
Persistent Large Granular Lymphocyte Clonal Expansions: “The Root of Many Evils”—And of Some Goodness

 , , and
, , and
Abstract
:Simple Summary
Abstract
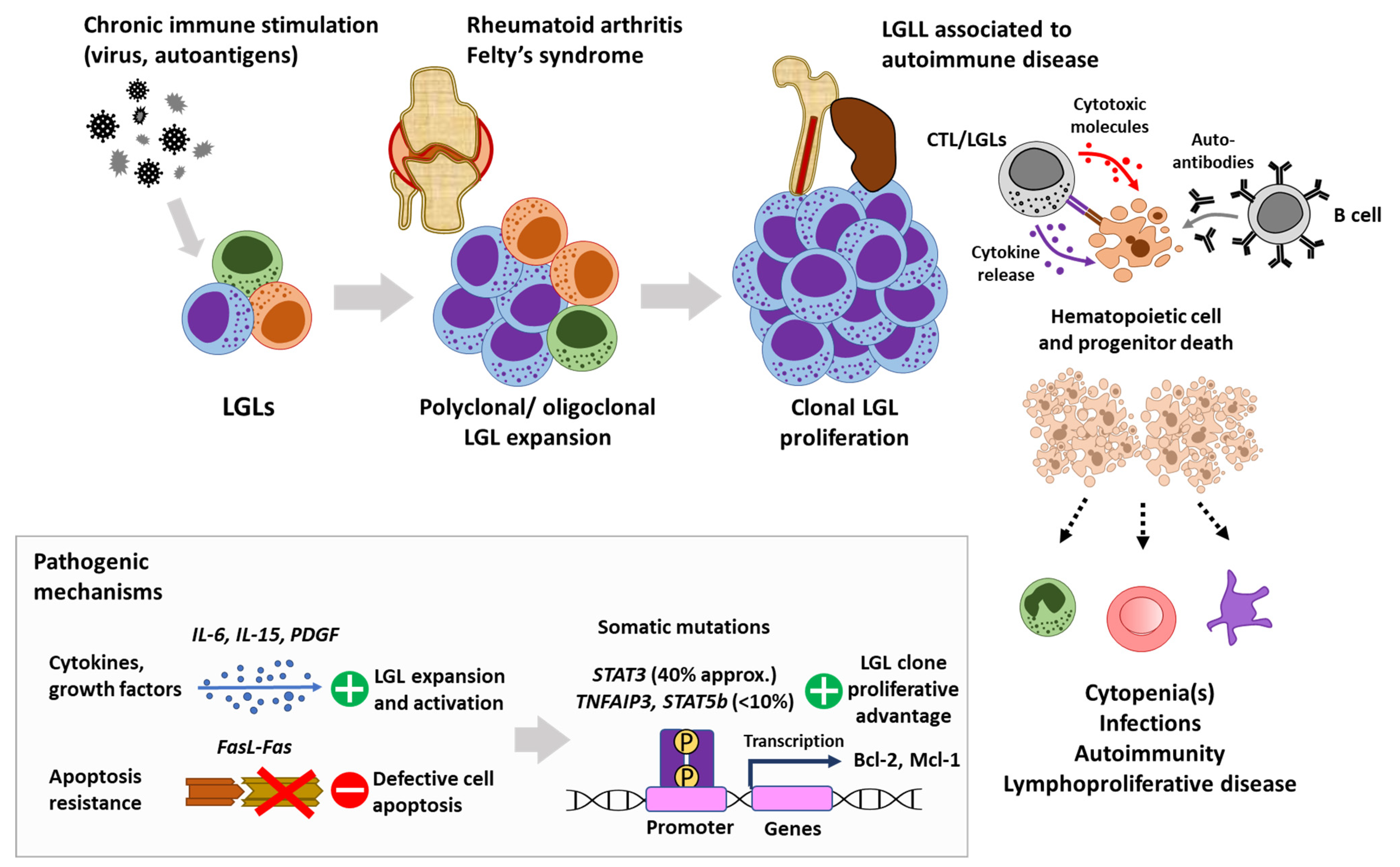
1. Introduction
2. Autoimmune Manifestations
2.1. Cytopenia(s), Underlying Immune-Mediated Mechanisms, and Prevalence of Peripheral Autoimmune Cytopenia(s)
2.2. Rheumatoid Arthritis and Felty’s Syndrome
2.3. Other Autoimmune Diseases Associated with LGLL
3. Bone Marrow Failure and Other Hematologic Neoplasms
3.1. Myelodysplastic Syndromes
Aplastic Anemia
3.2. Pure Red Cell Aplasia
3.3. Paroxysmal Nocturnal Hemoglobinuria
3.4. Other Hematological Neoplasms
4. Solid Neoplasms
5. Solid Organ Transplantation
6. Post-Allogeneic Hematopoietic Stem Cell Transplantation
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lamy, T.; Loughran, T.P.J. Clinical Features of Large Granular Lymphocyte Leukemia. Semin. Hematol. 2003, 40, 185–195. [Google Scholar] [CrossRef]
- Mustjoki, S.; Young, N.S. Somatic Mutations in “Benign” Disease. N. Engl. J. Med. 2021, 384, 2039–2052. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Myllymäki, M.; Kankainen, M.; Jarvinen, T.; Park, G.; Bruhn, R.; Murphy, E.L.; Mustjoki, S. Somatic STAT3 Mutations in CD8+ T Cells of Healthy Blood Donors Carrying Human T-Cell Leukemia Virus Type 2. Haematologica 2022, 107, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Loughran, T.P.; Kadin, M.E.; Starkebaum, G.; Abkowitz, J.L.; Clark, E.A.; Disteche, C.; Lum, L.G.; Slichter, S.J. Leukemia of Large Granular Lymphocytes: Association with Clonal Chromosomal Abnormalities and Autoimmune Neutropenia, Thrombocytopenia, and Hemolytic Anemia. Ann. Intern. Med. 1985, 102, 169–175. [Google Scholar] [CrossRef]
- Wallis, W.J.; Loughran, T.P.J.; Kadin, M.E.; Clark, E.A.; Starkebaum, G.A. Polyarthritis and Neutropenia Associated with Circulating Large Granular Lymphocytes. Ann. Intern. Med. 1985, 103, 357–362. [Google Scholar] [CrossRef]
- Semenzato, G.; Zambello, R.; Starkebaum, G.; Oshimi, K.; Loughran, T.P., Jr. The Lymphoproliferative Disease of Granular Lymphocytes: Updated Criteria for Diagnosis. Blood 1997, 89, 256–260. [Google Scholar] [CrossRef]
- Go, R.S.; Li, C.Y.; Tefferi, A.; Phyliky, R.L. Acquired Pure Red Cell Aplasia Associated with Lymphoproliferative Disease of Granular T Lymphocytes. Blood 2001, 98, 483–485. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Loughran, T.P.J. The Spectrum of Large Granular Lymphocyte Leukemia and Felty’s Syndrome. Curr. Opin. Hematol. 2011, 18, 254–259. [Google Scholar] [CrossRef] [Green Version]
- Sood, R.; Stewart, C.C.; Aplan, P.D.; Murai, H.; Ward, P.; Barcos, M.; Baer, M.R. Neutropenia Associated with T-Cell Large Granular Lymphocyte Leukemia: Long-Term Response to Cyclosporine Therapy despite Persistence of Abnormal Cells. Blood 1998, 91, 3372–3378. [Google Scholar] [CrossRef] [Green Version]
- Bockorny, B.; Codreanu, I.; Dasanu, C.A. Prevalence of Autoimmune Hematologic and Non-Hematologic Conditions in Large Granular Lymphocytic Leukemia: Exploratory Analysis of a Series of Consecutive Patients. Leuk. Lymphoma 2014, 55, 1399–1401. [Google Scholar] [CrossRef] [PubMed]
- Bareau, B.; Rey, J.; Hamidou, M.; Donadieu, J.; Morcet, J.; Reman, O.; Schleinitz, N.; Tournilhac, O.; Roussel, M.; Fest, T.; et al. Analysis of a French Cohort of Patients with Large Granular Lymphocyte Leukemia: A Report on 229 Cases. Haematologica 2010, 95, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Castillo Tokumori, F.; Isenalumhe, L.; Zhang, Y.; Tandon, A.; Knepper, T.C.; Mo, Q.; Shao, H.; Zhang, L.; Sokol, L. Large Granular Lymphocytic Leukemia - A Retrospective Study of 319 Cases. Am. J. Hematol. 2021, 96, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Sanikommu, S.R.; Clemente, M.J.; Chomczynski, P.; Afable, M.G., 2nd; Jerez, A.; Thota, S.; Patel, B.; Hirsch, C.; Nazha, A.; Desamito, J.; et al. Clinical Features and Treatment Outcomes in Large Granular Lymphocytic Leukemia (LGLL). Leuk. Lymphoma 2018, 59, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Breedveld, F.C.; Lafeber, G.J.; de Vries, E.; van Krieken, J.H.; Cats, A. Immune Complexes and the Pathogenesis of Neutropenia in Felty’s Syndrome. Ann. Rheum. Dis. 1986, 45, 696–702. [Google Scholar] [CrossRef] [Green Version]
- Morice, W.G.; Kurtin, P.J.; Tefferi, A.; Hanson, C.A. Distinct Bone Marrow Findings in T-Cell Granular Lymphocytic Leukemia Revealed by Paraffin Section Immunoperoxidase Stains for CD8, TIA-1, and Granzyme B. Blood 2002, 99, 268–274. [Google Scholar] [CrossRef]
- Prochorec-Sobieszek, M.; Rymkiewicz, G.; Makuch-Łasica, H.; Majewski, M.; Michalak, K.; Rupiński, R.; Warzocha, K.; Maryniak, R. Characteristics of T-Cell Large Granular Lymphocyte Proliferations Associated with Neutropenia and Inflammatory Arthropathy. Arthritis. Res. Ther. 2008, 10, R55. [Google Scholar] [CrossRef] [Green Version]
- Osuji, N.; Matutes, E.; Catovsky, D.; Lampert, I.; Wotherspoon, A. Histopathology of the Spleen in T-Cell Large Granular Lymphocyte Leukemia and T-Cell Prolymphocytic Leukemia: A Comparative Review. Am. J. Surg. Pathol. 2005, 29, 935–941. [Google Scholar] [CrossRef]
- Oshimi, K.; Shinkai, Y.; Okumura, K.; Oshimi, Y.; Mizoguchi, H. Perforin Gene Expression in Granular Lymphocyte Proliferative Disorders. Blood 1990, 75, 704–708. [Google Scholar] [CrossRef] [Green Version]
- Kothapalli, R.; Nyland, S.B.; Kusmartseva, I.; Bailey, R.D.; McKeown, T.M.; Loughran, T.P.J. Constitutive Production of Proinflammatory Cytokines RANTES, MIP-1beta and IL-18 Characterizes LGL Leukemia. Int. J. Oncol. 2005, 26, 529–535. [Google Scholar]
- Lamy, T.; Liu, J.H.; Landowski, T.H.; Dalton, W.S.; Loughran, T.P.J. Dysregulation of CD95/CD95 Ligand-Apoptotic Pathway in CD3(+) Large Granular Lymphocyte Leukemia. Blood 1998, 92, 4771–4777. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Wei, S.; Lamy, T.; Epling-Burnette, P.K.; Starkebaum, G.; Djeu, J.Y.; Loughran, T.P. Chronic Neutropenia Mediated by Fas Ligand. Blood 2000, 95, 3219–3222. [Google Scholar] [CrossRef] [PubMed]
- Giudice, V.; Cardamone, C.; Triggiani, M.; Selleri, C. Bone Marrow Failure Syndromes, Overlapping Diseases with a Common Cytokine Signature. Int. J. Mol. Sci. 2021, 22, 705. [Google Scholar] [CrossRef] [PubMed]
- Nixon, C.P.; Sweeney, J.D. Autoimmune Cytopenias: Diagnosis & Management. Rhode Isl. Med. J. 2016, 99, 36–40. [Google Scholar]
- Maciejewski, J.P.; O’Keefe, C.; Gondek, L.; Tiu, R. Immune-Mediated Bone Marrow Failure Syndromes of Progenitor and Stem Cells: Molecular Analysis of Cytotoxic T Cell Clones. Folia Histochem. Cytobiol. 2007, 45, 5–14. [Google Scholar]
- Rogers, K.A.; Woyach, J.A. Secondary Autoimmune Cytopenias in Chronic Lymphocytic Leukemia. Semin. Oncol. 2016, 43, 300–310. [Google Scholar] [CrossRef]
- Lamy, T.; Moignet, A.; Loughran, T.P. LGL Leukemia: From Pathogenesis to Treatment. Blood 2017, 129, 1082–1094. [Google Scholar] [CrossRef]
- Lamy, T.; Loughran, T.P.J. How I Treat LGL Leukemia. Blood 2011, 117, 2764–2774. [Google Scholar] [CrossRef]
