
CK1 Is a Druggable Regulator of Microtubule Dynamics and Microtubule-Associated Processes

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
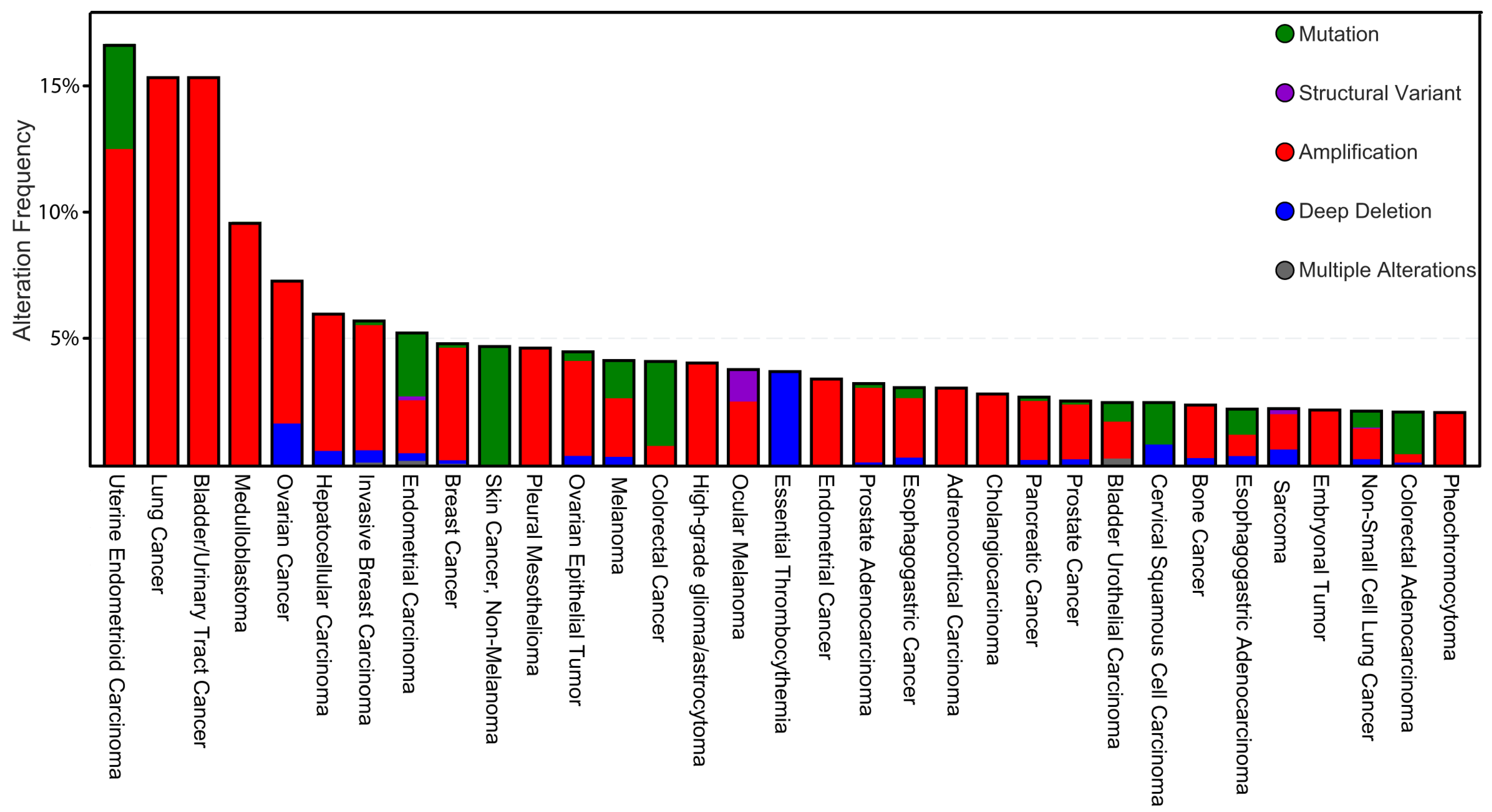
2. Participation of CK1 in Tumorigenesis and Tumor Progression
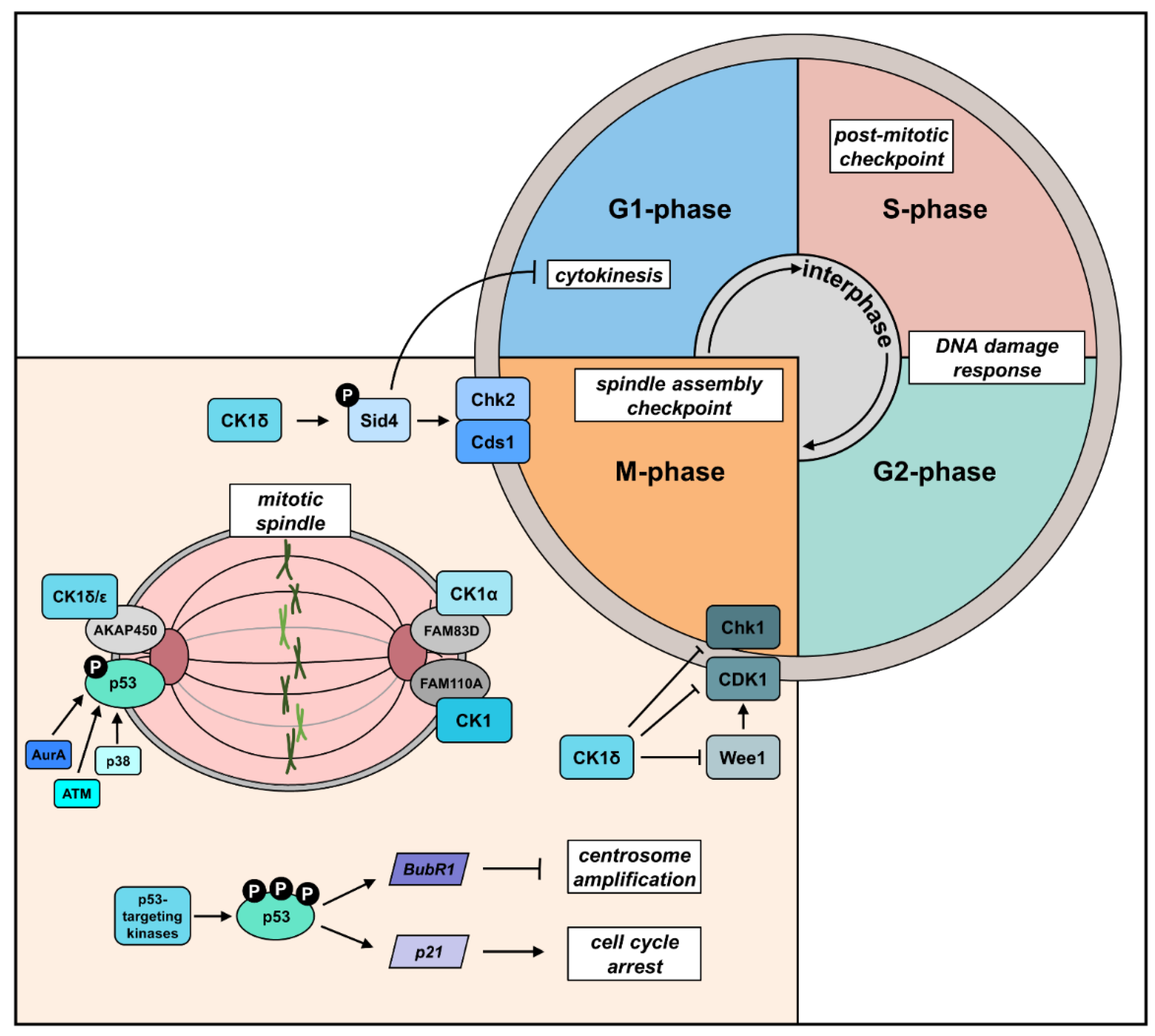
3. The Role of CK1 in Cell Cycle Progression

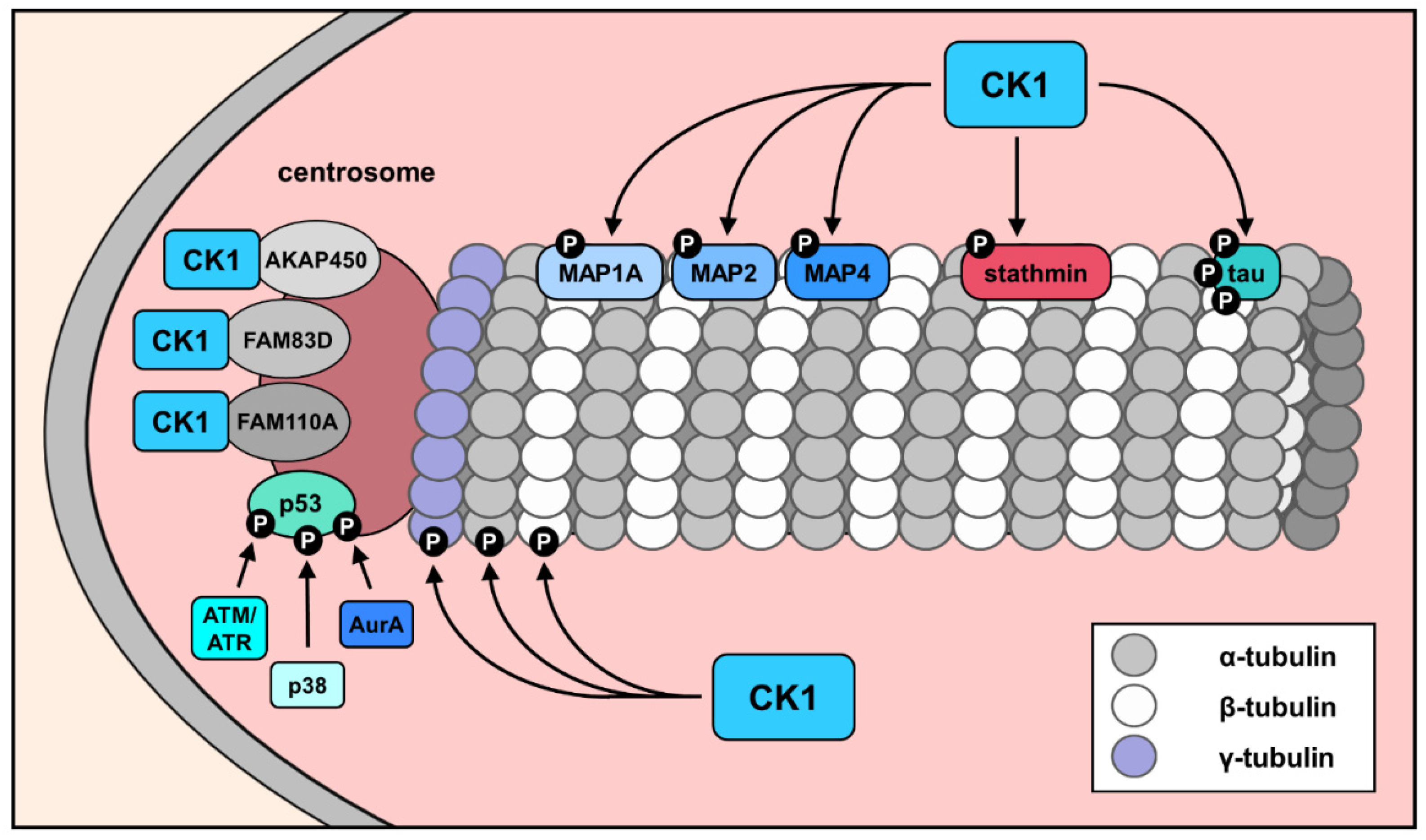
4. The Contribution of CK1 to the Modulation of Cytoskeleton Components
5. CK1-Associated Functions in Microtubule Transport
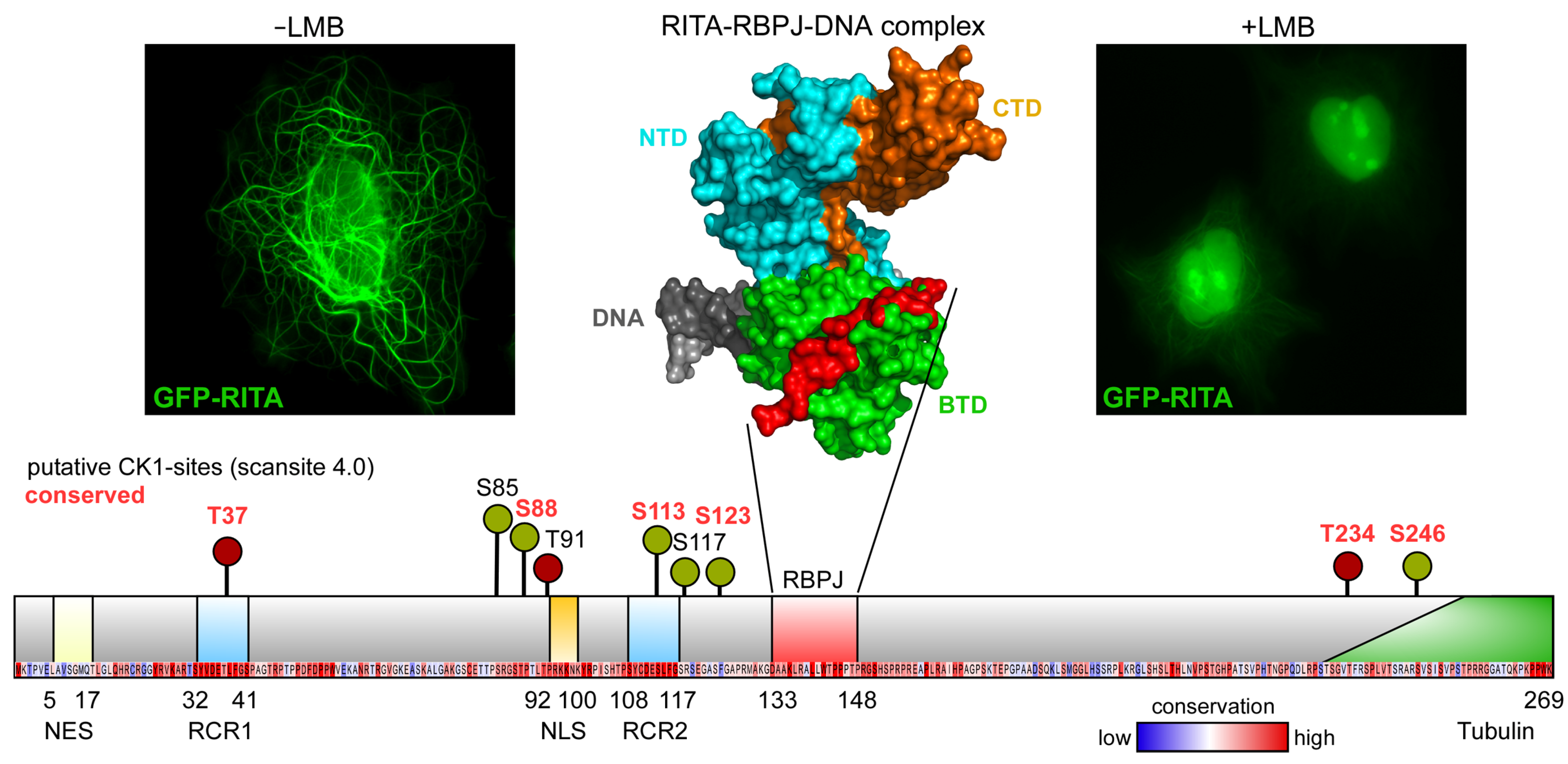
6. The MAP RITA as a Putative Target for CK1
7. Addressing Inhibition of Cell Division in Cancer Therapy
7.1. Microtubule-Targeting Agents
7.2. Inhibitors Targeting the Mitotic Kinome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Target | Molecular/Therapeutic Effect | Tumor Entity | Investigation Phase | Ref. |
|---|---|---|---|---|---|
| MSA-Taxol-domain binder | |||||
| Paclitaxel | β-tubulin | Increase the lateral interactions between the tubulin heterodimers resulting in increased polymerization and stabilization of microtubules | Ovarian and breast cancer | Approved | [160] |
| Ixabepilone | Breast cancer | Approved | [126,127] | ||
| MDA-Vinca-domain binder | |||||
| Vincristine | β-tubulin | Lead to microtubule depolymerization by decreasing or inhibiting longitudinal interactions between tubulin heterodimers | Breast cancer, lymphomas | Approved | [161] |
| Vinblastine | Lymphomas, solid tumors | Approved | [161] | ||
| MDA-Colchicine-domain binder | |||||
| Ombra- bulin | Interface of α-/β-tubulin | Lead to microtubule depolymerization by decreasing or inhibiting longitudinal interactions between tubulin heterodimers | Ovarian cancer | Stopped in phase III | [124] |
| ABT-751 | Lung cancer, colon cancer | Phase II | [128,129,162] | ||
| Protein kinase inhibitors | |||||
| Alisertib | AurA | Induce cell cycle arrest in G2/M phase, apoptosis, and autophagy; prevents AurA-induced stabilization of N-Myc | Leukemia, solid tumors | Phase III | [134,135,136,137,138,139,140,141] |
| AurkinA | AurA | Bind to the Tpx2-binding surface of AurA and consequently displacing AurA from the mitotic spindle | - | Preclinical | [146] |
| Barasertib | AurB | Decrease histone phosphorylation resulting in accumulation of aneuploidy cells and induction of apoptosis; associated with stimulation of ROS | Leukemia, solid tumors | Phase II | [142,143,144,145] |
| Volasertib | Plk1 | Arrests cells in the G2/M phase and subsequently induces apoptosis | Leukemia | Phase III | [147,148,149] |
| CK1-specific inhibitors | |||||
| IC261 | Initially designed for CK1; tubulin | Binds to tubulin resulting in direct inhibition of microtubule polymerization | Pancreatic cancer | Preclinical | [37,46,154,155,156,157,158,159] |
| D4476 | CK1α/δ | Inhibition of CK1α/δ activity; sensitizes colorectal cancer cells to 5-fluorouracil | Colorectal cancer | Preclinical | [163] |
| PF-670462 | CK1δ/ε | Selective inhibition of CK1δ/ε activity | Leukemia | Preclinical | [164,165] |
| SR-3029 | CK1δ/ε | Inhibition of overexpressed CK1δ/ε | Breast cancer, skin tumor | Preclinical | [166,167] |
| IWP-2/IWP-4 | CK1δ | Selective inhibition of CK1δ | Pancreatic, colon cancer cell lines | Preclinical | [168] |
| BTX-A51 | CK1α/δ/ε; CDK7/9 | Inhibition of CK1α and activation of p53-dependent cell death; inhibition of CDK7/9 | Leukemia | Phase I | [169] |
| Lenalidomide | CRL4CRBN E3 ubiquitin ligase; indirectly CK1α | Induces ubiquitination and degradation of CK1α | Leukemia | Approved | [170] |
| Umbralisib | PI3Kδ; CK1ε | Block the phosphorylation of eukaryotic translation initiation factor 4E binding protein (4E-BPI), leading to the inhibition of c-Myc translation and cell death | Lymphoma | Approved | [171,172] |
7.3. Combination of MTAs with Additional Anticancer Agents: Advantages of Dual-Specific Inhibitors
7.4. Modulation of the CK1 Activity with Biologicals
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Janke, C.; Magiera, M.M. The tubulin code and its role in controlling microtubule properties and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Goodson, H.V.; Jonasson, E.M. Microtubules and Microtubule-Associated Proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a022608. [Google Scholar] [CrossRef] [PubMed]
- Petry, S.; Vale, R.D. Microtubule nucleation at the centrosome and beyond. Nat. Cell Biol. 2015, 17, 1089–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornens, M. Centrosome organization and functions. Curr. Opin. Struct. Biol. 2021, 66, 199–206. [Google Scholar] [CrossRef]
