
Multiple Mechanisms of NOTCH1 Activation in Chronic Lymphocytic Leukemia: NOTCH1 Mutations and Beyond

 , and
, and 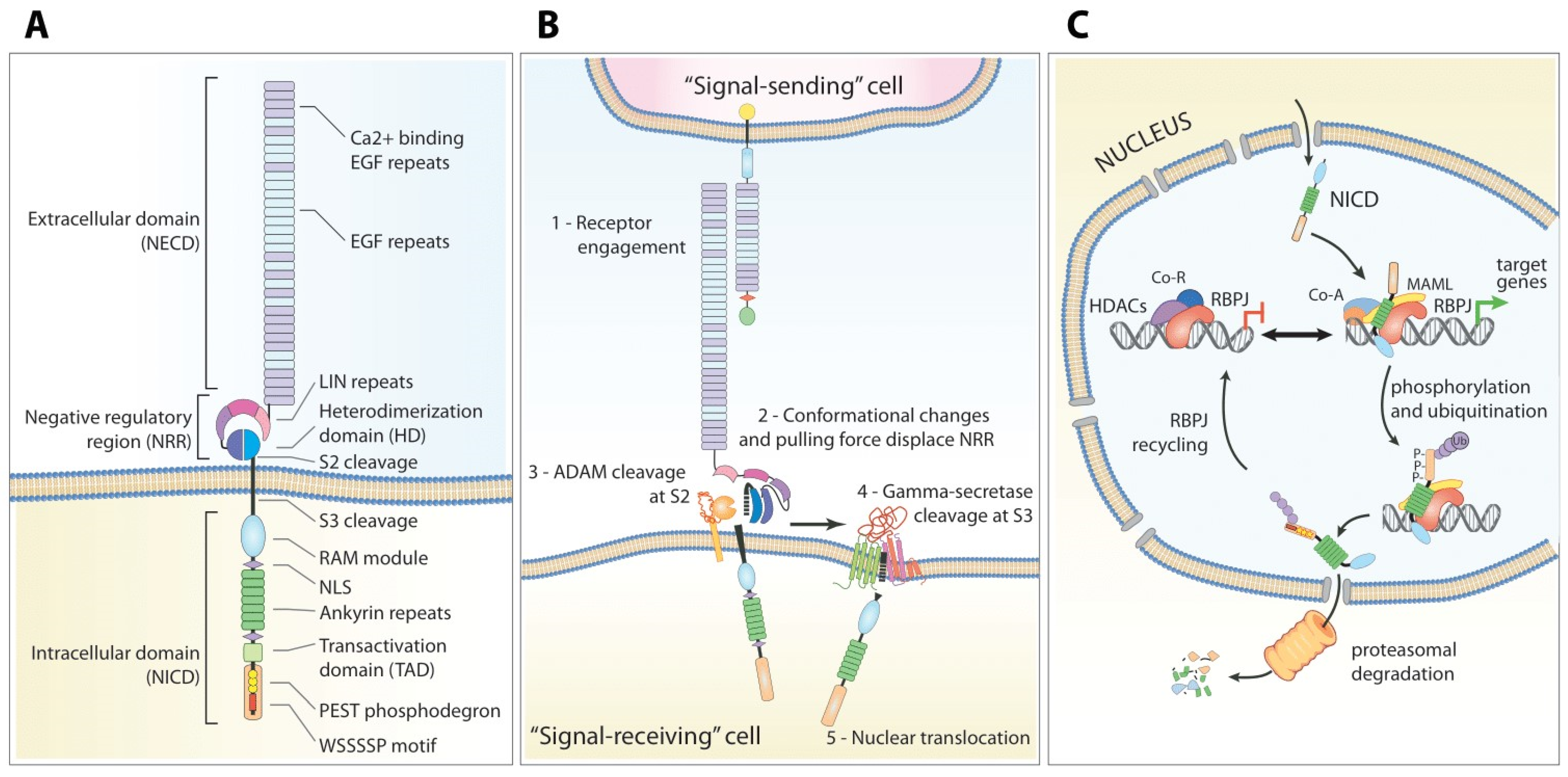{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. The Notch Pathway
2.1. Structure of the Notch Receptors
2.2. Maturation and Processing
2.3. Ligand-Induced Signaling Transduction
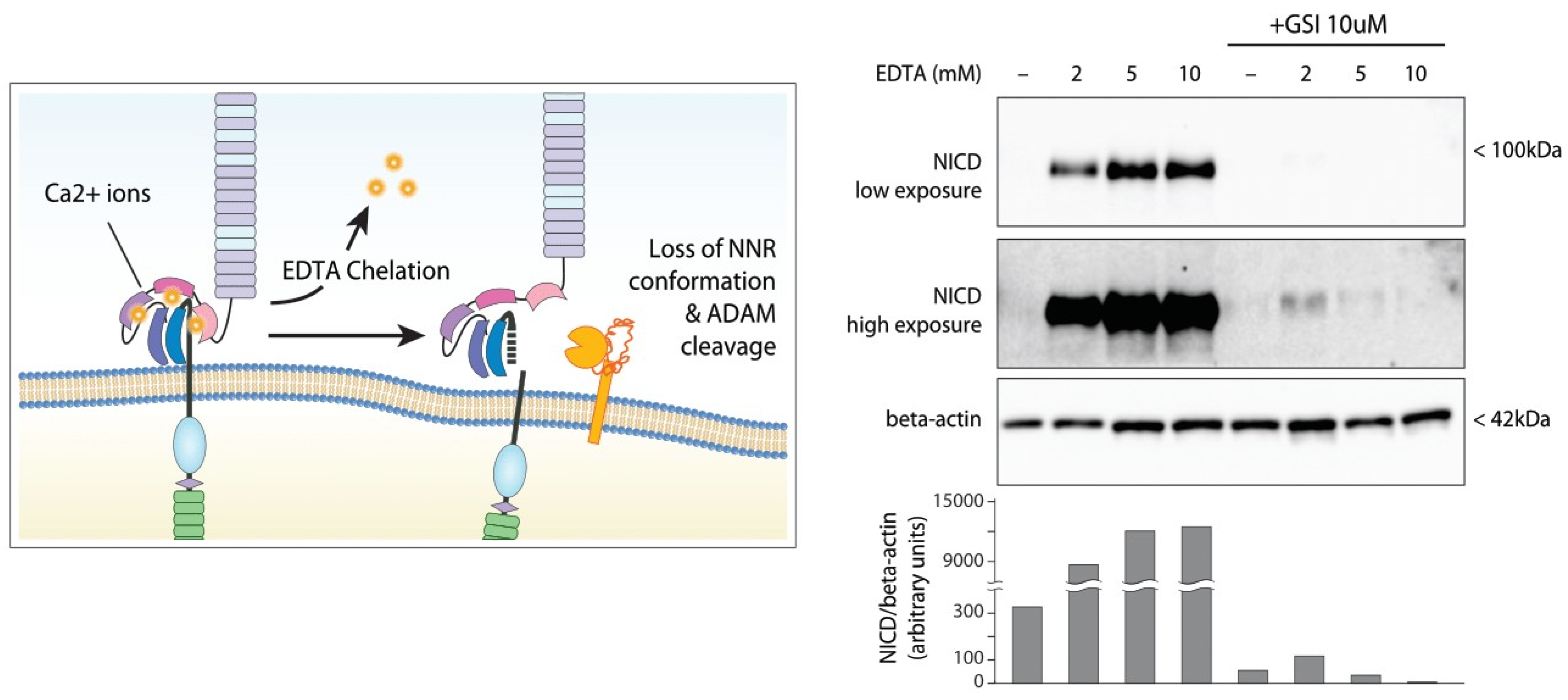
2.4. Ligand-Independent NOTCH1 Signaling
3. NOTCH1 Mutations in CLL
3.1. General Features
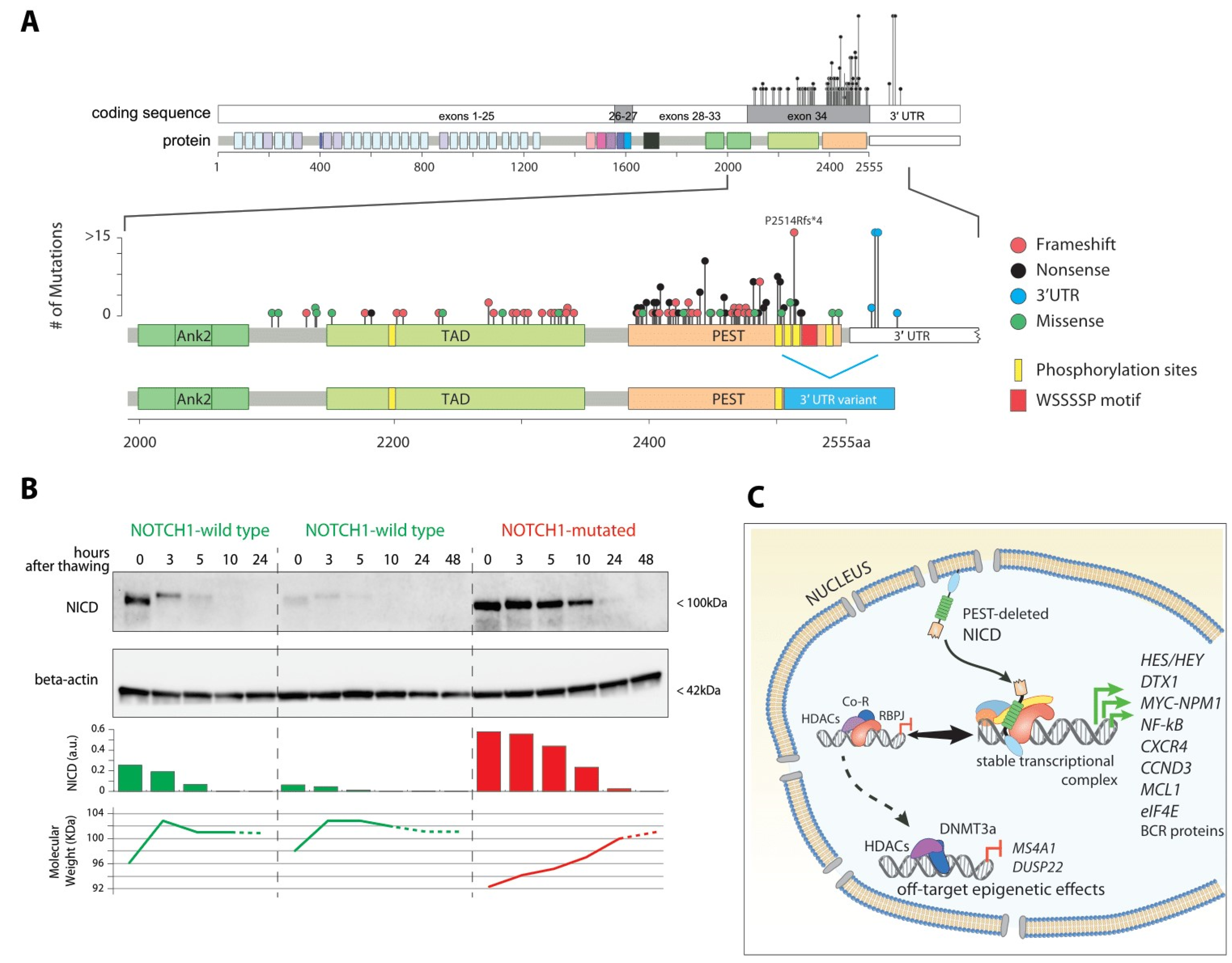
3.2. Coding Mutations
3.3. Non-Coding Mutations
4. Functional Consequences of NOTCH1 Mutation in CLL
4.1. Proliferation and Metabolism: A Story of MYC
4.2. The Complex Interplay with NF-kappaB
4.3. Nuclear Rewiring Triggers Epigenetic Downregulation of CD20: Clinical Implications for Anti-CD20 Immunotherapy
5. Other Mechanisms of NOTCH1 Activation in CLL
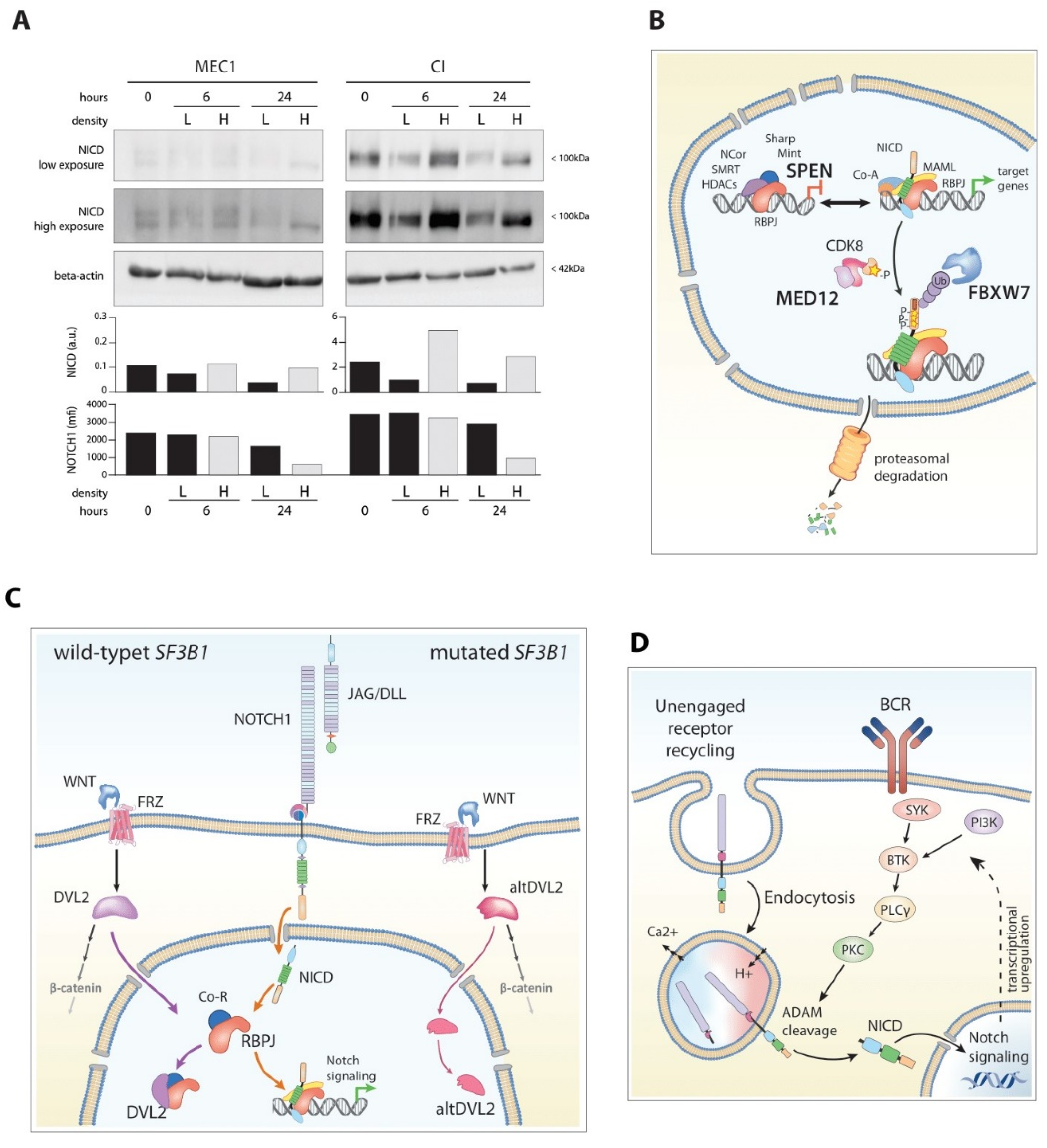
5.1. Evidence of NOTCH1 Sustained Activation in the Absence of Genetic Mutations
5.2. Recurrent Mutations of Modulators of NOTCH1 Signaling
5.2.1. FBXW7
5.2.2. MED-12
5.2.3. SPEN
5.3. Mutations of SF3B1: An Interplay with the Wnt Pathway
5.4. DNMT3a
5.5. Bidirectional Interplay between NOTCH1 and BCR Pathways
5.6. AKT towards Richter Syndrome
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Primers 2017, 3, 16096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Rasi, S.; Spina, V.; Bruscaggin, A.; Monti, S.; Ciardullo, C.; Deambrogi, C.; Khiabanian, H.; Serra, R.; Bertoni, F.; et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013, 121, 1403–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubmann, R.; Schwarzmeier, J.D.; Shehata, M.; Hilgarth, M.; Duechler, M.; Dettke, M.; Berger, R. Notch2 is involved in the overexpression of CD23 in B-cell chronic lymphocytic leukemia. Blood 2002, 99, 3742–3747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duechler, M.; Shehata, M.; Schwarzmeier, J.D.; Hoelbl, A.; Hilgarth, M.; Hubmann, R. Induction of apoptosis by proteasome inhibitors in B-CLL cells is associated with downregulation of CD23 and inactivation of Notch2. Leukemia 2005, 19, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Hajdu, M.; Sebestyen, A.; Barna, G.; Reiniger, L.; Janosi, J.; Sreter, L.; Varkonyi, J.; Demeter, J.; Kopper, L. Activity of the notch-signalling pathway in circulating human chronic lymphocytic leukaemia cells. Scand. J. Immunol. 2007, 65, 271–275. [Google Scholar] [CrossRef]
- Rosati, E.; Sabatini, R.; Rampino, G.; Tabilio, A.; Di Ianni, M.; Fettucciari, K.; Bartoli, A.; Coaccioli, S.; Screpanti, I.; Marconi, P. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood 2009, 113, 856–865. [Google Scholar] [CrossRef]
- Di Ianni, M.; Baldoni, S.; Rosati, E.; Ciurnelli, R.; Cavalli, L.; Martelli, M.F.; Marconi, P.; Screpanti, I.; Falzetti, F. A new genetic lesion in B-CLL: A NOTCH1 PEST domain mutation. Br. J. Haematol. 2009, 146, 689–691. [Google Scholar] [CrossRef]
- Fabbri, G.; Rasi, S.; Rossi, D.; Trifonov, V.; Khiabanian, H.; Ma, J.; Grunn, A.; Fangazio, M.; Capello, D.; Monti, S.; et al. Analysis of the chronic lymphocytic leukemia coding genome: Role of NOTCH1 mutational activation. J. Exp. Med. 2011, 208, 1389–1401. [Google Scholar] [CrossRef] [Green Version]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordonez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Bea, S.; Gonzalez-Diaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Nadeu, F.; Delgado, J.; Royo, C.; Baumann, T.; Stankovic, T.; Pinyol, M.; Jares, P.; Navarro, A.; Martin-Garcia, D.; Bea, S.; et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood 2016, 127, 2122–2130. [Google Scholar] [CrossRef]
- D’Agaro, T.; Bittolo, T.; Bravin, V.; Dal Bo, M.; Pozzo, F.; Bulian, P.; Rossi, F.M.; Zucchetto, A.; Degan, M.; D’Arena, G.; et al. NOTCH1 mutational status in chronic lymphocytic leukaemia: Clinical relevance of subclonal mutations and mutation types. Br. J. Haematol. 2018, 182, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Spina, V.; Gaidano, G. Biology and treatment of Richter syndrome. Blood 2018, 131, 2761–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klintman, J.; Appleby, N.; Stamatopoulos, B.; Ridout, K.; Eyre, T.A.; Robbe, P.; Pascua, L.L.; Knight, S.J.L.; Dreau, H.; Cabes, M.; et al. Genomic and transcriptomic correlates of Richter transformation in chronic lymphocytic leukemia. Blood 2021, 137, 2800–2816. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Rasi, S.; Fabbri, G.; Spina, V.; Fangazio, M.; Forconi, F.; Marasca, R.; Laurenti, L.; Bruscaggin, A.; Cerri, M.; et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 2012, 119, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Sportoletti, P.; Baldoni, S.; Cavalli, L.; Del Papa, B.; Bonifacio, E.; Ciurnelli, R.; Bell, A.S.; Di Tommaso, A.; Rosati, E.; Crescenzi, B.; et al. NOTCH1 PEST domain mutation is an adverse prognostic factor in B-CLL. Br. J. Haematol. 2010, 151, 404–406. [Google Scholar] [CrossRef]
- Stilgenbauer, S.; Schnaiter, A.; Paschka, P.; Zenz, T.; Rossi, M.; Dohner, K.; Buhler, A.; Bottcher, S.; Ritgen, M.; Kneba, M.; et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: Results from the CLL8 trial. Blood 2014, 123, 3247–3254. [Google Scholar] [CrossRef] [Green Version]
- Bo, M.D.; Del Principe, M.I.; Pozzo, F.; Ragusa, D.; Bulian, P.; Rossi, D.; Capelli, G.; Rossi, F.M.; Niscola, P.; Buccisano, F.; et al. NOTCH1 mutations identify a chronic lymphocytic leukemia patient subset with worse prognosis in the setting of a rituximab-based induction and consolidation treatment. Ann. Hematol. 2014, 93, 1765–1774. [Google Scholar] [CrossRef]
- Fabbri, G.; Holmes, A.B.; Viganotti, M.; Scuoppo, C.; Belver, L.; Herranz, D.; Yan, X.J.; Kieso, Y.; Rossi, D.; Gaidano, G.; et al. Common nonmutational NOTCH1 activation in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2017, 114, E2911–E2919. [Google Scholar] [CrossRef] [Green Version]
- Gazave, E.; Lapébie, P.; Richards, G.S.; Brunet, F.; Ereskovsky, A.V.; Degnan, B.M.; Borchiellini, C.; Vervoort, M.; Renard, E. Origin and evolution of the Notch signalling pathway: An overview from eukaryotic genomes. BMC Evol. Biol. 2009, 9, 249. [Google Scholar] [CrossRef] [Green Version]
- Fortini, M.E. Introduction--Notch in development and disease. Semin. Cell Dev. Biol. 2012, 23, 419–420. [Google Scholar] [CrossRef]
- Sottoriva, K.; Pajcini, K.V. Notch Signaling in the Bone Marrow Lymphopoietic Niche. Front. Immunol. 2021, 12, 723055. [Google Scholar] [CrossRef] [PubMed]
- Thambyrajah, R.; Bigas, A. Notch Signaling in HSC Emergence: When, Why and How. Cells 2022, 11, 358. [Google Scholar] [CrossRef] [PubMed]
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [Green Version]
- Gridley, T. Notch signaling in vertebrate development and disease. Mol. Cell. Neurosci. 1997, 9, 103–108. [Google Scholar] [CrossRef]
- Gridley, T. Notch signaling and inherited disease syndromes. Hum. Mol. Genet. 2003, 12, R9–R13. [Google Scholar] [CrossRef]
- Bocci, F.; Onuchic, J.N.; Jolly, M.K. Understanding the Principles of Pattern Formation Driven by Notch Signaling by Integrating Experiments and Theoretical Models. Front. Physiol. 2020, 11, 929. [Google Scholar] [CrossRef]
- LaFoya, B.; Munroe, J.A.; Mia, M.M.; Detweiler, M.A.; Crow, J.J.; Wood, T.; Roth, S.; Sharma, B.; Albig, A.R. Notch: A multi-functional integrating system of microenvironmental signals. Dev. Biol. 2016, 418, 227–241. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell. Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- Krejci, A.; Bernard, F.; Housden, B.E.; Collins, S.; Bray, S.J. Direct response to Notch activation: Signaling crosstalk and incoherent logic. Sci. Signal 2009, 2, ra1. [Google Scholar] [CrossRef]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarter, A.C.; Wang, Q.; Chiang, M. Notch in Leukemia. Adv. Exp. Med. Biol. 2018, 1066, 355–394. [Google Scholar] [CrossRef] [PubMed]