- Loughran, T.P.J.; Zickl, L.; Olson, T.L.; Wang, V.; Zhang, D.; Rajala, H.L.M.; Hasanali, Z.; Bennett, J.M.; Lazarus, H.M.; Litzow, M.R.; et al. Immunosuppressive Therapy of LGL Leukemia: Prospective Multicenter Phase II Study by the Eastern Cooperative Oncology Group (E5998). Leukemia 2015, 29, 886–894. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, D.P. T-Cell Large Granular Leukemia and Related Proliferations. Am. J. Clin. Pathol. 2007, 127, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Yu, Y.; Yan, S.; Wang, R.; Liu, X.; Chen, C. Pure Red Cell Aplasia and Autoimmune Hemolytic Anemia Sequentially Occurring in a Patient with Large Granular T-Lymphocytic Leukemia. Intern. Med. 2016, 55, 1491–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, D.W.; Loughran, T.P.J.; Maciejewski, J.P.; Sasu, S.; Song, S.X.; Epling-Burnette, P.K.; Paquette, R.L. Acquired Amegakaryocytic Thrombocytopenia and Pure Red Cell Aplasia Associated with an Occult Large Granular Lymphocyte Leukemia. Leuk. Res. 2008, 32, 823–827. [Google Scholar] [CrossRef]
- Brinkman, K.; van Dongen, J.J.; van Lom, K.; Groeneveld, K.; Miseré, J.F.; van der Heul, C. Induction of Clinical Remission in T-Large Granular Lymphocyte Leukemia with Cyclosporin A, Monitored by Use of Immunophenotyping with Vbeta Antibodies. Leukemia 1998, 12, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, A.M.; Orozco Niño, J.A.; Seehaus, C.M.; Giménez Conca, A.D.; Chuliber, F.A.; Lo Giudice, L.F.; Bendek, G.E. Evans syndrome and rheumatoid arthritis as autoimmune manifestations of large granular T-cell leukemia. Medicina 2021, 81, 1060–1064. [Google Scholar]
- Helmick, C.G.; Felson, D.T.; Lawrence, R.C.; Gabriel, S.; Hirsch, R.; Kwoh, C.K.; Liang, M.H.; Kremers, H.M.; Mayes, M.D.; Merkel, P.A.; et al. Estimates of the Prevalence of Arthritis and Other Rheumatic Conditions in the United States. Part I. Arthritis Rheum. 2008, 58, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Mielants, H.; Van den Bosch, F. Extra-Articular Manifestations. Clin. Exp. Rheumatol. 2009, 27, S56–S61. [Google Scholar] [PubMed]
- Bowman, S.J. Hematological Manifestations of Rheumatoid Arthritis. Scand. J. Rheumatol. 2002, 31, 251–259. [Google Scholar] [CrossRef]
- Felty, A. Chronic Arthritis in the Adult, Associated with Splenomegaly and Leukopenia. Bull. Johns Hopkins Hosp. 1924, 35, 16. [Google Scholar]
- Schwaneck, E.C.; Renner, R.; Junker, L.; Einsele, H.; Gadeholt, O.; Geissinger, E.; Kleinert, S.; Gernert, M.; Tony, H.-P.; Schmalzing, M. Prevalence and Characteristics of Persistent Clonal T Cell Large Granular Lymphocyte Expansions in Rheumatoid Arthritis: A Comprehensive Analysis of 529 Patients. Arthritis Rheumatol. 2018, 70, 1914–1922. [Google Scholar] [CrossRef] [Green Version]
- Savola, P.; Brück, O.; Olson, T.; Kelkka, T.; Kauppi, M.J.; Kovanen, P.E.; Kytölä, S.; Sokka-Isler, T.; Loughran, T.P.; Leirisalo-Repo, M.; et al. Somatic STAT3 Mutations in Felty Syndrome: An Implication for a Common Pathogenesis with Large Granular Lymphocyte Leukemia. Haematologica 2018, 103, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Bowman, S.J.; Bhavnani, M.; Geddes, G.C.; Corrigall, V.; Boylston, A.W.; Panayi, G.S.; Lanchbury, J.S. Large Granular Lymphocyte Expansions in Patients with Felty’s Syndrome: Analysis Using Anti-T Cell Receptor V Beta-Specific Monoclonal Antibodies. Clin. Exp. Immunol. 1995, 101, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Schattner, A.; Shvidel, L.; Berrebi, A. Characterization of T-Cell Large Granular Lymphocyte Leukemia Associated with Sjogren’s Syndrome-an Important but under-Recognized Association. Semin. Arthritis. Rheum. 2006, 35, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Marlton, P.; Taylor, K.; Elliott, S.; McCormack, J. Monoclonal Large Granular Lymphocyte Proliferation in SLE with HTLV-I Seroreactivity. Aust. N. Z. J. Med. 1992, 22, 54–55. [Google Scholar] [CrossRef]
- Ogata, A.; Kitano, M.; Fukamizu, M.; Hamano, T.; Sano, H. Increased Serum Interleukin-18 in a Patient with Systemic Lupus Erythematosus and T-Cell Large Granular Lymphocytic Leukemia. Mod. Rheumatol. 2004, 14, 267–270. [Google Scholar] [CrossRef]
- Okamoto, M.; Ichinose, K.; Sato, S.; Imadome, K.-I.; Furukawa, K.; Kawakami, A. A Case of Γδ T-Cell Large Granular Lymphocytic Leukemia Due to a Chronic Active Epstein-Barr Virus Infection in Systemic Lupus Erythematosus. Clin. Immunol. 2020, 214, 108378. [Google Scholar] [CrossRef]
- Olde Bekkink, M.; Ahmed-Ousenkova, Y.M.; Netea, M.G.; van der Velden, W.J.; Berden, J.H. Coexistence of Systemic Lupus Erythematosus, Tuberous Sclerosis and Aggressive Natural Killer-Cell Leukaemia: Coincidence or Correlated? Lupus 2016, 25, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Audemard, A.; Lamy, T.; Bareau, B.; Sicre, F.; Suarez, F.; Truquet, F.; Salaun, V.; Macro, M.; Verneuil, L.; Lobbedez, T.; et al. Vasculitis Associated with Large Granular Lymphocyte (LGL) Leukemia: Presentation and Treatment Outcomes of 11 Cases. Semin. Arthritis. Rheum. 2013, 43, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Matsushima, T.; Kaneko, Y.; Yokohama, A.; Handa, H.; Tsukamoto, N.; Karasawa, M.; Nojima, Y.; Murakami, H. T Cell Large Granular Lymphocyte (LGL) Leukemia Associated with Behcet’s Disease: High Expression of SFasL and IL-18 of CD8 LGL. Ann. Hematol. 2008, 87, 585–586. [Google Scholar] [CrossRef]
- Greenberg, S.A.; Pinkus, J.L.; Amato, A.A.; Kristensen, T.; Dorfman, D.M. Association of Inclusion Body Myositis with T Cell Large Granular Lymphocytic Leukaemia. Brain 2016, 139, 1348–1360. [Google Scholar] [CrossRef] [Green Version]
- O’Keefe, C.L.; Plasilova, M.; Wlodarski, M.; Risitano, A.M.; Rodriguez, A.R.; Howe, E.; Young, N.S.; Hsi, E.; Maciejewski, J.P. Molecular Analysis of TCR Clonotypes in LGL: A Clonal Model for Polyclonal Responses. J. Immunol. 2004, 172, 1960–1969. [Google Scholar] [CrossRef] [Green Version]
- Dhodapkar, M.V.; Li, C.Y.; Lust, J.A.; Tefferi, A.; Phyliky, R.L. Clinical Spectrum of Clonal Proliferations of T-Large Granular Lymphocytes: A T-Cell Clonopathy of Undetermined Significance? Blood 1994, 84, 1620–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Nasa, G.; Littera, R.; Cocco, E.; Battistini, L.; Marrosu, M.G.; Contu, L. Allogeneic Hematopoietic Stem Cell Transplantation in a Patient Affected by Large Granular Lymphocyte Leukemia and Multiple Sclerosis. Ann. Hematol. 2004, 83, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Cherel, B.; Humbert, M.; LeBlanc, F.R.; Zambello, R.; Hamidou, M.; Lifermann, F.; Montani, D.; Leoncin, M.; Decaux, O.; Pastoret, C.; et al. Large Granular Lymphocyte Leukemia and Precapillary Pulmonary Hypertension. Chest 2020, 158, 2602–2609. [Google Scholar] [CrossRef] [PubMed]
- Koskela, H.L.M.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmäki, H.; Andersson, E.I.; Lagström, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 Mutations in Large Granular Lymphocytic Leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helm, K.F.; Peters, M.S.; Tefferi, A.; Leiferman, K.M. Pyoderma Gangrenosum-like Ulcer in a Patient with Large Granular Lymphocytic Leukemia. J. Am. Acad. Dermatol. 1992, 27, 868–871. [Google Scholar] [CrossRef]
- Atieh, T.; Lichtin, A. An Unusual Cause of Bruising. Cleve. Clin. J. Med. 2019, 86, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, N.R.; Chaudhari, K.S.; Rao, V.; Sharma, V.R. Nuances in the Management of Acquired Hemophilia A in an Elderly Patient with Large Granular Lymphocytic Leukemia. Blood Res. 2020, 55, 180–181. [Google Scholar] [CrossRef]