- Bettencourt-Dias, M.; Glover, D.M. Centrosome biogenesis and function: Centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol. 2007, 8, 451–463. [Google Scholar] [CrossRef]
- Petry, S. Mechanisms of Mitotic Spindle Assembly. Annu. Rev. Biochem. 2016, 85, 659–683. [Google Scholar] [CrossRef] [Green Version]
- Crncec, A.; Hochegger, H. Triggering mitosis. FEBS Lett. 2019, 593, 2868–2888. [Google Scholar] [CrossRef] [Green Version]
- Dephoure, N.; Zhou, C.; Villén, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.T.; Poon, R.Y.C. How protein kinases co-ordinate mitosis in animal cells. Biochem. J. 2011, 435, 17–31. [Google Scholar] [CrossRef]
- Magnaghi-Jaulin, L.; Eot-Houllier, G.; Gallaud, E.; Giet, R. Aurora A Protein Kinase: To the Centrosome and Beyond. Biomolecules 2019, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef]
- Ahmed, A.; Shamsi, A.; Mohammad, T.; Hasan, G.M.; Islam, A.; Hassan, M.I. Aurora B kinase: A potential drug target for cancer therapy. J. Cancer Res. Clin. Oncol. 2021, 147, 2187–2198. [Google Scholar] [CrossRef] [PubMed]
- Borah, N.A.; Reddy, M.M. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules 2021, 26, 1981. [Google Scholar] [CrossRef] [PubMed]
- Habedanck, R.; Stierhof, Y.-D.; Wilkinson, C.J.; Nigg, E.A. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 2005, 7, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Maniswami, R.R.; Prashanth, S.; Karanth, A.V.; Koushik, S.; Govindaraj, H.; Mullangi, R.; Rajagopal, S.; Jegatheesan, S.K. PLK4: A link between centriole biogenesis and cancer. Expert Opin. Ther. Targets 2018, 22, 59–73. [Google Scholar] [CrossRef]
- Yang, X.; Li, H.; Zhou, Z.; Wang, W.-H.; Deng, A.; Andrisani, O.; Liu, X. Plk1-mediated phosphorylation of Topors regulates p53 stability. J. Biol. Chem. 2009, 284, 18588–18592. [Google Scholar] [CrossRef] [Green Version]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A.M. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Bloom, J.; Cross, F.R. Multiple levels of cyclin specificity in cell-cycle control. Nat. Rev. Mol. Cell Biol. 2007, 8, 149–160. [Google Scholar] [CrossRef]
- Melo-Hanchuk, T.D.; Slepicka, P.F.; Meirelles, G.V.; Basei, F.L.; Lovato, D.V.; Granato, D.C.; Pauletti, B.A.; Domingues, R.R.; Leme, A.F.P.; Pelegrini, A.L.; et al. NEK1 kinase domain structure and its dynamic protein interactome after exposure to Cisplatin. Sci. Rep. 2017, 7, 5445. [Google Scholar] [CrossRef]
- Melo-Hanchuk, T.D.; Slepicka, P.F.; Pelegrini, A.L.; Menck, C.F.M.; Kobarg, J. NEK5 interacts with topoisomerase IIβ and is involved in the DNA damage response induced by etoposide. J. Cell. Biochem. 2019, 120, 16853–16866. [Google Scholar] [CrossRef]
- O’Regan, L.; Fry, A.M. The Nek6 and Nek7 protein kinases are required for robust mitotic spindle formation and cytokinesis. Mol. Cell. Biol. 2009, 29, 3975–3990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, E.E.; Meirelles, G.V.; Godoy, B.B.; Perez, A.M.; Smetana, J.H.C.; Doxsey, S.J.; McComb, M.E.; Costello, C.E.; Whelan, S.A.; Kobarg, J. Characterization of the human NEK7 interactome suggests catalytic and regulatory properties distinct from those of NEK6. J. Proteome Res. 2014, 13, 4074–4090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser, S.L.; Sahota, N.K.; Pelletier, L.; Morrison, C.G.; Fry, A.M. Nek5 promotes centrosome integrity in interphase and loss of centrosome cohesion in mitosis. J. Cell Biol. 2015, 209, 339–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meirelles, G.V.; Perez, A.M.; de Souza, E.E.; Basei, F.L.; Papa, P.F.; Melo Hanchuk, T.D.; Cardoso, V.B.; Kobarg, J. “Stop Ne(c)king around”: How interactomics contributes to functionally characterize Nek family kinases. World J. Biol. Chem. 2014, 5, 141–160. [Google Scholar] [CrossRef]
- Schittek, B.; Sinnberg, T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol. Cancer 2014, 13, 231. [Google Scholar] [CrossRef] [Green Version]
- Knippschild, U.; Gocht, A.; Wolff, S.; Huber, N.; Löhler, J.; Stöter, M. The casein kinase 1 family: Participation in multiple cellular processes in eukaryotes. Cell. Signal. 2005, 17, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikezu, S.; Ikezu, T. Tau-tubulin kinase. Front. Mol. Neurosci. 2014, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Nayak, A.; Melendez, R.A.; Wynn, D.T.; Jackson, J.; Lee, E.; Ahmed, Y.; Robbins, D.J. Casein Kinase 1α as a Regulator of Wnt-Driven Cancer. Int. J. Mol. Sci. 2020, 21, 5940. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-C.; Chen, M.-C.; Hsieh, T.-H.; Liou, J.-P.; Chen, C.-H. CK1δ as a potential therapeutic target to treat bladder cancer. Aging 2020, 12, 5764–5780. [Google Scholar] [CrossRef] [PubMed]
