
Polymerase Epsilon-Associated Ultramutagenesis in Cancer

1
Department of Cancer Biology and Genetics, James Comprehensive Cancer Center, The Ohio State University Wexner Medical Center, Columbus, OH 43210, USA
2
Division of Medical Oncology, Department of Internal Medicine, James Comprehensive Cancer Center, The Ohio State University Wexner Medical Center, Columbus, OH 43210, USA
*
Authors to whom correspondence should be addressed.
Cancers 2022, 14(6), 1467; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061467
Submission received: 31 January 2022
/
Revised: 9 March 2022
/
Accepted: 9 March 2022
/
Published: 12 March 2022
Abstract
:Simple Summary
DNA polymerase epsilon is implicated to play a major role in DNA synthesis of the leading strand. In some cancer types, especially colorectal and endometrial cancers, polymerase epsilon is mutated at several hotspots, causing large amounts of mutations, termed ultramutation. The aim of this article is to describe the characteristics of polymerase epsilon mutations including their mutation sites and signatures, elucidate the underlying mechanisms of its ultramutagenesis, discuss its good prognosis and favorable responses to immunotherapies, and speculate on possible strategies to improve treatment of ultramutated cancers.
Abstract
With advances in next generation sequencing (NGS) technologies, efforts have been made to develop personalized medicine, targeting the specific genetic makeup of an individual. Somatic or germline DNA Polymerase epsilon (PolE) mutations cause ultramutated (>100 mutations/Mb) cancer. In contrast to mismatch repair-deficient hypermutated (>10 mutations/Mb) cancer, PolE-associated cancer is primarily microsatellite stable (MSS) In this article, we provide a comprehensive review of this PolE-associated ultramutated tumor. We describe its molecular characteristics, including the mutation sites and mutation signature of this type of tumor and the mechanism of its ultramutagenesis. We discuss its good clinical prognosis and elucidate the mechanism for enhanced immunogenicity with a high tumor mutation burden, increased neoantigen load, and enriched tumor-infiltrating lymphocytes. We also provide the rationale for immune checkpoint inhibitors in PolE-mutated tumors.
1. Introduction
Colorectal cancer (CRC) is a very common cancer worldwide. In the United States, CRC is the third most diagnosed cancer in men and women, and the second-leading cause of cancer deaths when men and women are combined. It is estimated that there were 149,500 new cases of CRC and 52,980 deaths in 2021. While the localized CRC tumor is largely curable with surgery followed by radiotherapy, in some cases metastatic CRC is treated by chemotherapy and immunotherapy. More and more patients benefit from precision medicine, based on a molecular genetic diagnosis.
Previously explored precision treatments have focused on targeting oncogenes (e.g, KRAS, PIK3CA, and BRAF). Currently, the attention toward CRC has shifted to clonal–stromal–immune perspectives, highlighting tumor immunity microenvironment heterogeneity.
Transcriptomic analysis identified four consensual molecular subtypes (CMS) of CRC: CMS1—microsatellite instability (MSI-H)/immunogenic tumors, accounting for 14% of all CRC cases and hypermutated with strong immune activation; CMS2—canonical tumors (37% of CRC cases), epithelial and exhibiting MYC and WNT signaling pathway activation; CMS3—metabolic (13% of CRC cases), epithelial, and displaying evident metabolic dysregulation; and CMS4—mesenchymal (23% of all CRC) featuring stromal invasion, transforming growth factor-beta activation, and angiogenesis [1]. DNA mismatch repair (MMR) defects lead to the CMS1 subset of CRC and the hypermutated phenotype. Immune checkpoint inhibition is a new therapeutic strategy that has shown promising efficacy in many cancer types. Significant immunity response and prolonged progression-free survival (PFS) were observed in MMR-deficient metastatic CRC [2].
Notably, another type of so-called “ultramutated” CRC, with more than 100 mutations/Mb, also presents a high neoantigen load, tumor-infiltrating lymphocytes, and predicts a favorable response to immune checkpoint inhibition [3]. Different from MMR-deficient hypermutated CRC, several unique point mutations in the proofreading domain of replicative DNA polymerase epsilon (PolE) were found in these CRC and the tumor is characterized as being microsatellite stable (MSS) [4].
In this review, we provide a comprehensive overview of this PolE-associated ultramutated tumor. We describe its molecular characteristics, including the mutant sites in PolE, the mutation signature of this type of tumor, and the progress in the molecular mechanism of its ultramutagenesis and carcinogenicity. We describe the clinical features and explain the potential reasons for its good prognosis, i.e., tumor-infiltrating lymphocytes and error threshold. Finally, we provide histological and clinical data on its immunity checkpoint blockade, addressing the favorable benefit from anti-PD-1 immunotherapy.
2. PolE Mutations Generate Ultramutation Genotype in Cancer
Genome instability is one of the hallmarks of cancer. In CRC, DNA MMR defects caused by mutations in repair enzymes have been found to be responsible for a high level of MSI-H and germline mutations of MMR genes are responsible for Lynch syndrome, also called hereditary nonpolyposis colorectal cancer (HNPCC) [5,6]. Clinically, CRC patients with MSI-H tumors have better prognoses and show more response to immunotherapy than those with MSS tumors [2,7]. On the other hand, although Larry Leob and his colleagues hypothesized in the 1970s that defective DNA polymerases could lead to genome instability [8], there were few reports of mutations in DNA polymerases [9,10,11,12].
In 2012, data from a comprehensive genomic study of colorectal carcinoma were made available by The Cancer Genome Atlas (TCGA), including exome sequencing of 224 tumor samples. The data show that while most hypermutated tumors (>10 mutations/Mb) exhibited MSI-H or MMR deficiency, a separate class of ultramutated tumors (>100 mutations/Mb) is surprisingly MSS and bears somatic mutations in PolE [4], which is responsible for DNA synthesis in the leading strand during DNA replication [13]. However, somatic mutations in the PolD1 gene encoding the catalytic subunit of Pol δ, another DNA replicative polymerase responsible for DNA synthesis in the lagging strand [14], are not detected in ultramutated tumors. Instead, they are found in sporadic hypermutated tumors together with MMR defects [15]. Pol δ not only proofreads the errors generated by itself in the lagging strand, but also extrinsically corrects the errors produced by Polε in the leading strand [16]. Soon after, germline mutations in PolE were reported to create a predisposition to colorectal adenomas and carcinomas [17]. The mutations are mainly located in the proofreading exonuclease domain of PolE [15]. P286R and V411L are the two most common somatic mutations in PolE. Additionally, P286H/S, F367S, S459F, and other mutations also appear in sporadic tumors but at a lower frequency, whereas the V424L mutation occurs in germline tumors [15]. While most of these mutations affect highly conserved residues in or adjacent to the exonuclease motifs at the DNA-binding interface, V411L is located beyond the exonuclease catalytic core [15,18]. Notably, the vast majority of PolE mutations are heterozygous [15]. A genetic study in yeast showed that, except for V411L, a vast majority of PolE mutations at the DNA binding cleft have elevated spontaneous mutation rates with varying degrees [19]. PolE mutations are most frequently found in colorectal cancer (1–3%) and endometrial cancer (6–12%), and rarely found in lung, breast, brain, prostate, kidney, ovarian, bone, and gastric cancers [3]. In addition, other somatic point mutations in the polymerase domain of PolE (R567C, K593C, S595P, E611K, and L621F) and a deletion frameshift in the middle region of the PolE gene (V1446fs *3) are possible pathogenic factors in cancer [20,21]. Mutations in non-catalytic subunits of Polε, PolE2, PolE3 and PolE4, are rare in cancer and their involvement in ultramutagenesis is not clear.