- Lazarou, I.; Petitpierre, N.; Auger, I.; Reber, G.; Roux-Lombard, P.; Boehlen, F.; Villard, J. Felty’s Syndrome and Hypofibrinogenemia: An Unusual Target for Anti-Cyclic Citrullinated Peptide Antibodies? Mod. Rheumatol. 2015, 25, 790–793. [Google Scholar] [CrossRef]
- Murphy, P.W.; Brett, L.K.; Verla-Tebit, E.; Macik, B.G.; Loughran, T.P.J. Acquired Inhibitors to Factor VIII and Fibrinogen in the Setting of T-Cell Large Granular Lymphocyte Leukemia: A Case Report and Review of the Literature. Blood Coagul. Fibrinolysis 2015, 26, 211–213. [Google Scholar] [CrossRef]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Koskela, H.; LeBlanc, F.; Peng Ng, K.; Olson, T.; Przychodzen, B.; Afable, M.; Gomez-Segui, I.; et al. STAT3 Mutations Unify the Pathogenesis of Chronic Lymphoproliferative Disorders of NK Cells and T-Cell Large Granular Lymphocyte Leukemia. Blood 2012, 120, 3048–3057. [Google Scholar] [CrossRef]
- Poullot, E.; Zambello, R.; Leblanc, F.; Bareau, B.; De March, E.; Roussel, M.; Boulland, M.L.; Houot, R.; Renault, A.; Fest, T.; et al. Chronic Natural Killer Lymphoproliferative Disorders: Characteristics of an International Cohort of 70 Patients. Ann. Oncol. 2014, 25, 2030–2035. [Google Scholar] [CrossRef] [PubMed]
- Gorodetskiy, V.R.; Sidorova, Y.V.; Kupryshina, N.A.; Vasilyev, V.I.; Probatova, N.A.; Ryzhikova, N.V.; Sudarikov, A.B. Analysis of a Single-Institution Cohort of Patients with Felty’s Syndrome and T-Cell Large Granular Lymphocytic Leukemia in the Setting of Rheumatoid Arthritis. Rheumatol. Int. 2021, 41, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Rajala, H.L.M.; Olson, T.; Clemente, M.J.; Lagström, S.; Ellonen, P.; Lundan, T.; Hamm, D.E.; Zaman, S.A.U.; Lopez Marti, J.M.; Andersson, E.I.; et al. The Analysis of Clonal Diversity and Therapy Responses Using STAT3 Mutations as a Molecular Marker in Large Granular Lymphocytic Leukemia. Haematologica 2015, 100, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, S.J.; Hall, M.A.; Panayi, G.S.; Lanchbury, J.S. T Cell Receptor Alpha-Chain and Beta-Chain Junctional Region Homology in Clonal CD3+, CD8+ T Lymphocyte Expansions in Felty’s Syndrome. Arthritis Rheum. 1997, 40, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Bigouret, V.; Hoffmann, T.; Arlettaz, L.; Villard, J.; Colonna, M.; Ticheli, A.; Gratwohl, A.; Samii, K.; Chapuis, B.; Rufer, N.; et al. Monoclonal T-Cell Expansions in Asymptomatic Individuals and in Patients with Large Granular Leukemia Consist of Cytotoxic Effector T Cells Expressing the Activating CD94:NKG2C/E and NKD2D Killer Cell Receptors. Blood 2003, 101, 3198–3204. [Google Scholar] [CrossRef] [Green Version]
- Namekawa, T.; Snyder, M.R.; Yen, J.H.; Goehring, B.E.; Leibson, P.J.; Weyand, C.M.; Goronzy, J.J. Killer Cell Activating Receptors Function as Costimulatory Molecules on CD4+CD28null T Cells Clonally Expanded in Rheumatoid Arthritis. J. Immunol. 2000, 165, 1138–1145. [Google Scholar] [CrossRef] [Green Version]
- Gulan, G.; Ravlic-Gulan, J.; Strbo, N.; Sotosek, V.; Nemec, B.; Matovinovic, D.; Rubinic, D.; Podack, E.R.; Rukavina, D. Systemic and Local Expression of Perforin in Lymphocyte Subsets in Acute and Chronic Rheumatoid Arthritis. J. Rheumatol. 2003, 30, 660–670. [Google Scholar]
- Raza, K.; Falciani, F.; Curnow, S.J.; Ross, E.J.; Lee, C.-Y.; Akbar, A.N.; Lord, J.M.; Gordon, C.; Buckley, C.D.; Salmon, M. Early Rheumatoid Arthritis Is Characterized by a Distinct and Transient Synovial Fluid Cytokine Profile of T Cell and Stromal Cell Origin. Arthritis. Res. Ther. 2005, 7, R784–R795. [Google Scholar] [CrossRef] [Green Version]
- Bommarito, D.; Hall, C.; Taams, L.S.; Corrigall, V.M. Inflammatory Cytokines Compromise Programmed Cell Death-1 (PD-1)-Mediated T Cell Suppression in Inflammatory Arthritis through up-Regulation of Soluble PD-1. Clin. Exp. Immunol. 2017, 188, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Melenhorst, J.J.; Brümmendorf, T.H.; Kirby, M.; Lansdorp, P.M.; Barrett, A.J. CD8+ T Cells in Large Granular Lymphocyte Leukemia Are Not Defective in Activation- and Replication-Related Apoptosis. Leuk. Res. 2001, 25, 699–708. [Google Scholar] [CrossRef]
- Schirmer, M.; Vallejo, A.N.; Weyand, C.M.; Goronzy, J.J. Resistance to Apoptosis and Elevated Expression of Bcl-2 in Clonally Expanded CD4+CD28- T Cells from Rheumatoid Arthritis Patients. J. Immunol. 1998, 161, 1018–1025. [Google Scholar] [PubMed]
- Battiwalla, M.; Melenhorst, J.; Saunthararajah, Y.; Nakamura, R.; Molldrem, J.; Young, N.S.; Barrett, A.J. HLA-DR4 Predicts Haematological Response to Cyclosporine in T-Large Granular Lymphocyte Lymphoproliferative Disorders. Br. J. Haematol. 2003, 123, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Starkebaum, G.; Loughran, T.P.J.; Gaur, L.K.; Davis, P.; Nepom, B.S. Immunogenetic Similarities between Patients with Felty’s Syndrome and Those with Clonal Expansions of Large Granular Lymphocytes in Rheumatoid Arthritis. Arthritis Rheum. 1997, 40, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Burks, E.J.; Loughran, T.P.J. Pathogenesis of Neutropenia in Large Granular Lymphocyte Leukemia and Felty Syndrome. Blood Rev. 2006, 20, 245–266. [Google Scholar] [CrossRef] [PubMed]
- Gorodetskiy, V.; Probatova, N.; Sidorova, Y.; Kupryshina, N.; Obukhova, T.; Vasilyev, V.; Ryzhikova, N.; Sudarikov, A. The Non-Leukemic T Cell Large Granular Lymphocytic Leukemia Variant with Marked Splenomegaly and Neutropenia in the Setting of Rheumatoid Arthritis - Felty Syndrome and Hepatosplenic T Cell Lymphoma Mask. Am. J. Blood Res. 2021, 11, 227–237. [Google Scholar]
- Molad, Y.; Okon, E.; Stark, P.; Prokocimer, M. Sjögren’s Syndrome Associated T Cell Large Granular Lymphocyte Leukemia: A Possible Common Etiopathogenesis. J. Rheumatol. 2001, 28, 2551–2552. [Google Scholar]
- Go, R.S.; Tefferi, A.; Li, C.-Y.; Lust, J.A.; Phyliky, R.L. Lymphoproliferative Disease of Granular T Lymphocytes Presenting as Aplastic Anemia. Blood 2000, 96, 3644–3646. [Google Scholar] [CrossRef]
- Risitano, A.M.; Maciejewski, J.P.; Muranski, P.; Wlodarski, M.; O’Keefe, C.; Sloand, E.M.; Young, N.S. Large Granular Lymphocyte (LGL)-like Clonal Expansions in Paroxysmal Nocturnal Hemoglobinuria (PNH) Patients. Leukemia 2005, 19, 217–222. [Google Scholar] [CrossRef]
- Saunthararajah, Y.; Molldrem, J.L.; Rivera, M.; Williams, A.; Stetler-Stevenson, M.; Sorbara, L.; Young, N.S.; Barrett, J.A. Coincident Myelodysplastic Syndrome and T-Cell Large Granular Lymphocytic Disease: Clinical and Pathophysiological Features. Br. J. Haematol. 2001, 112, 195–200. [Google Scholar] [CrossRef]
- Kwong, Y.L.; Wong, K.F. Association of Pure Red Cell Aplasia with T Large Granular Lymphocyte Leukaemia. J. Clin. Pathol. 1998, 51, 672–675. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Rajala, H.; Gómez-Seguí, I.; Olson, T.; McGraw, K.; Przychodzen, B.; Kulasekararaj, A.; Afable, M.; et al. STAT3 Mutations Indicate the Presence of Subclinical T-Cell Clones in a Subset of Aplastic Anemia and Myelodysplastic Syndrome Patients. Blood 2013, 122, 2453–2459. [Google Scholar] [CrossRef] [PubMed]
- Durrani, J.; Awada, H.; Kishtagari, A.; Visconte, V.; Kerr, C.; Adema, V.; Nagata, Y.; Kuzmanovic, T.; Hong, S.; Patel, B.; et al. Large Granular Lymphocytic Leukemia Coexists with Myeloid Clones and Myelodysplastic Syndrome. Leukemia 2020, 34, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Komrokji, R.S.; Ali, N.A.; Sallman, D.; Padron, E.; Lancet, J.; Sokol, L.; Varnadoe, C.; Burnette, P.K.; List, A. Characterization of Myelodysplastic Syndromes (MDS) with T-Cell Large Granular Lymphocyte Proliferations (LGL). Leukemia 2020, 34, 3097–3099. [Google Scholar] [CrossRef]
