
Dialogue among Lymphocytes and Microglia in Glioblastoma Microenvironment


Department of Physiology and Pharmacology, Sapienza University, 00185 Rome, Italy
*
Author to whom correspondence should be addressed.
Cancers 2022, 14(11), 2632; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14112632
Submission received: 28 April 2022
/
Revised: 24 May 2022
/
Accepted: 25 May 2022
/
Published: 26 May 2022
(This article belongs to the Special Issue Engineering the Tumor Immune Microenvironment)
{kind=link}
{kind=link}
Abstract
:Simple Summary
In this review, we summarize in vitro and in vivo studies related to glioblastoma models and human patients to outline the symbiotic bidirectional interaction between microglia, lymphocytes, and tumor cells that develops during tumor progression. Particularly, we highlight the current experimental therapeutic approaches that aim to shape these interplays, such as adeno-associated virus (AAV) delivery and CAR-T and -NK cell infusion, and to modulate the tumor microenvironment in an anti-tumoral way, thus counteracting glioblastoma growth.
Abstract
Microglia and lymphocytes are fundamental constituents of the glioblastoma microenvironment. In this review, we summarize the current state-of-the-art knowledge of the microglial role played in promoting the development and aggressive hallmarks of this deadly brain tumor. Particularly, we report in vitro and in vivo studies related to glioblastoma models and human patients to outline the symbiotic bidirectional interaction between microglia, lymphocytes, and tumor cells that develops during tumor progression. Furthermore, we highlight the current experimental therapeutic approaches that aim to shape these interplays, such as adeno-associated virus (AAV) delivery and CAR-T and -NK cell infusion, and to modulate the tumor microenvironment in an anti-tumoral way, thus counteracting glioblastoma growth.
1. Glioblastoma: The Most Aggressive Brain Tumor
Glioblastoma (GBM) is a high-grade glioma, representing the most common and lethal primary brain tumor in adults [1,2], characterized by high heterogeneity in terms of genetic and epigenetic modifications, histological hallmarks, and response to treatment [3,4]. GBM cells show a high ability to proliferate and invade the brain parenchyma, and the peculiar localization, shielded by the blood–brain barrier (BBB), enhances the resistance to several chemotherapeutic drugs [5], giving this tumor a poor prognosis despite the scientific progress and combination of surgery, chemotherapy, and radiotherapy [6,7,8]. The preferential routes of GBM cells to invade the parenchyma are along the blood vessels and white matter [9]. To efficiently migrate, GBM remodels the extracellular matrix through the expression of secreted proteases, including the MMP membrane types MMP1/14, MMP2, and MMP9, the serine protease uPA, and cell surface proteases of the ADAM family [9]. Interestingly, apart from the high invasion in the brain, GBM rarely forms metastasis out of the primary site, probably due to a particular tropism for brain areas [10,11,12,13,14,15,16].
Another pivotal hallmark of this brain tumor is the uncontrolled proliferation of GBM cells as a result of deregulation in several molecular pathways, such as p53 signaling. Loss-of-function mutations on the p53 protein have been found in many tumor malignancies, including GBM. The function of p53 can be deregulated by gain-of-function mutations of negative p53 regulators, such as MDM2 and MDM4, or loss-of-function mutations of its activators, such as p14ARF [17,18]. Furthermore, mutations in the PTEN protein, an inhibitor and part of the mTOR pathway, are epigenetically silenced or genetically mutated in almost 60% of GBM [19], and the deletion of chromosome 13, containing the gene Rb1 [7], results in uncontrolled tumor cell divisions [7,19]. Moreover, GBM supports its own growth by increasing angiogenesis, the ability to build up new blood vessels that are able to feed the tumor mass [20]. Recently, it was reported that angiogenesis is associated with the expression of the hypoxia inducible factor (HIF-1) in response to the hypoxia present in the tumor environment to produce pro-angiogenic factors such as vascular endothelial growth factor (VEGF) [21].
The genetic complexity shown in GBM involves several genetic and epigenetic modifications that result in the loss of tumor suppressor gene function (CDKN2A/B and PTEN) or the activation of oncogenic pathways (CDK4, p21–RAS, and MDM2) [22,23,24]. This genetic heterogeneity is accompanied by a high diversity in the cell populations forming the GBM microenvironment (GME), such as resident and peripheral immune cells, endothelial cells, mesenchymal cells, and glioma stem cells (GSCs) [25]. GSCs are characterized by the ability to differentiate into different cell lineages to reconstitute the tumor mass. This characteristic was first demonstrated with the identification of CD133+ GBM cells that were able to initiate the tumor process in vivo [26]. Furthermore, GSC cells show multiple drug resistance: it has been shown that CD133+ GBM cell fractions in the tumoral mass increase after exposure to radiation due to the activation of the DNA damage checkpoint response and an increase in DNA repair capacity [27]. Moreover, GBM cooperates with parenchymal cells in multiple ways: among them are soluble molecules [28,29,30,31,32,33,34,35,36], direct synaptic interactions [37,38,39], and extracellular vesicles [40], promoting tumor proliferation, angiogenesis, immunosuppression, degradation of the extracellular matrix, and invasion.
Improving the knowledge of the pathways driving the interactions between GBM, infiltrating cells, and microglial cells may provide new perspectives to manage GBM growth and development, highlighting the way for new therapeutical approaches and targets.
2. GBM–Microglia Symbiosis
In the last decades, our vision of tumor mass has been radically changed. Nowadays, it is well described that tumoral mass is not solely constituted by clonal cancer cells; indeed, there is great heterogeneity between the cancer cells inside the mass. Among them, glioma-associated resident microglia and peripheral-invading monocyte-derived macrophages (called GAMs) represent from 30% up to 50% of total cells in the tumor microenvironment [41,42,43,44], with several potentially overlapping functions [45]. Initially, during GBM development, the main population of GAMs cells is represented by microglia. Subsequently, with tumor development, there is a progressive increase in the number of infiltrating macrophages/monocytes, in response to molecular signals secreted by GBM, that weaken the BBB to recruit peripheral immune cells [46,47]. For a long time, the lack of specific markers has made the distinction between microglia and brain-infiltrating monocyte-derived macrophages difficult. Furthermore, the first experimental approaches used to deplete the bone marrow progenitors that induced damage to the BBB and consequent monocyte infiltration into the brain [48,49,50,51], such as whole-body irradiation, helped complicate this distinction. To date, this problem is partially overcome with head shielding during irradiation [52] and the enhancement of experimental techniques, such as RNA sequencing (RNA-seq), mapping studies, and single-cell RNA-seq [53,54,55,56], allowing us to investigate the different profiles inside the GAMs.
Once recruited, GAMs are educated by GBM cells toward an anti-inflammatory/pro-tumoral phenotype that releases a plethora of soluble molecules with pro-tumoral effects [57,58,59,60,61], sustaining the GME, the tumor growth [57,58,59], and angiogenesis [60]. Nevertheless, depletion experiments in mouse models have demonstrated that GAMs do not participate in gliomagenesis [62]. The CSF1-R (signaling pathway fundamental for microglia and macrophage survival) inhibition reduces GAMs’ recruitment in the tumor core, resulting in a reduction of tumor cell proliferation and invasion [63,64,65]. However, a CSF-1R inhibitor-based therapy failed to reach significant results in a phase II study for GBM patients [66].
The pattern of molecules produced by GBM cells, such as toll-like receptors (TLRs), GDNF, CXCRL1, and TGF-β, attracts and affects GAMs functions, supporting tumor growth [67,68,69,70,71,72,73]. Interestingly, the isocitrate dehydrogenase (IDH) mutation affects the production of these factors, resulting in different GMEs [29,30]. Indeed, non-mutated IDH1 supports an immunosuppressive ground through the activation of the Wnt/β-catenin pathway in GAMs, which shows different gene expression signatures with respect to IDH-mutant GBMs [74].