- Nowell, C.S.; Radtke, F. Notch as a tumour suppressor. Nat. Rev. Cancer 2017, 17, 145–159. [Google Scholar] [CrossRef]
- Arruga, F.; Vaisitti, T.; Deaglio, S. The NOTCH Pathway and Its Mutations in Mature B Cell Malignancies. Front. Oncol. 2018, 8, 550. [Google Scholar] [CrossRef] [Green Version]
- Pui, J.C.; Allman, D.; Xu, L.; DeRocco, S.; Karnell, F.G.; Bakkour, S.; Lee, J.Y.; Kadesch, T.; Hardy, R.R.; Aster, J.C.; et al. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity 1999, 11, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Kumano, K.; Chiba, S.; Kunisato, A.; Sata, M.; Saito, T.; Nakagami-Yamaguchi, E.; Yamaguchi, T.; Masuda, S.; Shimizu, K.; Takahashi, T.; et al. Notch1 but not Notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity 2003, 18, 699–711. [Google Scholar] [CrossRef]
- Pillai, S.; Cariappa, A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat. Rev. Immunol. 2009, 9, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, M.N.; Ulgiati, D. The role of notch signaling in the development of a normal B-cell repertoire. Immunol. Cell Biol. 2010, 88, 117–124. [Google Scholar] [CrossRef]
- Bertrand, F.E.; Eckfeldt, C.E.; Lysholm, A.S.; LeBien, T.W. Notch-1 and Notch-2 exhibit unique patterns of expression in human B-lineage cells. Leukemia 2000, 14, 2095–2102. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Chiba, S.; Ichikawa, M.; Kunisato, A.; Asai, T.; Shimizu, K.; Yamaguchi, T.; Yamamoto, G.; Seo, S.; Kumano, K.; et al. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity 2003, 18, 675–685. [Google Scholar] [CrossRef] [Green Version]
- Tanigaki, K.; Han, H.; Yamamoto, N.; Tashiro, K.; Ikegawa, M.; Kuroda, K.; Suzuki, A.; Nakano, T.; Honjo, T. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat. Immunol. 2002, 3, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.; Dalla-Favera, R. Chronic lymphocytic leukaemia: From genetics to treatment. Nat. Rev. Clin. Oncol. 2019, 16, 684–701. [Google Scholar] [CrossRef] [PubMed]
- Apelqvist, A.; Li, H.; Sommer, L.; Beatus, P.; Anderson, D.J.; Honjo, T.; Hrabe de Angelis, M.; Lendahl, U.; Edlund, H. Notch signalling controls pancreatic cell differentiation. Nature 1999, 400, 877–881. [Google Scholar] [CrossRef]
- Fan, X.; Mikolaenko, I.; Elhassan, I.; Ni, X.; Wang, Y.; Ball, D.; Brat, D.J.; Perry, A.; Eberhart, C.G. Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res. 2004, 64, 7787–7793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Gust, K.M.; Wyatt, A.W.; Goriki, A.; Jäger, W.; Awrey, S.; Li, N.; Oo, H.Z.; Altamirano-Dimas, M.; Buttyan, R.; et al. Not all NOTCH Is Created Equal: The Oncogenic Role of NOTCH2 in Bladder Cancer and Its Implications for Targeted Therapy. Clin. Cancer Res. 2016, 22, 2981–2992. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Brunskill, E.; Varnum-Finney, B.; Zhang, C.; Zhang, A.; Jay, P.Y.; Bernstein, I.; Morimoto, M.; Kopan, R. The intracellular domains of Notch1 and Notch2 are functionally equivalent during development and carcinogenesis. Development 2015, 142, 2452–2463. [Google Scholar] [CrossRef] [Green Version]
- de La Coste, A.; Freitas, A.A. Notch signaling: Distinct ligands induce specific signals during lymphocyte development and maturation. Immunol. Lett. 2006, 102, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Nalin, A.P.; Kowalski, J.J.; Sprague, A.C.; Schumacher, B.K.; Gerhardt, A.G.; Youssef, Y.; Vedantam, K.V.; Zhang, X.; Siebel, C.W.; Mace, E.M.; et al. Notch Regulates Innate Lymphoid Cell Plasticity during Human NK Cell Development. J. Immunol. 2020, 205, 2679–2693. [Google Scholar] [CrossRef]
- Oh, S.J.; Ahn, S.; Jin, Y.H.; Ishifune, C.; Kim, J.H.; Yasutomo, K.; Chung, D.H. Notch 1 and Notch 2 synergistically regulate the differentiation and function of invariant NKT cells. J. Leukoc. Biol. 2015, 98, 781–789. [Google Scholar] [CrossRef] [Green Version]
- Garis, M.; Garrett-Sinha, L.A. Notch Signaling in B Cell Immune Responses. Front. Immunol. 2020, 11, 609324. [Google Scholar] [CrossRef]
- Ohishi, K.; Varnum-Finney, B.; Flowers, D.; Anasetti, C.; Myerson, D.; Bernstein, I.D. Monocytes express high amounts of Notch and undergo cytokine specific apoptosis following interaction with the Notch ligand, Delta-1. Blood 2000, 95, 2847–2854. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.O.; Zhang, X.; Berner, P.; Blom, B.; Choi, Y.S. Notch ligands expressed by follicular dendritic cells protect germinal center B cells from apoptosis. J. Immunol. 2009, 183, 352–358. [Google Scholar] [CrossRef] [Green Version]
- Nwabo Kamdje, A.H.; Bassi, G.; Pacelli, L.; Malpeli, G.; Amati, E.; Nichele, I.; Pizzolo, G.; Krampera, M. Role of stromal cell-mediated Notch signaling in CLL resistance to chemotherapy. Blood Cancer J. 2012, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Gordon, W.R.; Vardar-Ulu, D.; Histen, G.; Sanchez-Irizarry, C.; Aster, J.C.; Blacklow, S.C. Structural basis for autoinhibition of Notch. Nat. Struct. Mol. Biol. 2007, 14, 295–300. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, M.Y.; Park, H.S. Phosphorylation-dependent regulation of Notch1 signaling: The fulcrum of Notch1 signaling. BMB Rep. 2015, 48, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [Green Version]
- Close, V.; Close, W.; Kugler, S.J.; Reichenzeller, M.; Yosifov, D.Y.; Bloehdorn, J.; Pan, L.; Tausch, E.; Westhoff, M.A.; Dohner, H.; et al. FBXW7 mutations reduce binding of NOTCH1, leading to cleaved NOTCH1 accumulation and target gene activation in CLL. Blood 2019, 133, 830–839. [Google Scholar] [CrossRef]
- Moloney, D.J.; Panin, V.M.; Johnston, S.H.; Chen, J.; Shao, L.; Wilson, R.; Wang, Y.; Stanley, P.; Irvine, K.D.; Haltiwanger, R.S.; et al. Fringe is a glycosyltransferase that modifies Notch. Nature 2000, 406, 369–375. [Google Scholar] [CrossRef]
- Kakuda, S.; Haltiwanger, R.S. Deciphering the Fringe-Mediated Notch Code: Identification of Activating and Inhibiting Sites Allowing Discrimination between Ligands. Dev. Cell 2017, 40, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Kovall, R.A.; Gebelein, B.; Sprinzak, D.; Kopan, R. The Canonical Notch Signaling Pathway: Structural and Biochemical Insights into Shape, Sugar, and Force. Dev. Cell 2017, 41, 228–241. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.; Niknejad, N.; Jafar-Nejad, H. Multifaceted regulation of Notch signaling by glycosylation. Glycobiology 2021, 31, 8–28. [Google Scholar] [CrossRef] [PubMed]
- Rand, M.D.; Lindblom, A.; Carlson, J.; Villoutreix, B.O.; Stenflo, J. Calcium binding to tandem repeats of EGF-like modules. Expression and characterization of the EGF-like modules of human Notch-1 implicated in receptor-ligand interactions. Protein Sci. 1997, 6, 2059–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetrivel, K.S.; Cheng, H.; Kim, S.-H.; Chen, Y.; Barnes, N.Y.; Parent, A.T.; Sisodia, S.S.; Thinakaran, G. Spatial Segregation of gamma-secretase and Substrates in Distinct Membrane Domains. J. Biol. Chem. 2005, 280, 25892–25900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaccari, T.; Lu, H.; Kanwar, R.; Fortini, M.E.; Bilder, D. Endosomal entry regulates Notch receptor activation in Drosophila melanogaster. J. Cell Biol. 2008, 180, 755–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakata, T.; Sakaguchi, H.; Tsuda, L.; Higashitani, A.; Aigaki, T.; Matsuno, K.; Hayashi, S. Drosophila Nedd4 regulates endocytosis of notch and suppresses its ligand-independent activation. Curr. Biol. 2004, 14, 2228–2236. [Google Scholar] [CrossRef] [Green Version]
- Chastagner, P.; Israël, A.; Brou, C. AIP4/Itch regulates Notch receptor degradation in the absence of ligand. PLoS ONE 2008, 3, e2735. [Google Scholar] [CrossRef] [Green Version]
- Schnute, B.; Troost, T.; Klein, T. Endocytic Trafficking of the Notch Receptor. Adv. Exp. Med. Biol. 2018, 1066, 99–122. [Google Scholar] [CrossRef]