- Ai, K.; Li, M.; Wu, P.; Deng, C.; Huang, X.; Ling, W.; Xu, R.; Geng, S.; Sun, Q.; Weng, J.; et al. Concurrence of Myelodysplastic Syndromes and Large Granular Lymphocyte Leukemia: Clinicopathological Features, Mutational Profile and Gene Ontology Analysis in a Single Center. Am. J. Cancer Res. 2021, 11, 1616–1631. [Google Scholar]
- Fasan, A.; Kern, W.; Grossmann, V.; Haferlach, C.; Haferlach, T.; Schnittger, S. STAT3 Mutations Are Highly Specific for Large Granular Lymphocytic Leukemia. Leukemia 2013, 27, 1598–1600. [Google Scholar] [CrossRef]
- Andersson, E.; Kuusanmäki, H.; Bortoluzzi, S.; Lagström, S.; Parsons, A.; Rajala, H.; van Adrichem, A.; Eldfors, S.; Olson, T.; Clemente, M.J.; et al. Activating Somatic Mutations Outside the SH2-Domain of STAT3 in LGL Leukemia. Leukemia 2016, 30, 1204–1208. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-García, N.; Jara-Acevedo, M.; Caldas, C.; Bárcena, P.; López, A.; Puig, N.; Alcoceba, M.; Fernández, P.; Villamor, N.; Flores-Montero, J.A.; et al. STAT3 and STAT5B Mutations in T/NK-Cell Chronic Lymphoproliferative Disorders of Large Granular Lymphocytes (LGL): Association with Disease Features. Cancers 2020, 12, 3508. [Google Scholar] [CrossRef]
- Johansson, P.; Bergmann, A.; Rahmann, S.; Wohlers, I.; Scholtysik, R.; Przekopowitz, M.; Seifert, M.; Tschurtschenthaler, G.; Webersinke, G.; Jäger, U.; et al. Recurrent Alterations of TNFAIP3 (A20) in T-Cell Large Granular Lymphocytic Leukemia. Int. J. Cancer 2016, 138, 121–124. [Google Scholar] [CrossRef]
- Andersson, E.I.; Rajala, H.L.M.; Eldfors, S.; Ellonen, P.; Olson, T.; Jerez, A.; Clemente, M.J.; Kallioniemi, O.; Porkka, K.; Heckman, C.; et al. Novel Somatic Mutations in Large Granular Lymphocytic Leukemia Affecting the STAT-Pathway and T-Cell Activation. Blood Cancer J. 2013, 3, e168. [Google Scholar] [CrossRef] [Green Version]
- Olson, T.L.; Cheon, H.; Xing, J.C.; Olson, K.C.; Paila, U.; Hamele, C.E.; Neelamraju, Y.; Shemo, B.C.; Schmachtenberg, M.; Sundararaman, S.K.; et al. Frequent Somatic TET2 Mutations in Chronic NK-LGL Leukemia with Distinct Patterns of Cytopenias. Blood 2021, 138, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Pastoret, C.; Desmots, F.; Drillet, G.; Le Gallou, S.; Boulland, M.-L.; Thannberger, A.; Doncker, A.-V.; Salaun, V.; Damaj, G.L.; Veyrat-Masson, R.; et al. Linking the KIR Phenotype with STAT3 and TET2 Mutations to Identify Chronic Lymphoproliferative Disorders of NK Cells. Blood 2021, 137, 3237–3250. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.A.; Lee, M.N.; DeAngelo, D.J.; Steensma, D.P.; Stone, R.M.; Kuo, F.C.; Aster, J.C.; Gibson, C.J.; Lindsley, R.C. Systematic STAT3 Sequencing in Patients with Unexplained Cytopenias Identifies Unsuspected Large Granular Lymphocytic Leukemia. Blood Adv. 2017, 1, 1786–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thol, F.; Platzbecker, U. Do Next-Generation Sequencing Results Drive Diagnostic and Therapeutic Decisions in MDS? Blood Adv. 2019, 3, 3449–3453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novikov, N.D.; Griffin, G.K.; Dudley, G.; Drew, M.; Rojas-Rudilla, V.; Lindeman, N.I.; Dorfman, D.M. Utility of a Simple and Robust Flow Cytometry Assay for Rapid Clonality Testing in Mature Peripheral T-Cell Lymphomas. Am. J. Clin. Pathol. 2019, 151, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Olteanu, H.; Jevremovic, D.; He, R.; Viswanatha, D.; Corley, H.; Horna, P. T-Cell Clones of Uncertain Significance Are Highly Prevalent and Show Close Resemblance to T-Cell Large Granular Lymphocytic Leukemia. Implications for Laboratory Diagnostics. Mod. Pathol. 2020, 33, 2046–2057. [Google Scholar] [CrossRef]
- Go, R.S.; Lust, J.A.; Phyliky, R.L. Aplastic Anemia and Pure Red Cell Aplasia Associated with Large Granular Lymphocyte Leukemia. Semin. Hematol. 2003, 40, 196–200. [Google Scholar] [CrossRef]
- Young, N.S.; Calado, R.T.; Scheinberg, P. Current Concepts in the Pathophysiology and Treatment of Aplastic Anemia. Blood 2006, 108, 2509–2519. [Google Scholar] [CrossRef]
- Young, N.S. Aplastic Anemia. N. Engl. J. Med. 2018, 379, 1643–1656. [Google Scholar] [CrossRef]
- Risitano, A.M.; Maciejewski, J.P.; Green, S.; Plasilova, M.; Zeng, W.; Young, N.S. In-Vivo Dominant Immune Responses in Aplastic Anaemia: Molecular Tracking of Putatively Pathogenetic T-Cell Clones by TCR Beta-CDR3 Sequencing. Lancet 2004, 364, 355–364. [Google Scholar] [CrossRef]
- Giudice, V.; Feng, X.; Lin, Z.; Hu, W.; Zhang, F.; Qiao, W.; Ibanez, M.D.P.F.; Rios, O.; Young, N.S. Deep Sequencing and Flow Cytometric Characterization of Expanded Effector Memory CD8+CD57+ T Cells Frequently Reveals T-Cell Receptor Vβ Oligoclonality and CDR3 Homology in Acquired Aplastic Anemia. Haematologica 2018, 103, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Liu, S.; Niu, Y.; Fang, S.; Wu, X.; Yu, Z.; Chen, S.; Yang, L.; Li, Y. Altered Expression of the TCR Signaling Related Genes CD3 and FcεRIγ in Patients with Aplastic Anemia. J. Hematol. Oncol. 2012, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-F.; Huang, Z.-D.; Wu, X.-R.; Li, Q.; Yu, Z.-F. Comparison of T Lymphocyte Subsets in Aplastic Anemia and Hypoplastic Myelodysplastic Syndromes. Life Sci. 2017, 189, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, S.; Keränen, M.A.I.; Kankainen, M.; Huuhtanen, J.; Walldin, G.; Kerr, C.M.; Clemente, M.; Ebeling, F.; Rajala, H.; Brück, O.; et al. Somatic Mutations in Lymphocytes in Patients with Immune-Mediated Aplastic Anemia. Leukemia 2021, 35, 1365–1379. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Holland, S.M.; Staudt, L.M. JAKs and STATs in Immunity, Immunodeficiency, and Cancer. N. Engl. J. Med. 2013, 368, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Al-Haidari, A.; Sun, J.; Kazi, J.U. T Cell Receptor (TCR) Signaling in Health and Disease. Signal. Transduct. Target. Ther. 2021, 6, 412. [Google Scholar] [CrossRef] [PubMed]
- Holmström, T.H.; Schmitz, I.; Söderström, T.S.; Poukkula, M.; Johnson, V.L.; Chow, S.C.; Krammer, P.H.; Eriksson, J.E. MAPK/ERK Signaling in Activated T Cells Inhibits CD95/Fas-Mediated Apoptosis Downstream of DISC Assembly. EMBO J. 2000, 19, 5418–5428. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ding, S.; Liu, C.; Chen, T.; Liu, H.; Li, L.; Shao, Z.; Fu, R. Abnormalities of Quantities and Functions of CD56bright Natural Killer Cells in Non-Severe Aplastic Anemia. Hematology 2019, 24, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Sawada, K.; Fujishima, N.; Hirokawa, M. Acquired Pure Red Cell Aplasia: Updated Review of Treatment. Br. J. Haematol. 2008, 142, 505–514. [Google Scholar] [CrossRef]
- Means, R.T. Pure Red Cell Aplasia. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Fisch, P.; Handgretinger, R.; Schaefer, H.E. Pure Red Cell Aplasia. Br. J. Haematol. 2000, 111, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Partanen, S.; Ruutu, T.; Vuopio, P.; Andersson, L.C. Acquired Pure Red-Cell Aplasia: A Consequence of Increased Natural Killer Cell Activity? Leuk. Res. 1984, 8, 117–122. [Google Scholar] [CrossRef]
- Tefferi, A.; Windebank, K.P.; Veeder, M.H.; Kiely, J.M. Steroid-Responsive Pure Red Cell Aplasia Associated with Natural Killer Cell Lymphocytosis. Am. J. Hematol. 1989, 31, 211–212. [Google Scholar] [CrossRef]