- Bowman, B.M.; Sebolt, K.A.; Hoff, B.A.; Boes, J.L.; Daniels, D.L.; Heist, K.A.; Galbán, C.J.; Patel, R.M.; Zhang, J.; Beer, D.G.; et al. Phosphorylation of FADD by the kinase CK1α promotes KRASG12D-induced lung cancer. Sci. Signal. 2015, 8, ra9. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhou, L.; Wang, Y.; Peng, Q.; Li, H.; Zhang, X.; Su, Z.; Song, J.; Sun, Q.; Sayed, S.; et al. The CK1δ/ε-AES axis regulates tumorigenesis and metastasis in colorectal cancer. Theranostics 2021, 11, 4421–4435. [Google Scholar] [CrossRef]
- Shin, S.; Wolgamott, L.; Roux, P.P.; Yoon, S.-O. Casein kinase 1ε promotes cell proliferation by regulating mRNA translation. Cancer Res. 2014, 74, 201–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockschmidt, C.; Hirner, H.; Huber, N.; Eismann, T.; Hillenbrand, A.; Giamas, G.; Radunsky, B.; Ammerpohl, O.; Bohm, B.; Henne-Bruns, D.; et al. Anti-apoptotic and growth-stimulatory functions of CK1 delta and epsilon in ductal adenocarcinoma of the pancreas are inhibited by IC261 in vitro and in vivo. Gut 2008, 57, 799–806. [Google Scholar] [CrossRef]
- Kaucká, M.; Plevová, K.; Pavlová, S.; Janovská, P.; Mishra, A.; Verner, J.; Procházková, J.; Krejcí, P.; Kotasková, J.; Ovesná, P.; et al. The planar cell polarity pathway drives pathogenesis of chronic lymphocytic leukemia by the regulation of B-lymphocyte migration. Cancer Res. 2013, 73, 1491–1501. [Google Scholar] [CrossRef] [Green Version]
- Janovská, P.; Normant, E.; Miskin, H.; Bryja, V. Targeting Casein Kinase 1 (CK1) in Hematological Cancers. Int. J. Mol. Sci. 2020, 21, 9026. [Google Scholar] [CrossRef]
- Spinello, Z.; Fregnani, A.; Quotti Tubi, L.; Trentin, L.; Piazza, F.; Manni, S. Targeting Protein Kinases in Blood Cancer: Focusing on CK1α and CK2. Int. J. Mol. Sci. 2021, 22, 3716. [Google Scholar] [CrossRef]
- Xu, P.; Ianes, C.; Gärtner, F.; Liu, C.; Burster, T.; Bakulev, V.; Rachidi, N.; Knippschild, U.; Bischof, J. Structure, regulation, and (patho-)physiological functions of the stress-induced protein kinase CK1 delta (CSNK1D). Gene 2019, 715, 144005. [Google Scholar] [CrossRef] [PubMed]
- Knippschild, U.; Krüger, M.; Richter, J.; Xu, P.; García-Reyes, B.; Peifer, C.; Halekotte, J.; Bakulev, V.; Bischof, J. The CK1 Family: Contribution to Cellular Stress Response and Its Role in Carcinogenesis. Front. Oncol. 2014, 4, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrend, L.; Milne, D.M.; Stöter, M.; Deppert, W.; Campbell, L.E.; Meek, D.W.; Knippschild, U. IC261, a specific inhibitor of the protein kinases casein kinase 1-delta and -epsilon, triggers the mitotic checkpoint and induces p53-dependent postmitotic effects. Oncogene 2000, 19, 5303–5313. [Google Scholar] [CrossRef] [Green Version]
- Behrend, L.; Stöter, M.; Kurth, M.; Rutter, G.; Heukeshoven, J.; Deppert, W.; Knippschild, U. Interaction of casein kinase 1 delta (CK1δ) with post-Golgi structures, microtubules and the spindle apparatus. Eur. J. Cell Biol. 2000, 79, 240–251. [Google Scholar] [CrossRef]
- Sillibourne, J.E.; Milne, D.M.; Takahashi, M.; Ono, Y.; Meek, D.W. Centrosomal anchoring of the protein kinase CK1delta mediated by attachment to the large, coiled-coil scaffolding protein CG-NAP/AKAP450. J. Mol. Biol. 2002, 322, 785–797. [Google Scholar] [CrossRef]
- Stöter, M.; Bamberger, A.-M.; Aslan, B.; Kurth, M.; Speidel, D.; Löning, T.; Frank, H.-G.; Kaufmann, P.; Löhler, J.; Henne-Bruns, D.; et al. Inhibition of casein kinase I delta alters mitotic spindle formation and induces apoptosis in trophoblast cells. Oncogene 2005, 24, 7964–7975. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.E.; Chen, J.-S.; Gould, K.L. CK1 is required for a mitotic checkpoint that delays cytokinesis. Curr. Biol. 2013, 23, 1920–1926. [Google Scholar] [CrossRef] [Green Version]
- Greer, Y.E.; Westlake, C.J.; Gao, B.; Bharti, K.; Shiba, Y.; Xavier, C.P.; Pazour, G.J.; Yang, Y.; Rubin, J.S. Casein kinase 1δ functions at the centrosome and Golgi to promote ciliogenesis. Mol. Biol. Cell 2014, 25, 1629–1640. [Google Scholar] [CrossRef] [Green Version]
- Penas, C.; Govek, E.-E.; Fang, Y.; Ramachandran, V.; Daniel, M.; Wang, W.; Maloof, M.E.; Rahaim, R.J.; Bibian, M.; Kawauchi, D.; et al. Casein kinase 1δ is an APC/C(Cdh1) substrate that regulates cerebellar granule cell neurogenesis. Cell Rep. 2015, 11, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Penas, C.; Ramachandran, V.; Simanski, S.; Lee, C.; Madoux, F.; Rahaim, R.J.; Chauhan, R.; Barnaby, O.; Schurer, S.; Hodder, P.; et al. Casein kinase 1δ-dependent Wee1 protein degradation. J. Biol. Chem. 2014, 289, 18893–18903. [Google Scholar] [CrossRef] [Green Version]