- Hounjet, J.; Vooijs, M. The Role of Intracellular Trafficking of Notch Receptors in Ligand-Independent Notch Activation. Biomolecules 2021, 11, 1369. [Google Scholar] [CrossRef]
- Berdnik, D.; Török, T.; González-Gaitán, M.; Knoblich, J.A. The endocytic protein alpha-Adaptin is required for numb-mediated asymmetric cell division in Drosophila. Dev. Cell 2002, 3, 221–231. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, B.; Miyamoto, A.; Weinmaster, G. The many facets of Notch ligands. Oncogene 2008, 27, 5148–5167. [Google Scholar] [CrossRef] [Green Version]
- Jaleco, A.C.; Neves, H.; Hooijberg, E.; Gameiro, P.; Clode, N.; Haury, M.; Henrique, D.; Parreira, L. Differential effects of Notch ligands Delta-1 and Jagged-1 in human lymphoid differentiation. J. Exp. Med. 2001, 194, 991–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehar, S.M.; Dooley, J.; Farr, A.G.; Bevan, M.J. Notch ligands Delta 1 and Jagged1 transmit distinct signals to T-cell precursors. Blood 2005, 105, 1440–1447. [Google Scholar] [CrossRef] [Green Version]
- Felli, M.P.; Maroder, M.; Mitsiadis, T.A.; Campese, A.F.; Bellavia, D.; Vacca, A.; Mann, R.S.; Frati, L.; Lendahl, U.; Gulino, A.; et al. Expression pattern of notch1, 2 and 3 and Jagged1 and 2 in lymphoid and stromal thymus components: Distinct ligand-receptor interactions in intrathymic T cell development. Int. Immunol. 1999, 11, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Tokoyoda, K.; Egawa, T.; Sugiyama, T.; Choi, B.I.; Nagasawa, T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 2004, 20, 707–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Walle, I.; De Smet, G.; Gärtner, M.; De Smedt, M.; Waegemans, E.; Vandekerckhove, B.; Leclercq, G.; Plum, J.; Aster, J.C.; Bernstein, I.D.; et al. Jagged2 acts as a Delta-like Notch ligand during early hematopoietic cell fate decisions. Blood 2011, 117, 4449–4459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luca, V.C.; Jude, K.M.; Pierce, N.W.; Nachury, M.V.; Fischer, S.; Garcia, K.C. Structural biology. Structural basis for Notch1 engagement of Delta-like 4. Science 2015, 347, 847–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luca, V.C.; Kim Byoung, C.; Ge, C.; Kakuda, S.; Wu, D.; Roein-Peikar, M.; Haltiwanger Robert, S.; Zhu, C.; Ha, T.; Garcia, K.C. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science 2017, 355, 1320–1324. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, F.; Li, I.T.; Ngo, T.T.; Leslie, B.J.; Kim, B.C.; Sokoloski, J.E.; Weiland, E.; Wang, X.; Chemla, Y.R.; Lohman, T.M.; et al. Defining Single Molecular Forces Required for Notch Activation Using Nano Yoyo. Nano Lett. 2016, 16, 3892–3897. [Google Scholar] [CrossRef] [Green Version]
- Chapman, G.; Major, J.A.; Iyer, K.; James, A.C.; Pursglove, S.E.; Moreau, J.L.M.; Dunwoodie, S.L. Notch1 endocytosis is induced by ligand and is required for signal transduction. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Meloty-Kapella, L.; Shergill, B.; Kuon, J.; Botvinick, E.; Weinmaster, G. Notch ligand endocytosis generates mechanical pulling force dependent on dynamin, epsins, and actin. Dev. Cell 2012, 22, 1299–1312. [Google Scholar] [CrossRef] [Green Version]
- Gordon, W.R.; Zimmerman, B.; He, L.; Miles, L.J.; Huang, J.; Tiyanont, K.; McArthur, D.G.; Aster, J.C.; Perrimon, N.; Loparo, J.J.; et al. Mechanical Allostery: Evidence for a Force Requirement in the Proteolytic Activation of Notch. Dev. Cell 2015, 33, 729–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephenson, N.L.; Avis, J.M. Direct observation of proteolytic cleavage at the S2 site upon forced unfolding of the Notch negative regulatory region. Proc. Natl. Acad. Sci. USA 2012, 109, E2757–E2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brou, C.; Logeat, F.; Gupta, N.; Bessia, C.; LeBail, O.; Doedens, J.R.; Cumano, A.; Roux, P.; Black, R.A.; Israel, A. A novel proteolytic cleavage involved in Notch signaling: The role of the disintegrin-metalloprotease TACE. Mol. Cell 2000, 5, 207–216. [Google Scholar] [CrossRef]
- Bozkulak, E.C.; Weinmaster, G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol. Cell. Biol. 2009, 29, 5679–5695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arruga, F.; Gizdic, B.; Serra, S.; Vaisitti, T.; Ciardullo, C.; Coscia, M.; Laurenti, L.; D’Arena, G.; Jaksic, O.; Inghirami, G.; et al. Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia. Leukemia 2014, 28, 1060–1070. [Google Scholar] [CrossRef]
- Pozzo, F.; Bittolo, T.; Vendramini, E.; Bomben, R.; Bulian, P.; Rossi, F.M.; Zucchetto, A.; Tissino, E.; Degan, M.; D’Arena, G.; et al. NOTCH1-mutated chronic lymphocytic leukemia cells are characterized by a MYC-related overexpression of nucleophosmin 1 and ribosome-associated components. Leukemia 2017, 31, 2407–2415. [Google Scholar] [CrossRef]
- Wu, B.; Slabicki, M.; Sellner, L.; Dietrich, S.; Liu, X.; Jethwa, A.; Hullein, J.; Walther, T.; Wagner, L.; Huang, Z.; et al. MED12 mutations and NOTCH signalling in chronic lymphocytic leukaemia. Br. J. Haematol. 2017, 179, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Arruga, F.; Bracciama, V.; Vitale, N.; Vaisitti, T.; Gizzi, K.; Yeomans, A.; Coscia, M.; D’Arena, G.; Gaidano, G.; Allan, J.N.; et al. Bidirectional linkage between the B-cell receptor and NOTCH1 in chronic lymphocytic leukemia and in Richter’s syndrome: Therapeutic implications. Leukemia 2020, 34, 462–477. [Google Scholar] [CrossRef]
- Kluk, M.J.; Ashworth, T.; Wang, H.; Knoechel, B.; Mason, E.F.; Morgan, E.A.; Dorfman, D.; Pinkus, G.; Weigert, O.; Hornick, J.L.; et al. Gauging NOTCH1 Activation in Cancer Using Immunohistochemistry. PLoS ONE 2013, 8, e67306. [Google Scholar] [CrossRef] [Green Version]
- Diez, P.; Lorenzo, S.; Degano, R.M.; Ibarrola, N.; Gonzalez-Gonzalez, M.; Nieto, W.; Almeida, J.; Gonzalez, M.; Orfao, A.; Fuentes, M. Multipronged functional proteomics approaches for global identification of altered cell signalling pathways in B-cell chronic lymphocytic leukaemia. Proteomics 2016, 16, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, E.; Itoh, M.; Tojo, N.; Koyama, T.; Nara, N.; Tohda, S. Flow cytometric analysis of Notch1 and Jagged1 expression in normal blood cells and leukemia cells. Exp. Ther. Med. 2012, 4, 397–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Ianni, M.; Baldoni, S.; Del Papa, B.; Aureli, P.; Dorillo, E.; De Falco, F.; Albi, E.; Varasano, E.; Di Tommaso, A.; Giancola, R.; et al. NOTCH1 Is Aberrantly Activated in Chronic Lymphocytic Leukemia Hematopoietic Stem Cells. Front. Oncol. 2018, 8, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998, 393, 382–386. [Google Scholar] [CrossRef]
- Struhl, G.; Greenwald, I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature 1999, 398, 522–525. [Google Scholar] [CrossRef]
- Castel, D.; Mourikis, P.; Bartels, S.J.; Brinkman, A.B.; Tajbakhsh, S.; Stunnenberg, H.G. Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 2013, 27, 1059–1071. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Lamarca, M.J.; Falo-Sanjuan, J.; Stojnic, R.; Abdul Rehman, S.; Muresan, L.; Jones, M.L.; Pillidge, Z.; Cerda-Moya, G.; Yuan, Z.; Baloul, S.; et al. Activation of the Notch Signaling Pathway In Vivo Elicits Changes in CSL Nuclear Dynamics. Dev. Cell 2018, 44, 611–623.e617. [Google Scholar] [CrossRef] [Green Version]