3. GAM Interactions with Lymphocytes in the GBM Microenvironment
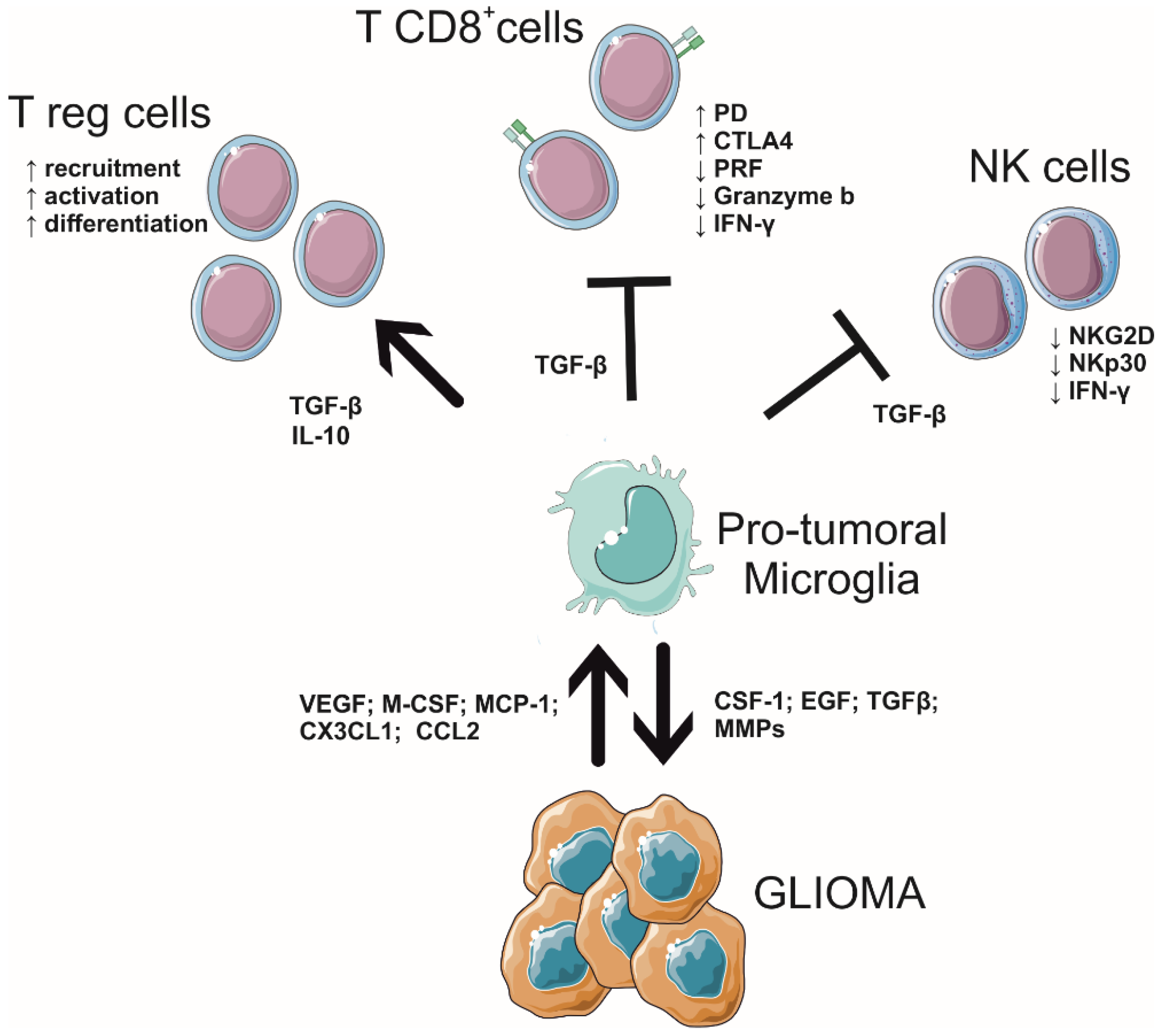
A key element facilitating GBM growth is its ability to promote an immunosuppressive ground that hampers the reaction of immune cells against tumor cells. Consistently, GBMs show a weak infiltration of cytotoxic lymphocytes and a rare patient’s responsiveness to checkpoint inhibitor immunotherapy, classifying this brain tumor among the so-called “cold” cancers [4,76,77,78]. Indeed, the lymphocytic level positively correlates with increased survival in brain tumor patients, but no results have been produced in GBM patients [79,80,81,82]. Further, a comparative study of GBM patients exhibited lower T cytotoxic cell activity and higher Treg cell activation compared with healthy volunteers [83]. In this scenario, GAM interactions are not mainly restricted to dialogue with GBM cells; contrarily, GAMs orchestrate the immunosuppressive GME by communicating with brain parenchymal cells and infiltrated lymphocytes recruited by GBM and brain metastasis derived from extracranial cancers [79,80].
Both CD8+ and CD4+ T-lymphocytes, besides T helper, FoxP3+ Treg, myeloid suppressor cells, and natural killer (NK) cells, invade GBM [81,82,83,84,85,86,87]. Interestingly, mutated IDH 1 and 2 astrocytomas, which have a better prognosis in comparison with wild-type IDH, are related to a reduced number of cytotoxic lymphocytes in the tumor core [88,89]. To date, it is well described that GAM and T lymphocyte interactions drive GBM development, infiltration, and differentiation, and the complexity of this interplay forms the heterogeneity of GBM tissue across different patients. Particularly, the immunosuppressive ground created by GAMs, which inhibits the cytotoxic activity of T-cells and explains why GBM patients do not respond to immune therapy [90], is mainly due to the expression of PD ligands PD-L1/2, and the cytotoxic T lymphocyte-associated protein 4 (CTLA-4) ligands CD80 and CD86 [91]. Further, GAMs release TGF-β, a key signal that inhibits the anti-tumoral effects of T-cells [92], downregulating the expression of the proteins responsible for lymphocyte cytotoxicity, such as perforin, granzyme, and interferon (IFN)-γ; consistently, in vivo studies in GBM-bearing mice have shown that the neutralization of TGF-β upregulates the expression of these genes in CD8+ T-cells [92]. In contrast, GAM-originated TGF-β, with the support of the IL-10, stimulates the differentiation of naïve T-cells into regulatory T (Treg) cells, which suppress CD8+ T-cells in the GME [93,94]. Moreover, TGF-β induces downregulation of NKp30 and NKG2D activating receptors on NK cells [95]. These molecules also promote GBM angiogenesis, growth, and invasion and the reduction of T-cell cytotoxic activity [96,97,98,99,100,101].
GAMs are also able to regulate the infiltration of lymphocytes in malignant tumors. GAMs release the chemokine CCL2, which is essential for the recruitment of regulatory T-cells and myeloid-derived suppressor cells [102]. Furthermore, GAMs control extracellular matrix stiffness and collagen deposition, regulating the movement of T-cells across the GME [103]. Consistently, GAM depletion has been reported to increase CD8+ T-cell migration and infiltration [104], helping to overcome immunosuppression.
Among the patterns of GAM’s molecules released to create the immunosuppressive microenvironment for GBM, neuropilin-1 (NRP-1), expressed by various types of cells, including microglia and macrophages, plays a pivotal role [105,106]. NRP-1 increases angiogenesis (enhancing the production of pro-vascularization signals) and boosts the infiltration of Treg in the tumor mass while decreasing the number of T CD8+ lymphocytes and driving GAM polarization toward a pro-tumoral way [107,108,109]. Consistently, the depletion of NRP-1 from microglia in glioma-bearing mice leads to a reduction in GBM volume, increasing the number of T CD8+ cells in the tumor mass and shaping GAM polarization [109].
The GAM’s role in driving immune responses against the GBM makes these cells a juicy target for several experimental immunotherapeutic studies that aim to reprogram microglia or macrophages to counteract tumor development.
3.1. Activating Lymphocytes to Modulate Microglia-GBM Cross-Talk
Given the evidence on the tumoricidal role of microglial cells when they are activated toward a pro-inflammatory phenotype, in the last years, one big effort of biomedical research has aimed to re-educate microglia against tumor cells. Consistently targeting the immune tumor microenvironment appears to be a promising therapeutic strategy to counteract GBM progression [5,33,110]. The switch to a specific phenotype correlates with prognosis, and the pathological assessment of a specific microglial setting can predict a patient’s outcome [111]. Microglia polarization is mediated by complex pathways involving cross-talk with GBM and immune cells. In this scenario, both environmental and peripheral stimuli seem to play a central role. In particular, recently, evidence has demonstrated that activated lymphocytes can modulate GAM phenotypes, highlighting a new potential target able to drive microglia against GBM.
3.1.1. Engineered Microglia Boost Lymphocyte Functions against GBM
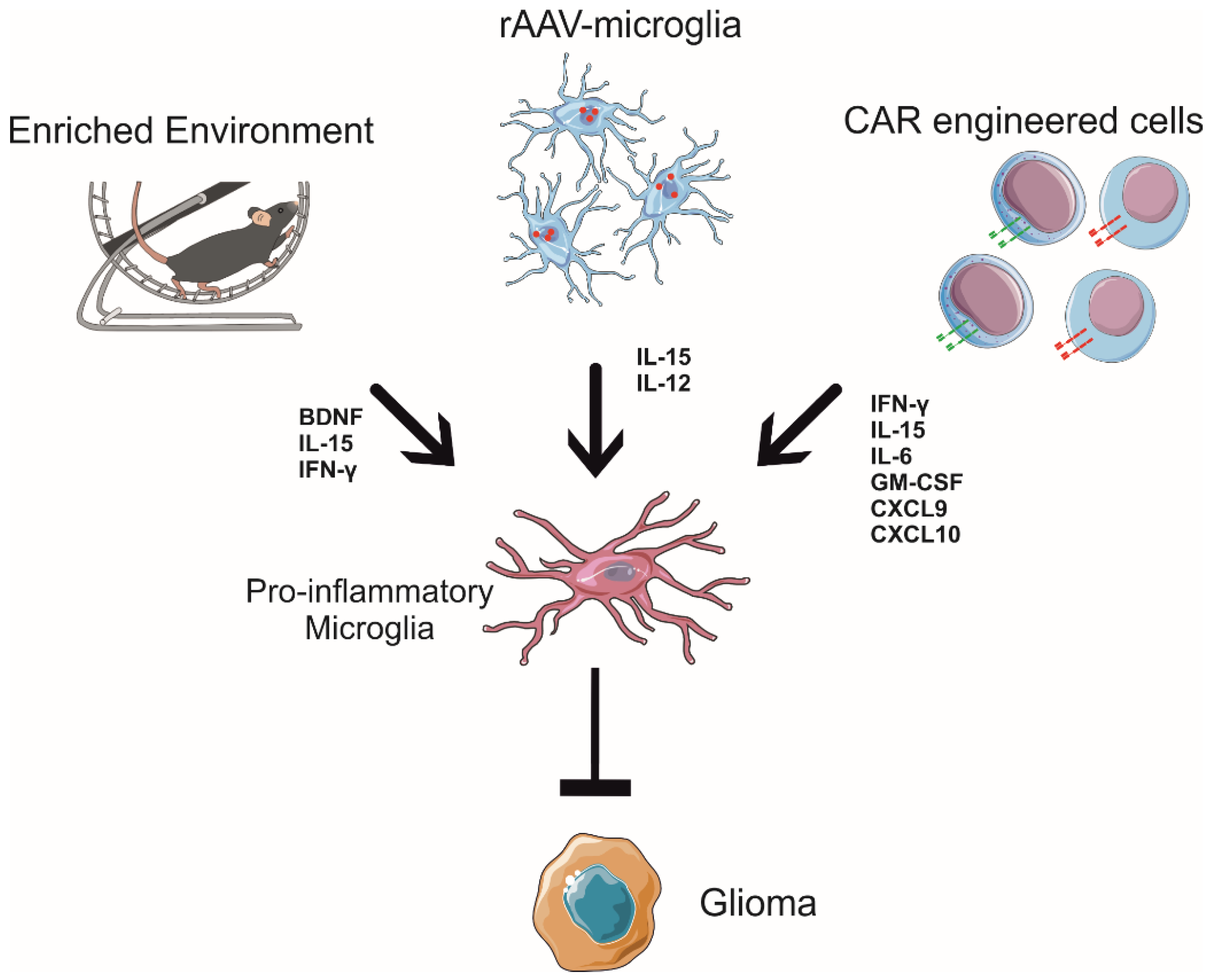
Gene therapies for GBM are being developed in clinical trials; particularly, in recent years, more and more studies have aimed to genetically manipulate microglial cells as a new effective therapeutical approach to defeating several neurodegenerative diseases [112,113,114]. The use of recombinant viruses such as adeno-associated viruses (AAVs), a small and non-pathogenic defective parvovirus, is a promising tool due to their characteristics, such as high titers, broad host range, efficient infection of quiescent cells, and vector integration [115]. In this way, AAVs represent an efficient vector system, determining long-lasting changes in gene expression even if the limited gene transfer to GBM cells hampers its use [116]. Consistently, AAVs are considered safe for human gene therapy and have been successfully used to target several cell types within the central (CNS) and peripheral (PNS) nervous systems, including neurons, oligodendrocytes, astrocytes, Müller glia, and Schwann cells [117]. On the other side, the AAV transduction of microglia is rare and challenging: indeed, in vivo, less than 20% of efficiency seems to be achieved, although some cases of microglial transductions, both in vitro and in vivo, have recently been reported, thanks to advances in the new strategies designed for recombinant viral vectors [113,118,119]. Furthermore, engineered microglial cells could be destined as a biologically active vehicle for the delivery of anti-tumoral molecules. Indeed, recently, the potential use of microglia engineered to express IL-15 upon infection with a recombinant AAV serotype 2 (rAAV2) carrying IL-15 (rAAV2-IL-15) was explored to counteract GBM growth in mouse models [119]. IL-15 is a crucial cytokine for the development, maturation, and activation of NK cells and CD8+ T-cells, with no effect on the expansion of the T regulatory cell population involved in suppressing immune responses, highlighting a potential therapeutic use in cancer immunotherapy [120]. Furthermore, IL-15 enhances the anti-tumor efficacy of the extracellular vesicles derived from NK cells, showing higher cytolytic activity against GBM [121]. Microglia infected with rAAV2-IL-15 functionally induce the release of IL-15, increasing the viability of NK cells without affecting their activation state in vitro [119]. In vivo, the rAAV2-IL-15 microglial cells infiltrate GBM mass and increase the recruitment of IFN-γ+ NK cells in GBM-bearing mice, with effects on tumor growth [32,33,119], highlighting the fundamental role of IL-15 in the tumor core to boost immune reaction. Moreover, rAAV2-IL-15 microglia consistently modulate the GAM state, with a reduction in arginase levels and an increased number of branches, and cover the parenchymal region [119], suggesting the switch to an anti-tumoral phenotype [122]. These data indicate that the recruited NK cells in the tumor core are activated and release pro-inflammatory cytokines (i.e., IFN-γ), thus explaining the modulation of the GAM phenotype [33].