- Phadnis, N.; Cipak, L.; Polakova, S.; Hyppa, R.W.; Cipakova, I.; Anrather, D.; Karvaiova, L.; Mechtler, K.; Smith, G.R.; Gregan, J. Casein Kinase 1 and Phosphorylation of Cohesin Subunit Rec11 (SA3) Promote Meiotic Recombination through Linear Element Formation. PLoS Genet. 2015, 11, e1005225. [Google Scholar] [CrossRef] [Green Version]
- Sakuno, T.; Watanabe, Y. Phosphorylation of cohesin Rec11/SA3 by casein kinase 1 promotes homologous recombination by assembling the meiotic chromosome axis. Dev. Cell 2015, 32, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.Y.; Alonso-Nuñez, M.; Grallert, A.; Tanaka, K.; Connolly, Y.; Smith, D.L.; Hagan, I.M. Dialogue between centrosomal entrance and exit scaffold pathways regulates mitotic commitment. J. Cell Biol. 2017, 216, 2795–2812. [Google Scholar] [CrossRef]
- Greer, Y.E.; Gao, B.; Yang, Y.; Nussenzweig, A.; Rubin, J.S. Lack of Casein Kinase 1 Delta Promotes Genomic Instability—The Accumulation of DNA Damage and Down-Regulation of Checkpoint Kinase 1. PLoS ONE 2017, 12, e0170903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoekstra, M.F.; Liskay, R.M.; Ou, A.C.; DeMaggio, A.J.; Burbee, D.G.; Heffron, F. HRR25, a putative protein kinase from budding yeast: Association with repair of damaged DNA. Science 1991, 253, 1031–1034. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Moritz, M.; Han, X.; Giddings, T.H.; Lyon, A.; Kollman, J.; Winey, M.; Yates, J.; Agard, D.A.; Drubin, D.G.; et al. Interaction of CK1δ with γTuSC ensures proper microtubule assembly and spindle positioning. Mol. Biol. Cell 2015, 26, 2505–2518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shi, Q.; Varia, S.N.; Xing, S.; Klett, B.M.; Cook, L.A.; Herman, P.K. The Activity-Dependent Regulation of Protein Kinase Stability by the Localization to P-Bodies. Genetics 2016, 203, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Butler, A.M.; Shi, Q.; Xing, S.; Herman, P.K. P-Body Localization of the Hrr25/Casein Kinase 1 Protein Kinase Is Required for the Completion of Meiosis. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, Z.C.; Guillen, R.X.; Gould, K.L. The kinase domain of CK1 enzymes contains the localization cue essential for compartmentalized signaling at the spindle pole. Mol. Biol. Cell 2018, 29, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Zyss, D.; Ebrahimi, H.; Gergely, F. Casein kinase I delta controls centrosome positioning during T cell activation. J. Cell Biol. 2011, 195, 781–797. [Google Scholar] [CrossRef]
- Bischof, J.; Randoll, S.-J.; Süßner, N.; Henne-Bruns, D.; Pinna, L.A.; Knippschild, U. CK1δ kinase activity is modulated by Chk1-mediated phosphorylation. PLoS ONE 2013, 8, e68803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockman, J.L.; Gross, S.D.; Sussman, M.R.; Anderson, R.A. Cell cycle-dependent localization of casein kinase I to mitotic spindles. Proc. Natl. Acad. Sci. USA 1992, 89, 9454–9458. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lu, A.; Zhou, H.-X.; Sun, R.; Zhao, J.; Zhou, C.-J.; Shen, J.-P.; Wu, S.-N.; Liang, C.-G. Casein kinase 1 alpha regulates chromosome congression and separation during mouse oocyte meiotic maturation and early embryo development. PLoS ONE 2013, 8, e63173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulcher, L.J.; Sapkota, G.P. Mitotic kinase anchoring proteins: The navigators of cell division. Cell Cycle 2020, 19, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Fulcher, L.J.; He, Z.; Mei, L.; Macartney, T.J.; Wood, N.T.; Prescott, A.R.; Whigham, A.J.; Varghese, J.; Gourlay, R.; Ball, G.; et al. FAM83D directs protein kinase CK1α to the mitotic spindle for proper spindle positioning. EMBO Rep. 2019, 20, e47495. [Google Scholar] [CrossRef]
- Fulcher, L.J.; Bozatzi, P.; Tachie-Menson, T.; Wu, K.Z.L.; Cummins, T.D.; Bufton, J.C.; Pinkas, D.M.; Dunbar, K.; Shrestha, S.; Wood, N.T.; et al. The DUF1669 domain of FAM83 family proteins anchor casein kinase 1 isoforms. Sci. Signal. 2018, 11, eaao2341. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.Z.L.; Jones, R.A.; Tachie-Menson, T.; Macartney, T.J.; Wood, N.T.; Varghese, J.; Gourlay, R.; Soares, R.F.; Smith, J.C.; Sapkota, G.P. Pathogenic FAM83G palmoplantar keratoderma mutations inhibit the PAWS1:CK1α association and attenuate Wnt signalling. Wellcome Open Res. 2019, 4, 133. [Google Scholar] [CrossRef]
- Aquino Perez, C.; Burocziova, M.; Jenikova, G.; Macurek, L. CK1-mediated phosphorylation of FAM110A promotes its interaction with mitotic spindle and controls chromosomal alignment. EMBO Rep. 2021, 22, e51847. [Google Scholar] [CrossRef]
- Servier Medical Art. SMART-Servier Medical ART. Available online: https://smart.servier.com/ (accessed on 25 January 2022).