Studies in yeast and mouse models mimicking human P286R mutation supported the hypothesis that ultramutation observed in tumors was driven by a single base alteration in human PolE [22,23]. This analogous substitution in yeast yielded the most powerful known PolE mutator phenotype, which twice surpassed that of proofreading-deficient PolE mutants [22]. Two very recent studies explored how PolE-P286R affects polymerase function [24,25]. Shcherbakova’s lab showed that yeast Pol2-P301R mutant polymerase has increased activation on standard DNA templates, as well as an increased ability for bypassing hairpin structures and extension. A mispaired primer termini mutant Arg residue, as demonstrated by the crystal structure, enters the primer binding cleft of the exonuclease active site which then blocks primer partitioning [25]. The authors hypothesized that the enzyme is kept in a state of hyperactive polymerization by the mutant, which results in mutagenesis. However, purified Polε-P301R displays a relatively low error rate in vitro, as compared to exonuclease-deficient Pol2, suggesting that additional factors must enhance the impact of its effect on ultra-mutagenesis in vivo [24]. MMRs and the proofreading of DNA polymerase δ prevent catastrophic errors made by Pol2-P301R in vivo [26]. Similarly, monoallelic PolE proofreading deficiency in MLH-1 (a human MMR gene) deficient HCT-116 cells caused a rapid increase in mutations, while the re-introduction of wild-type MLH-1 suppressed the mutation accumulation [27]. Deletion of translesion synthesis (TLS) polymerases, Polκ, suppressed the ultramutation generated by PolE-P286R in a fission yeast model [28]. Shcherbakova’s lab also characterized 13 other cancer-associated PolE mutations in the yeast model and found that only mutations that directly alter the exonuclease domain of the DNA binding cleft increase the mutation rate. Of these, the frequently occurring mutants were more powerful mutators than the less commonly occurring ones, which supports the idea that the mutator phenotype plays a causative part in tumorigenesis [19]. Whole exome sequencing (WES) data in yeast also showed similar results [29,30].
The heterozygous and homozygous proofreading deficient mice, PolEexo−/+ and PolEexo−/−, were constructed with alanine substitutions at residues D272 and E274. Approximately half of PolEexo−/−, but not PolEexo−/+, mice developed adenocarcinomas in the small intestine [31]. In comparison, heterozygous PolEP286R/+ mice are highly tumorigenic, producing tumors in multiple organs including the lung, thymus, shoulder, leg, and ovary [23]. An estimate of 1.6 (average of 0.9, 1.6, and 2.1 in three studied MEF cell lines) nucleotide substitutions per Mb per cell division was observed by whole genomic sequencing (WGS), which is at least 3 orders of magnitude higher than the intrinsic DNA replication error rate. WGS of seven primary tumors from PolEP286R/+ mice, including three lung adenocarcinomas, three T-cell lymphomas, one cutaneous squamous cell carcinomas, and four tumors from two PoleP286R LSL-PolEP286R (hemizygous) mice, revealed a very high frequency of mutations, in the range of 10 to 100 mutations/Mb [23]. PolEP286R/+ is reported to be the first genetically engineered mouse model to repeat the clonal variation, high mutation burden, and heterogeneity characteristics of human cancer [32].
3. Mutational Signatures of PolE-Associated Tumors
In 2013, Alexandrov et al. discovered over twenty distinct mutational signatures upon analyzing 4,938,362 mutations from 7042 tumors. The tenth signature among these was linked to PolE mutations found in colorectal cancer and endometrial cancer [33]. This type of signature, termed single base substitution 10 (SBS10), is not transcriptional strand biased and the landscape of this mutational signature is characterized predominately by TCG→TTG (>20%) transitions, TCT→TAT (>20%) transversions, and TTT→TGT (~7%) transversions, three subtypes named SBS10a, SBS10b and SBS28, respectively [33,34]. The frequency of TCT→TAT mutation is slightly higher in PolE V411L than in PolE P286R [35]. Japanese researchers explored the WES of 2141 solid tumor samples derived from 2042 cancer patients [36]. Their work confirmed the landscape of the mutational signature of PolE mutations with regard to number and frequency. These are, in descending order, TCT→TAT, TCG→TTG, and TTT→TGT. Further, they analyzed the mutation patterns of individual somatic tumors with different PolE mutations, including P286R (six samples), P286R&L120I (one sample), V411L (one sample), A189D&F367V (one sample), and K717N (one sample). Tumors with P286R or V411L mutations showed a mutation pattern consistent with that of PolE-associated tumors, although their genetic characteristics were found to be dramatically different in yeasts [19]. Interestingly, the pattern described above was enriched in SACS, LRP2, WDR87, and XIRP2 genes, but not in POLE and PTEN. The HCC2998 colon cancer cell line, harboring a PolE-P286R mutation, also showed a disproportionally large increase in GC→TA transversion, with a particular preference for AGA/TCT sequence context [15]. In vitro, a lacZ forward mutation assay with purified N terminal 1–1189 of the catalytic subunit of mutant human PolE-P286R showed a bias for TCT→TAT transversion over the complementary AGA→ACA transversion [37]. Different from somatic PolE mutations, ultramutated glioblastoma with the germline PolE V424L mutation is predominated by C > G transversion [38].
In yeast models, the mutation spectrum of the CAN1 forward assay and the lacZ forward assay showed an increase in the GC→AT transition/GC→TA transversion ratio, supporting the idea that mutations in Pol2-P301R mutant cells were produced by the Polε-P301R protein during DNA replication. Notably, the predominant base substitution in yeast is GC→AT transition, whereas it is GC→TA transversion in human tumors, possibly due to the slight difference in the exonuclease domain of PolE between yeast and humans [24].
4. Altered Pathways and Responses to Chemotherapy in PolE-Associated Cancer
Since point mutations in PolE produce ultramutation genotypes in tumors, one question is whether these mutations occur in specific oncogenes and/or tumor suppressor genes. Nebot-Bral and his colleagues analyzed altered oncogenic pathways in ultramutated EC and CRC, finding that the TP53 and TGF-β pathways are inactivated whereas the WNT, PI3K and RTK/RAS pathways are activated [3], not significantly different from MMR deficient CRC/EC tumors. However, unique alterations in certain pathways were found in PolE-associated tumors [36]. For example, dysfunction of the PTEN pathways and the activation of double-strand break repair, homologous recombination (HR), and non-homologous end-joining (NHEJ) pathways specifically occurred in PolE-associated ultramutated tumors, but not in hypermutated MSI-H tumors. A recent study also reported truncations of the homologous recombination repair gene, BRCA1/2, in PolE mutated CRC and EC [39,40]. Another study found that truncated TP53 R213* was specifically enriched in PolE P286R mutant CRC [35]. Additionally, there are two other pathways specifically mutated in PolE-associated tumors: the transcriptional regulation pathway, mediated by hypoxia inducible factor 1 alpha (HIF1α), and the p160 steroid receptor co-activator (SRC) pathway [36]. The mechanisms of the dysregulation of these pathways are not clear and need further investigation.