An elegant approach to modulating microglial functions in GBM using AAV delivery is the intracranial injection of rAAV2 that encodes IL-12 in rat models. rAAV2-IL-12 increases the expression of IL-12 and IFN-γ in the brain, potent cytokines that enhance microglial activity [118,123,124]. Consistently, the use of rAAV2-IL-12 increases microglial infiltration in GBM and the expression of the activation markers ED1 and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), turning in the acquisition of an anti-tumoral phenotype by microglia, which is associated with a reduction in tumor volume and longer survival time in rat models [118].
These data demonstrate the potential for improved AAV-based gene therapy for GBM-targeting microglial cells as a vehicle and tool to translate the anti-tumoral signals inside the tumor mass, boosting the lymphocytes’ tumoricidal activity and offering a new perspective to use them as Trojan horses to modify the tumor microenvironment (Figure 2).
3.1.2. Environmental Stimuli Boost the Interplay between Lymphocytes and Microglia, Reducing GBM Growth
Lifestyle, which includes many aspects of interactions with the environment, from nourishment and education to physical activity and quality of sleep, is one of the most powerful instruments shaping mankind. Exposure to different environments affects brain functions and cognitive performance [125,126,127]. Clinical studies have demonstrated that depression, feelings of loneliness, and low sociability represent important risks for the development of several types of cancers [128]. On the contrary, in humans, positive stimuli such as motor activity, social interaction, and cognitive stimulation related to, for example, art or music can boost neuronal connectivity and counteract cancer development [125,126,127,128], supporting the idea that patients should benefit from an improved lifestyle.
In mouse models, the enriched environment refers to housing animals in larger cages with various possibilities of physical activities and exploration, using objectives such as ladders, running wheels, plastic tubes, and other toys [129]. Enriched environment exposure has beneficial effects on several neuronal activities in mice, improving spatial memory, increasing dendritic arborization and the density of dendritic spines on cortical neurons [130], and exhibiting higher hippocampal neurogenesis in adults [131]. In particular, physical exercise, exposure to an enriched environment, and dieting act through complex modifications of microglial cells, which change their phenotype and modulate their functional activity [132]. All these environmental stimuli are able to be converted into molecular signals in the brain that educate microglial cells to remodel brain homeostasis and shape neural plasticity, enhancing neuroprotection and counteracting the development of several pathologies [133,134,135]. Among the potential candidates for this communication, brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) are key cerebral mediators of these phenomena [136,137,138,139,140]. With regard to cancer, clinical studies demonstrate that specific distressing stimuli, such as depression, feelings of loneliness, and lack of social relationships, represent important risk factors for tumor development and progression [128]. In contrast, it is known that living in environments that are enriched with sensorial, physical, and social stimuli affects the levels of hormones linked to the hypothalamic–pituitary axis, such as norepinephrine, BDNF, and glucocorticoids, regulating the growth of several types of tumors in both humans and mouse models [141,142,143]. In GBM, environmental stimulation shapes microglial toward an anti-tumoral profile [32,33]. Indeed, housing animals in an enriched environment deeply modifies GAM phenotypes, in particular the microglial phenotype, as shown by gene expression profiling: myeloid cells, isolated from the brain of glioma-bearing mice, show a reduction in genes related to the pro-tumoral phenotype, but only microglia increase the expression of pro-inflammatory genes, indicating an anti-tumoral state [33,58]. Furthermore, environmental stimuli modify the morphology of GAMs infiltrated in tumor mass, reducing cell body roundness and increasing the length and number of cell branches, the speed of process movement towards ATP (which mimics an injury signal), and the expression of P2ry12 mRNA, thus suggesting the re-establishment of a more efficient homeostatic and patrolling activity of these cells [144]. Interestingly, P2RY12 is specifically expressed by microglia and is associated with ATP-dependent process patrolling [145] and better survival of patients with astrocytoma [146]. Lastly, exposure to environmental enrichment modulates microglia phagocytic activity [33]. This effect of the environment on microglial cells in GBM is mediated by the NK cells. In fact, during enriched environment exposure, the NK cells more efficiently colonize the brain, producing IFN-γ and degranulating against GBM cells [32]. Moreover, upon housing in an enriched environment, there was a significant increase in direct contact between GBM and NK cells, and the NK cell depletion completely abolished the effect of the environment on pro- and anti-inflammatory gene expression in GAMs [33]. The interplay between microglia and NK cells was, at least in part, orchestrated by the IFN-γ released by the recruited NK cells and the IL-15 released by microglial cells upon environmental stimuli exposure [33]. Consistently, the BDNF produced in the brain of glioma-bearing mice after enriched environment exposure stimulates the production of IL-15 by microglial cells, which, in turn, stimulates NK cells to produce IFN-γ, with effects on GAM phenotypes, switching them towards an anti-tumoral state (see Figure 2), which explains the protective effects of the environment.
3.1.3. CAR Technology in GBM
Recently, chimeric antigen receptor (CAR) technology has been shown to be a valid approach to counteract the growth of several types of cancers [147,148]. This technology takes advantage of direct immune cells, particularly T-lymphocytes, against tumors. In detail, isolated T-lymphocytes from patients are engineered to express a chimeric receptor directed against tumoral antigens. Once generated, these CAR-T cells are expanded in vitro and, subsequently, are re-infused into the donor patient [149]. One of the main advantages of CAR technology is that the chimeric receptor has a higher affinity to its target compared to the T-cell receptor (TCR) expressed on the membrane of lymphocytes [150]. Furthermore, the binding of the receptor with the antigen is not mediated by major histocompatibility complexes (MHCs); in this way, CAR-T cells are insensitive to the loss of MHCs used by tumors as an immunoescaping strategy [150]. Indeed, CAR-T cell-based therapy showed great clinical success in fighting hematological malignancies [151,152,153,154], and several clinical trials were conducted exploring the use of CAR-T cells against solid tumors, including GBM [155,156,157,158].