- Brouhard, G.J.; Rice, L.M. Microtubule dynamics: An interplay of biochemistry and mechanics. Nat. Rev. Mol. Cell Biol. 2018, 19, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Gadoth, A.; Kryzer, T.J.; Fryer, J.; McKeon, A.; Lennon, V.A.; Pittock, S.J. Microtubule-associated protein 1B: Novel paraneoplastic biomarker. Ann. Neurol. 2017, 81, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Hu, Q.; Gou, M.; Liu, X.; Qin, Y.; Zhu, J.; Cai, C.; Tian, T.; Tu, Z.; Du, Y.; et al. Expression of Microtubule-Associated Proteins in Relation to Prognosis and Efficacy of Immunotherapy in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 680402. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; He, C.; Wu, A.; Zhou, J.; Wu, J. Microtubule-Associated Protein 4 Is a Prognostic Factor and Promotes Tumor Progression in Lung Adenocarcinoma. Dis. Markers 2018, 2018, 8956072. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.; Xiao, Z.; Wittau, M.; Süssner, N.; Stöter, M.; Knippschild, U. Interaction of casein kinase 1 delta (CK1 delta) with the light chain LC2 of microtubule associated protein 1A (MAP1A). Biochim. Biophys. Acta 2005, 1745, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Kuret, J.; Johnson, G.S.; Cha, D.; Christenson, E.R.; DeMaggio, A.J.; Hoekstra, M.F. Casein kinase 1 is tightly associated with paired-helical filaments isolated from Alzheimer’s disease brain. J. Neurochem. 1997, 69, 2506–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of tau protein by casein kinase-1 converts it to an abnormal Alzheimer-like state. J. Neurochem. 1995, 64, 1420–1423. [Google Scholar] [CrossRef]
- Hanger, D.P.; Byers, H.L.; Wray, S.; Leung, K.-Y.; Saxton, M.J.; Seereeram, A.; Reynolds, C.H.; Ward, M.A.; Anderton, B.H. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J. Biol. Chem. 2007, 282, 23645–23654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papin, S.; Paganetti, P. Emerging Evidences for an Implication of the Neurodegeneration-Associated Protein TAU in Cancer. Brain Sci. 2020, 10, 862. [Google Scholar] [CrossRef] [PubMed]
- Huda, M.N.; Da Kim, H.; Erdene-Ochir, E.; Kim, Y.S.; Pan, C.-H. Expression, phosphorylation, localization, and microtubule binding of tau in colorectal cell lines. Appl. Biol. Chem. 2016, 59, 807–812. [Google Scholar] [CrossRef]
- Kit, O.I.; Vodolazhsky, D.I.; Kutilin, D.S.; Enin, Y.S.; Gevorkyan, Y.A.; Zolotukhin, P.V.; Boumber, Y.; Kharin, L.V.; Panina, S.B. A Proteomics Analysis Reveals 9 Up-Regulated Proteins Associated with Altered Cell Signaling in Colon Cancer Patients. Protein J. 2017, 36, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Souter, S.; Lee, G. Microtubule-associated protein tau in human prostate cancer cells: Isoforms, phosphorylation, and interactions. J. Cell. Biochem. 2009, 108, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Wahl, A.F.; Donaldson, K.L.; Fairchild, C.; Lee, F.Y.; Foster, S.A.; Demers, G.W.; Galloway, D.A. Loss of normal p53 function confers sensitization to Taxol by increasing G2/M arrest and apoptosis. Nat. Med. 1996, 2, 72–79. [Google Scholar] [CrossRef]
- Meek, D.W. The role of p53 in the response to mitotic spindle damage. Pathol. Biol. 2000, 48, 246–254. [Google Scholar] [PubMed]
- Tarapore, P.; Fukasawa, K. Loss of p53 and centrosome hyperamplification. Oncogene 2002, 21, 6234–6240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, C.A.M.; Mesquita, M.; Cunha, A.I.; Cardoso, J.; Carapeta, S.; Laranjeira, C.; Pinto, A.E.; Pereira-Leal, J.B.; Dias-Pereira, A.; Bettencourt-Dias, M.; et al. Centrosome amplification arises before neoplasia and increases upon p53 loss in tumorigenesis. J. Cell Biol. 2018, 217, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.; Tan, B.X.; Lane, D. How the Other Half Lives: What p53 Does When It Is Not Being a Transcription Factor. Int. J. Mol. Sci. 2019, 21, 13. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Dai, T.; Xiong, H.; Lin, C.; Lin, H.; Shi, T.; Li, J. Inhibition of centriole duplication by centrobin depletion leads to p38–p53 mediated cell-cycle arrest. Cell. Signal. 2010, 22, 857–864. [Google Scholar] [CrossRef]
- Shinmura, K.; Bennett, R.A.; Tarapore, P.; Fukasawa, K. Direct evidence for the role of centrosomally localized p53 in the regulation of centrosome duplication. Oncogene 2007, 26, 2939–2944. [Google Scholar] [CrossRef] [Green Version]
- Tarapore, P.; Horn, H.F.; Tokuyama, Y.; Fukasawa, K. Direct regulation of the centrosome duplication cycle by the p53-p21Waf1/Cip1 pathway. Oncogene 2001, 20, 3173–3184. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, T.; Okuda, M.; Ma, Z.; Goorha, R.; Tsujimoto, H.; Inokuma, H.; Fukasawa, K. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol. Cell. Biol. 2005, 25, 4046–4061. [Google Scholar] [CrossRef] [Green Version]
- Ciciarello, M.; Mangiacasale, R.; Casenghi, M.; Zaira Limongi, M.; D’Angelo, M.; Soddu, S.; Lavia, P.; Cundari, E. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J. Biol. Chem. 2001, 276, 19205–19213. [Google Scholar] [CrossRef] [Green Version]
- Tarapore, P.; Tokuyama, Y.; Horn, H.F.; Fukasawa, K. Difference in the centrosome duplication regulatory activity among p53 ‘hot spot’ mutants: Potential role of Ser 315 phosphorylation-dependent centrosome binding of p53. Oncogene 2001, 20, 6851–6863. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Kaneko, S.; Yang, L.; Feldman, R.I.; Nicosia, S.V.; Chen, J.; Cheng, J.Q. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J. Biol. Chem. 2004, 279, 52175–52182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tritarelli, A.; Oricchio, E.; Ciciarello, M.; Mangiacasale, R.; Palena, A.; Lavia, P.; Soddu, S.; Cundari, E. p53 localization at centrosomes during mitosis and postmitotic checkpoint are ATM-dependent and require serine 15 phosphorylation. Mol. Biol. Cell 2004, 15, 3751–3757. [Google Scholar] [CrossRef] [Green Version]
- Mikule, K.; Delaval, B.; Kaldis, P.; Jurcyzk, A.; Hergert, P.; Doxsey, S. Loss of centrosome integrity induces p38–p53–p21-dependent G1–S arrest. Nat. Cell Biol. 2007, 9, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Suhail, T.V.; Singh, P.; Manna, T.K. Suppression of centrosome protein TACC3 induces G1 arrest and cell death through activation of p38–p53–p21 stress signaling pathway. Eur. J. Cell Biol. 2015, 94, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Knippschild, U.; Milne, D.M.; Campbell, L.E.; DeMaggio, A.J.; Christenson, E.; Hoekstra, M.F.; Meek, D.W. p53 is phosphorylated in vitro and in vivo by the delta and epsilon isoforms of casein kinase 1 and enhances the level of casein kinase 1 delta in response to topoisomerase-directed drugs. Oncogene 1997, 15, 1727–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Sale, W.S. Casein kinase I is anchored on axonemal doublet microtubules and regulates flagellar dynein phosphorylation and activity. J. Biol. Chem. 2000, 275, 18905–18912. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Zhapparova, O.; Brodsky, I.; Semenova, I.; Tirnauer, J.S.; Zaliapin, I.; Rodionov, V. CK1 activates minus-end-directed transport of membrane organelles along microtubules. Mol. Biol. Cell 2011, 22, 1321–1329. [Google Scholar] [CrossRef]
- Gokhale, A.; Wirschell, M.; Sale, W.S. Regulation of dynein-driven microtubule sliding by the axonemal protein kinase CK1 in Chlamydomonas flagella. J. Cell Biol. 2009, 186, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Wacker, S.A.; Alvarado, C.; Von Wichert, G.; Knippschild, U.; Wiedenmann, J.; Clauss, K.; Nienhaus, G.U.; Hameister, H.; Baumann, B.; Borggrefe, T.; et al. RITA, a novel modulator of Notch signalling, acts via nuclear export of RBP-J. EMBO J. 2011, 30, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Oswald, F.; Kovall, R.A. CSL-Associated Corepressor and Coactivator Complexes. Adv. Exp. Med. Biol. 2018, 1066, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell. Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef] [Green Version]
- Nam, Y.; Sliz, P.; Song, L.; Aster, J.C.; Blacklow, S.C. Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell 2006, 124, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Tabaja, N.; Yuan, Z.; Oswald, F.; Kovall, R.A. Structure-function analysis of RBP-J-interacting and tubulin-associated (RITA) reveals regions critical for repression of Notch target genes. J. Biol. Chem. 2017, 292, 10549–10563. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, Z.; Liu, C.; Huang, S.; Wang, H.; Chen, Y.; Chen, G. RBP-J-interacting and tubulin-associated protein induces apoptosis and cell cycle arrest in human hepatocellular carcinoma by activating the p53–Fbxw7 pathway. Biochem. Biophys. Res. Commun. 2014, 454, 71–77. [Google Scholar] [CrossRef]
- Wang, H.; Chen, G.; Wang, H.; Liu, C. RITA inhibits growth of human hepatocellular carcinoma through induction of apoptosis. Oncol. Res. 2013, 20, 437–445. [Google Scholar] [CrossRef]
- Steinhäuser, K.; Klöble, P.; Kreis, N.-N.; Ritter, A.; Friemel, A.; Roth, S.; Reichel, J.M.; Michaelis, J.; Rieger, M.A.; Louwen, F.; et al. Deficiency of RITA results in multiple mitotic defects by affecting microtubule dynamics. Oncogene 2017, 36, 2146–2159. [Google Scholar] [CrossRef]
- Kreis, N.-N.; Steinhäuser, K.; Ritter, A.; Klöble, P.; Hoock, S.C.; Roth, S.; Louwen, F.; Oswald, F.; Yuan, J. Potential involvement of RITA in the activation of Aurora A at spindle poles during mitosis. Oncogene 2019, 38, 4199–4214. [Google Scholar] [CrossRef]
- Hoock, S.C.; Ritter, A.; Steinhäuser, K.; Roth, S.; Behrends, C.; Oswald, F.; Solbach, C.; Louwen, F.; Kreis, N.-N.; Yuan, J. RITA modulates cell migration and invasion by affecting focal adhesion dynamics. Mol. Oncol. 2019, 13, 2121–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krismer, K.; Bernwinkler, T.; Ehrenberger, T. Home-Scansite 4. Available online: https://scansite4.mit.edu/#home (accessed on 25 January 2022).