- Tabata, R.; Tabata, C. Distinct Effect of Cyclophosphamide and Cyclosporine on Pure Red Cell Aplasia Associated with T-Cell Large Granular Lymphocyte Leukemia. Int. Immunopharmacol. 2014, 23, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Handgretinger, R.; Geiselhart, A.; Moris, A.; Grau, R.; Teuffel, O.; Bethge, W.; Kanz, L.; Fisch, P. Pure Red-Cell Aplasia Associated with Clonal Expansion of Granular Lymphocytes Expressing Killer-Cell Inhibitory Receptors. N. Engl. J. Med. 1999, 340, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Ishida, F.; Matsuda, K.; Sekiguchi, N.; Makishima, H.; Taira, C.; Momose, K.; Nishina, S.; Senoo, N.; Sakai, H.; Ito, T.; et al. STAT3 Gene Mutations and Their Association with Pure Red Cell Aplasia in Large Granular Lymphocyte Leukemia. Cancer Sci. 2014, 105, 342–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, T.; Sekiguchi, N.; Kobayashi, J.; Imi, T.; Matsuda, K.; Yamane, T.; Nishina, S.; Senoo, Y.; Sakai, H.; Ito, T.; et al. Frequent STAT3 Mutations in CD8+ T Cells from Patients with Pure Red Cell Aplasia. Blood Adv. 2018, 2, 2704–2712. [Google Scholar] [CrossRef]
- Qiu, Z.-Y.; Fan, L.; Wang, R.; Gale, R.P.; Liang, H.-J.; Wang, M.; Wang, L.; Wu, Y.-J.; Qiao, C.; Chen, Y.-Y.; et al. Methotrexate Therapy of T-Cell Large Granular Lymphocytic Leukemia Impact of STAT3 Mutation. Oncotarget 2016, 7, 61419–61425. [Google Scholar] [CrossRef] [Green Version]
- Schützinger, C.; Gaiger, A.; Thalhammer, R.; Vesely, M.; Fritsche-Polanz, R.; Schwarzinger, I.; Ohler, L.; Simonitsch-Klupp, I.; Reinhard, F.; Jäger, U. Remission of Pure Red Cell Aplasia in T-Cell Receptor Gammadelta-Large Granular Lymphocyte Leukemia after Therapy with Low-Dose Alemtuzumab. Leukemia 2005, 19, 2005–2008. [Google Scholar] [CrossRef] [Green Version]
- Ru, X.; Liebman, H.A. Successful Treatment of Refractory Pure Red Cell Aplasia Associated with Lymphoproliferative Disorders with the Anti-CD52 Monoclonal Antibody Alemtuzumab (Campath-1H). Br. J. Haematol. 2003, 123, 278–281. [Google Scholar] [CrossRef]
- Osuji, N.; Matutes, E.; Wotherspoon, A.; Catovsky, D. Lessons from a Case of T-Cell Large Granular Lymphocytic Leukaemia Suggesting That Immunomodulatory Therapy Is More Effective than Intensive Treatment. Leuk. Res. 2005, 29, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Au, W.-Y.; Lam, C.C.K.; Chim, C.-S.; Pang, A.W.K.; Kwong, Y.-L. Alemtuzumab Induced Complete Remission of Therapy-Resistant Pure Red Cell Aplasia. Leuk. Res. 2005, 29, 1213–1215. [Google Scholar] [CrossRef] [PubMed]
- Lacy, M.Q.; Kurtin, P.J.; Tefferi, A. Pure Red Cell Aplasia: Association with Large Granular Lymphocyte Leukemia and the Prognostic Value of Cytogenetic Abnormalities. Blood 1996, 87, 3000–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujishima, N.; Sawada, K.; Hirokawa, M.; Oshimi, K.; Sugimoto, K.; Matsuda, A.; Teramura, M.; Karasawa, M.; Arai, A.; Yonemura, Y.; et al. Long-Term Responses and Outcomes Following Immunosuppressive Therapy in Large Granular Lymphocyte Leukemia-Associated Pure Red Cell Aplasia: A Nationwide Cohort Study in Japan for the PRCA Collaborative Study Group. Haematologica 2008, 93, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; DeZern, A.E.; Kinoshita, T.; Brodsky, R.A. Paroxysmal Nocturnal Haemoglobinuria. Nat. Rev. Dis. Primers 2017, 3, 17028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodsky, R.A. How I Treat Paroxysmal Nocturnal Hemoglobinuria. Blood 2021, 137, 1304–1309. [Google Scholar] [CrossRef]
- Kinoshita, T.; Fujita, M. Biosynthesis of GPI-Anchored Proteins: Special Emphasis on GPI Lipid Remodeling. J. Lipid Res. 2016, 57, 6–24. [Google Scholar] [CrossRef] [Green Version]
- Karadimitris, A.; Li, K.; Notaro, R.; Araten, D.J.; Nafa, K.; Thertulien, R.; Ladanyi, M.; Stevens, A.E.; Rosenfeld, C.S.; Roberts, I.A.G.; et al. Association of Clonal T-Cell Large Granular Lymphocyte Disease and Paroxysmal Nocturnal Haemoglobinuria (PNH): Further Evidence for a Pathogenetic Link between T Cells, Aplastic Anaemia and PNH. Br. J. Haematol 2001, 115, 1010–1014. [Google Scholar] [CrossRef]
- Fukumoto, J.S.; Gotlib, J. A Patient with Paroxysmal Nocturnal Hemoglobinuria, T Cell Large Granular Lymphocyte Clonal Expansion, and Monoclonal Gammopathy of Undetermined Significance. Am. J. Hematol. 2006, 81, 870–874. [Google Scholar] [CrossRef]
- Boyer, T.; Grardel, N.; Copin, M.-C.; Roumier, C.; Touzart, A.; Buchdahl, A.-L.; Corm, S.; Peffault de Latour, R.; Preudhomme, C.; Terriou, L. Paroxysmal Nocturnal Hemoglobinuria (PNH) and T Cell Large Granular Lymphocyte (LGL) Leukemia--an Unusual Association: Another Cause of Cytopenia in PNH. Ann. Hematol. 2015, 94, 1759–1760. [Google Scholar] [CrossRef]
- Hillmen, P.; Young, N.S.; Schubert, J.; Brodsky, R.A.; Socié, G.; Muus, P.; Röth, A.; Szer, J.; Elebute, M.O.; Nakamura, R.; et al. The Complement Inhibitor Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N. Engl. J. Med. 2006, 355, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Sicre de Fontbrune, F.; Wong Lee Lee, L.; Pessoa, V.; Gualandro, S.; Füreder, W.; Ptushkin, V.; Rottinghaus, S.T.; Volles, L.; Shafner, L.; et al. Ravulizumab (ALXN1210) vs Eculizumab in Adult Patients with PNH Naive to Complement Inhibitors: The 301 Study. Blood 2019, 133, 530–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoda, Y.; Abe, T.; Mitamura, K.; Saito, K.; Kawada, K.; Onozawa, Y.; Adachi, Y.; Nomura, T. Deficient Natural Killer (NK) Cell Activity in Paroxysmal Nocturnal Haemoglobinuria (PNH). Br. J. Haematol. 1982, 52, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Y oda, Y.; Abe, T. Deficient Natural Killer (NK) Cells in Paroxysmal Nocturnal Haemoglobinuria (PNH): Studies of Lymphoid Cells Fractionated by Discontinuous Density Gradient Centrifugation. Br. J. Haematol. 1985, 60, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Howe, E.C.; Wlodarski, M.; Ball, E.J.; Rybicki, L.; Maciejewski, J.P. Killer Immunoglobulin-like Receptor Genotype in Immune-Mediated Bone Marrow Failure Syndromes. Exp. Hematol. 2005, 33, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbiny, Y.M.; Doody, G.M.; Kelly, R.J.; Hill, A.; Hillmen, P.; Cook, G.P. Natural Killer (NK) Cell Function in Paroxysmal Nocturnal Hemoglobinuria: A Deficiency of NK Cells, but Not an NK Cell Deficiency. Blood 2015, 125, 1351–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvan, S.R.; Sheehy, P.F.; Heinemann, F.S.; Anbuganapathi, S. Bone Marrow Failure Due to T-Cell Large Granular Lymphocytic Leukemia in a Patient with Essential Thrombocythemia. Leuk. Res. 2011, 35, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Malani, A.K.; Gupta, C.; Rangineni, R.; Singh, J.; Ammar, H. Concomitant Presentation of Acute Myeloid Leukemia with T-Cell Large Granular Lymphocytic Leukemia. Acta Oncol. 2007, 46, 247–249. [Google Scholar] [CrossRef]
- Reda, G.; Fattizzo, B.; Cassin, R.; Flospergher, E.; Orofino, N.; Gianelli, U.; Barcellini, W.; Cortelezzi, A. Multifactorial Neutropenia in a Patient with Acute Promyelocytic Leukemia and Associated Large Granular Lymphocyte Expansion: A Case Report. Oncol. Lett. 2017, 13, 1307–1310. [Google Scholar] [CrossRef]