Chemotherapy is the primary treatment for CRC patients. Van Gool and colleagues investigated whether mutated PolE changes the sensitivity to chemotherapeutic agents in mouse-derived embryonic stem (mES) cells. Two nucleoside analogs, cytarabine and fludarabine, were found to show a higher potency in inhibiting cell viability in cell lines with the PolE mutations P286R, S297F, and V411L than in the wild-type PolE cell line. However, other chemotherapeutic agents, 5-fluorouracil, cisplatin, paclitaxel, doxorubicin, etoposide, and methotrexate, did not display any difference in efficacy between the wild type and mutant PolE cell lines [41]. Mechanistically, exonuclease deficient Polε efficiently incorporates cytarabine but has difficulty extending it, resulting in a high sensitivity to cytarabine-induced cellular cytotoxicity. Additionally, the Vitamin B6 compound, pyridoxal (PL), converts to pyridoxal 5′-phosphate, inhibits PolE-mediated DNA synthesis by competing with nucleotide substrates and thus decreases the cell viability of HeLa cells [42]. These results indicate that nucleoside analogs, which inhibit DNA polymerases, have the potential to treat PolE-associated tumors effectively. Sulphoquinovosyl diacylglycerol (SQDG) was reported as the first mammalian PolE-specific inhibitor [43], which might be a potential drug candidate.
5. Good Prognosis in Patients Harboring PolE Mutations
It has been shown that PolE mutations are identified in pre-malignant lesions in EC and CRC with prominent CD8+ T-cell infiltration. It suggests that somatic PolE mutations are early events and may contribute to tumor initiation and good prognosis. PolE-associated ultramutated CRC and EC and high-grade gliomas have been shown to have good prognoses [42,43,44,45], although mutation rates in those cancers are mostly higher than 100 per Mb.
In univariable analysis, PolE-mutated EC patients have significantly improved outcomes in terms of PFS, disease-specific survival (DSS), and overall survival (OS), as compared with patients with wild-type PolE. However, in multivariable analysis, PolE mutations are only significantly associated with improved PFS. Although no definite conclusions as to the effects of adjuvant treatment on PolE-mutated cases can be drawn, a meta-analysis showed that PolE mutations were associated with disease-free survival (DFS) and improved progression-free survival (PFS), featuring pooled hazard ratios of 0.34 (95% confidence interval (CI), 0.15–0.73) and 0.35 (95% CI, 0.13–0.92) in treated and untreated patients, respectively [46].
Another study on ECs also showed that patients with PolE mutations have improved PFS (100% after 90 months), as compared to PFS of patients with other types of tumors (<80% after 50 months) [42]. In addition, a European research group reported that women with PolE-associated ECs have fewer recurrences (6.2% vs. 14.1%) and EC deaths (2.3% vs. 9.7%) than other ECs [43]. Other studies have also shown similar findings [47,48,49,50,51,52].
Colorectal cancer patients with PolE mutations are typically diagnosed younger than patients with wild-type PolE (median of 54.5 years vs. 67.2 years, p < 0.0001) and at an earlier disease stage (p = 0.006, x2 test for trend). In addition, PolE mutations are more common in male than female patients and occur more frequently in the right colon than the left [44]. In stage II/III tumors, only four of 59 (8%) PolE-mutated patients experienced recurrence after surgery (median duration of 4.7 years), compared to 130 of 689 patients (18.9%) with MSI-H CRC and 1074 of 3905 patients (27.5%) with MSS CRC. In stage II patients, one recurrence out of 34 cases with PolE mutations (2.9%) was noted, compared to 61 of 426 MSI-H (14.3%) and 360 of 1735 MSS (20.7%) cases. This indicates that PolE mutations are associated with a greatly reduced risk of recurrence. For stage III diseases, however, the effect of PolE mutations on recurrence was insignificant [44]. Other studies also showed that CRC patients with PolE mutations have favorable prognoses [53,54,55,56,57,58,59]. Interestingly, one study reported that all the patients who were diagnosed with PolE-mutated CRC at younger than 50 years of age were found to harbor P286R mutation [58].
Together, these studies indicate that cancer patients with PolE mutations have good prognoses and favorable outcomes, in general. However, the underlying mechanisms are not completely understood. One potential mechanism could involve increased immune cell infiltration and enhanced immune surveillance, as shown in Figure 1.
Church et al. investigated the interplay between PolE-associated endometrial cancer and immunogenicity [60]. The preliminary data revealed that PolE-associated EC often presents a prominent lymphocytic infiltration and lymphocytic reaction similar to Crohn’s disease. Later studies showed that CD8+ T cells were more enriched in the center and at the invasive tumor front, in comparison with the MSI-H or MSS counterparts in endometrial cancer. T-cell infiltration in PolE-associated tumors was also enhanced by the expression of CD3 and cytolytic marker TIA-1. TIA-1 expression in CD8+ lymphocytes shows that these T cells may contribute to the mediation of anti-tumor immune response. The observation that nearly all tumor infiltrated lymphocytes (TILs) are CD8+ in PolE-associated EC tumors increasingly supports TILs’ enhanced antitumor effects [61].
Analysis of TCGA data in EC found that PolE-associated tumors display the enrichment of a 200 gene signature which corresponds to T-cell infiltration, including the upregulation of genes in T-cell-mediated cytotoxicity; cytotoxic T-cell differentiation and activation markers; CD8A and IFNγ; Eomes; T-bet; granzymes B, H, K, and M; perforin; and IFNγ-induced cytokines, CXCL9 and CXCL10. Notably, PolE-associated endometrial tumors also show upregulation of the T-follicular helper genes, CXCL13 and CXCR5, which were recently shown to be powerfully predictive of a positive outcome in CRC [62]. As a comparison, in MSI-H tumors, only CD8A and IFNγ were upregulated, and no difference of the expression of cytotoxic T-cell differentiation and activation markers was detected [60]. In addition, in some cases, despite limited numbers, expression of cytotoxic T cell markers and IFNγ-induced cytokines (e.g., PRF, GZMH, CXCL 9/10) is significantly higher in tumors bearing PolE mutations, when compared to MSI-H tumors [62]. These studies support the conclusion that PolE-mutant tumors exhibit higher T-lymphocyte infiltration as compared to other endometrial tumors, and that these lymphocytes have the capacity to show antitumor activity.
