarian cancer proliferation and invasion via targeting PLXNB2. Reprod. Biol. 2018, 18, 218–224. [Google Scholar] [CrossRef]
- Zhang, X.; Shao, S.; Li, L. Characterization of class-3 semaphorin receptors, neuropilins and plexins, as therapeutic targets in a pan-cancer study. Cancers 2020, 12, 1816. [Google Scholar] [CrossRef]
- Conrotto, P.; Corso, S.; Gamberini, S.; Comoglio, P.M.; Giordano, S. Interplay between scatter factor receptors and B plexins controls invasive growth. Oncogene 2004, 23, 5131–5137. [Google Scholar] [CrossRef] [PubMed]
- Perrot, V.; Vázquez-Prado, J.; Silvio Gutkind, J. Plexin B regulates Rho through the guanine nucleotide exchange factors leukemia-associated Rho GEF (LARG) and PDZ-RhoGEF. J. Biol. Chem. 2002, 277, 43115–43120. [Google Scholar] [CrossRef]
- Roney, K.E.; O’Connor, B.P.; Wen, H.; Holl, E.K.; Guthrie, E.H.; Davis, B.K.; Jones, S.W.; Jha, S.; Sharek, L.; Garcia-Mata, R.; et al. Plexin-B2 negatively regulates macrophage motility, Rac, and Cdc42 activation. PLoS ONE 2011, 6, e24795. [Google Scholar] [CrossRef]
- Ghosh, M.; Shen, Z.; Fahey, J.V.; Cu-Uvin, S.; Mayer, K.; Wira, C.R. Trappin-2/Elafin: A novel innate anti-human immunodeficiency virus-1 molecule of the human female reproductive tract. Immunology 2010, 129, 207–219. [Google Scholar] [CrossRef]
- Caruso, J.A.; Karakas, C.; Zhang, J.; Yi, M.; Albarracin, C.; Sahin, A.; Bondy, M.; Liu, J.; Hunt, K.K.; Keyomarsi, K. Elafin is downregulated during breast and ovarian tumorigenesis but its residual expression predicts recurrence. Breast Cancer Res. 2014, 16, 1–14. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.; Yang, B.; Wei, J.; Zhen, C.; Feng, L. Clinical significance of PI3 and HLA-DOB as potential prognostic predicators for ovarian cancer. Transl. Cancer Res. 2020, 9, 466–476. [Google Scholar] [CrossRef]
- Zheng, M.; Mullikin, H.; Hester, A.; Czogalla, B.; Heidegger, H.; Vilsmaier, T.; Vattai, A.; Chelariu-raicu, A.; Jeschke, U.; Trillsch, F.; et al. Development and validation of a novel 11-gene prognostic model for serous ovarian carcinomas based on lipid metabolism expression profile. Int. J. Mol. Sci. 2020, 21, 9169. [Google Scholar] [CrossRef] [PubMed]
- PLXNA1 Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000114554-PLXNA1 (accessed on 31 December 2021).
- Shen, J.; Liu, T.; Lv, J.; Xu, S. Identification of an Immune-Related Prognostic Gene CLEC5A Based on Immune Microenvironment and Risk Modeling of Ovarian Cancer. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- CLEC5A Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000258227-CLEC5A (accessed on 31 December 2021).