- Steinmetz, M.O.; Prota, A.E. Microtubule-Targeting Agents: Strategies to Hijack the Cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Čermák, V.; Dostál, V.; Jelínek, M.; Libusová, L.; Kovář, J.; Rösel, D.; Brábek, J. Microtubule-targeting agents and their impact on cancer treatment. Eur. J. Cell Biol. 2020, 99, 151075. [Google Scholar] [CrossRef]
- Gallego-Jara, J.; Lozano-Terol, G.; Sola-Martínez, R.A.; Cánovas-Díaz, M.; de Diego Puente, T. A Compressive Review about Taxol®: History and Future Challenges. Molecules 2020, 25, 5986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Kanakkanthara, A. Beyond the Paclitaxel and Vinca Alkaloids: Next Generation of Plant-Derived Microtubule-Targeting Agents with Potential Anticancer Activity. Cancers 2020, 12, 1721. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Seetharamalu, P.; Schwarz, P.M.; Hausheer, F.H.; Ludueña, R.F. The interaction of the B-ring of colchicine with alpha-tubulin: A novel footprinting approach. J. Mol. Biol. 2000, 303, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Borys, F.; Joachimiak, E.; Krawczyk, H.; Fabczak, H. Intrinsic and Extrinsic Factors Affecting Microtubule Dynamics in Normal and Cancer Cells. Molecules 2020, 25, 3705. [Google Scholar] [CrossRef]
- McLoughlin, E.C.; O’Boyle, N.M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Ley, C.D.; Horsman, M.R.; Kristjansen, P.E.G. Early effects of combretastatin-A4 disodium phosphate on tumor perfusion and interstitial fluid pressure. Neoplasia 2007, 9, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Torijano-Gutiérrez, S.; Díaz-Oltra, S.; Falomir, E.; Murga, J.; Carda, M.; Marco, J.A. Synthesis of combretastatin A-4 O-alkyl derivatives and evaluation of their cytotoxic, antiangiogenic and antitelomerase activity. Bioorg. Med. Chem. 2013, 21, 7267–7274. [Google Scholar] [CrossRef] [Green Version]
- Abma, E.; Daminet, S.; Smets, P.; Ni, Y.; de Rooster, H. Combretastatin A4-phosphate and its potential in veterinary oncology: A review. Vet. Comp. Oncol. 2017, 15, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.J.; Ravoori, M.; Landen, C.N.; Kamat, A.A.; Han, L.Y.; Lu, C.; Lin, Y.G.; Merritt, W.M.; Jennings, N.; Spannuth, W.A.; et al. Antitumor and antivascular effects of AVE8062 in ovarian carcinoma. Cancer Res. 2007, 67, 9337–9345. [Google Scholar] [CrossRef] [Green Version]
- Long, R.; Yang, W.; Huang, G. Preparation and separation of epothilones with anticancer activity. Chem. Biol. Drug Des. 2020, 96, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Bollag, D.M.; McQueney, P.A.; Zhu, J.; Hensens, O.; Koupal, L.; Liesch, J.; Goetz, M.; Lazarides, E.; Woods, C.M. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 1995, 55, 2325–2333. [Google Scholar] [PubMed]
- Puhalla, S.; Brufsky, A. Ixabepilone: A new chemotherapeutic option for refractory metastatic breast cancer. Biologics 2008, 2, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Kamal, A.; Hussaini, S.M.A.; Nayak, V.L.; Malik, M.S.; Sucharitha, M.L.; Shaik, T.B.; Ashraf, M.; Bagul, C. Synthesis of 2-anilinopyridine dimers as microtubule targeting and apoptosis inducing agents. Bioorg. Med. Chem. 2014, 22, 6755–6767. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Khan, M.N.A.; Srinivasa Reddy, K.; Rohini, K. Synthesis of a new class of 2-anilino substituted nicotinyl arylsulfonylhydrazides as potential anticancer and antibacterial agents. Bioorg. Med. Chem. 2007, 15, 1004–1013. [Google Scholar] [CrossRef]
- Patel, F.; Spassieva, S.D. Side Effects in Cancer Therapy: Are Sphingolipids to Blame? Adv. Cancer Res. 2018, 140, 367–388. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Nawara, H.M.; Afify, S.M.; Hassan, G.; Zahra, M.H.; Seno, A.; Seno, M. Paclitaxel-Based Chemotherapy Targeting Cancer Stem Cells from Mono- to Combination Therapy. Biomedicines 2021, 9, 500. [Google Scholar] [CrossRef]
- Tataroglu Ozyukseler, D.; Basak, M.; Ay, S.; Koseoglu, A.; Arıcı, S.; Oyman, A.; Sürmeli, H.; Turan, M.; Turan, N.; Odabaş, H.; et al. Prognostic factors of ado-trastuzumab emtansine treatment in patients with metastatic HER-2 positive breast cancer. J. Oncol. Pharm. Pract. 2021, 27, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Sells, T.B.; Chau, R.; Ecsedy, J.A.; Gershman, R.E.; Hoar, K.; Huck, J.; Janowick, D.A.; Kadambi, V.J.; LeRoy, P.J.; Stirling, M.; et al. MLN8054 and Alisertib (MLN8237): Discovery of Selective Oral Aurora A Inhibitors. ACS Med. Chem. Lett. 2015, 6, 630–634. [Google Scholar] [CrossRef]
- Li, J.-P.; Yang, Y.-X.; Liu, Q.-L.; Pan, S.-T.; He, Z.-X.; Zhang, X.; Yang, T.; Chen, X.-W.; Wang, D.; Qiu, J.-X.; et al. The investigational Aurora kinase A inhibitor alisertib (MLN8237) induces cell cycle G2/M arrest, apoptosis, and autophagy via p38 MAPK and Akt/mTOR signaling pathways in human breast cancer cells. Drug Des. Devel. Ther. 2015, 9, 1627–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Zhang, Y.; Gao, M.; Quan, L.; Gui, R.; Liu, J. Alisertib induces apoptosis and autophagy through targeting the AKT/mTOR/AMPK/p38 pathway in leukemic cells. Mol. Med. Rep. 2016, 14, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.-J.; Zhou, Z.-W.; Zhu, D.-J.; Ju, Y.-L.; Wu, J.-H.; Ouyang, M.-Z.; Chen, X.-W.; Zhou, S.-F. Alisertib Induces Cell Cycle Arrest, Apoptosis, Autophagy and Suppresses EMT in HT29 and Caco-2 Cells. Int. J. Mol. Sci. 2015, 17, 41. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schüttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Niu, H.; Manfredi, M.; Ecsedy, J.A. Scientific Rationale Supporting the Clinical Development Strategy for the Investigational Aurora A Kinase Inhibitor Alisertib in Cancer. Front. Oncol. 2015, 5, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, O.A.; Özcan, M.; Jacobsen, E.D.; Roncero, J.M.; Trotman, J.; Demeter, J.; Masszi, T.; Pereira, J.; Ramchandren, R.; Beaven, A.; et al. Randomized Phase III Study of Alisertib or Investigator’s Choice (Selected Single Agent) in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 613–623. [Google Scholar] [CrossRef]