- Mustjoki, S.; Ekblom, M.; Arstila, T.P.; Dybedal, I.; Epling-Burnette, P.K.; Guilhot, F.; Hjorth-Hansen, H.; Höglund, M.; Kovanen, P.; Laurinolli, T.; et al. Clonal Expansion of T/NK-Cells during Tyrosine Kinase Inhibitor Dasatinib Therapy. Leukemia 2009, 23, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Kreutzman, A.; Juvonen, V.; Kairisto, V.; Ekblom, M.; Stenke, L.; Seggewiss, R.; Porkka, K.; Mustjoki, S. Mono/Oligoclonal T and NK Cells Are Common in Chronic Myeloid Leukemia Patients at Diagnosis and Expand during Dasatinib Therapy. Blood 2010, 116, 772–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Nakashima, S.; Usuda, M. Rapid and Sustained Increase of Large Granular Lymphocytes and Rare Cytomegalovirus Reactivation during Dasatinib Treatment in Chronic Myelogenous Leukemia Patients. Int. J. Hematol. 2012, 96, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Valent, J.N.; Schiffer, C.A. Prevalence of Large Granular Lymphocytosis in Patients with Chronic Myelogenous Leukemia (CML) Treated with Dasatinib. Leuk. Res. 2011, 35, e1–e3. [Google Scholar] [CrossRef]
- Powers, J.J.; Dubovsky, J.A.; Epling-Burnette, P.; Moscinski, L.; Zhang, L.; Mustjoki, S.; Sotomayor, E.M.; Pinilla-Ibarz, J.A. A Molecular and Functional Analysis of Large Granular Lymphocyte Expansions in Chronic Myelogenous Leukemia Patients Treated With Tyrosine Kinase Inhibitors. Leuk. Lymphoma 2011, 52, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kamel-Reid, S.; Chang, H.; Sutherland, R.; Jung, C.W.; Kim, H.-J.; Lee, J.-J.; Lipton, J.H. Natural Killer or Natural Killer/T Cell Lineage Large Granular Lymphocytosis Associated with Dasatinib Therapy for Philadelphia Chromosome Positive Leukemia. Haematologica 2009, 94, 135–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagihara, M.; Hua, J.; Inoue, M.; Uchida, T.; Ide, S.; Ohara, S.; Takaku, T. Nilotinib Treatment Induced Large Granular Lymphocyte Expansion and Maintenance of Longitudinal Remission in a Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Int. J. Hematol. 2020, 111, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Song, S. A Case Report: Concurrent Chronic Myelomonocytic Leukemia and T-Cell Large Granular Lymphocytic Leukemia-Type Clonal Proliferation as Detected by Multiparametric Flow Cytometry. Cytometry B Clin. Cytom. 2011, 80, 126–129. [Google Scholar] [CrossRef]
- Viny, A.D.; Lichtin, A.; Pohlman, B.; Loughran, T.; Maciejewski, J. Chronic B-Cell Dyscrasias Are an Important Clinical Feature of T-LGL Leukemia. Leuk. Lymphoma 2008, 49, 932–938. [Google Scholar] [CrossRef]
- Lesesve, J.F.; Feugier, P.; Lamy, T.; Béné, M.C.; Grégoire, M.J.; Lenormand, B.; Loughran, T. Association of B-Chronic Lymphocytic Leukaemia and T-Large Granular Lymphocyte Leukaemia. Clin. Lab. Haematol. 2000, 22, 121–122. [Google Scholar] [CrossRef]
- Marolleau, J.P.; Henni, T.; Gaulard, P.; Le Couedic, J.P.; Gourdin, M.F.; Divine, M.; Katz, A.; Tulliez, M.; Goossens, M.; Reyes, F. Hairy Cell Leukemia Associated with Large Granular Lymphocyte Leukemia: Immunologic and Genomic Study, Effect of Interferon Treatment. Blood 1988, 72, 655–660. [Google Scholar] [CrossRef] [Green Version]
- Papadaki, T.; Stamatopoulos, K.; Stavroyianni, N.; Paterakis, G.; Phisphis, M.; Stefanoudaki-Sofianatou, K. Evidence for T-Large Granular Lymphocyte-Mediated Neutropenia in Rituximab-Treated Lymphoma Patients: Report of Two Cases. Leuk. Res. 2002, 26, 597–600. [Google Scholar] [CrossRef]
- Gorodetskiy, V.R.; Probatova, N.A.; Kupryshina, N.A.; Palshina, S.G.; Obukhova, T.N.; Sidorova, Y.V.; Ryzhikova, N.V.; Sudarikov, A.B. Simultaneous Presentation of Leukemic Non-Nodal Mantle Cell Lymphoma and Gamma-Delta T-Large Granular Lymphocytic Leukemia in a Patient with Rheumatoid Arthritis. Cancer Manag. Res. 2020, 12, 9449–9457. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Ni, Y.; Yin, G.; Wu, H.; Li, J.; Miao, K. Mantle Cell Lymphoma Concurrent with T-Large Granular Lymphocytic Leukemia: Report of a Case and Review of Literature. Int. J. Clin. Exp. Pathol. 2015, 8, 3365–3369. [Google Scholar] [PubMed]
- Moignet, A.; Lamy, T. Latest Advances in the Diagnosis and Treatment of Large Granular Lymphocytic Leukemia. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 616–625. [Google Scholar] [CrossRef]
- Viny, A.D.; Maciejewski, J.P. High Rate of Both Hematopoietic and Solid Tumors Associated with Large Granular Lymphocyte Leukemia. Leuk. Lymphoma 2015, 56, 503–504. [Google Scholar] [CrossRef]
- Tefferi, A.; Li, C.Y.; Witzig, T.E.; Dhodapkar, M.V.; Okuno, S.H.; Phyliky, R.L. Chronic Natural Killer Cell Lymphocytosis: A Descriptive Clinical Study. Blood 1994, 84, 2721–2725. [Google Scholar] [CrossRef] [Green Version]
- Tiede, C.; Maecker-Kolhoff, B.; Klein, C.; Kreipe, H.; Hussein, K. Risk Factors and Prognosis in T-Cell Posttransplantation Lymphoproliferative Diseases: Reevaluation of 163 Cases. Transplantation 2013, 95, 479–488. [Google Scholar] [CrossRef]
- Kataria, A.; Cohen, E.; Saad, E.; Atallah, E.; Bresnahan, B. Large Granular Lymphocytic Leukemia Presenting Late after Solid Organ Transplantation: A Case Series of Four Patients and Review of the Literature. Transplant. Proc. 2014, 46, 3278–3281. [Google Scholar] [CrossRef]
- Feher, O.; Barilla, D.; Locker, J.; Oliveri, D.; Melhem, M.; Winkelstein, A. T-Cell Large Granular Lymphocytic Leukemia Following Orthotopic Liver Transplantation. Am. J. Hematol. 1995, 49, 216–220. [Google Scholar] [CrossRef]
- Masuda, M.; Arai, Y.; Nishina, H.; Fuchinoue, S.; Mizoguchi, H. Large Granular Lymphocyte Leukemia with Pure Red Cell Aplasia in a Renal Transplant Recipient. Am. J. Hematol. 1998, 57, 72–76. [Google Scholar] [CrossRef]
- Stamatopoulos, K.; Economidou, D.; Papadaki, T.; Vadikolia, C.; Papathanasiou, M.; Memmos, D.; Fassas, A. Large Granular Lymphocyte Leukemia after Renal Transplantation: An Immunologic, Immunohistochemical, and Genotypic Study. Transplantation 2007, 83, 102–103. [Google Scholar] [CrossRef] [PubMed]
- Alfano, G.; Ferrari, A.; Fontana, F.; Damiano, F.; Solazzo, A.; Mori, G.; Cappelli, G. Hemolytic Anemia as Presentation of T-Cell Large Granular Lymphocytic Leukemia After Kidney Transplantation: A Case Report. Transplant. Proc. 2020, 52, 1617–1618. [Google Scholar] [CrossRef] [PubMed]
- Awada, H.; Mahfouz, R.Z.; Durrani, J.; Kishtagari, A.; Jagadeesh, D.; Lichtin, A.E.; Hill, B.T.; Hamilton, B.K.; Carraway, H.E.; Nazha, A.; et al. Large Granular Lymphocytic Leukaemia after Solid Organ and Haematopoietic Stem Cell Transplantation. Br. J. Haematol. 2020, 189, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Alfano, G.; Fontana, F.; Colaci, E.; Mori, G.; Cerami, C.; Messerotti, A.; Potenza, L.; Luppi, M.; Cappelli, G. T-Cell Large Granular Lymphocyte Leukemia in Solid Organ Transplant Recipients: Case Series and Review of the Literature. Int. J. Hematol. 2019, 110, 313–321. [Google Scholar] [CrossRef]
- Sabnani, I.; Zucker, M.J.; Tsang, P.; Palekar, S. Clonal T-Large Granular Lymphocyte Proliferation in Solid Organ Transplant Recipients. Transplant. Proc. 2006, 38, 3437–3440. [Google Scholar] [CrossRef]
- Gabor, E.P.; Mishalani, S.; Lee, S. Rapid Response to Cyclosporine Therapy and Sustained Remission in Large Granular Lymphocyte Leukemia. Blood 1996, 87, 1199–1200. [Google Scholar] [CrossRef] [Green Version]
- Loh, E.; Couch, F.J.; Hendricksen, C.; Farid, L.; Kelly, P.F.; Acker, M.A.; Tomaszewski, J.E.; Malkowicz, S.B.; Weber, B.L. Development of Donor-Derived Prostate Cancer in a Recipient Following Orthotopic Heart Transplantation. JAMA 1997, 277, 133–137. [Google Scholar] [CrossRef]