One of the initial targets of CAR-T cells in GBM therapy was IL-13 receptor IL13Rα2, which has been found to be overexpressed in human GBM samples [159]. The first generation of IL13Rα2 CAR-T cells is able to discriminate GBM cells from normal cells and selectively exert cytolytic activity in vitro and in GBM-bearing mice [160]. A second generation of IL13Rα2 CAR-T cells was generated to overcome the problem of persistence and to enhance biological activity in terms of cytotoxicity and pro-inflammatory cytokine production [161]. After the infusions, increased levels of pro-inflammatory cytokines were detected in cerebrospinal fluid (CSF), including IFN-γ, IL-15, IL-6, IL-10, GM-CSF, IL-2, IL-2Ra, IL-1RA, CXCL10, granzyme b [162], and, interestingly, the chemokines CXCL9 and CXCL10 [163]. These ligands for the CXCR3 receptor are expressed by macrophages and microglia and could modulate the activation state of GAMs [164]; consistently, CXCR3-lacking macrophages promote cancer growth [164]. Moreover, CXCL9 and CXCL10 regulate the recruitment of T and NK cells in GBM [165].
Another CAR-T cell target is mutated epidermal growth factor receptor variant III (EGFRvIII), overexpressed in a subset of GBM patients [166]. These engineered EGFRvIII CAR-T cells selectively recognize and kill GBM cell lines in vitro [166] and produce pro-inflammatory cytokines [166,167,168] that are able to increase the survival of human and mouse models [167]. Although these are promising results, EGFRvIII CAR-T cells must overcome the problem of the heterogeneous expression of the receptor in GBM samples, the secondary effects on other cells expressing the EGF receptor, and the increased expression of immunocheckpoints by tumor cells [168]. Recently, CAR technology investigated the possibility of targeting epidermal growth factor receptor 2 (HER2) and the integrin protein αVβ3, with promising results. These proteins are highly expressed by many solid tumors, including GBM and diffuse intrinsic pontine glioma (DIPG), while it is minimally expressed in physiological tissues [169,170]. In a preclinical study, HER2 and αVβ3 CAR-T cells induced death in human and murine GBM cells and, in CD133+ GSCs, increased the production of pro-inflammatory cytokines, such as IFN-γ, TNF-α, and IL-2, in co-culture experiments in vitro. Furthermore, both HER2 and αVβ3 CAR-T cells significantly prolonged the survival of GBM xenograft mice, reducing tumor growth [170,171]. Although these are promising results, more studies are needed regarding their safety and efficacy in human GBM patients [149]. Moreover, αVβ3 CAR-T cells have been shown to develop memory and persist for a long term in mouse models [170]. The beneficial effects on the tumor mass can also be ascribed to re-educate GAMs toward an anti-tumoral phenotype.
All the CAR-T cells examined in this review showed increased production of IFN-γ and other pro-inflammatory cytokines. It has been demonstrated that IFN-γ can polarize microglia toward a pro-inflammatory phenotype with the upregulation of pro-inflammatory genes such as IL-1β, IL-6, TNF-α, NOS2, and CD86 [172]. The release of IFN-γ by CAR-T cells in the tumor mass can also affect the GAM population in the GME in a pro-inflammatory way.
With regard to NK cells, it is also interesting that these lymphocytes have been engineered using CAR technology to be efficient tools against GBM. Han and collaborators, in 2017, demonstrated that NK cell-targeting non-mutated EGFR and mutated EGFRvIII showed enhanced anti-tumor activity and increased production of IFN-γ in vitro. Furthermore, the intracranial administration of CAR-NK cells led to reduced tumor growth and increased glioma-bearing mice survival [173]. Furthermore, another CAR-NK cell target is ErbB2/HER2. These cells exhibit high cytotoxic activity on ErbB2+ GBM cells, in both in vitro and in vivo models. Moreover, immunocompetent mice showed resistance to tumor growth and development when re-challenged with successive GBM infusions, proof of the induction of long-lasting immunological memory [122]. ErbB2 CAR-NK cells actively produce IFN-γ, TNF-α, IL-10, and the chemokine macrophage inflammatory protein MIP-1α when co-cultured with ErbB2+ cells [174], possibly modulating microglial behavior [33].
These findings suggest that the use of CAR-T cells and CAR-NK cells has beneficial effects because of the direct cytotoxic activity on tumor cells and through the creation of an inflammatory microenvironment that can revert GAM phenotypes and behavior toward the anti-tumoral phenotype.
4. Conclusions
GBM represents 81% of primary brain tumors [175]. Despite the recent and accurate classification of all gliomas and the scientific findings regarding molecular mechanisms at the base of their properties, the GBM remains a devastating tumor. Furthermore, in GBM, recurrence is inevitable; the current improvement in surgery, chemotherapy, or radiotherapy increases the mean survival rate of GBM patients by only a few months, mainly due to treatment resistance and a lack of response to targeted therapies. The resistance to the therapies is due to GBM heterogeneity, hypermutation, and oncologically activated alternative molecular pathways that shape the tumor microenvironment to facilitate therapy failure [176]. Moreover, GBM promotes an immunosuppressive microenvironment, supported by infiltrated macrophages and brain resident microglia, that hampers an effective immune reaction against glioma cells, promoting immunotherapy failure [57,58,59]. In this scenario, microglial cells have dialogues with infiltrated lymphocytes, and these interactions play key roles in GBM progression.
Here, we review the state-of-the-art regarding this fascinating cellular communication, highlighting the current hypothesis that modulating this interaction could represent a promising therapeutical approach. The first approach is to engineer microglia using AAV delivery, with the aim of modifying the expression profiles of these cells in order to induce a pro-inflammatory microenvironment, contrasting tumor growth and recruiting competent immune cells that are able to exert cytotoxic activity [118,119]. The second strategy is to exploit environmental stimuli to re-educate microglia and infiltrated lymphocytes in an anti-tumoral interplay, with the release of cytokines that reinforce pro-inflammatory ground, thus creating a virtuous circle [33,143]. The last examined strategy is the direct engineering of T-lymphocytes and NK cells with CAR technology. The purpose of this method is to create personalized therapy that is selectively directed against GBM antigens [161,168,169,170].
In conclusion, it is crucial to keep improving the biological knowledge of GBM and the interplay with resident and infiltrating immune cells in order to understand cell-to-cell communication mechanisms and their role in driving tumor pathogenesis. The possibility of integrating these exciting discoveries with new combination therapies will open new tools for treating this devastating disease.
Author Contributions
A.M. writing—review and editing; S.G. writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by AIRC grant 22329 to S.G.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary Brain Tumours in Adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to Curing Primary Brain Tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Varghese, J.; Jain, R. Adult Glioma WHO Classification Update, Genomics, and Imaging: What the Radiologists Need to Know. Top. Magn. Reson. Imaging 2020, 29, 71–82. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current State of Immunotherapy for Glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Weller, M.; Belanger, K.; Bogdahn, U.; Ludwin, S.K.; Lacombe, D.; Mirimanoff, R.O. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Zeng, T.; Cui, D.; Gao, L. Glioma: An Overview of Current Classifications, Characteristics, Molecular Biology and Target Therapies. Front. Biosci. (Landmark Ed.) 2015, 20, 1104–1115. [Google Scholar] [CrossRef] [Green Version]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef] [Green Version]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A Neurocentric Perspective on Glioma Invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N. Molecular pathology of malignant gliomas. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 97–117. [Google Scholar] [CrossRef] [Green Version]