- Kim, B.J.; Fang, Y.; He, H.; Xu, B. Trypsin-Instructed Self-Assembly on Endoplasmic Reticulum for Selectively Inhibiting Cancer Cells. Adv. Healthc. Mater. 2021, 10, 2000416. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Tian, S.; Mi, W.; Zhang, Y.; Zhang, Y.; Zhang, Y.; Xu, F.; Zhang, C.; Lou, G. PRSS1 Upregulation Predicts Platinum Resistance in Ovarian Cancer Patients. Front. Cell Dev. Biol. 2021, 8. [Google Scholar] [CrossRef]
- Wang, L.; Sun, T.; Li, S.; Zhang, Z.; Jia, J.; Shan, B. Protein anabolism is key to long-term survival in high-grade serous ovarian cancer. Transl. Oncol. 2021, 14, 100885. [Google Scholar] [CrossRef] [PubMed]
- Feitosa, N.M.; Zhang, J.; Carney, T.J.; Metzger, M.; Korzh, V.; Bloch, W.; Hammerschmidt, M. Hemicentin 2 and Fibulin 1 are required for epidermal-dermal junction formation and fin mesenchymal cell migration during zebrafish development. Dev. Biol. 2012, 369, 235–248. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, X.; Li, T.; Wei, L.; Yang, A.; Lu, Y.; Zhang, J.; Li, L.; Wang, S.; Yin, F. Cross-validation of genes potentially associated with overall survival and drug resistance in ovarian cancer. Oncol. Rep. 2017, 37, 3084–3092. [Google Scholar] [CrossRef]
- NDNF Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000173376-NDNF (accessed on 31 December 2021).
- Papanastasiou, A.D.; Pampalakis, G.; Katsaros, D.; Sotiropoulou, G. Netrin-1 overexpression is predictive of ovarian malignancies. Oncotarget 2011, 2, 363–367. [Google Scholar] [CrossRef] [PubMed]
- NTN1 Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000065320-NTN1 (accessed on 31 December 2021).
- Mazelin, L.; Bernet, A.; Bonod-Bidaud, C.; Pays, L.; Arnaud, S.; Gespach, C.; Bredesen, D.E.; Scoazec, J.Y.; Mehlen, P. Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature 2004, 431, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Bruikman, C.S.; Zhang, H.; Kemper, A.M.; Van Gils, J.M.; Majerciak, V. Netrin Family: Role for Protein Isoforms in Cancer. J. Nucleic Acids 2019, 2019. [Google Scholar] [CrossRef]
- Ye, M.; Zhou, J.; Gao, Y.; Pan, S.; Zhu, X. The prognostic value of the lysyl oxidase family in ovarian cancer. J. Clin. Lab. Anal. 2020, 34, e23538. [Google Scholar] [CrossRef]
- LOXL4 Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000138131-LOXL4 (accessed on 31 December 2021).
- Guo, W.; He, X.; Ni, J.; Ma, L.; Cheng, X.; Wang, C.; Chen, X.; Wang, Y. High Serpin Family A Member 10 Expression Confers Platinum Sensitivity and Is Associated With Survival Benefit in High-Grade Serous Ovarian Cancer: Based on Quantitative Proteomic Analysis. Front. Oncol. 2021, 11, 4909. [Google Scholar] [CrossRef]
- TLL1 Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000038295-TLL1 (accessed on 31 December 2021).
- Wang, L.; Li, X. Identification of an energy metabolism-related gene signature in ovarian cancer prognosis. Oncol. Rep. 2020, 43, 1755–1770. [Google Scholar] [CrossRef]
- Liang, L.; Yu, J.; Li, J.; Li, N.; Liu, J.; Xiu, L.; Zeng, J.; Wang, T.; Wu, L. Integration of scRNA-Seq and Bulk RNA-Seq to Analyse the Heterogeneity of Ovarian Cancer Immune Cells and Establish a Molecular Risk Model. Front. Oncol. 2021, 11, 3734. [Google Scholar] [CrossRef] [PubMed]
- IL26 Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000111536-IL26 (accessed on 3 January 2022).
- NRG4 Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000169752-NRG4 (accessed on 3 January 2022).