In another study, PolE-mutant endometrial tumors were shown to have the highest expression of immune-associated genes in comparison to MSI and MSS tumors [63]. Similarly, PolE-mutant tumors also exhibit a high expression of T cell markers, including PD-1, CD8A, and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) [63], which may be indicative of T-cell infiltration. In addition, leukocyte subsets, as determined by CIBERSORT (https://cibersort.stanford.edu/, accessed on 31 January 2022), show that PolE-mutant endometrial tumors display much higher proportions of CD8+ T cells, T helper cells, M1 macrophages, and activated NK cells as compared to MSI-H and MSS tumors.
PolE-mutant EC shows a heightened cytotoxic T-cell response, reflected by more CD8+ TILs, alongside an increased expression of T-cell exhaustion markers, which is consistent with chronic antigen exposure [44,60,64]. In CRC, it has been shown that the number of CD8+ cytotoxic TIL in PolE-mutated CRC significantly exceeds that of MMR proficient tumors but is not different from that of MMR deficient tumors. Furthermore, expression of cytotoxic T-cell markers (CD8A, IFNG, and PRF), effector cytokines (CXCL9 and CXCL10), and immune checkpoint proteins (PD-1, PD-L1, and CTLA-4) is significantly increased in PolE-mutant CRC [44].
To determine the mechanisms of increased TILs in PolE-associated tumors, many studies evaluated the correlation of antigenic neoepitopes as a consequence of ultramutation in PolE-mutant tumors [44,45,46,47]. One study showed that approximately 5.9% (7880/134,473) of missense mutations in ECs were possibly antigenic. Of these, 73% (5767/7880) presented in PolE-associated tumors, which indicated a significantly higher number of antigenic mutations when compared with MSI-H or MSS tumors (median of 365.5 vs. 16 and 2 for MSI-H and MSS, respectively). This could contribute to an active T-cell response in those tumors [60]. Similarly, PolE-mutant CRC shows an increased neoantigen load and a high degree of TILs, as compared with PolE-WT CRC [46]. In addition, the neoantigen load in CRC correlates significantly with CD45RO+ memory T-cell density but not with that of CD8+ (cytotoxic), CD3+ (total), or FOXP3+ (regulatory) T cells [46].
Despite being able to mount an anti-tumor immune response, some patients with PolE mutation present with bulky tumors. Since there was no significant difference in HLA class I protein expression between PolE-mutant and MSI-H tumors, the likelihood of immune escape due to lack of antigen presentation may be low. Instead, there may be suppression of the adaptive immune system that could explain immune evasion in PolE-driven EC. Although expression of T-cell exhaustion markers, including LAG3, TIM-3, and TIGIT, and immune checkpoint proteins, PD-1 and CTLA-4, is strongly associated with CD8A expression in all subtypes of ECs, their expression in PolE mutant tumors is significantly and substantially upregulated, indicating prolonged antigen stimulation and increased adaptive immune resistance [60]. Consistent with this study, Church and colleagues demonstrated that PolE-mutant EC tumors express higher levels of the immune checkpoint-related proteins, PD-L1 and PD-L2, than MSI-H and MSS tumors [60].
Taken together, these studies demonstrate that PolE-mutant tumors are associated with increased cytolytic T-cell response and upregulated expression of immunosuppressive checkpoints [47], which may contribute to a good prognosis and an overall favorable outcome for PolE-mutant patients. Although it has been shown in PolEP286R/+ mice that CD3/CD19 thymus-based T cell lymphomas infiltrated the lungs aggressively [23], more studies are needed to establish a causative relationship between PolE mutations, enhanced immune surveillance and good prognosis. In addition, increased expression of immunosuppressive checkpoints in PolE-mutant tumors demonstrates an excellent potential to respond to anti-PD1/PD-L1 immunotherapy.
6. Response to Immunotherapy in Patients with PolE Mutations
The observations that PolE-mutant tumors display a high cytotoxic T-cell response and an upregulated expression of immune checkpoints implicates that patients with PolE mutations may likely have a good response to immunotherapy with checkpoint inhibitors. Table 1 summarizes the immunotherapy trials in PolE mutant cancers.
The first reported immunotherapy in PolE-mutant cancer was carried out in non–small cell lung cancer (NSCLC) [48]. In this trial, two patients responded well to an anti-PD-1 antibody pembrolizumab. When examining tumor samples, one sample showed a stop-gain mutation in PolE as well as mutations in DNA-dependent protein kinase catalytic subunit (PRKDC or DNA-PK). In the second sample, a deleterious mutation in PolE4 encoding the third non-catalytic subunit of DNA PolE was detected. Although these mutations were not point mutations in the exonuclease domain of PolE, the tumors still showed a high mutation burden and displayed a significant response to the anti-PD-1 antibody.
In a case report, a 53-year-old female EC patient with PolE-V411L and nonsense PolE-R114* mutations showed an exceptional response to pembrolizumab [49]. An estimated total tumor mutation burden (TMB) of 4500 and 6500 nonsynonymous mutations were detected by WES in primary and metastatic tumor samples, respectively. This is consistent with TCGA data which indicate that metastatic/recurrent ECs harboring mutant PolE also display a high mutational burden [52].
In another case report, a glioblastoma patient with germline PolE V424L mutation showed a good response to immunotherapy with pembrolizumab [38]. All tumors, including one primary and two metastases were ultramutated, with over half of 17,276 to 20,045 mutations nonsynonymous. Subsequent work identified 2040 to 3254 high-quality neoantigenic mutations per sample, in which one of the metastases carried the highest mutation burden. After three weeks of treatment with pembrolizumab, a decrease in the right frontal lesion was observed, but there was a perilesional increase in MRI signal that encompassed the frontal horn ependyma. After 13 weeks of treatment with pembrolizumab, the right frontal lesion continued to decrease in size, and there was no enhancement next to the left-side resection bed. A corresponding decrease in MRI signal in the right frontal horn was observed when compared with the previous signal. Immunohistochemistry (IHC) analysis of the resected primary tumors and metastases obtained before and three weeks after pembrolizumab treatment identified brisk CD3+, CD4+, and CD8+ T-cell infiltration in the post-treatment lesion.
In 2017, the first immunotherapy with pembrolizumab in metastatic CRC was reported [50]. An 81-year-old Hispanic male CRC patient was found to have PolE-V411L and RAF1-R256S mutations with a high TMB of 122 mutations per Mb. The tumor near the hepatic flexure significantly decreased in size after three and six cycles of treatment with 200 mg pembrolizumab every three weeks. IHC results showed large amounts of CD8+ tumor-infiltrating lymphocytes, >90% of which were PD-1 positive. Interestingly, >99% of PD-L1 expression occurred in the tumor microenvironment (TME).