- Kao, H.Y.; Ordentlich, P.; Koyano-Nakagawa, N.; Tang, Z.; Downes, M.; Kintner, C.R.; Evans, R.M.; Kadesch, T. A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev. 1998, 12, 2269–2277. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, K.; Han, H.; Tani, S.; Tanigaki, K.; Tun, T.; Furukawa, T.; Taniguchi, Y.; Kurooka, H.; Hamada, Y.; Toyokuni, S.; et al. Regulation of marginal zone B cell development by MINT, a suppressor of Notch/RBP-J signaling pathway. Immunity 2003, 18, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Yatim, A.; Benne, C.; Sobhian, B.; Laurent-Chabalier, S.; Deas, O.; Judde, J.G.; Lelievre, J.D.; Levy, Y.; Benkirane, M. NOTCH1 nuclear interactome reveals key regulators of its transcriptional activity and oncogenic function. Mol. Cell 2012, 48, 445–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, P.; Uosaki, H.; Shenje, L.T.; Kwon, C. Non-canonical Notch signaling: Emerging role and mechanism. Trends Cell Biol. 2012, 22, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbuck, M.P.; Winandy, S. A Review of Notch Processing With New Insights Into Ligand-Independent Notch Signaling in T-Cells. Front. Immunol. 2018, 9, 1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkin, M.; Tongngok, P.; Gensch, N.; Clemence, S.; Motoki, M.; Yamada, K.; Hori, K.; Taniguchi-Kanai, M.; Franklin, E.; Matsuno, K.; et al. Drosophila HOPS and AP-3 complex genes are required for a Deltex-regulated activation of notch in the endosomal trafficking pathway. Dev. Cell 2008, 15, 762–772. [Google Scholar] [CrossRef]
- Chastagner, P.; Rubinstein, E.; Brou, C. Ligand-activated Notch undergoes DTX4-mediated ubiquitylation and bilateral endocytosis before ADAM10 processing. Sci. Signal. 2017, 10, eaag2989. [Google Scholar] [CrossRef] [Green Version]
- Skovronsky, D.M.; Moore, D.B.; Milla, M.E.; Doms, R.W.; Lee, V.M. Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J. Biol. Chem. 2000, 275, 2568–2575. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Saunders, C.A.; Sorensen, E.B.; Waxmonsky, N.C.; Conner, S.D. Notch signaling from the endosome requires a conserved dileucine motif. Mol. Biol. Cell 2013, 24, 297–307. [Google Scholar] [CrossRef]
- Parsons, L.M.; Portela, M.; Grzeschik, N.A.; Richardson, H.E. Lgl regulates Notch signaling via endocytosis, independently of the apical aPKC-Par6-Baz polarity complex. Curr. Biol. 2014, 24, 2073–2084. [Google Scholar] [CrossRef] [Green Version]
- Hounjet, J.; Habets, R.; Schaaf, M.B.; Hendrickx, T.C.; Barbeau, L.M.O.; Yahyanejad, S.; Rouschop, K.M.; Groot, A.J.; Vooijs, M. The anti-malarial drug chloroquine sensitizes oncogenic NOTCH1 driven human T-ALL to γ-secretase inhibition. Oncogene 2019, 38, 5457–5468. [Google Scholar] [CrossRef]
- Aster, J.C.; Simms, W.B.; Zavala-Ruiz, Z.; Patriub, V.; North, C.L.; Blacklow, S.C. The folding and structural integrity of the first LIN-12 module of human Notch1 are calcium-dependent. Biochemistry 1999, 38, 4736–4742. [Google Scholar] [CrossRef]
- Rand, M.D.; Grimm, L.M.; Artavanis-Tsakonas, S.; Patriub, V.; Blacklow, S.C.; Sklar, J.; Aster, J.C. Calcium depletion dissociates and activates heterodimeric notch receptors. Mol. Cell. Biol. 2000, 20, 1825–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Tetering, G.; van Diest, P.; Verlaan, I.; van der Wall, E.; Kopan, R.; Vooijs, M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J. Biol. Chem. 2009, 284, 31018–31027. [Google Scholar] [CrossRef] [Green Version]
- Ran, Y.; Hossain, F.; Pannuti, A.; Lessard, C.B.; Ladd, G.Z.; Jung, J.I.; Minter, L.M.; Osborne, B.A.; Miele, L.; Golde, T.E. γ-Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol. Med. 2017, 9, 950–966. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Ries, R.E.; Ilagan, J.O.; Stirewalt, D.L.; Meshinchi, S.; Bradley, R.K. Sample processing obscures cancer-specific alterations in leukemic transcriptomes. Proc. Natl. Acad. Sci. USA 2014, 111, 16802–16807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelmann, J.; Holzmann, K.; Tausch, E.; Saunderson, E.A.; Jebaraj, B.M.C.; Steinbrecher, D.; Dolnik, A.; Blatte, T.J.; Landau, D.A.; Saub, J.; et al. Genomic alterations in high-risk chronic lymphocytic leukemia frequently affect cell cycle key regulators and NOTCH1-regulated transcription. Haematologica 2020, 105, 1379–1390. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, J.; Dokal, A.D.; Vilventhraraja, E.; Holzmann, K.; Britton, D.; Klymenko, T.; Döhner, H.; Cragg, M.; Braun, A.; Cutillas, P.; et al. Rituximab and obinutuzumab differentially hijack the B cell receptor and NOTCH1 signaling pathways. iScience 2021, 24, 102089. [Google Scholar] [CrossRef]
- Rossi, F.M.; Zucchetto, A.; Tissino, E.; Dal Bo, M.; Bomben, R.; Caldana, C.; Pozzo, F.; Del Poeta, G.; Rossi, D.; Gaidano, G.; et al. CD49d expression identifies a chronic-lymphocytic leukemia subset with high levels of mobilized circulating CD34(+) hemopoietic progenitors cells. Leukemia 2014, 28, 705–708. [Google Scholar] [CrossRef]
- Quijada-Alamo, M.; Hernandez-Sanchez, M.; Robledo, C.; Hernandez-Sanchez, J.M.; Benito, R.; Montano, A.; Rodriguez-Vicente, A.E.; Quwaider, D.; Martin, A.A.; Garcia-Alvarez, M.; et al. Next-generation sequencing and FISH studies reveal the appearance of gene mutations and chromosomal abnormalities in hematopoietic progenitors in chronic lymphocytic leukemia. J. Hematol. Oncol. 2017, 10, 83. [Google Scholar] [CrossRef]
- Wang, L.; Lawrence, M.S.; Wan, Y.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L.; et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 2011, 365, 2497–2506. [Google Scholar] [CrossRef]
- Mansour, M.R.; Linch, D.C.; Foroni, L.; Goldstone, A.H.; Gale, R.E. High incidence of Notch-1 mutations in adult patients with T-cell acute lymphoblastic leukemia. Leukemia 2006, 20, 537–539. [Google Scholar] [CrossRef]
- Barrio, S.; Shanafelt, T.D.; Ojha, J.; Chaffee, K.G.; Secreto, C.; Kortum, K.M.; Pathangey, S.; Van-Dyke, D.L.; Slager, S.L.; Fonseca, R.; et al. Genomic characterization of high-count MBL cases indicates that early detection of driver mutations and subclonal expansion are predictors of adverse clinical outcome. Leukemia 2017, 31, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Fabris, S.; Cutrona, G.; Agnelli, L.; Ciardullo, C.; Matis, S.; Ciceri, G.; Colombo, M.; Maura, F.; Mosca, L.; et al. High-throughput sequencing for the identification of NOTCH1 mutations in early stage chronic lymphocytic leukaemia: Biological and clinical implications. Br. J. Haematol. 2014, 165, 629–639. [Google Scholar] [CrossRef]
- Minervini, A.; Francesco Minervini, C.; Anelli, L.; Zagaria, A.; Casieri, P.; Coccaro, N.; Cumbo, C.; Tota, G.; Impera, L.; Orsini, P.; et al. Droplet digital PCR analysis of NOTCH1 gene mutations in chronic lymphocytic leukemia. Oncotarget 2016, 7, 86469–86479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoofd, C.; Huang, S.J.; Gusscott, S.; Lam, S.; Wong, R.; Johnston, A.; Ben-Neriah, S.; Steidl, C.; Scott, D.W.; Bruyere, H.; et al. Ultrasensitive Detection of NOTCH1 c.7544_7545delCT Mutations in Chronic Lymphocytic Leukemia by Droplet Digital PCR Reveals High Frequency of Subclonal Mutations and Predicts Clinical Outcome in Cases with Trisomy 12. J. Mol. Diagn. 2020, 22, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Skorka, K.; Chojnacki, M.; Masternak, M.; Karczmarczyk, A.; Subocz, E.; Wawrzyniak, E.; Giannopoulos, K. The Predominant Prognostic Significance of NOTCH1 Mutation Defined by Emulsion PCR in Chronic Lymphocytic Leukemia. Cancer Manag. Res. 2021, 13, 3663–3674. [Google Scholar] [CrossRef] [PubMed]
- Bomben, R.; Rossi, F.M.; Vit, F.; Bittolo, T.; D’Agaro, T.; Zucchetto, A.; Tissino, E.; Pozzo, F.; Vendramini, E.; Degan, M.; et al. TP53 Mutations with Low Variant Allele Frequency Predict Short Survival in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2021, 27, 5566–5575. [Google Scholar] [CrossRef]
- Puente, X.S.; Bea, S.; Valdes-Mas, R.; Villamor, N.; Gutierrez-Abril, J.; Martin-Subero, J.I.; Munar, M.; Rubio-Perez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef]
- Bittolo, T.; Pozzo, F.; Bomben, R.; D’Agaro, T.; Bravin, V.; Bulian, P.; Rossi, F.M.; Zucchetto, A.; Degan, M.; Macor, P.; et al. Mutations in the 3’ untranslated region of NOTCH1 are associated with low CD20 expression levels chronic lymphocytic leukemia. Haematologica 2017, 102, e305–e309. [Google Scholar] [CrossRef]