- Umphlett, M.; Shea, S.; Tome-Garcia, J.; Zhang, Y.; Hormigo, A.; Fowkes, M.; Tsankova, N.M.; Yong, R.L. Widely Metastatic Glioblastoma with BRCA1 and ARID1A Mutations: A Case Report. BMC Cancer 2020, 20, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broniscer, A.; Tatevossian, R.G.; Sabin, N.D.; Klimo, P.; Dalton, J.; Lee, R.; Gajjar, A.; Ellison, D.W. Clinical, Radiological, Histological and Molecular Characteristics of Paediatric Epithelioid Glioblastoma. Neuropathol. Appl. Neurobiol. 2014, 40, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lun, M.; Lok, E.; Gautam, S.; Wu, E.; Wong, E.T. The Natural History of Extracranial Metastasis from Glioblastoma Multiforme. J. Neurooncol. 2011, 105, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.D.; Rapp, M.; Schneiderhan, T.; Marcel Schneiderhan, T.; Sabel, M.; Hayman, A.; Scherer, A.; Kröpil, P.; Budach, W.; Gerber, P.; et al. Glioblastoma Multiforme Metastasis Outside the CNS: Three Case Reports and Possible Mechanisms of Escape. J. Clin. Oncol. 2014, 32, e80–e84. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha, M.L.V.; Maldaun, M.V.C. Metastasis from Glioblastoma Multiforme: A Meta-Analysis. Rev. Assoc. Med. Bras. 2019, 65, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, J.A.D.V.; Barbosa, C.C.; Feher, O.; Maldaun, M.V.C.; Camargo, V.P.D.; Moraes, F.Y.; Marta, G.N. Systemic Dissemination of Glioblastoma: Literature Review. Rev. Assoc. Med. Bras. 2019, 65, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Rubio, M.-P.; Correa, K.M.; Ueki, K.; Mohrenweiser, H.W.; Gusella, J.F.; von Deimling, A.; Louis, D.N. The Putative Glioma Tumor Suppressor Gene on Chromosome 19q Maps between APOC2 and HRC1. Cancer Res. 1994, 54, 4760–4763. [Google Scholar]
- Robertson, L.B.; Armstrong, G.N.; Olver, B.D.; Lloyd, A.L.; Shete, S.; Lau, C.; Claus, E.B.; Barnholtz-Sloan, J.; Lai, R.; Il’yasova, D.; et al. Survey of Familial Glioma and Role of Germline P16 INK4A /P14 ARF and P53 Mutation. Fam. Cancer 2010, 9, 413–421. [Google Scholar] [CrossRef]
- Koul, D. PTEN Signaling Pathways in Glioblastoma. Cancer Biol. Ther. 2008, 7, 1321–1325. [Google Scholar] [CrossRef]
- Das, S.; Marsden, P.A. Angiogenesis in Glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef] [Green Version]
- Fischer, I.; Gagner, J.-P.; Law, M.; Newcomb, E.W.; Zagzag, D. Angiogenesis in Gliomas: Biology and Molecular Pathophysiology. Brain Pathol. 2006, 15, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant Astrocytic Glioma: Genetics, Biology, and Paths to Treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068. [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of Human Brain Tumour Initiating Cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Klemm, F.; Joyce, J.A. Microenvironmental Regulation of Therapeutic Response in Cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [Green Version]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, S.; D’Alessandro, G.; Chece, G.; Brau, F.; Maggi, L.; Rosa, A.; Porzia, A.; Mainiero, F.; Esposito, V.; Lauro, C.; et al. Enriched Environment Reduces Glioma Growth through Immune and Non-Immune Mechanisms in Mice. Nat. Commun. 2015, 6, 6623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, S.; Porzia, A.; Mainiero, F.; Di Angelantonio, S.; Cortese, B.; Basilico, B.; Pagani, F.; Cignitti, G.; Chece, G.; Maggio, R.; et al. Environmental Stimuli Shape Microglial Plasticity in Glioma. eLife 2017, 6, e33415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boeck, A.; Ahn, B.Y.; D’Mello, C.; Lun, X.; Menon, S.V.; Alshehri, M.M.; Szulzewsky, F.; Shen, Y.; Khan, L.; Dang, N.H.; et al. Glioma-Derived IL-33 Orchestrates an Inflammatory Brain Tumor Microenvironment That Accelerates Glioma Progression. Nat. Commun. 2020, 11, 4997. [Google Scholar] [CrossRef]
- Alghamri, M.S.; McClellan, B.L.; Avvari, R.P.; Thalla, R.; Carney, S.; Hartlage, M.S.; Haase, S.; Ventosa, M.; Taher, A.; Kamran, N.; et al. G-CSF Secreted by Mutant IDH1 Glioma Stem Cells Abolishes Myeloid Cell Immunosuppression and Enhances the Efficacy of Immunotherapy. Sci. Adv. 2021, 7, eabh3243. [Google Scholar] [CrossRef]
- Mormino, A.; Cocozza, G.; Fontemaggi, G.; Valente, S.; Esposito, V.; Santoro, A.; Bernardini, G.; Santoni, A.; Fazi, F.; Mai, A.; et al. Histone-Deacetylase 8 Drives the Immune Response and the Growth of Glioma. Glia 2021, 69, 2682–2698. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting Neuronal Activity-Regulated Neuroligin-3 Dependency in High-Grade Glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and Synaptic Integration of Glioma into Neural Circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic Synaptic Input to Glioma Cells Drives Brain Tumour Progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Gourlay, J.; Morokoff, A.P.; Luwor, R.B.; Zhu, H.-J.; Kaye, A.H.; Stylli, S.S. The Emergent Role of Exosomes in Glioma. J. Clin. Neurosci. 2017, 35, 13–23. [Google Scholar] [CrossRef]
- Gutmann, D.H.; McLellan, M.D.; Hussain, I.; Wallis, J.W.; Fulton, L.L.; Fulton, R.S.; Magrini, V.; Demeter, R.; Wylie, T.; Kandoth, C.; et al. Somatic Neurofibromatosis Type 1 (NF1) Inactivation Characterizes NF1-Associated Pilocytic Astrocytoma. Genome Res. 2013, 23, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss, M.; Van Gassen, S.; Movahedi, K.; Saeys, Y.; Laoui, D. Myeloid Cell Heterogeneity in Cancer: Not a Single Cell Alike. Cell. Immunol. 2018, 330, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Masuda, T.; Jordão, M.J.C.; Prinz, M. Macrophages at CNS Interfaces: Ontogeny and Function in Health and Disease. Nat. Rev. Neurosci. 2019, 20, 547–562. [Google Scholar] [CrossRef]
- Guldner, I.H.; Wang, Q.; Yang, L.; Golomb, S.M.; Zhao, Z.; Lopez, J.A.; Brunory, A.; Howe, E.N.; Zhang, Y.; Palakurthi, B.; et al. CNS-Native Myeloid Cells Drive Immune Suppression in the Brain Metastatic Niche through Cxcl10. Cell 2020, 183, 1234–1248.e25. [Google Scholar] [CrossRef]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-Cell Profiling of Human Gliomas Reveals Macrophage Ontogeny as a Basis for Regional Differences in Macrophage Activation in the Tumor Microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef]
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P.-O. The Blood-Brain Barrier in Brain Homeostasis and Neurological Diseases. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 842–857. [Google Scholar] [CrossRef] [Green Version]
- Ling, E.-A.; Wong, W.-C. The Origin and Nature of Ramified and Amoeboid Microglia: A Historical Review and Current Concepts. Glia 1993, 7, 9–18. [Google Scholar] [CrossRef]
- Priller, J.; Flügel, A.; Wehner, T.; Boentert, M.; Haas, C.A.; Prinz, M.; Fernández-Klett, F.; Prass, K.; Bechmann, I.; de Boer, B.A.; et al. Targeting Gene-Modified Hematopoietic Cells to the Central Nervous System: Use of Green Fluorescent Protein Uncovers Microglial Engraftment. Nat. Med. 2001, 7, 1356–1361. [Google Scholar] [CrossRef]
- Hess, D.C.; Abe, T.; Hill, W.D.; Studdard, A.M.; Carothers, J.; Masuya, M.; Fleming, P.A.; Drake, C.J.; Ogawa, M. Hematopoietic Origin of Microglial and Perivascular Cells in Brain. Exp. Neurol. 2004, 186, 134–144. [Google Scholar] [CrossRef]
- Mildner, A.; Schmidt, H.; Nitsche, M.; Merkler, D.; Hanisch, U.-K.; Mack, M.; Heikenwalder, M.; Brück, W.; Priller, J.; Prinz, M. Microglia in the Adult Brain Arise from Ly-6ChiCCR2+ Monocytes Only under Defined Host Conditions. Nat. Neurosci. 2007, 10, 1544–1553. [Google Scholar] [CrossRef]
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M.V. Infiltrating Monocytes Trigger EAE Progression, but Do Not Contribute to the Resident Microglia Pool. Nat. Neurosci. 2011, 14, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, Fate and Dynamics of Macrophages at Central Nervous System Interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Szulzewsky, F.; Arora, S.; de Witte, L.; Ulas, T.; Markovic, D.; Schultze, J.L.; Holland, E.C.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Human Glioblastoma-Associated Microglia/Monocytes Express a Distinct RNA Profile Compared to Human Control and Murine Samples. Glia 2016, 64, 1416–1436. [Google Scholar] [CrossRef]
- Li, W.; Graeber, M.B. The Molecular Profile of Microglia under the Influence of Glioma. Neuro-Oncology 2012, 14, 958–978. [Google Scholar] [CrossRef] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Yu-Ju Wu, C.; Chen, C.-H.; Lin, C.-Y.; Feng, L.-Y.; Lin, Y.-C.; Wei, K.-C.; Huang, C.-Y.; Fang, J.-Y.; Chen, P.-Y. CCL5 of Glioma-Associated Microglia/Macrophages Regulates Glioma Migration and Invasion via Calcium-Dependent Matrix Metalloproteinase 2. Neuro-Oncology 2020, 22, 253–266. [Google Scholar] [CrossRef]