- An, Y.; Bi, F.; You, Y.; Liu, X.; Yang, Q. Development of a novel autophagy-related prognostic signature for serous ovarian cancer. J. Cancer 2018, 9, 4058–4071. [Google Scholar] [CrossRef]
- Wang, F.; Chang, J.T.H.; Kao, C.J.; Huang, R.S. High expression of MIR-532-5p, a tumor suppressor, leads to better prognosis in ovarian cancer both in vivo and in vitro. Mol. Cancer Ther. 2016, 15, 1123–1131. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Shu, Y.; Qin, M.; Song, Y.; Tang, Q.; Huang, Y.; Shen, P.; Lu, Y. M2 polarization of tumor-associated macrophages is dependent on integrin β3 via peroxisome proliferator-activated receptor-γ up-regulation in breast cancer. Immunology 2020, 160, 345–356. [Google Scholar] [CrossRef]
- Liu, R.; Hu, R.; Zeng, Y.; Zhang, W.; Zhou, H.H. Tumour immune cell infiltration and survival after platinum-based chemotherapy in high-grade serous ovarian cancer subtypes: A gene expression-based computational study. EBioMedicine 2020, 51, 102602. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Tarique, A.A.; Logan, J.; Thomas, E.; Holt, P.G.; Sly, P.D.; Fantino, E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am. J. Respir. Cell Mol. Biol. 2015, 53, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Tekin, C.; Aberson, H.L.; Bijlsma, M.F.; Spek, C.A. Early macrophage infiltrates impair pancreatic cancer cell growth by TNF-α secretion. BMC Cancer 2020, 13, e1002194. [Google Scholar] [CrossRef]
- Ali, H.R.; Chlon, L.; Pharoah, P.D.P.; Markowetz, F.; Caldas, C. Patterns of Immune Infiltration in Breast Cancer and Their Clinical Implications: A Gene-Expression-Based Retrospective Study. PLoS Med. 2016, 13, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Colvin, E.K. Tumor-associated macrophages contribute to tumor progression in ovarian cancer. Front. Oncol. 2014, 4, 137. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Klink, M. The Role of Tumor-Associated Macrophages in the Progression and Chemoresistance of Ovarian Cancer. Cells 2020, 9, 1299. [Google Scholar] [CrossRef]
- Yousefzadeh, Y.; Hallaj, S.; Baghi Moornani, M.; Asghary, A.; Azizi, G.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Jadidi-Niaragh, F. Tumor associated macrophages in the molecular pathogenesis of ovarian cancer. Int. Immunopharmacol. 2020, 84. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Shen, J.; Yu, S.; Fei, J.; Zhu, X.; Zhao, J.; Zhai, L.; Sadhukhan, A.; Zhou, J. Tumor-associated macrophages (Tams): A critical activator in ovarian cancer metastasis. Onco. Targets. Ther. 2019, 12, 8687–8699. [Google Scholar] [CrossRef]
- Bi, F.; Chen, Y.; Yang, Q. Significance of tumor mutation burden combined with immune infiltrates in the progression and prognosis of ovarian cancer. Cancer Cell Int. 2020, 20, 1–11. [Google Scholar] [CrossRef]
- Wouters, M.C.A.; Nelson, B.H. Prognostic significance of tumor-infiltrating B cells and plasma cells in human cancer. Clin. Cancer Res. 2018, 24, 6125–6135. [Google Scholar] [CrossRef]
- Buzdin, A.; Sorokin, M.; Garazha, A.; Glusker, A.; Aleshin, A.; Poddubskaya, E.; Sekacheva, M.; Kim, E.; Gaifullin, N.; Giese, A.; et al. RNA sequencing for research and diagnostics in clinical oncology. Semin. Cancer Biol. 2020, 60, 311–323. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belotti, Y.; Lim, E.H.; Lim, C.T. The Role of the Extracellular Matrix and Tumor-Infiltrating Immune Cells in the Prognostication of High-Grade Serous Ovarian Cancer. Cancers 2022, 14, 404. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020404
Belotti Y, Lim EH, Lim CT. The Role of the Extracellular Matrix and Tumor-Infiltrating Immune Cells in the Prognostication of High-Grade Serous Ovarian Cancer. Cancers. 2022; 14(2):404. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020404
Chicago/Turabian StyleBelotti, Yuri, Elaine Hsuen Lim, and Chwee Teck Lim. 2022. "The Role of the Extracellular Matrix and Tumor-Infiltrating Immune Cells in the Prognostication of High-Grade Serous Ovarian Cancer" Cancers 14, no. 2: 404. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14020404