In subsequent investigations, five patients with MSI-H and three patients with PolE-mutated (one PolE-V411L and two PolE-P286R) metastatic CRC were treated with a PD-1 inhibitor [51]. Four patients (three MSI-H and one PolE-mutated) responded to PD-1 blockade while the other four (two MSI-H and two PolE-mutated) were nonresponders. There were more CD8+ T cells, with the majority expressing PD-1, in the four responders than in nonresponders. In three out of the four responders, PD-1-expressing CD8+ T cells were mostly found in the tumor stroma. In addition, non-tumor PD-L1 expression was shown in CD68+ tumor-associated macrophages in all eight patients, whereas in only one responder with MSI-H, less than 5% of PD-L1 was observed on tumor cells. PD-L1 expression varied among individual responders (from ~600 to 3800 PD-L1 positive cells/cm2) and nonresponders (from ~400 to 3800 positive cells/cm2), but average levels of PD-L1 expression were not significantly different between the responder and nonresponder groups. Additionally, a large amount of CD4+ T-bet+ T-cells were detected within the TME of the responders as compared to that of the nonresponders.
Taken together, these clinical studies indicate that PolE mutations are associated with high TMB, abundant neoantigens, increased TILs in TME, and a good response to pembrolizumab treatment, as shown in Figure 1. Although there are increasing numbers of reported cases of cancer patients with PolE mutations showing good response to immune checkpoint blockade of PD-1 [38,49,50,51,53,54,55,56,57,58], some patients with PolE mutations still do not respond to the treatment. Investigating the mechanisms by which PolE-mutant tumors respond to immunotherapy would help identify ways to improve the efficacy.
7. Discussions
Since PolE-associated ultramutated tumors were discovered in 2012, there has been significant progress in pathogenicity and clinical application. Work in yeast indicates that the arginine substitution at the 286th amino acid residue of the catalytic subunit of Polε prevents single strand DNA from entering the proofreading core and generating the hyperactive polymerase [22,24,25]. Subsequent studies in a PolE-P286R mouse model confirm the pathogenicity of PolE mutation in cancer [23]. Other PolE-associated mutations have also been explored for their role in ultramutagenesis [19,37]. However, several questions still remain. PolE-V411L mutants are distinct from those of PolE-P286R, as shown by structural and genetic data [19]. V411 is located outside of the binding cleft in the exonuclease domain. Although PolE-V411L is found in ultramutated tumors, surprisingly, Pol2-V411L, a yeast analog of PolE-V411L, does not generate elevated mutagenesis in yeast. It is still not understood how PolE-V411L is involved in ultramutagenesis. A possible mechanism could be that PolE V411L may alter the interaction between PolE and other essential proteins involved in DNA replication and/or DNA damage repair, leading to an increased mutation rate. Thirdly, although PolE-P286R and PolE-V411L may affect the behavior of DNA Polε differently, the mutation signatures of tumors harboring PolE-P286R and PolE-V411L are very similar [35]. The underlying reasons are not known.
PolE-driven ultramutated CRC and EC have good prognosis and favorable response to immunotherapy, possibly due to enhanced anti-tumor immune response caused by significantly elevated antigenic tumor neopeptides [47,59]. This is consistent with the observations in MSI-H CRC, in which increased numbers of neoantigens are correlated with enhanced immune infiltration and better patient survival [46]. Predicted neoantigen load in PolE-associated tumors is higher than in MSS and MSI-H tumors [60]. Notably, in MSI-H CRC, frameshift indels generate more neoantigens than single-nucleotide variants (SNVs) [46,61]. It is not clearly demonstrated which neoantigens contribute to increased immune infiltration and what percentage of neoantigens are generated by indels or SNVs in PolE and MSI tumors.
In addition to neoantigen load, another possible mechanism of the favorable outcome of PolE-mutant cancer patients is the “error threshold” theory. If too many mutations occur in the genome beyond an “error threshold”, cell viability would dramatically decrease. This phenomenon was proven in a yeast model with a combination of defective DNA polymerase proofreading and a complete loss of MMR [62,63]. Combining Pol2-P301R mutation and the deletion of MSH6, yeast cells did not develop after microcolonies were formed [24]. MMR deficient or PolE-mutant tumors rapidly accumulate large amounts of mutations (~600 mutations/cell division), reaching but not exceeding, ~20,000 exon mutations in under six months, suggesting a threshold compatible with cancer cell survival [64]. Moreover, the mutation rate in the PolE P286R mutant mouse cell line is estimated to be ~110 exon mutations per cell division, still not beyond the threshold of cancer cell survival [23]. More studies are needed to validate the “error threshold” mechanism.
8. Conclusions
In summary, cancer patients bearing PolE mutations display hypermutation and demonstrate good prognosis and a favorable outcome. A large amount of progress has been made in determining how PolE mutations affect DNA replication and mutation accumulation. However, most of the studies in human cancer are correlative. Understanding the mechanisms of PolE mutations in cancer biology would provide insight to a causal relationship between genome instability and cancer progression. It would also help to develop new strategies for effective cancer treatment.
Author Contributions
X.X. drafted the manuscript. J.W. and X.X. produced the figure and table. J.W. and N.J. revised the content and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The work is supported by NIH grants R01CA212241, R01CA215389 and R01CA208063 to JW.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We thank Kamila Jaroniec, who edited the manuscript.
Conflicts of Interest
The authors declare that they have no competing interest.