- Abramenko, I.V.; Bilous, N.I.; Chumak, A.A.; Dyagil, I.S.; Martina, Z.V. Analysis of the 3’UTR region of the NOTCH1 gene in chronic lymphocytic leukemia patients. Exp. Oncol. 2018, 40, 211–217. [Google Scholar] [CrossRef]
- Larrayoz, M.; Rose-Zerilli, M.J.; Kadalayil, L.; Parker, H.; Blakemore, S.; Forster, J.; Davis, Z.; Steele, A.J.; Collins, A.; Else, M.; et al. Non-coding NOTCH1 mutations in chronic lymphocytic leukemia; their clinical impact in the UK CLL4 trial. Leukemia 2017, 31, 510–514. [Google Scholar] [CrossRef] [Green Version]
- Rosati, E.; Baldoni, S.; De Falco, F.; Del Papa, B.; Dorillo, E.; Rompietti, C.; Albi, E.; Falzetti, F.; Di Ianni, M.; Sportoletti, P. NOTCH1 Aberrations in Chronic Lymphocytic Leukemia. Front. Oncol. 2018, 8, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Falco, F.; Sabatini, R.; Del Papa, B.; Falzetti, F.; Di Ianni, M.; Sportoletti, P.; Baldoni, S.; Screpanti, I.; Marconi, P.; Rosati, E. Notch signaling sustains the expression of Mcl-1 and the activity of eIF4E to promote cell survival in CLL. Oncotarget 2015, 6, 16559–16572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jitschin, R.; Braun, M.; Qorraj, M.; Saul, D.; Le Blanc, K.; Zenz, T.; Mougiakakos, D. Stromal cell-mediated glycolytic switch in CLL cells involves Notch-c-Myc signaling. Blood 2015, 125, 3432–3436. [Google Scholar] [CrossRef] [Green Version]
- Pozzo, F.; Bittolo, T.; Arruga, F.; Bulian, P.; Macor, P.; Tissino, E.; Gizdic, B.; Rossi, F.M.; Bomben, R.; Zucchetto, A.; et al. NOTCH1 mutations associate with low CD20 level in chronic lymphocytic leukemia: Evidence for a NOTCH1 mutation-driven epigenetic dysregulation. Leukemia 2016, 30, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Arruga, F.; Gizdic, B.; Bologna, C.; Cignetto, S.; Buonincontri, R.; Serra, S.; Vaisitti, T.; Gizzi, K.; Vitale, N.; Garaffo, G.; et al. Mutations in NOTCH1 PEST domain orchestrate CCL19-driven homing of chronic lymphocytic leukemia cells by modulating the tumor suppressor gene DUSP22. Leukemia 2017, 31, 1882–1893. [Google Scholar] [CrossRef]
- Benedetti, D.; Tissino, E.; Pozzo, F.; Bittolo, T.; Caldana, C.; Perini, C.; Martorelli, D.; Bravin, V.; D’Agaro, T.; Rossi, F.M.; et al. NOTCH1 mutations are associated with high CD49d expression in chronic lymphocytic leukemia: Link between the NOTCH1 and the NF-kappaB pathways. Leukemia 2018, 32, 654–662. [Google Scholar] [CrossRef]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [Green Version]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.t.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.M.; Calvo, J.A.; Draheim, K.M.; Cunningham, L.A.; Hermance, N.; Beverly, L.; Krishnamoorthy, V.; Bhasin, M.; Capobianco, A.J.; Kelliher, M.A. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol. Cell. Biol. 2006, 26, 8022–8031. [Google Scholar] [CrossRef] [Green Version]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [Green Version]
- Klinakis, A.; Szabolcs, M.; Politi, K.; Kiaris, H.; Artavanis-Tsakonas, S.; Efstratiadis, A. Myc is a Notch1 transcriptional target and a requisite for Notch1-induced mammary tumorigenesis in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 9262–9267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yashiro-Ohtani, Y.; Wang, H.; Zang, C.; Arnett, K.L.; Bailis, W.; Ho, Y.; Knoechel, B.; Lanauze, C.; Louis, L.; Forsyth, K.S.; et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E4946–E4953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, M.; Bruns, H.; Völkl, S.; Lu, J.; Chartomatsidou, E.; Papakonstantinou, N.; Mentz, K.; Büttner-Herold, M.; Zenz, T.; Herling, M.; et al. Control of PD-L1 expression in CLL-cells by stromal triggering of the Notch-c-Myc-EZH2 oncogenic signaling axis. J. Immunother. Cancer 2021, 9, e001889. [Google Scholar] [CrossRef]
- Lopez-Guerra, M.; Xargay-Torrent, S.; Fuentes, P.; Roldan, J.; Gonzalez-Farre, B.; Rosich, L.; Silkenstedt, E.; Garcia-Leon, M.J.; Lee-Verges, E.; Gimenez, N.; et al. Specific NOTCH1 antibody targets DLL4-induced proliferation, migration, and angiogenesis in NOTCH1-mutated CLL cells. Oncogene 2020, 39, 1185–1197. [Google Scholar] [CrossRef]
- Ryan, R.J.H.; Petrovic, J.; Rausch, D.M.; Zhou, Y.; Lareau, C.A.; Kluk, M.J.; Christie, A.L.; Lee, W.Y.; Tarjan, D.R.; Guo, B.; et al. A B Cell Regulome Links Notch to Downstream Oncogenic Pathways in Small B Cell Lymphomas. Cell Rep. 2017, 21, 784–797. [Google Scholar] [CrossRef] [Green Version]
- Hassan, W.A.; Ito, T. Identifying specific Notch1 target proteins in lung carcinoma cells. Histol. Histopathol. 2021, 36, 69–76. [Google Scholar] [CrossRef]
- Feng, L.; Wang, K.; Tang, P.; Chen, S.; Liu, T.; Lei, J.; Yuan, R.; Hu, Z.; Li, W.; Yu, X. Deubiquitinase USP18 promotes the progression of pancreatic cancer via enhancing the Notch1-c-Myc axis. Aging (Albany NY) 2020, 12, 19273–19292. [Google Scholar] [CrossRef]
- Giaimo, B.D.; Gagliani, E.K.; Kovall, R.A.; Borggrefe, T. Transcription Factor RBPJ as a Molecular Switch in Regulating the Notch Response. Adv. Exp. Med. Biol. 2021, 1287, 9–30. [Google Scholar] [CrossRef]
- Chen, J.; Dong, Y.; Peng, J.; Zhang, J.; Gao, X.; Lu, A.; Shen, C. Notch signaling mitigates chemotherapy toxicity by accelerating hematopoietic stem cells proliferation via c-Myc. Am. J. Transl. Res. 2020, 12, 6723–6739. [Google Scholar] [PubMed]
- Ryan, R.J.; Drier, Y.; Whitton, H.; Cotton, M.J.; Kaur, J.; Issner, R.; Gillespie, S.; Epstein, C.B.; Nardi, V.; Sohani, A.R.; et al. Detection of Enhancer-Associated Rearrangements Reveals Mechanisms of Oncogene Dysregulation in B-cell Lymphoma. Cancer Discov. 2015, 5, 1058–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuní, S.; Pérez-Aciego, P.; Pérez-Chacón, G.; Vargas, J.A.; Sánchez, A.; Martín-Saavedra, F.M.; Ballester, S.; García-Marco, J.; Jordá, J.; Durántez, A. A sustained activation of PI3K/NF-kappaB pathway is critical for the survival of chronic lymphocytic leukemia B cells. Leukemia 2004, 18, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, C.; Celeghini, C.; Monasta, L.; Zauli, G. NF-κB pathways in hematological malignancies. Cell. Mol. Life Sci. 2014, 71, 2083–2102. [Google Scholar] [CrossRef]
- Mansouri, L.; Papakonstantinou, N.; Ntoufa, S.; Stamatopoulos, K.; Rosenquist, R. NF-κB activation in chronic lymphocytic leukemia: A point of convergence of external triggers and intrinsic lesions. Semin. Cancer Biol. 2016, 39, 40–48. [Google Scholar] [CrossRef]
- Espinosa, L.; Cathelin, S.; D’Altri, T.; Trimarchi, T.; Statnikov, A.; Guiu, J.; Rodilla, V.; Inglés-Esteve, J.; Nomdedeu, J.; Bellosillo, B.; et al. The Notch/Hes1 pathway sustains NF-κB activation through CYLD repression in T cell leukemia. Cancer Cell 2010, 18, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, R.; Dörken, B.; Jundt, F. Notch is an essential upstream regulator of NF-κB and is relevant for survival of Hodgkin and Reed-Sternberg cells. Leukemia 2012, 26, 806–813. [Google Scholar] [CrossRef]
- Moran, S.T.; Cariappa, A.; Liu, H.; Muir, B.; Sgroi, D.; Boboila, C.; Pillai, S. Synergism between NF-kappa B1/p50 and Notch2 during the development of marginal zone B lymphocytes. J. Immunol. 2007, 179, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Bash, J.; Zong, W.X.; Banga, S.; Rivera, A.; Ballard, D.W.; Ron, Y.; Gelinas, C. Rel/NF-kappaB can trigger the Notch signaling pathway by inducing the expression of Jagged1, a ligand for Notch receptors. EMBO J. 1999, 18, 2803–2811. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.S.; Zhang, J.S.; Zhang, J.Y.; Wu, S.Q.; Xiong, D.L.; Chen, H.J.; Chen, Z.Z.; Zhan, R. Constitutive activation of NF-kappaB signaling by NOTCH1 mutations in chronic lymphocytic leukemia. Oncol. Rep. 2015, 33, 1609–1614. [Google Scholar] [CrossRef] [Green Version]