- Wei, Q.; Singh, O.; Ekinci, C.; Gill, J.; Li, M.; Mamatjan, Y.; Karimi, S.; Bunda, S.; Mansouri, S.; Aldape, K.; et al. TNFα Secreted by Glioma Associated Macrophages Promotes Endothelial Activation and Resistance against Anti-Angiogenic Therapy. Acta Neuropathol. Commun. 2021, 9, 67. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional Communication in the Microenvirons of Glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- De, I.; Steffen, M.D.; Clark, P.A.; Patros, C.J.; Sokn, E.; Bishop, S.M.; Litscher, S.; Maklakova, V.I.; Kuo, J.S.; Rodriguez, F.J.; et al. CSF1 Overexpression Promotes High-Grade Glioma Formation without Impacting the Polarization Status of Glioma-Associated Microglia and Macrophages. Cancer Res. 2016, 76, 2552–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial Stimulation of Glioblastoma Invasion Involves Epidermal Growth Factor Receptor (EGFR) and Colony Stimulating Factor 1 Receptor (CSF-1R) Signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Van Overmeire, E.; Stijlemans, B.; Heymann, F.; Keirsse, J.; Morias, Y.; Elkrim, Y.; Brys, L.; Abels, C.; Lahmar, Q.; Ergen, C.; et al. M-CSF and GM-CSF Receptor Signaling Differentially Regulate Monocyte Maturation and Macrophage Polarization in the Tumor Microenvironment. Cancer Res. 2016, 76, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally Administered Colony Stimulating Factor 1 Receptor Inhibitor PLX3397 in Recurrent Glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium Phase II Study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.-Y.; Ling, X.; Caruso, H.; et al. Osteopontin Mediates Glioblastoma-Associated Macrophage Infiltration and Is a Potential Therapeutic Target. J. Clin. Investig. 2018, 129, 137–149. [Google Scholar] [CrossRef]
- Sielska, M.; Przanowski, P.; Wylot, B.; Gabrusiewicz, K.; Maleszewska, M.; Kijewska, M.; Zawadzka, M.; Kucharska, J.; Vinnakota, K.; Kettenmann, H.; et al. Distinct Roles of CSF Family Cytokines in Macrophage Infiltration and Activation in Glioma Progression and Injury Response: GM-CSF in Glioma Pathology. J. Pathol. 2013, 230, 310–321. [Google Scholar] [CrossRef]
- Okada, M.; Saio, M.; Kito, Y.; Ohe, N.; Yano, H.; Yoshimura, S.; Iwama, T.; Takami, T. Tumor-Associated Macrophage/Microglia Infiltration in Human Gliomas Is Correlated with MCP-3, but Not MCP-1. Int. J. Oncol. 2009, 34, 1621–1627. [Google Scholar] [CrossRef] [Green Version]
- Held-Feindt, J.; Hattermann, K.; Müerköster, S.S.; Wedderkopp, H.; Knerlich-Lukoschus, F.; Ungefroren, H.; Mehdorn, H.M.; Mentlein, R. CX3CR1 Promotes Recruitment of Human Glioma-Infiltrating Microglia/Macrophages (GIMs). Exp. Cell Res. 2010, 316, 1553–1566. [Google Scholar] [CrossRef]
- Ku, M.-C.; Wolf, S.A.; Respondek, D.; Matyash, V.; Pohlmann, A.; Waiczies, S.; Waiczies, H.; Niendorf, T.; Synowitz, M.; Glass, R.; et al. GDNF Mediates Glioblastoma-Induced Microglia Attraction but Not Astrogliosis. Acta Neuropathol. 2013, 125, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Vinnakota, K.; Hu, F.; Ku, M.-C.; Georgieva, P.B.; Szulzewsky, F.; Pohlmann, A.; Waiczies, S.; Waiczies, H.; Niendorf, T.; Lehnardt, S.; et al. Toll-like Receptor 2 Mediates Microglia/Brain Macrophage MT1-MMP Expression and Glioma Expansion. Neuro-Oncology 2013, 15, 1457–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Zhang, Q.; Lubas, M.; Yuan, Y.; Yalcin, F.; Efe, I.E.; Xia, P.; Motta, E.; Buonfiglioli, A.; Lehnardt, S.; et al. Synergistic Toll-like Receptor 3/9 Signaling Affects Properties and Impairs Glioma-Promoting Activity of Microglia. J. Neurosci. 2020, 40, 6428–6443. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Yue, Q.; Chen, J.; Wang, C.; Yu, R.; Jin, Z.; Yin, S.; Wang, Q.; Chen, L.; Liao, X.; et al. Reprogramming the Immunosuppressive Microenvironment of IDH1 Wild-Type Glioblastoma by Blocking Wnt Signaling between Microglia and Cancer Cells. OncoImmunology 2021, 10, 1932061. [Google Scholar] [CrossRef]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-Cell Profiling of Myeloid Cells in Glioblastoma across Species and Disease Stage Reveals Macrophage Competition and Specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling Genetics, Lineages, and Microenvironment in IDH-Mutant Gliomas by Single-Cell RNA-Seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, M.; Sankowski, R.; Bunse, L.; Kilian, M.; Green, E.; Ramallo Guevara, C.; Pusch, S.; Poschet, G.; Sanghvi, K.; Hahn, M.; et al. Tryptophan Metabolism Drives Dynamic Immunosuppressive Myeloid States in IDH-Mutant Gliomas. Nat. Cancer 2021, 2, 723–740. [Google Scholar] [CrossRef]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; Heiland, D.H.; et al. Mapping Microglia States in the Human Brain through the Integration of High-Dimensional Techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef]
- Amit, M.; Laider-Trejo, L.; Shalom, V.; Shabtay-Orbach, A.; Krelin, Y.; Gil, Z. Characterization of the Melanoma Brain Metastatic Niche in Mice and Humans. Cancer Med. 2013, 2, 155–163. [Google Scholar] [CrossRef]
- Bieńkowski, M.; Preusser, M. Prognostic Role of Tumour-Infiltrating Inflammatory Cells in Brain Tumours: Literature Review. Curr. Opin. Neurol. 2015, 28, 647–658. [Google Scholar] [CrossRef]
- Jacobs, N.L.; Holtan, S.G.; Porrata, L.F.; Markovic, S.N.; Tefferi, A.; Steensma, D.P. Host Immunity Affects Survival in Myelodysplastic Syndromes: Independent Prognostic Value of the Absolute Lymphocyte Count. Am. J. Hematol. 2010, 85, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Kuppner, M.C.; Hamou, M.-F.; Bodmer, S.; Fontana, A.; De Tribolet, N. The Glioblastoma-Derived T-Cell Suppressor Factor/Transforming Growth Factor Beta2 Inhibits the Generation of Lymphokineactivated Killer (LAK) Cells. Int. J. Cancer 1988, 42, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Learn, C.A.; Fecci, P.E.; Schmittling, R.J.; Xie, W.; Karikari, I.; Mitchell, D.A.; Archer, G.E.; Wei, Z.; Dressman, H.; Sampson, J.H. Profiling of CD4+, CD8+, and CD4+CD25+CD45RO+FoxP3+ T Cells in Patients with Malignant Glioma Reveals Differential Expression of the Immunologic Transcriptome Compared with T Cells from Healthy Volunteers. Clin. Cancer Res. 2006, 12, 7306–7315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohr, J.; Ratliff, T.; Huppertz, A.; Ge, Y.; Dictus, C.; Ahmadi, R.; Grau, S.; Hiraoka, N.; Eckstein, V.; Ecker, R.C.; et al. Effector T-Cell Infiltration Positively Impacts Survival of Glioblastoma Patients and Is Impaired by Tumor-Derived TGF-β. Clin. Cancer Res. 2011, 17, 4296–4308. [Google Scholar] [CrossRef] [Green Version]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E.; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased Regulatory T-Cell Fraction Amidst a Diminished CD4 Compartment Explains Cellular Immune Defects in Patients with Malignant Glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef] [Green Version]
- Poli, A.; Wang, J.; Domingues, O.; Planagumà, J.; Yan, T.; Rygh, C.B.; Skaftnesmo, K.O.; Thorsen, F.; McCormack, E.; Hentges, F.; et al. Targeting Glioblastoma with NK Cells and MAb against NG2/CSPG4 Prolongs Animal Survival. Oncotarget 2013, 4, 1527–1546. [Google Scholar] [CrossRef] [Green Version]
- Carpentier, A.F.; Meng, Y. Recent Advances in Immunotherapy for Human Glioma. Curr. Opin. Oncol. 2006, 18, 631–636. [Google Scholar] [CrossRef]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of Antitumor T Cell Immunity by the Oncometabolite (R)-2-Hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate Dehydrogenase Mutations Suppress STAT1 and CD8+ T Cell Accumulation in Gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of Immunotherapy Resistance: Lessons from Glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without Ipilimumab in Patients with Recurrent Glioblastoma: Results from Exploratory Phase I Cohorts of CheckMate 143. Neuro-Oncology 2018, 20, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-β Directly Targets Cytotoxic T Cell Functions during Tumor Evasion of Immune Surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proto, J.D.; Doran, A.C.; Gusarova, G.; Yurdagul, A.; Sozen, E.; Subramanian, M.; Islam, M.N.; Rymond, C.C.; Du, J.; Hook, J.; et al. Regulatory T Cells Promote Macrophage Efferocytosis during Inflammation Resolution. Immunity 2018, 49, 666–677.e6. [Google Scholar] [CrossRef] [Green Version]
- Castriconi, R.; Cantoni, C.; Chiesa, M.D.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming Growth Factor β1 Inhibits Expression of NKp30 and NKG2D Receptors: Consequences for the NK-Mediated Killing of Dendritic Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Burguillos, M.A.; Osman, A.M.; Frijhoff, J.; Carrillo-Jiménez, A.; Kanatani, S.; Augsten, M.; Saidi, D.; Rodhe, J.; Kavanagh, E.; et al. Glioma-Induced Inhibition of Caspase-3 in Microglia Promotes a Tumor-Supportive Phenotype. Nat. Immunol. 2016, 17, 1282–1290. [Google Scholar] [CrossRef]
- Wagner, S.; Czub, S.; Greif, M.; Vince, G.H.; Süss, N.; Kerkau, S.; Rieckmann, P.; Roggendorf, W.; Roosen, K.; Tonn, J.-C. Microglial/Macrophage Expression of Interleukin 10 in Human Glioblastomas. Int. J. Cancer 1999, 82, 12–16. [Google Scholar] [CrossRef]
- Ye, X.; Xu, S.; Xin, Y.; Yu, S.; Ping, Y.; Chen, L.; Xiao, H.; Wang, B.; Yi, L.; Wang, Q.; et al. Tumor-Associated Microglia/Macrophages Enhance the Invasion of Glioma Stem-like Cells via TGF-Β1 Signaling Pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [Green Version]