- Shah, H.A.; Fischer, J.H.; Venepalli, N.K.; Danciu, O.C.; Christian, S.; Russell, M.J.; Liu, L.C.; Zacny, J.P.; Dudek, A.Z. Phase I Study of Aurora A Kinase Inhibitor Alisertib (MLN8237) in Combination With Selective VEGFR Inhibitor Pazopanib for Therapy of Advanced Solid Tumors. Am. J. Clin. Oncol. 2019, 42, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Mortlock, A.A.; Foote, K.M.; Heron, N.M.; Jung, F.H.; Pasquet, G.; Lohmann, J.-J.M.; Warin, N.; Renaud, F.; de Savi, C.; Roberts, N.J.; et al. Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J. Med. Chem. 2007, 50, 2213–2224. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, R.W.; Odedra, R.; Heaton, S.P.; Wedge, S.R.; Keen, N.J.; Crafter, C.; Foster, J.R.; Brady, M.C.; Bigley, A.; Brown, E.; et al. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin. Cancer Res. 2007, 13, 3682–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zekri, A.; Mesbahi, Y.; Ghanizadeh-Vesali, S.; Alimoghaddam, K.; Ghavamzadeh, A.; Ghaffari, S.H. Reactive oxygen species generation and increase in mitochondrial copy number: New insight into the potential mechanism of cytotoxicity induced by aurora kinase inhibitor, AZD1152-HQPA. Anti-Cancer Drugs 2017, 28, 841–851. [Google Scholar] [CrossRef]
- Ashton, S.; Song, Y.H.; Nolan, J.; Cadogan, E.; Murray, J.; Odedra, R.; Foster, J.; Hall, P.A.; Low, S.; Taylor, P.; et al. Aurora kinase inhibitor nanoparticles target tumors with favorable therapeutic index in vivo. Sci. Transl. Med. 2016, 8, 325ra17. [Google Scholar] [CrossRef] [PubMed]
- Janeček, M.; Rossmann, M.; Sharma, P.; Emery, A.; Huggins, D.J.; Stockwell, S.R.; Stokes, J.E.; Tan, Y.S.; Almeida, E.G.; Hardwick, B.; et al. Allosteric modulation of AURKA kinase activity by a small-molecule inhibitor of its protein-protein interaction with TPX2. Sci. Rep. 2016, 6, 28528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottmann, O.G.; Müller-Tidow, C.; Krämer, A.; Schlenk, R.F.; Lübbert, M.; Bug, G.; Krug, U.; Bochtler, T.; Voss, F.; Taube, T.; et al. Phase I dose-escalation trial investigating volasertib as monotherapy or in combination with cytarabine in patients with relapsed/refractory acute myeloid leukaemia. Br. J. Haematol. 2019, 184, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjertsen, B.T.; Schöffski, P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 2015, 29, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yin, H.; Kuret, J. Casein kinase 1 delta phosphorylates tau and disrupts its binding to microtubules. J. Biol. Chem. 2004, 279, 15938–15945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.-S.; Dong, Y.-H.; Liu, Z.-P. Recent Advances in the Development of Casein Kinase 1 Inhibitors. Curr. Med. Chem. 2021, 28, 1585–1604. [Google Scholar] [CrossRef]
- Ikeda, H.; Taira, N.; Hara, F.; Fujita, T.; Yamamoto, H.; Soh, J.; Toyooka, S.; Nogami, T.; Shien, T.; Doihara, H.; et al. The estrogen receptor influences microtubule-associated protein tau (MAPT) expression and the selective estrogen receptor inhibitor fulvestrant downregulates MAPT and increases the sensitivity to taxane in breast cancer cells. Breast Cancer Res. 2010, 12, R43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, P.; Wang, B.; Clark, E.; Lee, H.; Rouzier, R.; Pusztai, L. Microtubule Associated Protein (MAP)-Tau: A novel mediator of paclitaxel sensitivity in vitro and in vivo. Cell Cycle 2005, 4, 1149–1152. [Google Scholar] [CrossRef] [PubMed]
- Mashhoon, N.; DeMaggio, A.J.; Tereshko, V.; Bergmeier, S.C.; Egli, M.; Hoekstra, M.F.; Kuret, J. Crystal structure of a conformation-selective casein kinase-1 inhibitor. J. Biol. Chem. 2000, 275, 20052–20060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoshima, M.; Howie, H.L.; Imakura, M.; Walsh, R.M.; Annis, J.E.; Chang, A.N.; Frazier, J.; Chau, B.N.; Loboda, A.; Linsley, P.S.; et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 9545–9550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, J.K.; Nguyen, T.H.; Wang, H.; Tan, P.; Voorhoeve, P.M.; Lee, S.H.; Virshup, D.M. IC261 induces cell cycle arrest and apoptosis of human cancer cells via CK1δ/ɛ and Wnt/β-catenin independent inhibition of mitotic spindle formation. Oncogene 2011, 30, 2558–2569. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Li, D.; Guo, M.; Fang, T.; Sun, J.; Qi, F.; Rao, Q.; Zhao, Z.; Huang, P.; Yang, B.; et al. IC261 suppresses progression of hepatocellular carcinoma in a casein kinase 1 δ/ε independent manner. Biochem. Biophys. Res. Commun. 2020, 523, 809–815. [Google Scholar] [CrossRef]
- Stöter, M.; Krüger, M.; Banting, G.; Henne-Bruns, D.; Knippschild, U. Microtubules depolymerization caused by the CK1 inhibitor IC261 may be not mediated by CK1 blockage. PLoS ONE 2014, 9, e100090. [Google Scholar] [CrossRef] [Green Version]
- Xian, J.; Bu, F.; Wang, Y.; Long, F.; Zhang, Z.; Wu, C.; Tao, Y.; Wang, T.; Wang, G. A Rationale for Drug Design Provided by Co-Crystal Structure of IC261 in Complex with Tubulin. Molecules 2021, 26, 946. [Google Scholar] [CrossRef]
- Choy, H. Taxanes in combined modality therapy for solid tumors. Crit. Rev. Oncol. Hematol. 2001, 37, 237–247. [Google Scholar] [CrossRef]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Gridelli, C.; Rossi, A.; Maione, P.; Rossi, E.; Castaldo, V.; Sacco, P.C.; Colantuoni, G. Vascular disrupting agents: A novel mechanism of action in the battle against non-small cell lung cancer. Oncologist 2009, 14, 612–620. [Google Scholar] [CrossRef]
- Siri, M.; Behrouj, H.; Dastghaib, S.; Zamani, M.; Likus, W.; Rezaie, S.; Hudecki, J.; Khazayel, S.; Łos, M.J.; Mokarram, P.; et al. Casein Kinase-1-Alpha Inhibitor (D4476) Sensitizes Microsatellite Instable Colorectal Cancer Cells to 5-Fluorouracil via Authophagy Flux Inhibition. Arch. Immunol. Ther. Exp. 2021, 69, 26. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.M.; Fisher, K.; Rubitski, D.; Marconi, M.; Meng, Q.-J.; Sládek, M.; Adams, J.; Bass, M.; Chandrasekaran, R.; Butler, T.; et al. Selective inhibition of casein kinase 1 epsilon minimally alters circadian clock period. J. Pharmacol. Exp. Ther. 2009, 330, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Janovska, P.; Verner, J.; Kohoutek, J.; Bryjova, L.; Gregorova, M.; Dzimkova, M.; Skabrahova, H.; Radaszkiewicz, T.; Ovesna, P.; Vondalova Blanarova, O.; et al. Casein kinase 1 is a therapeutic target in chronic lymphocytic leukemia. Blood 2018, 131, 1206–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, L.H.; Lafitte, M.; Quereda, V.; Grant, W.; Chen, W.; Bibian, M.; Noguchi, Y.; Fallahi, M.; Yang, C.; Chang, J.C.; et al. Therapeutic targeting of casein kinase 1δ in breast cancer. Sci. Transl. Med. 2015, 7, 318ra202. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Song, J.; Wang, Z.; Zhou, L.; Xia, Y.; Yu, S.; Sun, Q.; Liu, S.-S.; Zhao, L.; Li, S.; et al. Tumor promoter TPA activates Wnt/β-catenin signaling in a casein kinase 1-dependent manner. Proc. Natl. Acad. Sci. USA 2018, 115, E7522–E7531. [Google Scholar] [CrossRef] [Green Version]