- Baldoni, S.; Del Papa, B.; Dorillo, E.; Aureli, P.; De Falco, F.; Rompietti, C.; Sorcini, D.; Varasano, E.; Cecchini, D.; Zei, T.; et al. Bepridil exhibits anti-leukemic activity associated with NOTCH1 pathway inhibition in chronic lymphocytic leukemia. Int. J. Cancer 2018, 143, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.M.; Minter, L.M.; Cho, O.H.; Gottipati, S.; Fauq, A.H.; Golde, T.E.; Sonenshein, G.E.; Osborne, B.A. Notch1 augments NF-kappaB activity by facilitating its nuclear retention. EMBO J. 2006, 25, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulian, P.; Shanafelt, T.D.; Fegan, C.; Zucchetto, A.; Cro, L.; Nuckel, H.; Baldini, L.; Kurtova, A.V.; Ferrajoli, A.; Burger, J.A.; et al. CD49d is the strongest flow cytometry-based predictor of overall survival in chronic lymphocytic leukemia. J. Clin. Oncol. 2014, 32, 897–904. [Google Scholar] [CrossRef]
- Tissino, E.; Pozzo, F.; Benedetti, D.; Caldana, C.; Bittolo, T.; Rossi, F.M.; Bomben, R.; Nanni, P.; Chivilo, H.; Cattarossi, I.; et al. CD49d promotes disease progression in chronic lymphocytic leukemia: New insights from CD49d bimodal expression. Blood 2020, 135, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, S.; Roller, A.; Jeromin, S.; Hernandez, M.; Abaigar, M.; Hernandez-Rivas, J.M.; Grossmann, V.; Haferlach, C.; Kern, W.; Haferlach, T.; et al. Prognostic impact and landscape of NOTCH1 mutations in chronic lymphocytic leukemia (CLL): A study on 852 patients. Leukemia 2013, 27, 2393–2396. [Google Scholar] [CrossRef] [Green Version]
- Willander, K.; Dutta, R.K.; Ungerback, J.; Gunnarsson, R.; Juliusson, G.; Fredrikson, M.; Linderholm, M.; Soderkvist, P. NOTCH1 mutations influence survival in chronic lymphocytic leukemia patients. BMC Cancer 2013, 13, 274. [Google Scholar] [CrossRef] [Green Version]
- Villamor, N.; Conde, L.; Martinez-Trillos, A.; Cazorla, M.; Navarro, A.; Bea, S.; Lopez, C.; Colomer, D.; Pinyol, M.; Aymerich, M.; et al. NOTCH1 mutations identify a genetic subgroup of chronic lymphocytic leukemia patients with high risk of transformation and poor outcome. Leukemia 2013, 27, 1100–1106. [Google Scholar] [CrossRef] [Green Version]
- Del Poeta, G.; Dal Bo, M.; Del Principe, M.I.; Pozzo, F.; Rossi, F.M.; Zucchetto, A.; Bomben, R.; Degan, M.; Rasi, S.; Rossi, D.; et al. Clinical significance of c.7544-7545 delCT NOTCH1 mutation in chronic lymphocytic leukaemia. Br. J. Haematol. 2013, 160, 415–418. [Google Scholar] [CrossRef]
- Jeromin, S.; Weissmann, S.; Haferlach, C.; Dicker, F.; Bayer, K.; Grossmann, V.; Alpermann, T.; Roller, A.; Kohlmann, A.; Haferlach, T.; et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia 2014, 28, 108–117. [Google Scholar] [CrossRef]
- Tausch, E.; Beck, P.; Schlenk, R.F.; Jebaraj, B.J.; Dolnik, A.; Yosifov, D.Y.; Hillmen, P.; Offner, F.; Janssens, A.; Babu, G.K.; et al. Prognostic and predictive role of gene mutations in chronic lymphocytic leukemia: Results from the pivotal phase III study COMPLEMENT1. Haematologica 2020, 105, 2440–2447. [Google Scholar] [CrossRef] [Green Version]
- Oscier, D.G.; Rose-Zerilli, M.J.; Winkelmann, N.; Gonzalez de Castro, D.; Gomez, B.; Forster, J.; Parker, H.; Parker, A.; Gardiner, A.; Collins, A.; et al. The clinical significance of NOTCH1 and SF3B1 mutations in the UK LRF CLL4 trial. Blood 2013, 121, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, S.; Marinelli, M.; Del Giudice, I.; Bonina, S.; Piciocchi, A.; Messina, M.; Vignetti, M.; Rossi, D.; Di Maio, V.; Mauro, F.R.; et al. NOTCH1, SF3B1, BIRC3 and TP53 mutations in patients with chronic lymphocytic leukemia undergoing first-line treatment: Correlation with biological parameters and response to treatment. Leuk Lymphoma 2014, 55, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Hillmen, P.; O’Brien, S.; Barrientos, J.C.; Reddy, N.M.; Coutre, S.E.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; Barr, P.M.; et al. Extended follow-up and impact of high-risk prognostic factors from the phase 3 RESONATE study in patients with previously treated CLL/SLL. Leukemia 2018, 32, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herling, C.D.; Klaumunzer, M.; Rocha, C.K.; Altmuller, J.; Thiele, H.; Bahlo, J.; Kluth, S.; Crispatzu, G.; Herling, M.; Schiller, J.; et al. Complex karyotypes and KRAS and POT1 mutations impact outcome in CLL after chlorambucil-based chemotherapy or chemoimmunotherapy. Blood 2016, 128, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzo, F.; Bittolo, T.; Tissino, E.; Vit, F.; Vendramini, E.; Laurenti, L.; D’Arena, G.; Olivieri, J.; Pozzato, G.; Zaja, F.; et al. SF3B1-mutated chronic lymphocytic leukemia shows evidence of NOTCH1 pathway activation including CD20 downregulation. Haematologica 2021, 106, 3125. [Google Scholar] [CrossRef]
- Del Giudice, I.; Rossi, D.; Chiaretti, S.; Marinelli, M.; Tavolaro, S.; Gabrielli, S.; Laurenti, L.; Marasca, R.; Rasi, S.; Fangazio, M.; et al. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica 2012, 97, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Tam, C.S.; Otero-Palacios, J.; Abruzzo, L.V.; Jorgensen, J.L.; Ferrajoli, A.; Wierda, W.G.; Lerner, S.; O’Brien, S.; Keating, M.J. Chronic lymphocytic leukaemia CD20 expression is dependent on the genetic subtype: A study of quantitative flow cytometry and fluorescent in-situ hybridization in 510 patients. Br. J. Haematol. 2008, 141, 36–40. [Google Scholar] [CrossRef]
- Onaindia, A.; Gomez, S.; Piris-Villaespesa, M.; Martinez-Laperche, C.; Cereceda, L.; Montes-Moreno, S.; Batlle, A.; de Villambrosia, S.G.; Pollan, M.; Martin-Acosta, P.; et al. Chronic lymphocytic leukemia cells in lymph nodes show frequent NOTCH1 activation. Haematologica 2015, 100, e200–e203. [Google Scholar] [CrossRef] [Green Version]
- Liefke, R.; Oswald, F.; Alvarado, C.; Ferres-Marco, D.; Mittler, G.; Rodriguez, P.; Dominguez, M.; Borggrefe, T. Histone demethylase KDM5A is an integral part of the core Notch-RBP-J repressor complex. Genes Dev. 2010, 24, 590–601. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, J.; Grim, J.; Strack, P.; Rao, S.; Tibbitts, D.; Winter, C.; Hardwick, J.; Welcker, M.; Meijerink, J.P.; Pieters, R.; et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J. Exp. Med. 2007, 204, 1813–1824. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, T.L.; Brennan, K.; Arias, A.M.; Muskavitch, M.A. Cis-interactions between Delta and Notch modulate neurogenic signalling in Drosophila. Development 1998, 125, 4531–4540. [Google Scholar] [CrossRef] [PubMed]
- Sprinzak, D.; Lakhanpal, A.; Lebon, L.; Santat, L.A.; Fontes, M.E.; Anderson, G.A.; Garcia-Ojalvo, J.; Elowitz, M.B. Cis-interactions between Notch and Delta generate mutually exclusive signalling states. Nature 2010, 465, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- del Álamo, D.; Rouault, H.; Schweisguth, F. Mechanism and significance of cis-inhibition in Notch signalling. Curr. Biol. 2011, 21, R40–R47. [Google Scholar] [CrossRef] [Green Version]
- Fleming, R.J. Ligand-Induced Cis-Inhibition of Notch Signaling: The Role of an Extracellular Region of Serrate. In Notch Signaling in Embryology and Cancer: Molecular Biology of Notch Signaling; Reichrath, J., Reichrath, S., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 29–49. [Google Scholar]
- Nandagopal, N.; Santat, L.A.; Elowitz, M.B. Cis-activation in the Notch signaling pathway. Elife 2019, 8, e37880. [Google Scholar] [CrossRef]
- Koepp, D.M.; Schaefer, L.K.; Ye, X.; Keyomarsi, K.; Chu, C.; Harper, J.W.; Elledge, S.J. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001, 294, 173–177. [Google Scholar] [CrossRef]
- Welcker, M.; Clurman, B.E. FBW7 ubiquitin ligase: A tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 2008, 8, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef]