Abbreviations
| PolE | Polymerase Epsilon |
| MSS | Microsatellite Stability |
| MSI | Microsatellite Instability |
| EC | Endometrial Cancer |
| CRC | Colorectal Cancer |
| CMS | Consensual Molecular Subtypes |
| HNPCC | Hereditary Nonpolyposis Colorectal Cancer |
| WGS | Whole Genomic Sequencing |
| SBS | Single Base Substitution |
| PTEN | Phosphatase and tensin homolog |
| HR | Homologous Recombination |
| NHEJ | Non-Homologous End-Joining |
| HIF1α | Hypoxia Inducible Factor 1 alpha |
| SRC | Steroid Receptor Co-activator |
| mES | mouse-derived Embryonic Stem |
| PL | Pyridoxal |
| SQDG | Sulphoquinovosyl diacylglycerol |
| PFS | progression-free survival |
| DSS | disease-specific survival |
| OS | Overall Survival (OS) |
| HRs | Hazard Ratios |
| CI | Confidence Interval |
| DNA-PK | DNA-dependent protein kinase |
| TME | Tumor Microenvironment |
References
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 268. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebot-Bral, L.; Brandao, D.; Verlingue, L.; Rouleau, E.; Caron, O.; Despras, E.; El-Dakdouki, Y.; Champiat, S.; Aoufouchi, S.; Leary, A.; et al. Hypermutated tumours in the era of immunotherapy: The paradigm of personalised medicine. Eur. J. Cancer 2017, 84, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Network, T.C.G.A. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Parsons, R.; Li, G.M.; Longley, M.J.; Fang, W.H.; Papadopoulos, N.; Jen, J.; de la Chapelle, A.; Kinzler, K.W.; Vogelstein, B.; Modrich, P. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 1993, 75, 1227–1236. [Google Scholar] [CrossRef]
- Peltomaki, P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J. Clin. Oncol. 2003, 21, 1174–1179. [Google Scholar] [CrossRef]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Springgate, C.F.; Battula, N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974, 34, 2311–2321. [Google Scholar] [PubMed]
- Da Costa, L.T.; Liu, B.; el-Deiry, W.; Hamilton, S.R.; Kinzler, K.W.; Vogelstein, B.; Markowitz, S.; Willson, J.K.; de la Chapelle, A.; Downey, K.M.; et al. Polymerase delta variants in RER colorectal tumours. Nat. Genet. 1995, 9, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Flohr, T.; Dai, J.C.; Buttner, J.; Popanda, O.; Hagmuller, E.; Thielmann, H.W. Detection of mutations in the DNA polymerase delta gene of human sporadic colorectal cancers and colon cancer cell lines. Int. J. Cancer 1999, 80, 919–929. [Google Scholar] [CrossRef]
- Daee, D.L.; Mertz, T.M.; Shcherbakova, P.V. A cancer-associated DNA polymerase delta variant modeled in yeast causes a catastrophic increase in genomic instability. Proc. Natl. Acad. Sci. USA 2010, 107, 157–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, R.; Miyashita, K.; Inoue, M.; Shimamoto, A.; Yan, Z.; Egashira, A.; Oki, E.; Kakeji, Y.; Oda, S.; Maehara, Y. Concurrent genetic alterations in DNA polymerase proofreading and mismatch repair in human colorectal cancer. Eur. J. Hum. Genet. 2011, 19, 320–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pursell, Z.F.; Isoz, I.; Lundstrom, E.B.; Johansson, E.; Kunkel, T.A. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science 2007, 317, 127–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nick McElhinny, S.A.; Gordenin, D.A.; Stith, C.M.; Burgers, P.M.; Kunkel, T.A. Division of labor at the eukaryotic replication fork. Mol. Cell 2008, 30, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Barbari, S.R.; Shcherbakova, P.V. Replicative DNA polymerase defects in human cancers: Consequences, mechanisms, and implications for therapy. DNA Repair 2017, 56, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Bulock, C.R.; Xing, X.; Shcherbakova, P.V. DNA polymerase delta proofreads errors made by DNA polymerase epsilon. Proc. Natl. Acad. Sci. USA 2020, 117, 6035–6041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Barbari, S.R.; Kane, D.P.; Moore, E.A.; Shcherbakova, P.V. Functional Analysis of Cancer-Associated DNA Polymerase epsilon Variants in Saccharomyces cerevisiae. G3 2018, 8, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Wei, L.; Devbhandari, S.; Zhang, T.; Xiang, J.; Remus, D.; Zhao, X. DNA polymerase ε relies on a unique domain for efficient replisome assembly and strand synthesis. Nat. Commun. 2020, 11, 2437. [Google Scholar] [CrossRef] [PubMed]
- Min, K.W.; Kim, W.S.; Kim, D.H.; Son, B.K.; Oh, Y.H.; Kwon, M.J.; Lee, H.S.; Lee, S.E.; Kim, I.A.; Moon, J.Y.; et al. High polymerase ε expression associated with increased CD8+T cells improves survival in patients with non-small cell lung cancer. PLoS ONE 2020, 15, e0233066. [Google Scholar] [CrossRef]
- Kane, D.P.; Shcherbakova, P.V. A common cancer-associated DNA polymerase epsilon mutation causes an exceptionally strong mutator phenotype, indicating fidelity defects distinct from loss of proofreading. Cancer Res. 2014, 74, 1895–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.D.; Cuevas, I.; Zhang, M.; Lu, C.; Alam, M.M.; Fu, Y.X.; You, M.J.; Akbay, E.A.; Zhang, H.; Castrillon, D.H. Polymerase-mediated ultramutagenesis in mice produces diverse cancers with high mutational load. J. Clin. Investig. 2018, 128, 4179–4191. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Kane, D.P.; Bulock, C.R.; Moore, E.A.; Sharma, S.; Chabes, A.; Shcherbakova, P.V. A recurrent cancer-associated substitution in DNA polymerase epsilon produces a hyperactive enzyme. Nat. Commun. 2019, 10, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkash, V.; Kulkarni, Y.; Ter Beek, J.; Shcherbakova, P.V.; Kamerlin, S.C.L.; Johansson, E. Structural consequence of the most frequently recurring cancer-associated substitution in DNA polymerase epsilon. Nat. Commun. 2019, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Bulock, C.R.; Xing, X.; Shcherbakova, P.V. Mismatch repair and DNA polymerase δ proofreading prevented catastrophic accumlation of leading strand errors in cellls expressing a cancer-assoicated DNA polymerase ε variant. PLoS Genet. 2020, 48, 9124–9134. [Google Scholar]