- García-Reyes, B.; Witt, L.; Jansen, B.; Karasu, E.; Gehring, T.; Leban, J.; Henne-Bruns, D.; Pichlo, C.; Brunstein, E.; Baumann, U.; et al. Discovery of Inhibitor of Wnt Production 2 (IWP-2) and Related Compounds As Selective ATP-Competitive Inhibitors of Casein Kinase 1 (CK1) δ/ε. J. Med. Chem. 2018, 61, 4087–4102. [Google Scholar] [CrossRef]
- Minzel, W.; Venkatachalam, A.; Fink, A.; Hung, E.; Brachya, G.; Burstain, I.; Shaham, M.; Rivlin, A.; Omer, I.; Zinger, A.; et al. Small Molecules Co-targeting CKIα and the Transcriptional Kinases CDK7/9 Control AML in Preclinical Models. Cell 2018, 175, 171–185.e25. [Google Scholar] [CrossRef] [Green Version]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Deng, C.; Lipstein, M.R.; Scotto, L.; Jirau Serrano, X.O.; Mangone, M.A.; Li, S.; Vendome, J.; Hao, Y.; Xu, X.; Deng, S.-X.; et al. Silencing c-Myc translation as a therapeutic strategy through targeting PI3Kδ and CK1ε in hematological malignancies. Blood 2017, 129, 88–99. [Google Scholar] [CrossRef]
- Dhillon, S.; Keam, S.J. Umbralisib: First Approval. Drugs 2021, 81, 857–866. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Poornima, P.; Kumar, J.D.; Zhao, Q.; Blunder, M.; Efferth, T. Network pharmacology of cancer: From understanding of complex interactomes to the design of multi-target specific therapeutics from nature. Pharmacol. Res. 2016, 111, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, J.T. The importance of predictive biomarkers in oncology drug development. Expert Rev. Mol. Diagn. 2016, 16, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Koutsoukas, A.; Simms, B.; Kirchmair, J.; Bond, P.J.; Whitmore, A.V.; Zimmer, S.; Young, M.P.; Jenkins, J.L.; Glick, M.; Glen, R.C.; et al. From in silico target prediction to multi-target drug design: Current databases, methods and applications. J. Proteom. 2011, 74, 2554–2574. [Google Scholar] [CrossRef] [PubMed]
- Moffat, J.G.; Rudolph, J.; Bailey, D. Phenotypic screening in cancer drug discovery-past, present and future. Nat. Rev. Drug Discov. 2014, 13, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Arnst, K.E.; Banerjee, S.; Chen, H.; Deng, S.; Hwang, D.-J.; Li, W.; Miller, D.D. Current advances of tubulin inhibitors as dual acting small molecules for cancer therapy. Med. Res. Rev. 2019, 39, 1398–1426. [Google Scholar] [CrossRef] [PubMed]
- Krüger, M.; Kalbacher, H.; Kastritis, P.L.; Bischof, J.; Barth, H.; Henne-Bruns, D.; Vorgias, C.; Sarno, S.; Pinna, L.A.; Knippschild, U. New potential peptide therapeutics perturbing CK1δ/α-tubulin interaction. Cancer Lett. 2016, 375, 375–383. [Google Scholar] [CrossRef]
- Dolde, C.; Bischof, J.; Grüter, S.; Montada, A.; Halekotte, J.; Peifer, C.; Kalbacher, H.; Baumann, U.; Knippschild, U.; Suter, B. A CK1 FRET biosensor reveals that DDX3X is an essential activator of CK1ε. J. Cell Sci. 2018, 131, jcs207316. [Google Scholar] [CrossRef] [Green Version]
- Cruciat, C.-M.; Dolde, C.; de Groot, R.E.A.; Ohkawara, B.; Reinhard, C.; Korswagen, H.C.; Niehrs, C. RNA helicase DDX3 is a regulatory subunit of casein kinase 1 in Wnt-β-catenin signaling. Science 2013, 339, 1436–1441. [Google Scholar] [CrossRef]
- Harnoš, J.; Ryneš, J.; Víšková, P.; Foldynová-Trantírková, S.; Bajard-Ešner, L.; Trantírek, L.; Bryja, V. Analysis of binding interfaces of the human scaffold protein AXIN1 by peptide microarrays. J. Biol. Chem. 2018, 293, 16337–16347. [Google Scholar] [CrossRef] [Green Version]
- Huart, A.-S.; MacLaine, N.J.; Narayan, V.; Hupp, T.R. Exploiting the MDM2-CK1α protein-protein interface to develop novel biologics that induce UBL-kinase-modification and inhibit cell growth. PLoS ONE 2012, 7, e43391. [Google Scholar] [CrossRef] [PubMed]




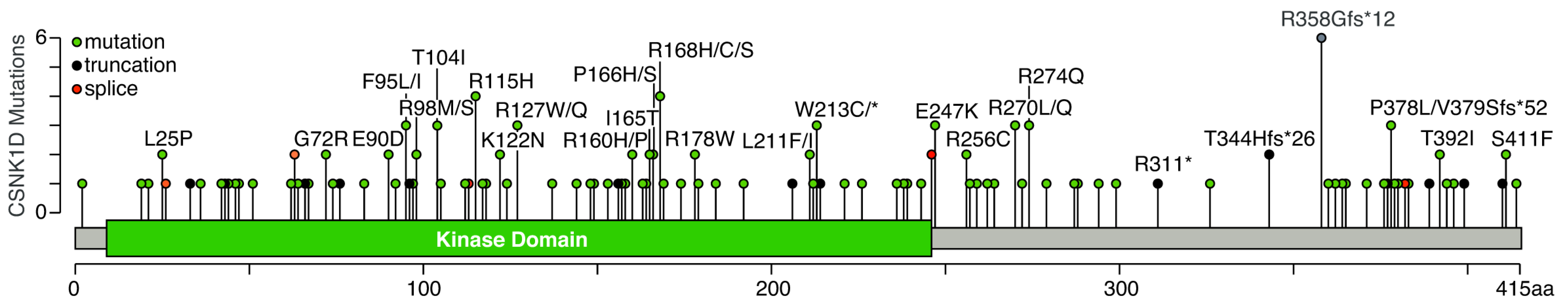
| Mutation | Cancer |
|---|---|
| L25P | Lung Adenocarcinoma, Stomach Adenocarcinoma |
| G72R | Uterine Endometrioid Carcinoma, Colorectal Adenocarcinoma |
| E90D | Uterine Endometrioid Carcinoma, Lung Adenocarcinoma |
| F95I F95L | Cutaneous Melanoma Mucinous Adenocarcinoma of the Colon and Rectum (2×) |
| R98M R98S | Mucinous Adenocarcinoma of the Colon and Rectum Serous Ovarian Cancer |
| T104I T104Pfs*9 | Cutaneous Squamous Cell Carcinoma, Skin Cancer, Non-Melanoma Uterine Endometrioid Carcinoma |
| R115H | Colon Adenocarcinoma, Head and Neck Squamous Cell Carcinoma, Uterine Endometrioid Carcinoma, Colorectal Adenocarcinoma |
| K122N | Endometrial Carcinoma, Lung Adenocarcinoma |
| R127W R127Q | Cervical Squamous Cell Carcinoma, Colorectal Adenocarcinoma Bladder Urothelial Carcinoma |
| R160P R160H | Colon Adenocarcinoma Colorectal Adenocarcinoma |
| I165T | Colorectal Adenocarcinoma, Intestinal Type Stomach Adenocarcinoma |
| P166H P166S | Cutaneous Melanoma Glioblastoma |
| R168S R168C R168H | Acute Myeloid Leukemia Skin Cancer, Non-Melanoma Uterine Endometrioid Carcinoma, Melanoma |
| R178W | Prostate, Colorectal Adenocarcinoma |
| L211F L211I | Lung Adenocarcinoma Uterine Serous Carcinoma/Uterine Papillary Serous Carcinoma |
| W213C W213* | Cutaneous Squamous Cell Carcinoma, Melanoma Lung Adenocarcinoma |
| S246= | Breast Invasive Lobular Carcinoma, Cutaneous Squamous Cell Carcinoma |
| E247K | Rectal Adenocarcinoma, Uterine Endometrioid Carcinoma (2×) |
| R256C | Angiosarcoma, Intestinal Type Stomach Adenocarcinoma |
| R270L R270Q | Cutaneous Melanoma (2×) Cutaneous Melanoma |
| R274Q | Colon Adenocarcinoma, Uterine Serous Carcinoma/Uterine Papillary Serous Carcinoma, Uterine Endometrioid Carcinoma |
| T344Hfs*26 | Colon Adenocarcinoma, Mucinous Adenocarcinoma of the Colon and Rectum |
| R358Gfs*12 | Colon Adenocarcinoma (3×), Uterine Endometrioid Carcinoma (2×), Cervical Squamous Cell Carcinoma |
| P378L | Glioblastoma, Skin Cancer, Non-Melanoma |
| V379Sfs*52 | Breast Invasive Lobular Carcinoma |
| T392I | Stomach Adenocarcinoma (2×) |
| S411F | Cutaneous Squamous Cell Carcinoma, Bladder Urothelial Carcinoma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roth, A.; Gihring, A.; Bischof, J.; Pan, L.; Oswald, F.; Knippschild, U. CK1 Is a Druggable Regulator of Microtubule Dynamics and Microtubule-Associated Processes. Cancers 2022, 14, 1345. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051345
Roth A, Gihring A, Bischof J, Pan L, Oswald F, Knippschild U. CK1 Is a Druggable Regulator of Microtubule Dynamics and Microtubule-Associated Processes. Cancers. 2022; 14(5):1345. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051345
Chicago/Turabian StyleRoth, Aileen, Adrian Gihring, Joachim Bischof, Leiling Pan, Franz Oswald, and Uwe Knippschild. 2022. "CK1 Is a Druggable Regulator of Microtubule Dynamics and Microtubule-Associated Processes" Cancers 14, no. 5: 1345. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14051345