- Malyukova, A.; Dohda, T.; von der Lehr, N.; Akhoondi, S.; Corcoran, M.; Heyman, M.; Spruck, C.; Grandér, D.; Lendahl, U.; Sangfelt, O. The tumor suppressor gene hCDC4 is frequently mutated in human T-cell acute lymphoblastic leukemia with functional consequences for Notch signaling. Cancer Res. 2007, 67, 5611–5616. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.J.; Welcker, M.; Clurman, B.E. Tumor suppression by the Fbw7 ubiquitin ligase: Mechanisms and opportunities. Cancer Cell 2014, 26, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Kar, R.; Jha, S.K.; Ojha, S.; Sharma, A.; Dholpuria, S.; Raju, V.S.R.; Prasher, P.; Chellappan, D.K.; Gupta, G.; Kumar Singh, S.; et al. The FBXW7-NOTCH interactome: A ubiquitin proteasomal system-induced crosstalk modulating oncogenic transformation in human tissues. Cancer Rep. 2021, 4, e1369. [Google Scholar] [CrossRef]
- Lan, H.; Sun, Y. Tumor Suppressor FBXW7 and Its Regulation of DNA Damage Response and Repair. Front. Cell Dev. Biol. 2021, 9, 751574. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Yan, C.; Liu, R.; Chen, L.; Cheng, D.; Hao, L.; Yuan, W.; Chen, J.; Yang, H. Clinical significance of FBXW7 tumor suppressor gene mutations and expression in human colorectal cancer: A systemic review and meta-analysis. BMC Cancer 2021, 21, 770. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Li, L.; Zhou, L.; Li, D.; Liu, M.; Han, S.; Zheng, B. Critical roles of the E3 ubiquitin ligase FBW7 in B-cell response and the pathogenesis of experimental autoimmune arthritis. Immunology 2021, 164, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Falisi, E.; Novella, E.; Visco, C.; Guercini, N.; Maura, F.; Giaretta, I.; Pomponi, F.; Nichele, I.; Finotto, S.; Montaldi, A.; et al. B-cell receptor configuration and mutational analysis of patients with chronic lymphocytic leukaemia and trisomy 12 reveal recurrent molecular abnormalities. Hematol. Oncol. 2014, 32, 22–30. [Google Scholar] [CrossRef]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Rigolin, G.M.; Saccenti, E.; Bassi, C.; Lupini, L.; Quaglia, F.M.; Cavallari, M.; Martinelli, S.; Formigaro, L.; Lista, E.; Bardi, M.A.; et al. Extensive next-generation sequencing analysis in chronic lymphocytic leukemia at diagnosis: Clinical and biological correlations. J. Hematol. Oncol. 2016, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Helbig, D.R.; Abu-Zeinah, G.; Bhavsar, E.; Christos, P.J.; Furman, R.R.; Allan, J.N. Outcomes in CLL patients with NOTCH1 regulatory pathway mutations. Am. J. Hematol. 2021, 96, E187–E189. [Google Scholar] [CrossRef]
- Arabi, A.; Ullah, K.; Branca, R.M.; Johansson, J.; Bandarra, D.; Haneklaus, M.; Fu, J.; Ariës, I.; Nilsson, P.; Den Boer, M.L.; et al. Proteomic screen reveals Fbw7 as a modulator of the NF-κB pathway. Nat. Commun. 2012, 3, 976. [Google Scholar] [CrossRef] [Green Version]
- Welcker, M.; Orian, A.; Grim, J.E.; Eisenman, R.N.; Clurman, B.E. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr. Biol. 2004, 14, 1852–1857. [Google Scholar] [CrossRef] [Green Version]
- Decker, T.; Hipp, S.; Hahntow, I.; Schneller, F.; Peschel, C. Expression of cyclin E in resting and activated B-chronic lymphocytic leukaemia cells: Cyclin E/cdk2 as a potential therapeutic target. Br. J. Haematol. 2004, 125, 141–148. [Google Scholar] [CrossRef]
- Harper, T.M.; Taatjes, D.J. The complex structure and function of Mediator. J Biol. Chem. 2018, 293, 13778–13785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.D.; Oldenbroek, M.; Boyer, T.G. Mediator kinase module and human tumorigenesis. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 393–426. [Google Scholar] [CrossRef] [PubMed]
- Fryer, C.J.; White, J.B.; Jones, K.A. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 2004, 16, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Fassl, A.; Chick, J.; Inuzuka, H.; Li, X.; Mansour, M.R.; Liu, L.; Wang, H.; King, B.; Shaik, S.; et al. Cyclin C is a haploinsufficient tumour suppressor. Nat. Cell Biol. 2014, 16, 1080–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damm, F.; Mylonas, E.; Cosson, A.; Yoshida, K.; Della Valle, V.; Mouly, E.; Diop, M.; Scourzic, L.; Shiraishi, Y.; Chiba, K.; et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov. 2014, 4, 1088–1101. [Google Scholar] [CrossRef] [Green Version]
- Kämpjärvi, K.; Järvinen, T.M.; Heikkinen, T.; Ruppert, A.S.; Senter, L.; Hoag, K.W.; Dufva, O.; Kontro, M.; Rassenti, L.; Hertlein, E.; et al. Somatic MED12 mutations are associated with poor prognosis markers in chronic lymphocytic leukemia. Oncotarget 2015, 6, 1884–1888. [Google Scholar] [CrossRef] [Green Version]
- Guieze, R.; Robbe, P.; Clifford, R.; de Guibert, S.; Pereira, B.; Timbs, A.; Dilhuydy, M.S.; Cabes, M.; Ysebaert, L.; Burns, A.; et al. Presence of multiple recurrent mutations confers poor trial outcome of relapsed/refractory CLL. Blood 2015, 126, 2110–2117. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.A.; Hwang, B.; Park, S.N.; Huh, S.; Im, K.; Choi, S.; Chung, H.Y.; Huh, J.; Seo, E.J.; Lee, J.H.; et al. Genomic Profile of Chronic Lymphocytic Leukemia in Korea Identified by Targeted Sequencing. PLoS ONE 2016, 11, e0167641. [Google Scholar] [CrossRef]
- Machnicki, M.M.; Gorniak, P.; Pepek, M.; Szymczyk, A.; Iskierka-Jazdzewska, E.; Steckiewicz, P.; Bluszcz, A.; Rydzanicz, M.; Hus, M.; Płoski, R.; et al. Predictive significance of selected gene mutations in relapsed and refractory chronic lymphocytic leukemia patients treated with ibrutinib. Eur. J. Haematol. 2021, 106, 320–326. [Google Scholar] [CrossRef]
- Tsuji, M.; Shinkura, R.; Kuroda, K.; Yabe, D.; Honjo, T. Msx2-interacting nuclear target protein (Mint) deficiency reveals negative regulation of early thymocyte differentiation by Notch/RBP-J signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 1610–1615. [Google Scholar] [CrossRef] [Green Version]
- Yabe, D.; Fukuda, H.; Aoki, M.; Yamada, S.; Takebayashi, S.; Shinkura, R.; Yamamoto, N.; Honjo, T. Generation of a conditional knockout allele for mammalian Spen protein Mint/SHARP. Genesis 2007, 45, 300–306. [Google Scholar] [CrossRef]
- Hu, B.; Patel, K.P.; Chen, H.C.; Wang, X.; Wang, F.; Luthra, R.; Routbort, M.J.; Kanagal-Shamanna, R.; Medeiros, L.J.; Yin, C.C.; et al. Routine sequencing in CLL has prognostic implications and provides new insight into pathogenesis and targeted treatments. Br. J. Haematol. 2019, 185, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Quesada, V.; Conde, L.; Villamor, N.; Ordonez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Bea, S.; Pinyol, M.; Martinez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2011, 44, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Te Raa, G.D.; Derks, I.A.; Navrkalova, V.; Skowronska, A.; Moerland, P.D.; van Laar, J.; Oldreive, C.; Monsuur, H.; Trbusek, M.; Malcikova, J.; et al. The impact of SF3B1 mutations in CLL on the DNA-damage response. Leukemia 2015, 29, 1133–1142. [Google Scholar] [CrossRef]
- Wang, L.; Brooks, A.N.; Fan, J.; Wan, Y.; Gambe, R.; Li, S.; Hergert, S.; Yin, S.; Freeman, S.S.; Levin, J.Z.; et al. Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia. Cancer Cell 2016, 30, 750–763. [Google Scholar] [CrossRef] [Green Version]
- Pacholewska, A.; Grimm, C.; Herling, C.D.; Lienhard, M.; Konigs, A.; Timmermann, B.; Altmuller, J.; Mucke, O.; Reinhardt, H.C.; Plass, C.; et al. Altered DNA Methylation Profiles in SF3B1 Mutated CLL Patients. Int. J. Mol. Sci. 2021, 22, 9337. [Google Scholar] [CrossRef]
- Minoguchi, S.; Taniguchi, Y.; Kato, H.; Okazaki, T.; Strobl, L.J.; Zimber-Strobl, U.; Bornkamm, G.W.; Honjo, T. RBP-L, a transcription factor related to RBP-Jkappa. Mol. Cell. Biol. 1997, 17, 2679–2687. [Google Scholar] [CrossRef] [Green Version]
- Collu, G.M.; Hidalgo-Sastre, A.; Acar, A.; Bayston, L.; Gildea, C.; Leverentz, M.K.; Mills, C.G.; Owens, T.W.; Meurette, O.; Dorey, K.; et al. Dishevelled limits Notch signalling through inhibition of CSL. Development 2012, 139, 4405–4415. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, L.; Gundry, M.C.; Goodell, M.A. DNMT3A in Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7, a030320. [Google Scholar] [CrossRef] [PubMed]