- Park, V.S.; Pursell, Z.F. POLE proofreading defects: Contributions to mutagenesis and cancer. DNA Repair 2019, 76, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Soriano, I.; Vazquez, E.; De Leon, N.; Bertrand, S.; Heitzer, E.; Toumazou, S.; Bo, Z.; Palles, C.; Pai, C.C.; Humphrey, T.C.; et al. Expression of the cancer-associated DNA polymerase epsilon P286R in fission yeast leads to translesion synthesis polymerase dependent hypermutation and defective DNA replication. PLoS Genet. 2021, 17, e1009526. [Google Scholar] [CrossRef] [PubMed]
- Hodel, K.P.; de Borja, R.; Henninger, E.E.; Campbell, B.B.; Ungerleider, N.; Light, N.; Wu, T.; LeCompte, K.G.; Goksenin, A.Y.; Bunnell, B.A.; et al. Explosive mutation accumulation triggered by heterozygous human Pol epsilon proofreading-deficiency is driven by suppression of mismatch repair. Elife 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Herzog, M.; Alonso-Perez, E.; Salguero, I.; Warringer, J.; Adams, D.J.; Jackson, S.P.; Puddu, F. Mutagenic mechanisms of cancer-associated DNA polymerase ϵ alleles. Nucleic Acids Res. 2021, 49, 3919–3931. [Google Scholar] [CrossRef]
- Albertson, T.M.; Ogawa, M.; Bugni, J.M.; Hays, L.E.; Chen, Y.; Wang, Y.; Treuting, P.M.; Heddle, J.A.; Goldsby, R.E.; Preston, B.D. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 17101–17104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkel, T.A. A simple but profound mutation in mouse DNA polymerase epsilon drives tumorigenesis. J. Clin. Investig. 2018, 128, 3754–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.; Barbour, J.A.; Poulos, R.C.; Katainen, R.; Aaltonen, L.A.; Wong, J.W.H. Mutational processes of distinct POLE exonuclease domain mutants drive an enrichment of a specific TP53 mutation in colorectal cancer. PLoS Genet 2020, 16, e1008572. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, K.; Ohshima, K.; Nagashima, T.; Ohnami, S.; Ohnami, S.; Serizawa, M.; Shimoda, Y.; Maruyama, K.; Akiyama, Y.; Urakami, K.; et al. Molecular profiling and sequential somatic mutation shift in hypermutator tumours harbouring POLE mutations. Sci. Rep. 2018, 8, 8700. [Google Scholar] [CrossRef] [PubMed]
- Shinbrot, E.; Henninger, E.E.; Weinhold, N.; Covington, K.R.; Goksenin, A.Y.; Schultz, N.; Chao, H.; Doddapaneni, H.; Muzny, D.M.; Gibbs, R.A.; et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014, 24, 1740–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johanns, T.M.; Miller, C.A.; Dorward, I.G.; Tsien, C.; Chang, E.; Perry, A.; Uppaluri, R.; Ferguson, C.; Schmidt, R.E.; Dahiya, S.; et al. Immunogenomics of Hypermutated Glioblastoma: A Patient with Germline POLE Deficiency Treated with Checkpoint Blockade Immunotherapy. Cancer Discov. 2016, 6, 1230–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Eng, G.; Barbari, S.R.; Deshpande, V.; Shcherbakova, P.V.; Gala, M.K. Homologous Recombination Repair Truncations Predict Hypermutation in Microsatellite Stable Colorectal and Endometrial Tumors. Clin. Transl. Gastroenterol. 2020, 11, e00149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, M.; Gassner, F.J.; Parigger, T.; Neureiter, D.; Egle, A.; Geisberger, R.; Greil, R.; Zaborsky, N. A POLE Splice Site Deletion Detected in a Patient with Biclonal CLL and Prostate Cancer: A Case Report. Int. J. Mol. Sci. 2021, 22, 9410. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, I.C.; Rayner, E.; Osse, E.M.; Nout, R.A.; Creutzberg, C.L.; Tomlinson, I.P.M.; Church, D.N.; Smit, V.; de Wind, N.; Bosse, T.; et al. Adjuvant Treatment for POLE Proofreading Domain-Mutant Cancers: Sensitivity to Radiotherapy, Chemotherapy, and Nucleoside Analogues. Clin. Cancer Res. 2018, 24, 3197–3203. [Google Scholar] [CrossRef] [Green Version]
- Mizushina, Y.; Xu, X.; Matsubara, K.; Murakami, C.; Kuriyama, I.; Oshige, M.; Takemura, M.; Kato, N.; Yoshida, H.; Sakaguchi, K. Pyridoxal 5′-phosphate is a selective inhibitor in vivo of DNA polymerase alpha and epsilon. Biochem. Biophys. Res. Commun. 2003, 312, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Mizushina, Y.; Xu, X.; Asahara, H.; Takeuchi, R.; Oshige, M.; Shimazaki, N.; Takemura, M.; Yamaguchi, T.; Kuroda, K.; Linn, S.; et al. A sulphoquinovosyl diacylglycerol is a DNA polymerase epsilon inhibitor. Biochem. J. 2003, 370, 299–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temko, D.; Van Gool, I.C.; Rayner, E.; Glaire, M.; Makino, S.; Brown, M.; Chegwidden, L.; Palles, C.; Depreeuw, J.; Beggs, A.; et al. Somatic POLE exonuclease domain mutations are early events in sporadic endometrial and colorectal carcinogenesis, determining driver mutational landscape, clonal neoantigen burden and immune response. J. Pathol. 2018, 245, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gool, I.C.; Eggink, F.A.; Freeman-Mills, L.; Stelloo, E.; Marchi, E.; de Bruyn, M.; Palles, C.; Nout, R.A.; de Kroon, C.D.; Osse, E.M.; et al. POLE Proofreading Mutations Elicit an Antitumor Immune Response in Endometrial Cancer. Clin. Cancer Res. 2015, 21, 3347–3355. [Google Scholar] [CrossRef] [Green Version]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Van Gool, I.C.; Bosse, T.; Church, D.N. POLE proofreading mutation, immune response and prognosis in endometrial cancer. Oncoimmunology 2016, 5, e1072675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehnert, J.M.; Panda, A.; Zhong, H.; Hirshfield, K.; Damare, S.; Lane, K.; Sokol, L.; Stein, M.N.; Rodriguez-Rodriquez, L.; Kaufman, H.L.; et al. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J. Clin. Investig. 2016, 126, 2334–2340. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Wang, C.; Lee, P.P.; Chu, P.; Fakih, M. Response to PD-1 Blockade in Microsatellite Stable Metastatic Colorectal Cancer Harboring a POLE Mutation. J. Natl. Compr. Cancer Netw. 2017, 15, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Gong, J.; Tu, T.Y.; Lee, P.P.; Fakih, M. Immune profiling of microsatellite instability-high and polymerase epsilon (POLE)-mutated metastatic colorectal tumors identifies predictors of response to anti-PD-1 therapy. J. Gastrointest. Oncol. 2018, 9, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Momen, S.; Fassihi, H.; Davies, H.R.; Nikolaou, C.; Degasperi, A.; Stefanato, C.M.; Dias, J.M.L.; Dasgupta, D.; Craythorne, E.; Sarkany, R.; et al. Dramatic response of metastatic cutaneous angiosarcoma to an immune checkpoint inhibitor in a patient with xeroderma pigmentosum: Whole-genome sequencing aids treatment decision in end-stage disease. Mol. Case Stud. 2019, 5, a004408. [Google Scholar] [CrossRef] [PubMed]