- a Dzaye, O.D.; Hu, F.; Derkow, K.; Haage, V.; Euskirchen, P.; Harms, C.; Lehnardt, S.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Glioma Stem Cells but Not Bulk Glioma Cells Upregulate IL-6 Secretion in Microglia/Brain Macrophages via Toll-like Receptor 4 Signaling. J. Neuropathol. Exp. Neurol. 2016, 75, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Dumas, A.A.; Pomella, N.; Rosser, G.; Guglielmi, L.; Vinel, C.; Millner, T.O.; Rees, J.; Aley, N.; Sheer, D.; Wei, J.; et al. Microglia Promote Glioblastoma via MTOR-Mediated Immunosuppression of the Tumour Microenvironment. EMBO J. 2020, 39, e103790. [Google Scholar] [CrossRef]
- Brandenburg, S.; Müller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident Microglia Rather than Peripheral Macrophages Promote Vascularization in Brain Tumors and Are Source of Alternative Pro-Angiogenic Factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Hegde, S.; DeNardo, D.G. Tumor-Associated Fibrosis as a Regulator of Tumor Immunity and Response to Immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, V.; Rainer, C.; Nielsen, S.R.; Raymant, M.L.; Ahmed, M.S.; Engle, D.D.; Taylor, A.; Murray, T.; Campbell, F.; Palmer, D.H.; et al. Macrophage-Derived Granulin Drives Resistance to Immune Checkpoint Inhibition in Metastatic Pancreatic Cancer. Cancer Res. 2018, 78, 4253–4269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtin, J.F.; Candolfi, M.; Fakhouri, T.M.; Liu, C.; Alden, A.; Edwards, M.; Lowenstein, P.R.; Castro, M.G. Treg Depletion Inhibits Efficacy of Cancer Immunotherapy: Implications for Clinical Trials. PLoS ONE 2008, 3, e1983. [Google Scholar] [CrossRef] [Green Version]
- Bhondeley, M.K.; Mehra, R.D.; Mehra, N.K.; Mohapatra, A.K.; Tandon, P.N.; Roy, S.; Bijlani, V. Imbalances in T Cell Subpopulations in Human Gliomas. J. Neurosurg. 1988, 68, 589–593. [Google Scholar] [CrossRef]
- Glinka, Y.; Prud’homme, G.J. Neuropilin-1 Is a Receptor for Transforming Growth Factor Beta-1, Activates Its Latent Form, and Promotes Regulatory T Cell Activity. J. Leukoc. Biol. 2008, 84, 302–310. [Google Scholar] [CrossRef]
- Chaudhary, B.; Khaled, Y.S.; Ammori, B.J.; Elkord, E. Neuropilin 1: Function and Therapeutic Potential in Cancer. Cancer Immunol. Immunother. 2014, 63, 81–99. [Google Scholar] [CrossRef]
- Miyauchi, J.T.; Caponegro, M.D.; Chen, D.; Choi, M.K.; Li, M.; Tsirka, S.E. Deletion of Neuropilin 1 from Microglia or Bone Marrow–Derived Macrophages Slows Glioma Progression. Cancer Res. 2018, 78, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Borin, T.F.; Piranlioglu, R.; Ara, R.; Lebedyeva, I.; Angara, K.; Achyut, B.R.; Arbab, A.S.; Rashid, M.H. Changes in the Tumor Microenvironment and Outcome for TME-Targeting Therapy in Glioblastoma: A Pilot Study. PLoS ONE 2021, 16, e0246646. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune Microenvironment of Gliomas. Lab. Invest. 2017, 97, 498–518. [Google Scholar] [CrossRef] [Green Version]
- Goldmann, T.; Wieghofer, P.; Müller, P.F.; Wolf, Y.; Varol, D.; Yona, S.; Brendecke, S.M.; Kierdorf, K.; Staszewski, O.; Datta, M.; et al. A New Type of Microglia Gene Targeting Shows TAK1 to Be Pivotal in CNS Autoimmune Inflammation. Nat. Neurosci. 2013, 16, 1618–1626. [Google Scholar] [CrossRef]
- Maes, M.E.; Colombo, G.; Schulz, R.; Siegert, S. Targeting Microglia with Lentivirus and AAV: Recent Advances and Remaining Challenges. Neurosci. Lett. 2019, 707, 134310. [Google Scholar] [CrossRef]
- Griciuc, A.; Federico, A.N.; Natasan, J.; Forte, A.M.; McGinty, D.; Nguyen, H.; Volak, A.; LeRoy, S.; Gandhi, S.; Lerner, E.P.; et al. Gene Therapy for Alzheimer’s Disease Targeting CD33 Reduces Amyloid Beta Accumulation and Neuroinflammation. Hum. Mol. Genet. 2020, 29, 2920–2935. [Google Scholar] [CrossRef]
- Gonçalves, M.A. Adeno-Associated Virus: From Defective Virus to Effective Vector. Virol. J. 2005, 2, 43. [Google Scholar] [CrossRef] [Green Version]
- Zolotukhin, I.; Luo, D.; Gorbatyuk, O.; Hoffman, B.; Warrington, K.; Herzog, R.; Harrison, J.; Cao, O. Improved Adeno-Associated Viral Gene Transfer to Murine Glioma. J. Genet. Syndr. Gene 2013, 4, 12815. [Google Scholar] [CrossRef]
- O’Carroll, S.J.; Cook, W.H.; Young, D. AAV Targeting of Glial Cell Types in the Central and Peripheral Nervous System and Relevance to Human Gene Therapy. Front. Mol. Neurosci. 2021, 13, 256. [Google Scholar] [CrossRef]
- Chiu, T.-L.; Wang, M.-J.; Su, C.-C. The Treatment of Glioblastoma Multiforme through Activation of Microglia and TRAIL Induced by RAAV2-Mediated IL-12 in a Syngeneic Rat Model. J. Biomed. Sci. 2012, 19, 45. [Google Scholar] [CrossRef] [Green Version]
- Mormino, A.; Bernardini, G.; Cocozza, G.; Corbi, N.; Passananti, C.; Santoni, A.; Limatola, C.; Garofalo, S. Enriched Environment Cues Suggest a New Strategy to Counteract Glioma: Engineered RAAV2-IL-15 Microglia Modulate the Tumor Microenvironment. Front. Immunol. 2021, 12, 730128. [Google Scholar] [CrossRef]
- Waldmann, T.A. The Biology of Interleukin-2 and Interleukin-15: Implications for Cancer Therapy and Vaccine Design. Nat. Rev. Immunol. 2006, 6, 595–601. [Google Scholar] [CrossRef]
- Zhu, L.; Kalimuthu, S.; Oh, J.M.; Gangadaran, P.; Baek, S.H.; Jeong, S.Y.; Lee, S.-W.; Lee, J.; Ahn, B.-C. Enhancement of Antitumor Potency of Extracellular Vesicles Derived from Natural Killer Cells by IL-15 Priming. Biomaterials 2019, 190–191, 38–50. [Google Scholar] [CrossRef]
- Zhang, I.; Alizadeh, D.; Liang, J.; Zhang, L.; Gao, H.; Song, Y.; Ren, H.; Ouyang, M.; Wu, X.; D’Apuzzo, M.; et al. Characterization of Arginase Expression in Glioma-Associated Microglia and Macrophages. PLoS ONE 2016, 11, e0165118. [Google Scholar] [CrossRef] [Green Version]
- Taoufik, Y.; de Goër de Herve, M.G.; Giron-Michel, J.; Durali, D.; Cazes, E.; Tardieu, M.; Azzarone, B.; Delfraissy, J.F. Human Microglial Cells Express a Functional IL-12 Receptor and Produce IL-12 Following IL-12 Stimulation. Eur. J. Immunol. 2001, 31, 3228–3239. [Google Scholar] [CrossRef]
- Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Kato, H.; Wang, J.; Mitsuma, N.; Suzumura, A. Production of Interferon-γ by Microglia. Mult. Scler. 2006, 12, 558–564. [Google Scholar] [CrossRef]
- Kempermann, G.; Kuhn, H.G.; Gage, F.H. More Hippocampal Neurons in Adult Mice Living in an Enriched Environment. Nature 1997, 386, 493–495. [Google Scholar] [CrossRef]
- Naka, F.; Shiga, T.; Yaguchi, M.; Okado, N. An Enriched Environment Increases Noradrenaline Concentration in the Mouse Brain. Brain Res. 2002, 924, 124–126. [Google Scholar] [CrossRef]
- Moser, M.B.; Trommald, M.; Andersen, P. An Increase in Dendritic Spine Density on Hippocampal CA1 Pyramidal Cells Following Spatial Learning in Adult Rats Suggests the Formation of New Synapses. Proc. Natl. Acad. Sci. USA 1994, 91, 12673–12675. [Google Scholar] [CrossRef] [Green Version]
- Armaiz-Pena, G.N.; Lutgendorf, S.K.; Cole, S.W.; Sood, A.K. Neuroendocrine Modulation of Cancer Progression. Brain Behav. Immun. 2009, 23, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Slater, A.M.; Cao, L. A Protocol for Housing Mice in an Enriched Environment. JoVE 2015, 8, e52874. [Google Scholar] [CrossRef] [Green Version]
- Leggio, M.G.; Mandolesi, L.; Federico, F.; Spirito, F.; Ricci, B.; Gelfo, F.; Petrosini, L. Environmental Enrichment Promotes Improved Spatial Abilities and Enhanced Dendritic Growth in the Rat. Behav. Brain Res. 2005, 163, 78–90. [Google Scholar] [CrossRef]
- Kempermann, G.; Gage, F.H. Experienced-Dependent Regulation of Adult Hippocampal Neurogenesis: Effects of Long-Term Stimulation and Stimulus Withdrawal. Hippocampus 1999, 9, 321–332. [Google Scholar] [CrossRef]
- Augusto-Oliveira, M.; Verkhratsky, A. Lifestyle-Dependent Microglial Plasticity: Training the Brain Guardians. Biol. Direct 2021, 16, 12. [Google Scholar] [CrossRef]
- Guo, Y.-S.; Yuan, M.; Han, Y.; Shen, X.-Y.; Gao, Z.-K.; Bi, X. Effects of Enriched Environment on Microglia and Functional White Matter Recovery in Rats with Post Stroke Cognitive Impairment. Neurochem. Int. 2022, 154, 105295. [Google Scholar] [CrossRef]
- Chabry, J.; Nicolas, S.; Cazareth, J.; Murris, E.; Guyon, A.; Glaichenhaus, N.; Heurteaux, C.; Petit-Paitel, A. Enriched Environment Decreases Microglia and Brain Macrophages Inflammatory Phenotypes through Adiponectin-Dependent Mechanisms: Relevance to Depressive-like Behavior. Brain Behav. Immun. 2015, 50, 275–287. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Noristani, H.N.; Verkhratsky, A. Microglial Response to Alzheimer’s Disease Is Differentially Modulated by Voluntary Wheel Running and Enriched Environments. Brain Struct. Funct. 2015, 220, 941–953. [Google Scholar] [CrossRef]
- Branchi, I.; Francia, N.; Alleva, E. Epigenetic Control of Neurobehavioural Plasticity: The Role of Neurotrophins. Behav. Pharm. 2004, 15, 353–362. [Google Scholar] [CrossRef]