- Bagacean, C.; Tempescul, A.; Le Dantec, C.; Bordron, A.; Mohr, A.; Saad, H.; Olivier, V.; Zdrenghea, M.; Cristea, V.; Cartron, P.F.; et al. Alterations in DNA methylation/demethylation intermediates predict clinical outcome in chronic lymphocytic leukemia. Oncotarget 2017, 8, 65699–65716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.L.; Goswami, S.; Frissora, F.W.; Xie, Z.; Yan, P.S.; Bundschuh, R.; Walker, L.A.; Huang, X.; Mani, R.; Mo, X.M.; et al. ROR1-targeted delivery of miR-29b induces cell cycle arrest and therapeutic benefit in vivo in a CLL mouse model. Blood 2019, 134, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Palamarchuk, A.; Yan, P.S.; Zanesi, N.; Wang, L.; Rodrigues, B.; Murphy, M.; Balatti, V.; Bottoni, A.; Nazaryan, N.; Alder, H.; et al. Tcl1 protein functions as an inhibitor of de novo DNA methylation in B-cell chronic lymphocytic leukemia (CLL). Proc. Natl. Acad. Sci. USA 2012, 109, 2555–2560. [Google Scholar] [CrossRef] [Green Version]
- Haney, S.L.; Upchurch, G.M.; Opavska, J.; Klinkebiel, D.; Hlady, R.A.; Suresh, A.; Pirruccello, S.J.; Shukla, V.; Lu, R.; Costinean, S.; et al. Promoter Hypomethylation and Expression Is Conserved in Mouse Chronic Lymphocytic Leukemia Induced by Decreased or Inactivated Dnmt3a. Cell Rep. 2016, 15, 1190–1201. [Google Scholar] [CrossRef] [Green Version]
- Biran, A.; Yin, S.; Kretzmer, H.; Ten Hacken, E.; Parvin, S.; Lucas, F.; Uduman, M.; Gutierrez, C.; Dangle, N.; Billington, L.; et al. Activation of Notch and Myc signaling via B cell-restricted depletion of Dnmt3a generates a consistent murine model of chronic lymphocytic leukemia. Cancer Res. 2021, 81, 6117–6130. [Google Scholar] [CrossRef] [PubMed]
- Mazzarello, A.N.; Gentner-Göbel, E.; Dühren-von Minden, M.; Tarasenko, T.N.; Nicolò, A.; Ferrer, G.; Vergani, S.; Liu, Y.; Bagnara, D.; Rai, K.R.; et al. B cell receptor isotypes differentially associate with cell signaling, kinetics, and outcome in chronic lymphocytic leukemia. J. Clin. Investig. 2022, 132, e149308. [Google Scholar] [CrossRef]
- Schmid, V.K.; Khadour, A.; Ahmed, N.; Brandl, C.; Nitschke, L.; Rajewsky, K.; Jumaa, H.; Hobeika, E. B cell antigen receptor expression and phosphatidylinositol 3-kinase signaling regulate genesis and maintenance of mouse chronic lymphocytic leukemia. Haematologica 2022. [Google Scholar] [CrossRef]
- Kang, J.A.; Kim, W.S.; Park, S.G. Notch1 is an important mediator for enhancing of B-cell activation and antibody secretion by Notch ligand. Immunology 2014, 143, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Del Papa, B.; Baldoni, S.; Dorillo, E.; De Falco, F.; Rompietti, C.; Cecchini, D.; Cantelmi, M.G.; Sorcini, D.; Nogarotto, M.; Adamo, F.M.; et al. Decreased NOTCH1 Activation Correlates with Response to Ibrutinib in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2019, 25, 7540–7553. [Google Scholar] [CrossRef] [Green Version]
- Steinbuck, M.P.; Arakcheeva, K.; Winandy, S. Novel TCR-Mediated Mechanisms of Notch Activation and Signaling. J. Immunol. 2018, 200, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Del Poeta, G.; Biagi, A.; Laurenti, L.; Chiarenza, A.; Pozzo, F.; Innocenti, I.; Postorino, M.; Rossi, F.M.; Del Principe, M.I.; Bomben, R.; et al. Impaired nodal shrinkage and apoptosis define the independent adverse outcome of NOTCH1 mutated patients under ibrutinib therapy in chronic lymphocytic leukaemia. Haematologica 2021, 106, 2345–2353. [Google Scholar] [CrossRef]
- Rossi, D.; Spina, V.; Bomben, R.; Rasi, S.; Dal-Bo, M.; Bruscaggin, A.; Rossi, F.M.; Monti, S.; Degan, M.; Ciardullo, C.; et al. Association between molecular lesions and specific B-cell receptor subsets in chronic lymphocytic leukemia. Blood 2013, 121, 4902–4905. [Google Scholar] [CrossRef] [PubMed]
- Villegas, S.N.; Gombos, R.; Garcia-Lopez, L.; Gutierrez-Perez, I.; Garcia-Castillo, J.; Vallejo, D.M.; Da Ros, V.G.; Ballesta-Illan, E.; Mihaly, J.; Dominguez, M. PI3K/Akt Cooperates with Oncogenic Notch by Inducing Nitric Oxide-Dependent Inflammation. Cell Rep. 2018, 22, 2541–2549. [Google Scholar] [CrossRef] [Green Version]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlhaas, V.; Blakemore, S.J.; Al-Maarri, M.; Nickel, N.; Pal, M.; Roth, A.; Hovelmeyer, N.; Schafer, S.C.; Knittel, G.; Lohneis, P.; et al. Active Akt signaling triggers CLL toward Richter transformation via overactivation of Notch1. Blood 2021, 137, 646–660. [Google Scholar] [CrossRef]
- Rossi, D.; Rasi, S.; Spina, V.; Fangazio, M.; Monti, S.; Greco, M.; Ciardullo, C.; Famà, R.; Cresta, S.; Bruscaggin, A.; et al. Different impact of NOTCH1 and SF3B1 mutations on the risk of chronic lymphocytic leukemia transformation to Richter syndrome. Br. J. Haematol. 2012, 158, 426–429. [Google Scholar] [CrossRef]
- Condoluci, A.; Rossi, D. Biology and Treatment of Richter Transformation. Front Oncol. 2022, 12, 829983. [Google Scholar] [CrossRef]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Mangolini, M.; Maiques-Diaz, A.; Charalampopoulou, S.; Gerhard-Hartmann, E.; Bloehdorn, J.; Moore, A.; Lu, J.; Franklin, V.N.R.; Chilamakuri, C.S.R.; Moutsoupoulos, I.; et al. NOTCH1 drives immune-escape mechanisms in B cell malignancies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Matutes, E.; Owusu-Ankomah, K.; Morilla, R.; Garcia Marco, J.; Houlihan, A.; Que, T.H.; Catovsky, D. The immunological profile of B-cell disorders and proposal of a scoring system for the diagnosis of CLL. Leukemia 1994, 8, 1640–1645. [Google Scholar] [PubMed]
- Sorrentino, C.; Cuneo, A.; Roti, G. Therapeutic Targeting of Notch Signaling Pathway in Hematological Malignancies. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019037. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Guerra, M.; Xargay-Torrent, S.; Rosich, L.; Montraveta, A.; Roldan, J.; Matas-Cespedes, A.; Villamor, N.; Aymerich, M.; Lopez-Otin, C.; Perez-Galan, P.; et al. The gamma-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells. Leukemia 2015, 29, 96–106. [Google Scholar] [CrossRef]
- Silkenstedt, E.; Arenas, F.; Colom-Sanmarti, B.; Xargay-Torrent, S.; Higashi, M.; Giro, A.; Rodriguez, V.; Fuentes, P.; Aulitzky, W.E.; van der Kuip, H.; et al. Notch1 signaling in NOTCH1-mutated mantle cell lymphoma depends on Delta-Like ligand 4 and is a potential target for specific antibody therapy. J. Exp. Clin. Cancer Res. 2019, 38, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secchiero, P.; Voltan, R.; Rimondi, E.; Melloni, E.; Athanasakis, E.; Tisato, V.; Gallo, S.; Rigolin, G.M.; Zauli, G. The gamma-secretase inhibitors enhance the anti-leukemic activity of ibrutinib in B-CLL cells. Oncotarget 2017, 8, 59235–59245. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pozzo, F.; Bittolo, T.; Tissino, E.; Zucchetto, A.; Bomben, R.; Polcik, L.; Dannewitz Prosseda, S.; Hartmann, T.N.; Gattei, V. Multiple Mechanisms of NOTCH1 Activation in Chronic Lymphocytic Leukemia: NOTCH1 Mutations and Beyond. Cancers 2022, 14, 2997. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14122997
Pozzo F, Bittolo T, Tissino E, Zucchetto A, Bomben R, Polcik L, Dannewitz Prosseda S, Hartmann TN, Gattei V. Multiple Mechanisms of NOTCH1 Activation in Chronic Lymphocytic Leukemia: NOTCH1 Mutations and Beyond. Cancers. 2022; 14(12):2997. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14122997
Chicago/Turabian StylePozzo, Federico, Tamara Bittolo, Erika Tissino, Antonella Zucchetto, Riccardo Bomben, Laura Polcik, Svenja Dannewitz Prosseda, Tanja Nicole Hartmann, and Valter Gattei. 2022. "Multiple Mechanisms of NOTCH1 Activation in Chronic Lymphocytic Leukemia: NOTCH1 Mutations and Beyond" Cancers 14, no. 12: 2997. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14122997