- Veneris, J.T.; Lee, E.K.; Goebel, E.A.; Nucci, M.R.; Lindeman, N.; Horowitz, N.S.; Lee, L.; Raut, C.P.; Crotzer, D.; Matulonis, U.; et al. Diagnosis and management of a recurrent polymerase-epsilon (POLE)-mutated endometrial cancer. Gynecol. Oncol. 2019, 153, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, E.; He, T.; Goldstein, D.Y.; Halmos, B.; Chuy, J. DNA Polymerase varepsilon Deficiency Leading to an Ultramutator Phenotype: A Novel Clinically Relevant Entity. Oncologist 2017, 22, 497–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.; Wolchok, J.D. Successful Treatment of a Patient with Glioblastoma and a Germline POLE Mutation: Where Next? Cancer Discov. 2016, 6, 1210–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santin, A.D.; Bellone, S.; Buza, N.; Choi, J.; Schwartz, P.E.; Schlessinger, J.; Lifton, R.P. Regression of Chemotherapy-Resistant Polymerase ε (POLE) Ultra-Mutated and MSH6 Hyper-Mutated Endometrial Tumors with Nivolumab. Clin. Cancer Res. 2016, 22, 5682–5687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittica, G.; Ghisoni, E.; Giannone, G.; Aglietta, M.; Genta, S.; Valabrega, G. Checkpoint inhibitors in endometrial cancer: Preclinical rationale and clinical activity. Oncotarget 2017, 8, 90532–90544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, H.H.; Shi, Q.; Heying, E.N.; Muranyi, A.; Bredno, J.; Ough, F.; Djalilvand, A.; Clements, J.; Bowermaster, R.; Liu, W.W.; et al. Intertumoral Heterogeneity of CD3(+) and CD8(+) T-Cell Densities in the Microenvironment of DNA Mismatch-Repair-Deficient Colon Cancers: Implications for Prognosis. Clin. Cancer Res. 2019, 25, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Howitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.; Stover, E.; Strickland, K.C.; D’Andrea, A.D.; Wu, C.J.; et al. Association of Polymerase e-Mutated and Microsatellite-Instable Endometrial Cancers With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015, 1, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Mandal, R.; Samstein, R.M.; Lee, K.W.; Havel, J.J.; Wang, H.; Krishna, C.; Sabio, E.Y.; Makarov, V.; Kuo, F.; Blecua, P.; et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019, 364, 485–491. [Google Scholar] [CrossRef]
- Williams, L.N.; Herr, A.J.; Preston, B.D. Emergence of DNA polymerase epsilon antimutators that escape error-induced extinction in yeast. Genetics 2013, 193, 751–770. [Google Scholar] [CrossRef] [Green Version]
- Herr, A.J.; Ogawa, M.; Lawrence, N.A.; Williams, L.N.; Eggington, J.M.; Singh, M.; Smith, R.A.; Preston, B.D. Mutator suppression and escape from replication error-induced extinction in yeast. PLoS Genet. 2011, 7, e1002282. [Google Scholar] [CrossRef]
- Shlien, A.; Campbell, B.B.; de Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; Van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262. [Google Scholar] [CrossRef]
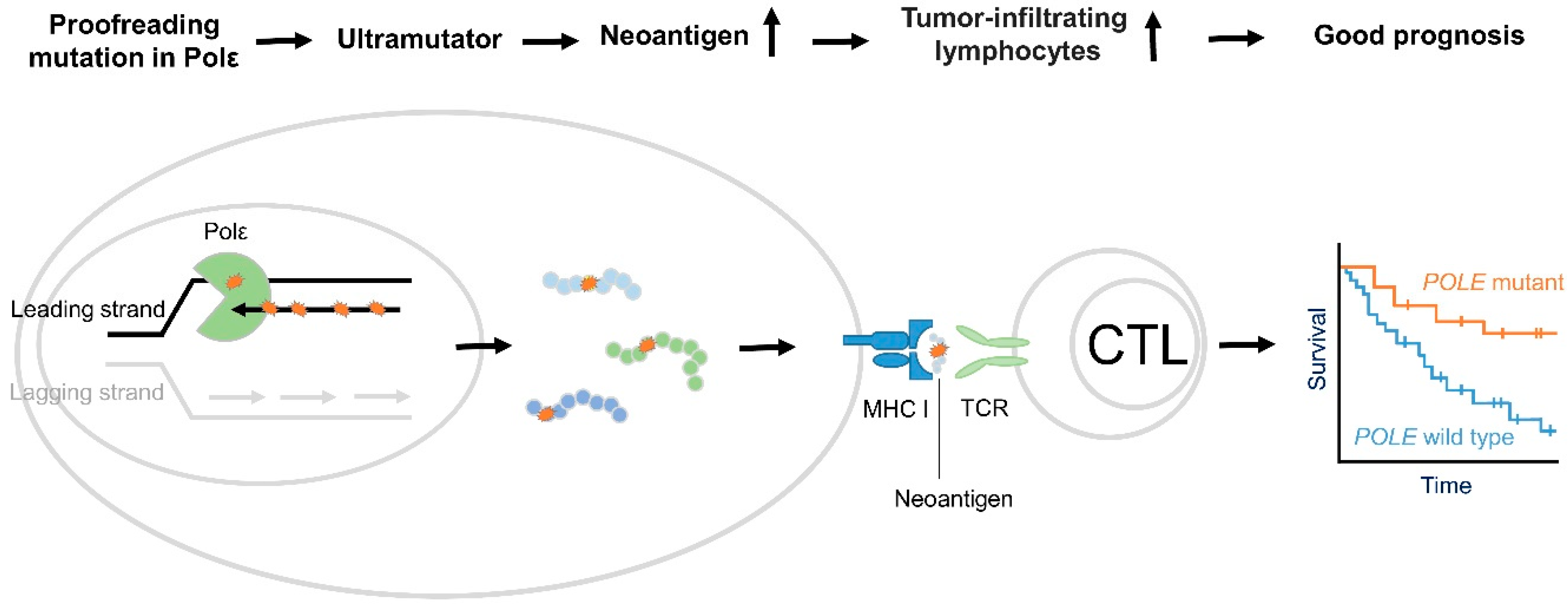
Figure 1.
Proposed mechanism of good prognosis in PolE associated cancer. PolE encodes the catalytic and proofreading exonuclease subunit of DNA polymerase ε (Pol ε), which is responsible for bulks of DNA synthesis in the leading strand during DNA replication. Cancer-associated PolE mutations in the exonuclease domain hinder proofreading activity and increase polymerase activity, resulting in ultramutation [22,23,24,25]. Subsequently, they lead to an increased neoantigen load [44,47]. When neoantigens are presented, the immune system recognizes them and ultimately generates an immune response, eliciting tumor suppressor function [44,47].
Figure 1.
Proposed mechanism of good prognosis in PolE associated cancer. PolE encodes the catalytic and proofreading exonuclease subunit of DNA polymerase ε (Pol ε), which is responsible for bulks of DNA synthesis in the leading strand during DNA replication. Cancer-associated PolE mutations in the exonuclease domain hinder proofreading activity and increase polymerase activity, resulting in ultramutation [22,23,24,25]. Subsequently, they lead to an increased neoantigen load [44,47]. When neoantigens are presented, the immune system recognizes them and ultimately generates an immune response, eliciting tumor suppressor function [44,47].


{kind=link}
Table 1.
Immunotherapy in PolE mutant cancers.
| PolE Mutation | Cancer Type | Tumor Mutation Burden | Immunotherapy | Overall Best Response | Reference |
|---|---|---|---|---|---|
| somatic V411L | metastatic EC | 4500 to 6500 nonsynonymous mutations | pembrolizumab | PR | [49] |
| germline V424L | glioblastoma | 17,276 to 20,045 mutations | pembrolizumab | PR | [38] |
| somatic V411L | metastatic CRC | 122 mutations per Mb | pembrolizumab | PR | [50] |
| somatic V411L | metastatic CRC | NA | pembrolizumab | CR | [51] |
| somatic P286R | metastatic CRC | NA | pembrolizumab | PD | [51] |
| somatic P286R | metastatic CRC | NA | pembrolizumab | SD | [51] |
EC, Endometrial Cancer; CRC, Colorectal Cancer; PR, Partial Response; CR, Complete Response; PD, Progressive Disease; SD, Stable Disease.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Xing, X.; Jin, N.; Wang, J. Polymerase Epsilon-Associated Ultramutagenesis in Cancer. Cancers 2022, 14, 1467. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061467
AMA Style
Xing X, Jin N, Wang J. Polymerase Epsilon-Associated Ultramutagenesis in Cancer. Cancers. 2022; 14(6):1467. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061467
Chicago/Turabian StyleXing, XuanXuan, Ning Jin, and Jing Wang. 2022. "Polymerase Epsilon-Associated Ultramutagenesis in Cancer" Cancers 14, no. 6: 1467. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14061467
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.