- Bodvarsson, S.; Burlingham, W.; Kusaka, S.; Hafez, G.R.; Becker, B.N.; Pintar, T.; Sollinger, H.W.; Albertini, M.R. Donor-Derived Small Cell Lung Carcinoma in a Kidney Transplant Recipient. Cancer 2001, 92, 2429–2434. [Google Scholar] [CrossRef]
- Desai, R.; Collett, D.; Watson, C.J.; Johnson, P.; Evans, T.; Neuberger, J. Cancer Transmission from Organ Donors-Unavoidable but Low Risk. Transplantation 2012, 94, 1200–1207. [Google Scholar] [CrossRef]
- Au, W.Y.; Lam, C.C.; Lie, A.K.; Pang, A.; Kwong, Y.L. T-Cell Large Granular Lymphocyte Leukemia of Donor Origin after Allogeneic Bone Marrow Transplantation. Am. J. Clin. Pathol. 2003, 120, 626–630. [Google Scholar] [CrossRef]
- Toubert, A.; Glauzy, S.; Douay, C.; Clave, E. Thymus and Immune Reconstitution after Allogeneic Hematopoietic Stem Cell Transplantation in Humans: Never Say Never Again. Tissue Antigens 2012, 79, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Mohty, M.; Faucher, C.; Vey, N.; Chabannon, C.; Sainty, D.; Arnoulet, C.; Gaugler, B.; Gastaut, J.A.; Maraninchi, D.; Olive, D.; et al. Features of Large Granular Lymphocytes (LGL) Expansion Following Allogeneic Stem Cell Transplantation: A Long-Term Analysis. Leukemia 2002, 16, 2129–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nann-Rütti, S.; Tzankov, A.; Cantoni, N.; Halter, J.; Heim, D.; Tsakiris, D.; Arber, C.; Buser, A.; Gratwohl, A.; Tichelli, A.; et al. Large Granular Lymphocyte Expansion after Allogeneic Hematopoietic Stem Cell Transplant Is Associated with a Cytomegalovirus Reactivation and Shows an Indolent Outcome. Biol. Blood Marrow Transplant. 2012, 18, 1765–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Al-Dawsari, G.; Chang, H.; Panzarella, T.; Gupta, V.; Kuruvilla, J.; Lipton, J.H.; Messner, H.A. Large Granular Lymphocytosis and Its Impact on Long-Term Clinical Outcomes Following Allo-SCT. Bone Marrow Transplant. 2013, 48, 1104–1111. [Google Scholar] [CrossRef] [Green Version]
- Gill, H.; Ip, A.H.W.; Leung, R.; So, J.C.C.; Pang, A.W.K.; Tse, E.; Leung, A.Y.H.; Lie, A.K.W.; Kwong, Y.-L. Indolent T-Cell Large Granular Lymphocyte Leukaemia after Haematopoietic SCT: A Clinicopathologic and Molecular Analysis. Bone Marrow Transplant. 2012, 47, 952–956. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Ballester, J.; Chen-Liang, T.H.; Hurtado, A.M.; Heras, I.; de Arriba, F.; García-Malo, M.D.; Iniesta, P.; Lozano, M.L.; Nieto, J.B.; Ortuño, F.J.; et al. Persistent Cytotoxic T Lymphocyte Expansions after Allogeneic Haematopoietic Stem Cell Transplantation: Kinetics, Clinical Impact and Absence of STAT3 Mutations. Br. J. Haematol. 2016, 172, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Poch Martell, M.; Hamad, N.; Shin, E.; Moon, J.H.; Sohn, S.K.; Uhm, J.; Michelis, F.V.; Viswabandya, A.; Lipton, J.H.; Messner, H.A.; et al. Distinctive Clinical Characteristics and Favorable Outcomes in Patients with Large Granular Lymphocytosis after Allo-HCT: 12-Year Follow-up Data. Eur. J. Haematol. 2017, 99, 160–168. [Google Scholar] [CrossRef]
- Le Bris, Y.; Guillaume, T.; Ménard, A.; Illiaquer, M.; Martin, J.; Malard, S.; Duquesne, A.; Peterlin, P.; Debord, C.; Robillard, N.; et al. Lymphocyte Expansion after Unrelated Cord Blood Allogeneic Stem Cell Transplantation in Adults. Bone Marrow Transplant. 2017, 52, 854–858. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Shi, Y.Y.; Zhang, G.X.; Zhai, W.H.; Pang, A.M.; Ma, Q.L.; Zhang, R.L.; Wei, J.L.; Huang, Y.; Yang, D.L.; et al. Clinical features and risk factors analyses of patients with T cell large granular lymphocytosis following allo-HSCT. Zhonghua Xue Ye Xue Za Zhi 2020, 41, 630–636. [Google Scholar] [CrossRef]
- Messmer, M.; Wake, L.; Tsai, H.-L.; Jones, R.J.; Varadhan, R.; Wagner-Johnston, N. Large Granular Lymphocytosis With Cytopenias After Allogeneic Blood or Marrow Transplantation: Clinical Characteristics and Response to Immunosuppressive Therapy. Transplant. Cell Ther. 2021, 27, 260.e1–260.e6. [Google Scholar] [CrossRef]
- Hidalgo Lopez, J.E.; Yabe, M.; Carballo-Zarate, A.A.; Wang, S.A.; Jorgensen, J.L.; Ahmed, S.; Lee, J.; Li, S.; Schlette, E.; McDonnell, T.; et al. Donor-Derived T-Cell Large Granular Lymphocytic Leukemia in a Patient With Peripheral T-Cell Lymphoma. J. Natl. Compr. Canc. Netw. 2016, 14, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolstra, H.; Preijers, F.; Van de Wiel-van Kemenade, E.; Schattenberg, A.; Galama, J.; de Witte, T. Expansion of CD8+CD57+ T Cells after Allogeneic BMT Is Related with a Low Incidence of Relapse and with Cytomegalovirus Infection. Br. J. Haematol. 1995, 90, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.C.; Moss, P.A.; Frodsham, P.; Lehner, P.J.; Bell, J.I.; Borysiewicz, L.K. CD8highCD57+ T Lymphocytes in Normal, Healthy Individuals Are Oligoclonal and Respond to Human Cytomegalovirus. J. Immunol. 1995, 155, 5046–5056. [Google Scholar] [PubMed]
- Mendes, A.V.A.; Kallas, E.G.; Benard, G.; Pannuti, C.S.; Menezes, R.; Dulley, F.L.; Evans, T.G.; Salomão, R.; Machado, C.M. Impact of Cytomegalovirus and Grafts versus Host Disease on the Dynamics of CD57+CD28-CD8+ T Cells after Bone Marrow Transplant. Clinics 2008, 63, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Loughran, T.P.; Zambello, R.; Ashley, R.; Guderian, J.; Pellenz, M.; Semenzato, G.; Starkebaum, G. Failure to Detect Epstein-Barr Virus DNA in Peripheral Blood Mononuclear Cells of Most Patients with Large Granular Lymphocyte Leukemia. Blood 1993, 81, 2723–2727. [Google Scholar] [CrossRef] [Green Version]
- Narumi, H.; Kojima, K.; Matsuo, Y.; Shikata, H.; Sekiya, K.; Niiya, T.; Bando, S.; Niiya, H.; Azuma, T.; Yakushijin, Y.; et al. T-Cell Large Granular Lymphocytic Leukemia Occurring after Autologous Peripheral Blood Stem Cell Transplantation. Bone Marrow Transplant. 2004, 33, 99–101. [Google Scholar] [CrossRef] [Green Version]
- Sajeva, M.R.; Greco, M.M.; Cascavilla, N.; D’Arena, G.; Scalzulli, P.; Melillo, L.; Minervini, M.M.; Bonini, A.; Di Mauro, L.; Carotenuto, M.; et al. Effective Autologous Peripheral Blood Stem Cell Transplantation in Plasma Cell Leukemia Followed by T-Large Granular Lymphocyte Expansion: A Case Report. Bone Marrow Transplant. 1996, 18, 225–227. [Google Scholar]
- Taylor, A.L.; Marcus, R.; Bradley, J.A. Post-Transplant Lymphoproliferative Disorders (PTLD) after Solid Organ Transplantation. Crit. Rev. Oncol. Hematol. 2005, 56, 155–167. [Google Scholar] [CrossRef]
- Morecki, S.; Gelfand, Y.; Nagler, A.; Or, R.; Naparstek, E.; Varadi, G.; Engelhard, D.; Akerstein, A.; Slavin, S. Immune Reconstitution Following Allogeneic Stem Cell Transplantation in Recipients Conditioned by Low Intensity vs Myeloablative Regimen. Bone Marrow Transplant. 2001, 28, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.S.; Quesada, A.E.; Goswami, M.; Johnston, P.K.; Brown, R.E.; Jaso, J.M. Phosphorylated STAT3 Expression in Hematopoietic Stem Cell Transplant-Associated Large Granular Lymphocytic Leukemia. Bone Marrow Transplant. 2016, 51, 741–743. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Cohort, Year | Bareu, 2010 [12] | Sanikommu, 2018 [14] | Dong, 2021 [13] |
|---|---|---|---|
| N of patients | 229 | 204 | 319 |
| Median age, y.o. | 59 | 63 | 65 |