- Birch, A.M.; McGarry, N.B.; Kelly, A.M. Short-Term Environmental Enrichment, in the Absence of Exercise, Improves Memory, and Increases NGF Concentration, Early Neuronal Survival, and Synaptogenesis in the Dentate Gyrus in a Time-Dependent Manner. Hippocampus 2013, 23, 437–450. [Google Scholar] [CrossRef]
- Sadegzadeh, F.; Sakhaie, N.; Isazadehfar, K.; Saadati, H. Effects of Exposure to Enriched Environment during Adolescence on Passive Avoidance Memory, Nociception, and Prefrontal BDNF Level in Adult Male and Female Rats. Neurosci. Lett. 2020, 732, 135133. [Google Scholar] [CrossRef]
- Xu, S.; Tang, L.; Li, X.; Fan, F.; Liu, Z. Immunotherapy for Glioma: Current Management and Future Application. Cancer Lett. 2020, 476, 1–12. [Google Scholar] [CrossRef]
- Garofalo, S.; Grimaldi, A.; Chece, G.; Porzia, A.; Morrone, S.; Mainiero, F.; D’Alessandro, G.; Esposito, V.; Cortese, B.; Di Angelantonio, S.; et al. The Glycoside Oleandrin Reduces Glioma Growth with Direct and Indirect Effects on Tumor Cells. J. Neurosci. 2017, 37, 3926–3939. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Liu, X.; Lin, E.-J.D.; Wang, C.; Choi, E.Y.; Riban, V.; Lin, B.; During, M.J. Environmental and Genetic Activation of a Brain-Adipocyte BDNF/Leptin Axis Causes Cancer Remission and Inhibition. Cell 2010, 142, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlqvist, P.; Zhao, L.; Johansson, I.-M.; Mattsson, B.; Johansson, B.B.; Seckl, J.R.; Olsson, T. Environmental Enrichment Alters Nerve Growth Factor-Induced Gene A and Glucocorticoid Receptor Messenger RNA Expression after Middle Cerebral Artery Occlusion in Rats. Neuroscience 1999, 93, 527–535. [Google Scholar] [CrossRef]
- Cao, W.; Duan, J.; Wang, X.; Zhong, X.; Hu, Z.; Huang, F.; Wang, H.; Zhang, J.; Li, F.; Zhang, J.; et al. Early Enriched Environment Induces an Increased Conversion of ProBDNF to BDNF in the Adult Rat’s Hippocampus. Behav. Brain Res. 2014, 265, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.-B.; Julius, D. The P2Y12 Receptor Regulates Microglial Activation by Extracellular Nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a Unique TGF-β–Dependent Molecular and Functional Signature in Microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Kros, J.M.; van der Weiden, M.; Zheng, P.; Cheng, C.; Mustafa, D.A.M. Expression Site of P2RY12 in Residential Microglial Cells in Astrocytomas Correlates with M1 and M2 Marker Expression and Tumor Grade. Acta Neuropathol. Commun. 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The Application of CAR-T Cell Therapy in Hematological Malignancies: Advantages and Challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [Green Version]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T Cells for Brain Tumors: Lessons Learned and Road Ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef] [Green Version]
- Stone, J.D.; Kranz, D.M. Role of T Cell Receptor Affinity in the Efficacy and Specificity of Adoptive T Cell Therapies. Front. Immunol. 2013, 4, 244. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wang, G.; Cheng, H.; Wei, C.; Qi, K.; Sang, W.; Zhenyu, L.; Shi, M.; Li, H.; Qiao, J.; et al. Potent Anti-Leukemia Activities of Humanized CD19-Targeted Chimeric Antigen Receptor T (CAR-T) Cells in Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia. Am. J. Hematol. 2018, 93, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heng, G.; Jia, J.; Li, S.; Fu, G.; Wang, M.; Qin, D.; Li, Y.; Pei, L.; Tian, X.; Zhang, J.; et al. Sustained Therapeutic Efficacy of Humanized Anti-CD19 Chimeric Antigen Receptor T Cells in Relapsed/Refractory Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2020, 26, 1606–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I Study of Chimeric Antigen Receptor Modified T Cells in Treating HER2-Positive Advanced Biliary Tract Cancers and Pancreatic Cancers. Protein Cell 2018, 9, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Posey, A.D.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [Green Version]
- Junghans, R.P.; Ma, Q.; Rathore, R.; Gomes, E.M.; Bais, A.J.; Lo, A.S.Y.; Abedi, M.; Davies, R.A.; Cabral, H.J.; Al-Homsi, A.S.; et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate 2016, 76, 1257–1270. [Google Scholar] [CrossRef]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [Green Version]
- Debinski, W.; Obiri, N.I.; Powers, S.K.; Pastan, I.; Puri, R.K. Human Glioma Cells Overexpress Receptors for Interleukin 13 and Are Extremely Sensitive to a Novel Chimeric Protein Composed of Interleukin 13 and Pseudomonas Exotoxin. Clin. Cancer Res. 1995, 1, 1253–1258. [Google Scholar]
- Kahlon, K.S.; Brown, C.; Cooper, L.J.N.; Raubitschek, A.; Forman, S.J.; Jensen, M.C. Specific Recognition and Killing of Glioblastoma Multiforme by Interleukin 13-Zetakine Redirected Cytolytic T Cells. Cancer Res. 2004, 64, 9160–9166. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.-C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-Tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gust, J.; Ponce, R.; Liles, W.C.; Garden, G.A.; Turtle, C.J. Cytokines in CAR T Cell–Associated Neurotoxicity. Front. Immunol. 2020, 11, 577027. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Cerny, C.; Bronger, H.; Davoodi, M.; Sharma, S.; Zhu, L.; Obana, S.; Sharma, J.; Ebrahimi, R.; St John, M.; Lee, J.M.; et al. The Role of CXCR3/Ligand Axis in Cancer. ITI 2015, 3, 46–52. [Google Scholar] [CrossRef]
- Liu, C.; Luo, D.; Reynolds, B.A.; Meher, G.; Katritzky, A.R.; Lu, B.; Gerard, C.J.; Bhadha, C.P.; Harrison, J.K. Chemokine Receptor CXCR3 Promotes Growth of Glioma. Carcinogenesis 2011, 32, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Johnson, L.A.; Davis, J.L.; Zheng, Z.; Woolard, K.D.; Reap, E.A.; Feldman, S.A.; Chinnasamy, N.; Kuan, C.-T.; Song, H.; et al. Recognition of Glioma Stem Cells by Genetically Modified T Cells Targeting EGFRvIII and Development of Adoptive Cell Therapy for Glioma. Hum. Gene Ther. 2012, 23, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Suryadevara, C.M.; Gedeon, P.C.; Herndon II, J.E.; Sanchez-Perez, L.; Bigner, D.D.; Sampson, J.H. Intracerebral Delivery of a Third Generation EGFRvIII-Specific Chimeric Antigen Receptor Is Efficacious against Human Glioma. J. Clin. Neurosci. 2014, 21, 189–190. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.T.C.; Meza, D.; Sanai, N. Trends in Fluorescence Image-Guided Surgery for Gliomas. Neurosurgery 2014, 75, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Cobb, D.A.; de Rossi, J.; Liu, L.; An, E.; Lee, D.W. Targeting of the Alpha v Beta 3 Integrin Complex by CAR-T Cells Leads to Rapid Regression of Diffuse Intrinsic Pontine Glioma and Glioblastoma. J. Immunother. Cancer 2022, 10, e003816. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-Specific T Cells Target Primary Glioblastoma Stem Cells and Induce Regression of Autologous Experimental Tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Chu, J.; Keung Chan, W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.; Grandi, P.; et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective Inhibition of Tumor Growth by Clonal NK Cells Expressing an ErbB2/HER2-Specific Chimeric Antigen Receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The Epidemiology of Glioma in Adults: A “State of the Science” Review. Neuro-Oncology 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goenka, A.; Tiek, D.; Song, X.; Huang, T.; Hu, B.; Cheng, S.-Y. The Many Facets of Therapy Resistance and Tumor Recurrence in Glioblastoma. Cells 2021, 10, 484. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Scheme of interplay among microglia, lymphocytes, and glioblastoma in the tumor microenvironment.
Figure 1.
Scheme of interplay among microglia, lymphocytes, and glioblastoma in the tumor microenvironment.

Figure 2.
Environmental stimuli, engineered microglia, and CAR-lymphocytes shape the glioblastoma microenvironment, educating microglial functions in an anti-tumoral way.
Figure 2.
Environmental stimuli, engineered microglia, and CAR-lymphocytes shape the glioblastoma microenvironment, educating microglial functions in an anti-tumoral way.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mormino, A.; Garofalo, S. Dialogue among Lymphocytes and Microglia in Glioblastoma Microenvironment. Cancers 2022, 14, 2632. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14112632
AMA Style
Mormino A, Garofalo S. Dialogue among Lymphocytes and Microglia in Glioblastoma Microenvironment. Cancers. 2022; 14(11):2632. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14112632
Chicago/Turabian StyleMormino, Alessandro, and Stefano Garofalo. 2022. "Dialogue among Lymphocytes and Microglia in Glioblastoma Microenvironment" Cancers 14, no. 11: 2632. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14112632
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.