| Sex, N females (%) | 125 (54.6%) | 94 (46.1%) | 149 (46.7%) |
| LGLL characterization | |||
| LGL count, mean (×109/L) | 1.71 | 1.74 | 0.94 |
| LGL immunophenotype | |||
| T-LGLL, N (%) | 201 (87.8%) | 183 (90.0%) | 295 (92.5%) |
| NK-LGLL, N (%) | 28 (12.2%) | 21 (10.0%) | 24 (7.5%) |
| STAT3 mutation, N (%) | - | 66/183 (36.0%) | 10/25 (40.0%) |
| Cytopenia(s) | |||
| Neutropenia, N (%) | 135 (59.0%) | 93 (46.0%) | 131 (41.1%) |
| Neutropenia < 0.5 × 109/L, N (%) | 56 (24.5%) | 36 (17.0%) | 4 (16.9%) |
| Anemia, N (%) | 55 (26.3%) | 79 (40.0%) | 132 (41.4%) |
| AIHA (DAT+), N (%) | 10 (4.4%) | - | 13 (4.1%) |
| Thrombocytopenia, N (%) | 40 (17.5%) | 59 (30.0%) | 83 (26.0%) |
| ITP, N (%) | 1 (0.4%) | - | 16 (5.0%) |
| Splenomegaly, N (%) | 55 (24.0%) | 49 (24.0%) | 91 (28.5%) |
| Recurrent infections, N (%) | 51 (22.3%) | - | - |
| Serologic autoimmune markers | |||
| Rheumatoid factor, N (%) | 33/79 (41.8%) | - | 41/106 (38.7%) |
| Antinuclear antibodies, N (%) | 39/78 (50.0%) | - | 25/114 (21.9%) |
| Associated autoimmune disease | |||
| Autoimmune disease, all N (%) | 74 (32.3%) | 51 (25.0%) | 83 (26.0%) |
| Rheumatoid arthritis, N (%) | 38 (16.6%) | 31 (15.2%) | 51 (16.0%) |
| Associated neoplasms | |||
| Hematological neoplasm, N (%) | 22 (9.6%) | 39 (19.1%) | 59 (18.5%) |
| Solid tumor, N (%) | 10 (4.4%) | 30 (14.7%) | 66 (20.7%) |
| Need for treatment, N (%) | 100 (44.0%) | 118 (58.0%) | 181 (56.7%) |
| Rheumatic Diseases | Prevalence | Reference |
|---|---|---|
| Rheumatoid arthritis | 11–36% | [6,11,12,13,14] |
| Felty’s syndrome | ~5% | [11,12,13] |
| Sjögren’s syndrome (and subclinical forms) | 2–27% | [12,42] |
| Systemic lupus erythematosus | ~2% | [43,44,45,46] |
| Systemic vasculitis | 2–3% | [47] |
| Behçet disease | Rare | [48] |
| Polymyalgia rheumatica | Rare | [11,13] |
| Rhizomelic pseudo polyarthritis | Rare | [12] |
| Inflammatory arthritis, not otherwise specified | Rare | [12] |
| Inclusion body myositis | Rare | [49] |
| Organ-specific autoimmune disease | ||
| Endocrinopathies | ||
| Hashimoto’s thyroiditis (and subclinical forms) | 2–14% | [11,12,50] |
| Grave’s disease | Rare | [51] |
| Cushing disease | Rare | [51] |
| Polyglandular autoimmune syndrome | Rare | [12,51] |
| Gastrointestinal tract diseases | ||
| Inflammatory bowel disease | 2–4% | [11,12,13] |
| Autoimmune gastritis (pernicious anemia) | Rare | [11] |
| Celiac disease | Rare | [12] |
| Neurologic diseases | ||
| Polyneuritis | 2–3% | [12,47] |
| Multiple sclerosis | Rare | [52] |
| Organ-specific vascular diseases | ||
| Precapillary pulmonary hypertension | <0.5% | [53] |
| Glomerulonephritis | Rare | [12,47] |
| Cutaneous inflammatory diseases | ||
| Leukocytoclastic vasculitis | Rare | [12,54] |
| Pyoderma gangrenosum | Rare | [55] |
| Acquired coagulopathy | ||
| Acquired hemophilia A (anti-FVIII) | Rare | [56,57] |
| Acquired hypofibrinogenemia (anti-FGN) | Rare | [58] |
| Multiple coagulation factor inhibitors | Rare | [59] |
| Study | Global Cohort (N) | Frequency of Concomitant Disease | STAT3 Sequencing Method | Mutation in STAT3 | Frequency of STAT3 Mutation (in Concomitant Cases) | Other Altered Genes |
|---|---|---|---|---|---|---|
| Saunthararajah et al. (2001) [79] | 100 | (9/100) 9% | NA | NA | NA | NA |
| Jerez et al. (2013) [82] | 367 | (24/367) 6.5% | ARMS-PCR | D661Y/Y640F | (9/24) 37.5% | NRAS (50%) |
| Durrani et al. (2020) [83] | 240 | (13/240) 5.4% | NGS | D661V | (2/13) 15.4% | CHIP (TET2, ASXL1 or DNMT3A, among others) |
| Komrokji et al. (2020) [84] | 1177 | (322/1177) 27.4% | NA | NA | NA | CHIP-related genes (TET2, SF3B1, ASXL1, among others) |
| Ai et al. (2021) [85] | 721 | (10/721) 1.4% | NGS | NA | (2/10) 20% | ASXL1 (30%) and STAG2 (30%) |
| Reference | (N) | HSCT Type | LGL Expansion Assessed (Definition) | LGL Expansion Incidence %, N | Time from Transplant to LGL Expansion Detection | TCR Clonality | LGL Count (109/L) Median (Range) | Impact on Outcome (In Multivariate Analysis) | Associations with Post-AlloHSCT Events | Symptoms/ Signs Attributed to LGL Expansion |
|---|---|---|---|---|---|---|---|---|---|---|
| Mohty. 2002. [172] | 201 | Allo | T-LGL lymphocytosis (≥2.0 × 109/L) | 3% (6) | 295 d. (75–450) | 33% oligo./ 67% poly. | 2.3 (2.0–4.1) | NA | CMV reactivation RIC | 4 of 6 autoimmune manifestations |
| Nann-Rütti. 2012. [173] | 215 | Allo | T-LGL lymphocytosis (>3 mo.) (≥3.0 × 109/L) & abnormal CD4/CD8 ratio (<1.0 or >1.5) | 7% (14) | 25 mo. (3–150) | 36% clonal/ 7% oligo./ 57% poly. | 2.9 (1.3–11.5) | NA | CMV reactivation | 5/14 ANA+ 2/14 Polyclonal hypergamma. 3/14 M-comp. |
| Kim. 2013. [174] | 418 | Allo | LGL lymphocytosis 2 of 3: (1) ≥3.0 × 109/L; (2) > 30%LGL; (3) Clonal TCR | 18.4% (73 T-LGL, 2 NK and 2 mixed NK/T) | 312 d. (26–1840) | 90% Clonal | 1.6 (range 0.6–2.7) | ↑ OS ↓ NRM ↓ Relapse | CMV reactivation cGVHD | 4/77 pancytopenia 2/77 proteinuria |
| Gill. 2012. [175] | 1675 | Allo (N = 1246) | LGL Leukemia | 0.3% (4) | 9 mo.(3–24) | Clonal | 3 (1.9–4.7) | NA | NA | Asymptomatic |
| Auto (N = 429) | LGL Leukemia | 0.7% (3) | 28 mo. (6–72) | Clonal | 2.4 (1.6–2.9) | NA | NA | |||
| Muñoz-Ballester. 2016. [176] | 154 | Allo | LGL lymphocytosis (>6 mo.) Relative (T CD8+/CD4+ ratio >1.5) | 49% (75 T-LGL) | NA | 77% clonal/16% oligo./7% poly. | NA | ⊜ OR ⊜ Relapse | aGVHD, CMV viremia | 1 of 75 (neutropenia) |
| LGL lymphocytosis (>6 mo.) Absolute (≥2.0 × 109/L) | 9% (14 T-LGL) | NA | All clonal | 2.6 (2.2–3.9) | ⊜ OR ↓ Relapse | cGVHD | 1 AIHA case, 50% MC and/or ANA+ | |||
| Martell. 2017. [177] | 826 | Allo | LGL lymphocytosis (>2–3 mo.) (≥3.0 × 109/L) | 14.5% (40 T-LGL, 14 mixed T/NK, 2 NK) | 306 d. (18–2175) | Clonal | 3.7 ± 0.08 Mean ± SD | ↑ OS ⊜ Relapse ↓ NRM | CMV viremia aGVHD cGVHD | 1 out of 121 (Anemia) |
| Le Bris. 2017. [178] | 85 | UCB Allo | LGL lymphocytosis (≥3.0 × 109/L) | 8.5% (9 T-LGL/1 NK) | 12.6 mo. (1.4–49) | 48% clonal/28% oligo./28%poly. | 4.8 (4–9.2) | NA | CMV reactivation | NA |
| Zhao. 2020. [179] | 359 | Allo | T-LGL lymphocytosis | 4.7% (17 T-LGL) | 175 d. (25–763) | 82% clonal | NA | ↑ OS ↓ NRM ↓ Relapse | CMV Reactivation EB viremia aGVHD | Asymptomatic 7 of 17 autoimmune-related parameters |
| Messmer. 2021- [180] | 1930 | Allo | LGL lymphocytosis in patients with unexplained cytopenias | 3.6% (65 T-LGL, 5 NK-LGL) | 6.4 mo. (1.4–81) | 22% clonal/53% oligo/4% poly/and 20% indeterminate | 1.1 (0.3–6.8) | ⊜ OR ⊜ Relapse | CMV viremia | 30 treated with IST (83% corticosteroids): effective at improving LGL associated neutropenia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bravo-Pérez, C.; Carrillo-Tornel, S.; García-Torralba, E.; Jerez, A. Persistent Large Granular Lymphocyte Clonal Expansions: “The Root of Many Evils”—And of Some Goodness. Cancers 2022, 14, 1340. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051340
Bravo-Pérez C, Carrillo-Tornel S, García-Torralba E, Jerez A. Persistent Large Granular Lymphocyte Clonal Expansions: “The Root of Many Evils”—And of Some Goodness. Cancers. 2022; 14(5):1340. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051340
Chicago/Turabian StyleBravo-Pérez, Carlos, Salvador Carrillo-Tornel, Esmeralda García-Torralba, and Andrés Jerez. 2022. "Persistent Large Granular Lymphocyte Clonal Expansions: “The Root of Many Evils”—And of Some Goodness" Cancers 14, no. 5: 1340. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051340